
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel tryptophan 2,3-dioxygenase-targeted ruthenium(II)-indole complex activates immunotherapy in vitro and in vivo†
Zheng-Qi
Shen‡
,
Binglian
Guo‡
,
Xiangyu
Dai‡
,
Hanxue
Liu
,
Meng
Ren
,
Peisen
Wang
,
Yating
Zhang
,
Yinuo
Xu
,
Zhi
Su
 ,
Xuling
Xue
* and
Hong-Ke
Liu
*
,
Xuling
Xue
* and
Hong-Ke
Liu
*
Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: xuexuling87@163.com; liuhongke@njnu.edu.cn
First published on 30th July 2025
Abstract
Immunotherapy targeting immune checkpoints has emerged as a promising strategy in cancer treatment; however, the heterogeneous and dynamic tumor microenvironment (TME) imposes critical constraints on therapeutic outcomes. Tryptophan 2,3-dioxygenase (TDO), often dysregulated in malignant tissues, plays a pivotal role in shaping an immunosuppressive milieu by depleting tryptophan, thereby hindering anti-tumor immune response. To counteract this immune evasion mechanism, we designed a novel indole-coordinated ruthenium(II) arene complex (In-Ru), aimed at bolstering tumor immunotherapy and thwarting immune evasion by targeting TDO expression. Our findings reveal that In-Ru exerts markedly potent anti-proliferative effects against HepG2 cells. It achieves this by specifically localizing to the cell nucleus, inducing DNA damage, and initiating a cascade of necroptosis as well as immunogenic cell death (ICD), thereby potentially enhancing the immune system's capacity to recognize and attack cancer cells. RNA sequencing and qRT-PCR analysis indicate that In-Ru modulates pathways linked to tryptophan metabolism and immune reprogramming, with specific degradation of TDO protein and reversal of tryptophan-mediated immunosuppression. Furthermore, TDO inhibition boosts ROS production and induces necroptosis via mitochondrial damage, triggering a strong immune response. The tumor vaccine experiment revealed that In-Ru significantly reduced TDO levels and triggered ICD effect in liver cancer animal models. By reversing the immunosuppressive microenvironment, In-Ru facilitated the maturation of dendritic cells (DCs) and promoted T-cell infiltration, thereby achieving robust anti-tumor efficacy and long-lasting immune protection. This study represents the first report of a metal-arene complex with dual functions of TDO inhibition and ICD induction. It not only enhances anti-tumor immunogenicity but also effectively mitigates the risk of immune overactivation, offering a precise regulatory paradigm for the development of metal-based complexes in tumor immunotherapy.
Introduction
Platinum complexes have achieved remarkable breakthroughs in biomedical research.1,2 Motivated by these advances, significant efforts have been devoted to exploring the biological mechanisms of other metal complexes.3–11 In recent years, extensive studies have demonstrated that metal-arene complexes (e.g., ruthenium, iridium, and rhodium) exhibit innovative anti-tumor mechanisms and enhanced safety profiles compared to traditional platinum drugs.12 These properties enable them to markedly reduce systemic toxicity and circumvent chemotherapy resistance. Then, metal-arene complexes possess superior lipid solubility and cell membrane permeability. Through structural optimization, they can facilitate extensive intracellular accumulation and precise subcellular organelle targeting.13 Building on our prior research,3,14–22 we introduced a metal–ligand synergistic enhancement (MLSE) strategy, which uses the cooperative effects of metal precursors and organic ligands to design and synthesize Ru/Ir complexes for tumor immunotherapy. Among these, Ir-Bet can evoke ferroptosis for synergistic enhancement of immunotherapy,14 whereas RuBTB can trigger immunogenic ferroptosis for reverse drug resistance.19 Furthermore, we successfully achieved the in situ synthesis of Ru(II) arene complex (Ru-rhein) within tumors, enabling the precise fabrication of metal anti-tumor complex and proposing a “bio-orthogonally catalyzed lethality” strategy.17 These findings highlight the therapeutic potential and unique advantages of metal-arene complexes in tumor immunotherapy, as well as their applicability for in situ drug synthesis.Hepatocellular carcinoma (HCC) is one of the most prevalent malignant tumors, marked by high incidence and mortality rates.23 Moreover, patients with HCC often exhibit a poor prognosis due to the propensity for secondary metastasis to occur in multiple organs, which ultimately contributes to treatment failure and death.24 Tryptophan 2,3-dioxygenase (TDO) serves as a rate-limiting enzyme in the conversion of L-tryptophan to L-kynurenine.25 It is aberrantly overexpressed in various cancers, particularly in HCC tissues, where its levels are significantly elevated compared to those in normal liver tissues.26 Clinical studies have demonstrated that TDO directly drives the progression of HCC by regulating metabolism, inducing the differentiation of regulatory T cells (Tregs), and suppressing the activity of effector T cells, thereby mediating tumor immune escape.27 Recent reports highlight that TDO represents a promising therapeutic target for HCC treatment. Reducing TDO levels in cancer cells not only alleviates tryptophan depletion but also potentiates anti-tumor immune response.28,29 Consequently, the development of novel TDO inhibitors for HCC therapy could effectively mitigate immune resistance during treatment, offering new opportunities for clinical intervention.
TDO serves as a prototypical immunosuppressive checkpoint that facilitates tumor immune evasion.30 It catalyzes the catabolism of tryptophan via the KYN pathway, leading to tryptophan depletion and the accumulation of downstream metabolites aryl hydrocarbon receptor (AhR).25,31 This metabolic shift promotes the differentiation of immunosuppressive DCs and Tregs, which collectively establish an immunosuppressive microenvironment that accelerates tumor immune escape. Dolusic pointed for the first time that indole compounds exhibit structural similarities to tryptophan, enabling them to act as inhibitors of TDO.32 This action reduces tryptophan catabolism and elevates local tryptophan concentrations, thereby enhancing the efficacy of immunotherapy. Although TDO and IDO are key enzymes in the tryptophan metabolic pathway and contribute to tumor immune escape,30 studies on metal complexes targeting TDO are relatively scarce, particularly metal-arene complexes, which have not yet been reported. Very few metal-based complexes incorporating indole ligands have been reported to function as TDO inhibitors. Gou reported a platinum(IV) complex coordinated by an indole ligand,33,34 which suppresses TDO expression, modulates T cell activation and proliferation only in vivo. It remains challenging to elucidate the correlation between TDO inhibition and immune activation by metal complexes in HCC animal models due to insufficient evaluation of immune effects at the in vitro level, and the lack of systematic investigations into their impacts on the TDO-related immune microenvironment.
To address the above challenges of TDO inhibition and immune activation by metal complexes in HCC therapy, this work innovatively designed and synthesized a novel indole-coordinated Ru(II) arene complex (In-Ru). By using the MLSE strategy, this complex aims to simultaneously exert chemotherapeutic effects and inhibit TDO expression, thereby enhancing the immunotherapeutic efficacy. The experiment results demonstrate that In-Ru accumulates significantly in the cell nucleus, by inhibiting the TDO pathway, potentiates necroptosis induced by ROS accumulation and mitochondrial membrane potential collapse. Necroptosis triggers cell membrane rupture and the release of damage-associated molecular patterns (DAMPs), thereby inducing ICD effect and subsequently activating systemic anti-tumor immune responses. RNA sequencing and qRT-PCR analysis demonstrated that In-Ru significantly suppresses TDO expression in HepG2 cells, concomitant with the inactivation of the AhR signaling pathway, thereby effectively inhibiting tryptophan metabolism-mediated immune escape. In vivo vaccine experiments revealed that In-Ru specifically attenuates TDO activity within mouse tumors, thereby enhancing anti-tumor immunity. Furthermore, the complex induces ICD effect, promoting DCs maturation and T-cell activation, which in turn triggers systemic immune protection and tumor ablation. As the first metal complex-based ICD inducer targeting the TDO pathway, this study systematically elucidated the molecular mechanism by which metal-arene complexes mediate the integration of chemotherapy and immunotherapy via the TDO-AhR axis. A novel “chemotherapy sensitization-immunoregulation” tumor treatment paradigm was successfully established, offering new perspectives for the immunotherapy of solid tumors (Fig. 1).
 | ||
| Fig. 1 By using the metal–ligand synergistic enhancement (MLSE) strategy, a novel TDO metal-based TDO inhibitor In-Ru was successfully developed. In-Ru reverses the immunosuppressive microenvironment through the inhibition of the TDO-KYN-AhR pathway, at the same time can induce cell necroptosis and enhances the efficacy of immunogenic cell death, and consequently activates relevant immune cells to augment the anti-tumor immune response. | ||
Results and discussion
Synthesis and characterization
Metal complexes In-Ru and In-Ir were synthesized following previously reported procedures.14 The functional ligand Indo and metal precursors (Ppy-Ru, Ppy-Ir) were synthesized with reference to previously reported literature.35 After the metal precursors (Ppy-Ru, Ppy-Ir) were dissolved and reacted with AgNO3 to remove Cl atom in methanol solution, the ligand Indo was added to the filtrate to form the final mononuclear complex In-Ru or In-Ir (Fig. 2A and Scheme S1†). All complexes were characterized by 1H and 13C NMR spectroscopy, and electrospray ionization mass spectra (ESI-MS), as shown in Fig. S1–S8.† Indo acts as a monodentate ligand and coordinates with Ppy-Ru or Ppy-Ir via its N atom in the final mononuclear complex In-Ru or In-Ir, that is further confirmed by distinct ESI-MS peaks at 628.2 ([M–H]+) for In-Ru or 720.3 ([M–H]+) for In-Ir.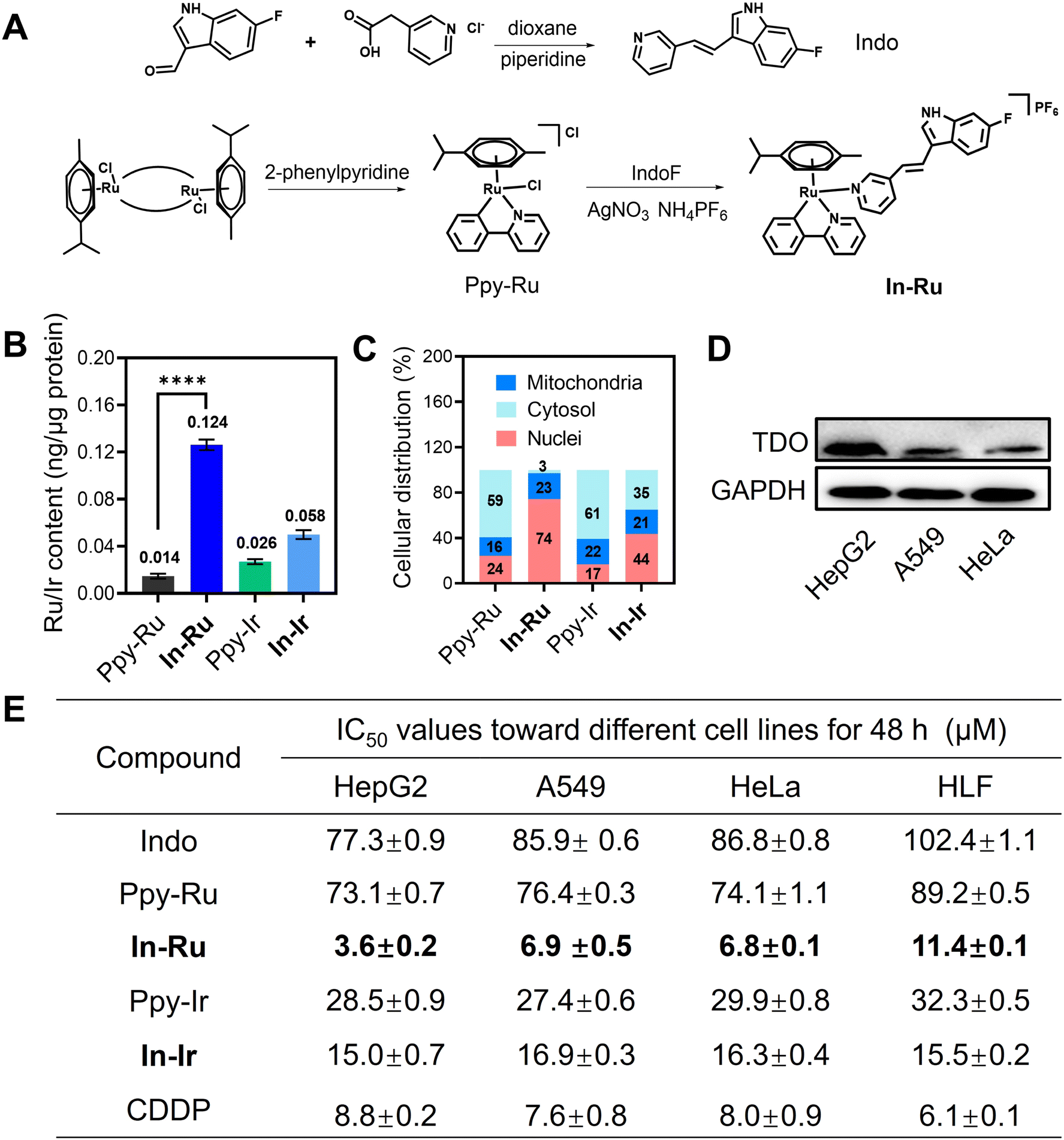 | ||
| Fig. 2 Chemical structure, synthetic route and anti-tumor performance of In-Ru. (A) Chemical structure and synthetic route of In-Ru. (B and C) Intracellular Ru/Ir uptake and metal distribution in HepG2 cells after exposure for 8 h to Ppy-Ru/Ppy-Ir/In-Ru/In-Ir, as measured by ICP-MS. (D) TDO protein expression levels in different cell lines (HepG2, A549, HeLa). (E) IC50 (μM) values of Indo, Ppy-Ru, Ppy-Ir, In-Ru, In-Ir and CDDP toward different cell lines after treated for 48 h. Error bars: S.D., n = 3. **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Stability, lipophilicity, and intracellular distribution
The stability of In-Ru and In-Ir in DMSO/H2O solution (5% DMSO) was investigated by UV-vis spectroscopy respectively. The time-dependent spectra and ESI-MS analysis reveal that these complexes remain stable under physiological conditions. This is primarily attributed to the η6-coordinated arene ligand moiety which stabilizes the oxidation state of the ruthenium metal cation. Additionally, the negligible changes in the UV-vis spectra provide further evidence of their structural stability (Fig. S9†).Lipophilicity (Log Po/w) constitutes one of the crucial parameters for assessing the cell membrane penetrating ability of drugs. The Log Po/w values determined by the octanol–water method were 1.33 and 0.65 for In-Ru or In-Ir, respectively. In contrast to metal precursors and Indo ligand, a high degree of lipophilicity is conducive to the accumulation of metal drugs in cancer cells (Fig. S10†). A positive correlation exists between intracellular drug accumulation and lipophilicity, and the cellular uptake capacity and subcellular organelle distribution of metal drugs were further determined by inductively coupled plasma mass-spectrometry (ICP-MS). After co-incubation of different complexes with HepG2 cells for 6 h, the content of Ru or Ir within the cells was determined. The results indicated that the introduction of lipophilic ligand was conducive to enhancing the cellular uptake of metal drugs. In both the groups of In-Ru and In-Ir, the metal ion content was observed significant increase, with the Ru content reaching 0.124 ng μg−1 protein, which was in accordance with the previously reported lipophilic properties of the complexes (Fig. 2B).19 The distinctive structural characteristics of metal complexes are capable of targeting various subcellular organelles, thereby influencing cell survival and development through diverse pathways and demonstrating their inherent pharmacological properties. As shown in Fig. 2C, over 70% of Ru or about 45% of Ir accumulated in the nucleus, which indicates that the involvement of mononuclear ligand has augmented the burden imposed by metal drugs on the nuclei of cancer cells, which might give rise to a treatment modality related to nuclear damage.
Anti-proliferative activity
The anti-proliferative activity of different compounds against human hepatocellular carcinoma (HepG2), human lung cancer (A549), human cervical epithelioid carcinoma (HeLa), and human lung fibroblast (HLF) cell lines was evaluated by the MTT assay. In-Ru and In-Ir demonstrate diverse degrees of antiproliferative activity against various cancer cell lines, with the most remarkable inhibition observed in hepatocellular carcinoma cells. By contrast, the cytotoxicity of metal precursors and Indo ligand is merely moderate or almost non-toxic (Fig. 2E). It is noteworthy that In-Ru displays outstanding anti-proliferative activity against diverse cancer cells, with the inhibitory effect (IC50 value) escalating in the following sequence: A549 (6.90 μM) < HeLa (6.80 μM) < HepG2 (3.64 μM). We used cisplatin (CDDP) as the reference compound. In-Ru demonstrated a stronger anti-proliferative effect compared to CDDP. At the same time, it showed lower toxicity to normal cell HLF, demonstrating that In-Ru has a more superior therapeutic index. We analyzed the expression levels of TDO protein across various cancer cell lines. Notably, HepG2 cells exhibited significantly higher TDO expression compared to A549 and HeLa cells (Fig. 2D). This finding correlates well with the observation that In-Ru demonstrated superior anti-proliferative activity specifically in HepG2 cells (Fig. 2D, E and S11†).36 The above results indicate that the anti-tumor activity of In-Ru and In-Ir is closely related to two factors: first, they exhibit better efficacy against cancer cells with high TDO expression, and second, their activity is positively correlated with cellular uptake in cancer cells. Since demonstrates significantly higher cellular uptake compared to In-Ir, the anti-tumor activity of In-Ru is much better that of In-Ir. Remarkably, the anti-tumor effect of In-Ru against HepG2 cells with much high TDO expression is superior to that observed in the other two tumor cell lines with low TDO expression. Based on the cytotoxicity characteristics, In-Ru was selected for further studies on HepG2 cells, while In-Ir was used as a control complex in some experiments.Cell necroptosis and DNA damage
Inhibition of TDO levels promotes an increase in intracellular ROS levels and impairs mitochondrial membrane potential, thereby triggering necroptosis in cancer cells.37,38 RIP3 (receptor-interacting protein 3) and NF-κB (p65) (RelA, a subunit of NF-κB) play critical roles in this process, contributing to cell membrane rupture and the release of intracellular contents.39 To examine cell death mechanism of In-Ru, different inhibitors for cell death modes were applied in HepG2 cells (Fig. S12†). After treated with different inhibitors, cells demonstrate diverse levels of viability. Neither Ferroptotic inhibitor Ferrostatin-1(Fer-1) nor autophagic inhibitor chloroquine (CQ) exerted a significant impact on cell viability. While after incubated with apoptotic inhibitor z-VAD-fmk and necrotic inhibitor Necrostatin-1 (Nec-1), cell viabilities were significantly improved, implying that In-Ru could induce cell death by necroptosis and apoptosis pathways. Additionally, the cell lethality treated with In-Ru was demonstrated by flow cytometry on HepG2 cells (Fig. 3A). With the concentration of In-Ru rose, the proportion of cell necroptosis rose linearly, far surpassing that of the other experimental groups. Even with complex concentration at 4 μM, it could induce approximately 27.1% of cell necroptosis. Subsequently, we investigated the changes in these related proteins. Upon co-incubation with In-Ru, RIP3 expression was upregulated by more than 7-fold, which strongly confirmed the occurrence of necroptosis in HepG2 cells. Additionally, we assessed the expression changes of p65 protein in HepG2 cells and found that after treatment with In-Ru, the p65 content decreased to approximately one-third of that in the control group (Fig. 3B and C). From the bright-field microscopic images, it is evident that treatment with In-Ru induces necroptosis in cells, leading to cell membrane disruption and the release of antigenic substance (Fig. 3D). These results indicate that In-Ru can induce necroptosis in HepG2 cells by suppressing TDO expression, thereby potentially enhancing the anti-tumor efficacy of the complex. | ||
| Fig. 3 Investigation into the mechanism of necroptosis and DNA damage in HepG2 cells induced by In-Ru. (A) Flow cytometry analysis of apoptosis and necroptosis evaluation treated with different compounds for 24 h by Annexin-FITC and PI assay. (B) Western blot analysis of the expression of DNA damage (γ-H2AX) and necroptosis (RIP3, p65) proteins treated with different compounds for 24 h. (C) Quantitative analysis for expressions of γ-H2AX, RIP3 and p65 proteins in (B). (D) Representative bright-field images of HepG2 cells treated with various compounds for 24 h are shown. In-Ru can induce cell necroptosis and promote antigen release. Scale bar: 20 μm. (E) Immunofluorescence images of HepG2 cells indicating the signal of γ-H2AX after incubation with Indo, Ppy-Ru, Ppy-Ir, In-Ru, In-Ir for 24 h. Scale bar: 20 μm. γ-H2AX antibody: λex = 488 nm, λem = 525 ± 20 nm; DAPI: λex = 405 nm, λem = 430 ± 20 nm. (F) Cell cycle arrest of HepG2 cells after incubation with different compounds at the same concentration for 24 h. Error bars: S.D., n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001. | ||
ROS generation and mitochondrial membrane potential (ΔΨm)
Cell necroptosis typically related to the disruption of the cellular oxidative-reduction homeostasis and impairment of mitochondrial function membrane potential (ΔΨm, MMP).40 Flow cytometry results indicated that ROS gradually accumulated in cells treated with diverse compound groups, with In-Ru at 8 μM eliciting a significant increase in ROS concentration, approximately five-fold that of the control group. The bright green fluorescence signal in confocal laser scanning microscope (CLSM) images likewise attests to the capacity of In-Ru to induce ROS production (Fig. S13†). The effect of In-Ru on the ΔΨm of HepG2 cells was tested using the JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazolylcarbocyanine iodide) assay kit. The alterations in ΔΨm were manifested by the increase of green fluorescence (JC-1 monomers) and the decrease of red fluorescence (JC-1 aggregates). Upon treatment of the cells with the different compounds, the red signal attenuated in these groups, concurrently with the emergence of the green signal. When In-Ru was introduced, the green fluorescence was conspicuously enhanced, while the red fluorescence almost vanished (Fig. S14†). The flow cytometry analysis indicated that 65% of the cells subjected to In-Ru displayed high JC-1 monomers signals, in contrast to 0.5% of the control cells. Overall, Indo-Ru significantly enhanced ROS levels, which might induce DNA and mitochondrial damage in cancer cells and ultimately result in cell apoptosis and necroptosis.Considering that In-Ru accumulates in the nucleus of cells in considerable quantities and is capable of inducing cell death through necroptosis and other pathways, the cytotoxicity of the complex might be triggered by nuclear damage and mediate cell apoptosis and necroptosis. The nucleus houses the genomic materials, regulates gene expression, and governs the replication of DNA during the cell cycle. DNA damage within the nucleus is succeeded by the phosphorylation of histone H2AX in recruiting and localizing the proteins for DNA damage repair. Thus, the expression of γ-H2AX in HepG2 cells was evaluated by western blotting to disclose the function of metal complexes in inducing DNA damage. As shown in Fig. 3B and C, In-Ru elicited a marked upregulation of γ-H2AX during 24 h, which indicated that complex could induce DNA damage. Nevertheless, In-Ir demonstrates a relatively low potential for DNA damage, which might be associated with its relatively low cellular uptake. Meanwhile, CLSM imaging was utilized to further substantiate the occurrence of DNA damage. In contrast to the control group and other compounds, the green fluorescence of γ-H2AX within the cells treated with In-Ru was markedly enhanced, disclosing the occurrence of DNA damage pattern (Fig. 3E). Cellular DNA damage could induce cell cycle arrest.41 Cell division cycles in HepG2 cells during treatment were recorded by flow cytometry after incubation with different compounds for 24 h. A negligible alteration in cell cycle distribution was detected in the control group. However, the G2 arrest was witnessed significantly after treatment with In-Ru, with the proportion of the G2 phase was increased by 44% compared to the control group (Fig. 3F). This indicates that In-Ru upregulates the expression of γ-H2AX protein and induces severe DNA damage, resulting in G2 arrest in HepG2 cells.
Autophagy
Autophagy plays a critical role in maintaining intracellular homeostasis, and the elevation of ROS levels can promote autophagy induction.42 We assessed the ability of In-Ru to induce autophagy using immunofluorescence imaging and western blot analysis. As shown in Fig. S15,† compared with the control group and other compound-treated groups, cells treated with In-Ru exhibited significantly enhanced green fluorescence signals corresponding to LC3 protein, indicating that In-Ru induces autophagosome formation. Furthermore, the western blot results confirmed these findings by demonstrating increased expression levels of LC3-II and decreased levels of p62, which are hallmarks of autophagy activation.RNA-sequence (seq) analysis
To further illuminate the specific anti-tumor mechanisms and the enhancement effect mediated by TDO inhibition of In-Ru, RNA-seq analysis was conducted on HepG2 cells treated with the complex for 24 h. As indicated in Table S2,† the proportion of ribosomal RNA is less than 5%, while the percentage of uniquely mapped reads exceeds 90%, aligning with the standard threshold (>70%). The average sequencing coverages for the control and In-Ru group is 94.53% and 93.62%, respectively. The correlation coefficient between replicate samples exceeds 0.747, and over 80% of the mapped reads are located within exonic regions. These findings confirm the reproducibility and reliability of the observed significant differences between the control and In-Ru group in the downstream analysis. Fig. 4A presents a volcano plot of 761 significantly differentially expressed genes (DEGs) between the In-Ru group and the control group, with |log2FC| > 1. Among these, 328 genes were upregulated and 433 genes were downregulated. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that In-Ru can modulate the tryptophan metabolic pathway, IL-17 signaling pathway, and NF-κB signaling pathway, TNF signaling pathway, and natural killer cell-mediated cytotoxicity (Fig. 4B). GO enrichment analysis further demonstrated that In-Ru effectively promotes the regulation of inflammatory responses, tryptophan metabolism, and activates immune therapy-related pathways through TDO inhibition (Fig. S16†). Additionally, In-Ru influences aryl hydrocarbon receptor signaling in the tryptophan metabolic pathway, negatively regulates gene expression, modulates T cell antigen processing and presentation, mediates metal ion binding in the nucleus and cytoplasm, facilitates DNA binding, and enhances the activity of DNA-binding transcription factors (Fig. S17 and 18†).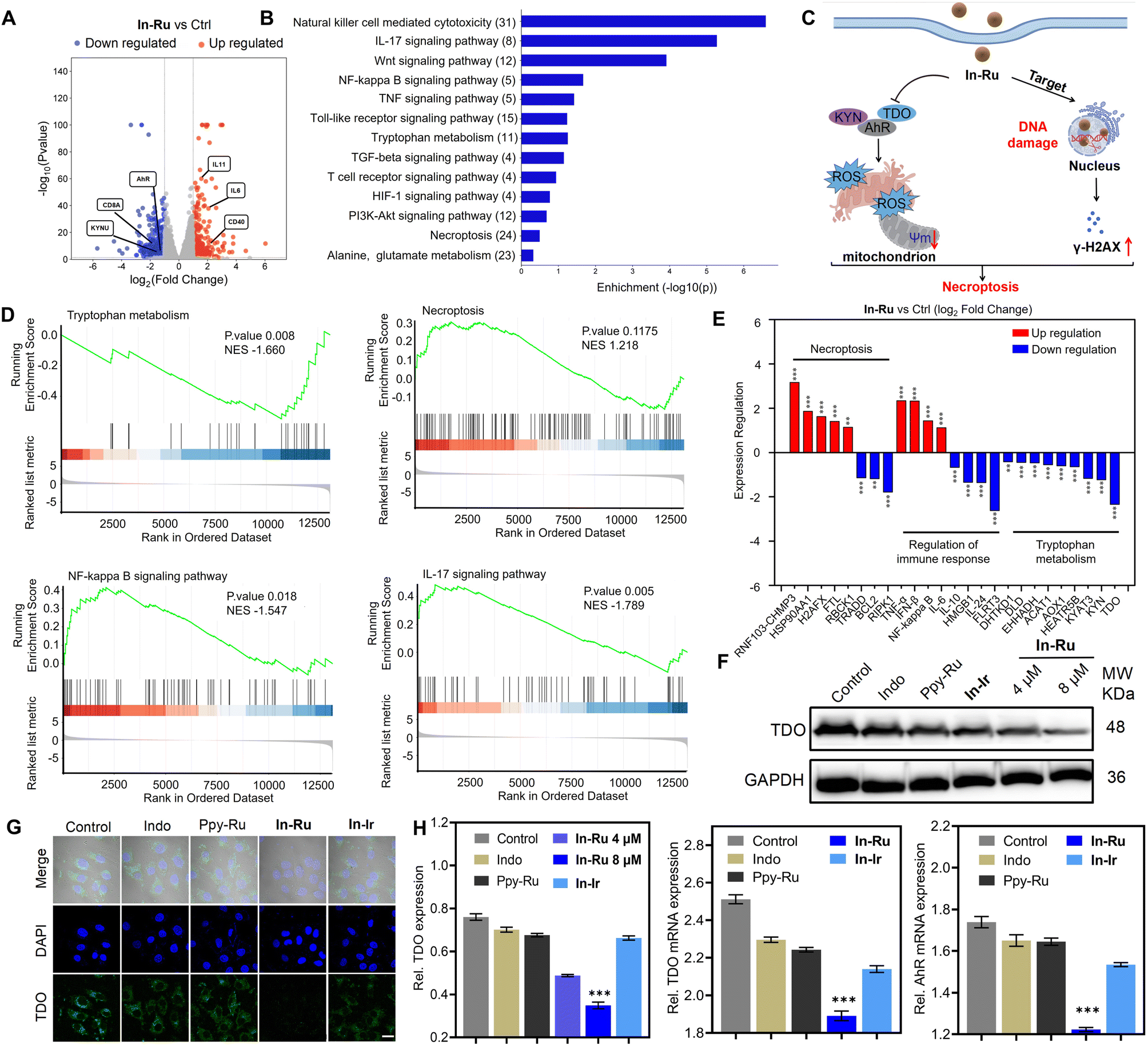 | ||
| Fig. 4 Assessment of TDO inhibition and ICD-induced immune activation. (A) Volcano plot showing the DEGs in HepG2 cells treated with In-Ru (4 μM) for 24 h. Standard: |log2FC| >1; q-value <0.05. (B) KEGG enrichment analysis of differentially expressed genes after treated by In-Ru (4 μM) for 24 h. (C) In-Ru molecule targets the cell nucleus, inducing DNA damage while simultaneously suppressing the expression of TDO. These combined effects lead to necroptosis in HepG2 cells. (D) GSEA reveals negative and positive enrichment of altered genes in cellular processes after treated by In-Ru (4 μM) for 24 h. Standard: observed genes > 2, fold change > 2 and FDR < 0.05 (Tryptophan metabolism, Necroptosis, NF-κB signaling pathway, IL-17 signaling pathway). (E) Expression level changes of the impacted genes involved in necroptosis, regulation of immune response and tryptophan metabolism in HepG2 cells treated with In-Ru. (F) Western blot analysis of TDO inhibition in HepG2 cells treated with various compounds for 24 h. (G) Confocal images of TDO signal in HepG2 cells treated with various compounds for 24 h. FITC: λex = 488 nm, λem = 525 ± 20 nm; DAPI: λex = 364 nm, λem = 454 ± 20 nm. Scale bar: 20 μm. (H) Quantitative analysis for expressions of TDO proteins in (F); statistical analysis of TDO and AhR mRNA expression changes following treatment with various compounds. Error bars: S.D., n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001. | ||
Treatment with In-Ru resulted in significant alterations in the tryptophan metabolic pathway within cells, which facilitates the reversal of immunosuppression and activation of T cells via the TDO pathway.25 Gene set enrichment analysis (GSEA) reveals that variations in gene expression are significantly correlated with TDO downregulation and immune signaling pathways (Fig. 4D and S19†). Furthermore, immune-related genes involved in innate and adaptive immune responses, including the T-cell receptor signaling pathway, TNF signaling pathway, NF-κB signaling pathway, and IL-17 signaling pathway,43,44 were significantly upregulated. Subsequently, a detailed analysis of the related genes in each pathway was conducted. Specifically, we examined genes associated with the TDO pathway (Fig. 4E). As expected, inhibitory genes such as KYN, DHTDK1, DLD, ACAT1, AOX1, KYAT3 were significantly downregulated, consistent with the negative regulation observed in the GSEA results.45–48 Additionally, the upregulation of immune-related genes (IL-6, IFN-β, TNF-α, NF-κB, and LIF) further confirmed an enhanced immune response in cells following In-Ru treatment. Significant changes in necroptosis-related genes (RNF103-CHMP3,49 HSP90AA1, FTL, BCL2, RIPK1)50 provided strong evidence of cell death. Collectively, these data suggest that In-Ru can enhance both innate and adaptive immunity by activating HepG2 cells through TDO pathway, thereby promoting an anti-tumor immune response.
Modulation of TDO and AhR signaling proteins
TDO is an enzyme that overexpressed in liver cancer tissues and functions as a critical species in the regulation of the activation of the aryl hydrocarbon receptor (AhR) and tumor immune escape.46 Thus, in light of the TDO inhibitory mechanism of Indo, we further examined the mechanism of In-Ru in the TDO immune response. Similar to other immune checkpoint inhibitors, suppressing the levels of TDO is conducive to facilitating the activation and proliferation of T cells, thereby strengthening the anti-tumor immune response.33 Then the ability of In-Ru to inhibit TDO protein expression in HepG2 cells was examined. As shown in Fig. 4F, after treated with In-Ru, the expression of TDO in the cells was significantly downregulated and demonstrated a dependence on the complex concentration, but that in the other treatment groups exhibited a more moderate trend, with no significant differences in alterations (Fig. 4F). The CLSM images (Fig. 4G) revealed more intense TDO-associated fluorescence signals in the control group, which completely disappeared following In-Ru treatment, indicating substantial suppression of TDO activity. In contrast, fluorescence signals for TDO exhibited varying degrees of attenuation after exposure to the other tested compounds. To further investigate the mechanism of In-Ruvia the TDO pathway, we evaluated the expression of TDO and AhR through quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) in HepG2 cells (Fig. 4H). Following incubation with In-Ru, the mRNA levels of TDO and AhR in cells were significantly reduced by approximately 24% or 30%, respectively, demonstrating inhibitory effects that were markedly stronger than those of other compounds (Fig. 4H and S20†). In-Ru could induce nuclear damage which further impede the accumulation of AhR in the nucleus and diminish its expression. The results suggest that In-Ru, as a novel TDO immune checkpoint inhibitor, is capable of inhibiting the expression of TDO protein and mediating the inactivation of the downstream AhR pathway to obstruct the TDO-AhR signaling pathway.ICD effect in vitro
In-Ru accumulates abundantly in the nucleus of cells and can induce necroptosis in liver cancer cells, which might evoke a potent immunogenicity.51 Considering the immune response elicited by In-Ru and the reversal of immune suppression, we subsequently assessed the efficacy of the TDO inhibitor based on the MLSE strategy against ICD, which were further investigated in HepG2 cells. ICD possesses three key signals, including high mobility group box-1 protein (HMGB1) release, adenosine triphosphate (ATP) secretion and calreticulin (CRT) exposure.14 As shown in Fig. 5A, the stimulation of In-Ru induced a decrease in HMGB1 expression in the nucleus as detected by CLSM images. Typically, HMGB1 is retained within the nucleus of normal cells, but released into the extracellular space in necrotic cells. The assay of intracellular HMGB1 by western blot and flow cytometry indicated that In-Ru caused the greatest leakage of HMGB1 compared to other groups (Fig. 5C–E), which also in accordance with the fact that In-Ru induces the most severe cell necroptosis. Furthermore, green strong emission signals of CRT antibody of the whole cell, excluding the nucleus, suggested its exposure after treated by In-Ru. Flow cytometry analysis indicated a dose-dependent increase of CRT level in the In-Ru group. Inducing ICD typically results in the substantial release of ATP from the cells. ATP release was detected by the ATP detection kit, the results demonstrate that In-Ru significantly promotes ATP secretion in a dose-dependent manner. Additionally, the marked decrease in intracellular ATP levels corroborates the release of ATP from the cytoplasm into the extracellular space (Fig. S21†). The experiment results demonstrate that In-Ru accumulates extensively in the cell nucleus, inducing robust necroptosis and promoting intracellular substance leakage, thereby significantly enhancing anti-tumor immunity. In contrast, at the same concentration, the ligand Indo and metal precursor Ppy-Ru exhibit limited cellular uptake and low anti-tumor activity, failing to induce effective ICD effect. Furthermore, inhibition of TDO expression directly promotes ROS generation and impairs mitochondrial function, thereby triggering more pronounced necroptosis. This novel metal complex not only enhances cytotoxicity but also effectively triggers ICD, achieving immunotherapeutic effects at low doses, thus facilitating further investigation into in vivo immune activation mechanisms. | ||
| Fig. 5 Detection and analysis of ICD marker signals in HepG2 cells treated by In-Ru. (A) Confocal images of HMGB1 release in HepG2 cells incubated with various compounds for 24 h (Indo, Ppy-Ru, In-Ru, In-Ir). Scale bar: 20 μm. HMGB1 antibody: λex = 488 nm, λem = 520 ± 20 nm; DAPI: λex = 405 nm, λem = 430 ± 20 nm. (B) Confocal images of CRT release in HepG2 cells incubated with various compounds for 24 h (Indo, Ppy-Ru, In-Ru, In-Ir). Scale bar: 20 μm. CRT antibody: λex = 488 nm, λem = 520 ± 20 nm; DAPI: λex = 405 nm, λem = 430 ± 20 nm. (C) Western blot analysis for the expression of CRT and HMGB1 proteins in HepG2 cells treated with Indo, Ppy-Ru, In-Ru, In-Ir for 24 h. (D) Quantitative analysis for expressions of CRT/HMGB1 proteins in (C). (E) Flow cytometry analysis of HMGB1 and CRT expression changes evaluation treated with different compounds for 24 h. (F) Quantitative analysis of CRT/HMGB1 intensity changes in (E). (G) Induction of ICD and associated immune responses by cell necroptosis. Error bars: S.D., n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001. | ||
Biological evaluation on 3D multicellular tumor spheroids
In preclinical basic research, 3D multicellular tumor spheroids (MCTSs) used as the widely utilized tumor model, were employed to assess the biological anti-proliferative effects of In-Ru. Compared to cultured cell lines, solid tumor models exhibit reduced sensitivity to chemotherapeutic drugs and can effectively simulate the microenvironment of solid tumors.52 The morphology and growth of 3D MCTSs were assessed every 48 h over a period of 8 days. As shown in Fig. 6A and B, the MCTS in the In-Ru group began to shrink in volume after 2 days of treatment. By the end of 8 days after treatment, the spheroids became loose and their boundaries disappeared, indicating that the In-Ru had severely affected the morphological growth and integrity of the MCTS. In the absence of drug intervention, the tumor spheres naturally grew to a diameter of approximately 600 μm and had a volume approximately four times that of the In-Ru treatment group. After incubation with In-Ir, the spheroids exhibited nearly complete growth arrest, demonstrating a certain degree of anti-proliferative activity. To further elucidate the impact of In-Ru on cell viability within MCTS, a dual-staining approach utilizing calcein AM (green fluorescence) for live cells and propidium iodide (red fluorescence) for dead cells was employed. In the control group, bright green fluorescence indicated that most cells were alive, while weak red signals representing cell death were observed in several treatment groups. In the MCTS treated with In-Ru, the strong red fluorescence signal indicated irreversible damage to the spheres and cell death (Fig. 6C). These results indicate that In-Ru not only demonstrated superior tumor penetration ability but also effectively inhibited the proliferation of HepG2 3D MCTSs compared to the control group. | ||
| Fig. 6 Evaluation of the cytotoxicity and anti-tumor activity of In-Ru on 3D HepG2 tumor cell spheroids. (A) Representative images of 3D tumor spheroids treated with various compounds every 48 h for 8 days, Indo (30 μM), Ppy-Ru (30 μM), In-Ru (30 μM), In-Ir (30 μM). Scale bar: 200 μm. (B) Curves of 3D tumor spheroids volume treated with PBS, Indo, Ppy-Ru, In-Ru and In-Ir. (C) Confocal images of the live/dead cells in the tumor spheroids after treatment with PBS, Indo, Ppy-Ru, In-Ru and In-Ir for 8 days, stained by Calcein AM/PI and photographed by confocal microscope. Scale bar: 200 μm. Error bars: S.D., n = 3. | ||
Activation of anti-tumor immunity in vivo
In-Ru can inhibit TDO expression in HepG2 cells and enhance the efficacy of immunotherapy. We further evaluated the potential of the complex in mediating TDO inhibition and immunotherapy in vivo through mouse vaccination experiments.14 As illustrated in Fig. 7A, healthy C57BL/6J mice received intravenous injections of H22 hepatocellular carcinoma cells, which had undergone various treatments (PBS, Indo, Ppy-Ru, In-Ru) on days −7, −5, and −3. Then, on day 0, 1 × 106 viable H22 cells were subcutaneously inoculated into each mouse, and tumor volume and body weight were monitored every two days over a period of 10 days. Analysis of the tumor growth trend in mice showed that the tumor growth in mice immunized with In-Ru-treated H22 cells was significantly inhibited, while in the other groups, the tumor growth rate showed a phased upward trend (Fig. 7B–D and S22†). Hematoxylin-eosin (H&E) staining analysis of tumor sections also showed large number of dead cells in group treated with In-Ru-treated H22 cells. Meanwhile, no significant changes were observed in body weight or major organ morphology across all groups of mice during the treatment period indicates the safety and stability of the therapeutic agents and vaccine regimens (Fig. S23†). In-Ru can inhibit TDO in vitro, which subsequently results in the down-regulation of AhR expression. We performed a series of in vivo experiments to determine whether the observed effects in vitro could be replicated within a living organism. Immunofluorescence staining analysis of tumor sites in mice revealed that, the fluorescence signal intensities of HMGB1 and TDO compared to the PBS group, were reduced by 4.5- and 2.4-fold after treatment, respectively (Fig. S24†). Compared with the reported platinum-based TDO inhibitor, In-Ru exhibits a more pronounced inhibitory effect on TDO.33 Concurrently, CRT signaling was significantly enhanced (Fig. 7E). These findings collectively demonstrate the capacity of In-Ru to suppress TDO expression and induce ICD activation in vivo. | ||
| Fig. 7 Vaccine administration and tumor suppression. (A) Schematic diagram of the vaccine administration experiment workflow. (B) Representative photographs of dissected tumors with various treatment conditions for 10 days as illustrated in (A). (C) Curves of H22 tumor volume treated with PBS, Indo, Ppy-Ru and In-Ru. (D) Tumor weight on day 10 in the various treatment groups. (E) Immunofluorescence analysis of specific protein expression (CRT, HMGB1 and TDO) from primary tumor tissues via staining with the corresponding probes. Scale bar: 200 μm. (F–H) Flow cytometry of DC maturation by determining CD11c+ CD80+ CD86+ cells in primary tumors. (I–K) The proportion of primary tumor infiltrating T lymphocytes (CD8+ and CD4+) measured with different treatments by flow cytometry. Error bars: S.D., n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001. | ||
The dead tumor cells caused by ICD effect, leading to the release of associated antigenic substances.53 This process subsequently stimulates various types of immune cells and ultimately promotes systemic immune activation.54 DCs as the primary antigen-presenting cells, play a crucial role in mediating communication between the innate and adaptive immune systems. ICD-induced tumor-associated substances promote the maturation of DCs. Subsequently, these antigens are captured by mature DCs and presented specifically to T cells, triggering a cellular immune response against homologous cancers. The increased proportion of CD80+ CD86+ cells, a hallmark of DCs maturation, rose from 0.6% to 20% in the tumor microenvironment of treated mice, which facilitated robust activation of adaptive immunity (Fig. 7F–H). Cytotoxic T lymphocytes (CTLs, CD3+ CD4+ CD8+) play a crucial role in tumor cytotoxicity and immune defense, while CD8+ T cells serve as the direct indicator of autoimmune activation and tumor killing.55 As shown in Fig. 7I–K, the percentage of CD8+ T cells increased approximately 20-fold compared to the untreated group. Notably, In-Ru demonstrated a more pronounced enhancement in CD8+ T cell activation, showing superior activation ratios compared to other ruthenium-based complexes (RuBTB: (≈3.9 fold); 6a: (≈2.0 fold); Ru2c@biotin-DNA cage: (≈1.5 fold)).19,56,57 Meanwhile, Ppy-Ru failed to promote T cell activation, whereas the TDO inhibitor Indo significantly increased the proportion of T cells, likely due to its ability to reverse the immunosuppressive microenvironment. CD4+ T cells possess the ability to assist CD8+ T cells in performing immune functions and participating in immune responses. The proportion of CD4+ T cells in In-Ru group increased approximately 6.5-fold, the value from 3% to 20.3%. Remarkably, In-Ru exhibited markedly superior enhancement in CD4+ T cell activation compared to other ruthenium complexes (RuBTB: (≈5.4 fold); Ru2c@biotin-DNA cage: (≈1.5 fold)).56,57In-Ru serves as an effective immunomodulator by inhibiting TDO to suppress kynurenine production, thereby reversing the immunosuppressive microenvironment in tumor tissues and enhancing the activation and proliferation of T cells. These critical results demonstrate that In-Ru can effectively ablate tumors in vivo and robustly activate the potent anti-tumor immune response through the synergistic effects of chemotherapy and immunotherapy, highlighting the potential of MLSE strategy in the treatment of solid tumors.
Conclusions
In conclusion, this study developed a Ru(II)-arene anti-tumor complex (In-Ru) that effectively inhibits TDO expression, thereby inducing ICD effect and reshaping the tumor immune microenvironment. In-Ru mediates the TDO/KYN/AhR metabolic pathway to alleviate tryptophan depletion, modulate the immune microenvironment, and promote T-cell proliferation and infiltration. Furthermore, In-Ru exhibits nuclear targeting properties, inducing severe DNA damage and thereby triggering necroptosis. This resulted in cell membrane rupture and antigen release, eliciting a systemic immune response. The in vivo vaccine experiments demonstrated that In-Ru effectively reduces TDO levels in solid tumors, activates the ICD effect, and enhances T cell-mediated immune cytotoxicity. This MLSE-based approach enables precise coordination between immune checkpoint inhibitors and metal-arene precursors, establishing a dual mechanism that synergistically augments chemotherapeutic and immunotherapeutic efficacy while concurrently diminishing therapeutic agent dosage requirements, thereby effectively mitigating associated adverse events. This paradigm not only systematically elucidates the anti-tumor immune mechanisms of metal-based TDO inhibitors but also offers new perspectives for developing safe and effective metal-based immunotherapies.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Nanjing Normal” University and approved by the Animal Ethics Committee of “Nanjing Normal University”.Data availability
The data that support the findings of this study are available in the ESI of this article.†Author contributions
Zheng-Qi Shen: writing – original draft, validation, methodology, investigation, formal analysis. Binglian Guo: methodology, formal analysis. Xiangyu Dai: formal analysis, data curation. Hanxue Liu: resources, methodology. Meng Ren: writing – review & editing, visualization. Peisen Wang: methodology, writing – review & editing, visualization. Yating Zhang: resources, formal analysis. Yinuo Xu: resources, formal analysis. Zhi Su: validation. Xuling Xue: writing – review & editing, visualization, resources. Hongke Liu: writing – review & editing, supervision, resources, funding acquisition, conceptualization.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22077066, No. 22477062), Original Exploration Program of National Natural Science Foundation of China (No. 22350001) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX24-1805).References
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- Q. Fu, S. Zhang, S. Shen, Z. Gu, J. Chen, D. Song, P. Sun, C. Wang, Z. Guo, Y. Xiao, Y. Q. Gao, Z. Guo and Z. Liu, Nat. Biomed. Eng., 2024, 8, 1425–1435 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264 RSC.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- K. Peng, Y. Zheng, W. Xia and Z.-W. Mao, Chem. Soc. Rev., 2023, 52, 2790–2832 RSC.
- C. Zhang, N. Montesdeoca, S. Tang, H. Liang, H. Cui, C. Xu, L. M. Servos, J. Bing, Z. Papadopoulos, F. Shen, H. Xiao, J. Yu and J. Karges, Nat. Commun., 2024, 15, 9405 CrossRef PubMed.
- S. Sen, S. Hufnagel, E. Y. Maier, I. Aguilar, J. Selvakumar, J. E. DeVore, V. M. Lynch, K. Arumugam, J. L. Sessler and J. F. Arambula, J. Am. Chem. Soc., 2020, 142, 20536–20541 CrossRef CAS PubMed.
- K. Shang, N. Montesdeoca, C. Zhang, E. Efanova, H. Liang, J. Ochs, J. karges, Q. Song and P. Zhang, J. Controlled Release, 2024, 373, 496–506 CrossRef PubMed.
- P. Kaur, A. Johnson, X. Lu and K. Suntharalingam, ChemBioChem, 2020, 21, 3618–3624 CrossRef CAS PubMed.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- M. Lv, Y. Zheng, J. Wu, Z. Shen, B. Guo, G. Hu, Y. Huang, J. Zhao, Y. Qian, Z. Su, C. Wu, X. Xue, H. Liu and Z. Mao, Angew. Chem., Int. Ed., 2023, 62, e202312897 CrossRef CAS PubMed.
- X. Xue, C. Qian, H. Fang, H. Liu, H. Yuan, Z. Guo, Y. Bai and W. He, Angew. Chem., Int. Ed., 2019, 58, 12661–12666 CrossRef CAS PubMed.
- H. Liu, S. J. Berners-Price, F. Wang, J. A. Parkinson, J. Xu, J. Bella and P. J. Sadler, Angew. Chem., Int. Ed., 2006, 45, 8153–8156 CrossRef CAS PubMed.
- X. Xue, C. Qian, Q. Tao, Y. Dai, M. Lv, J. Dong, Z. Su, Y. Qian, J. Zhao, H.-K. Liu and Z. Guo, Natl. Sci. Rev., 2021, 8, nwaa286 CrossRef CAS PubMed.
- H.-K. Liu, J. A. Parkinson, J. Bella, F. Wang and P. J. Sadler, Chem. Sci., 2010, 1, 258 RSC.
- M. Lv, Y. Zheng, X. Dai, J. Zhao, G. Hu, M. Ren, Z. Shen, Z. Su, C. Wu, H.-K. Liu, X. Xue and Z.-W. Mao, J. Med. Chem., 2024, 67, 20156–20171 CrossRef CAS PubMed.
- M. Wang, F. Xu, Y. Su, Y. Geng, X. Qian, X. Xue, Y. Kong, Z. Yu, H. Liu and Z. Su, Angew. Chem., Int. Ed., 2022, 61, e202203843 CrossRef CAS PubMed.
- N. Xu, G.-D. Zhang, Z.-Y. Xue, M.-M. Wang, Y. Su, H. Fang, Z.-H. Yu, H.-K. Liu, H. Lu and Z. Su, Chem. Eng. J., 2024, 497, 155022 CrossRef CAS.
- P. Wang, H. Fang, M. Wang, G. Zhang, N. Xu, Y. Su, H. Liu and Z. Su, Chin. Chem. Lett., 2025, 36, 110099 CrossRef CAS.
- C. Lu, D. Rong, B. Zhang, W. Zheng, X. Wang, Z. Chen and W. Tang, Mol. Cancer, 2019, 18, 130 CrossRef PubMed.
- J. Zheng, S. Wang, L. Xia, Z. Sun, K. M. Chan, R. Bernards, W. Qin, J. Chen, Q. Xia and H. Jin, Signal Transduct. Targeted Ther., 2025, 10, 35 CrossRef PubMed.
- J. E. Cheong and L. Sun, Trends Pharmacol. Sci., 2018, 39, 307–325 CrossRef CAS PubMed.
- S. I. Ilyas, J. Wang and A. B. El-Khoueiry, Hepatology, 2021, 73, 86–103 CrossRef PubMed.
- Y. Liu, X. Liang, W. Dong, Y. Fang, J. Lv, T. Zhang, R. Fiskesund, J. Xie, J. Liu, X. Yin, X. Jin, D. Chen, K. Tang, J. Ma, H. Zhang, J. Yu, J. Yan, H. Liang, S. Mo, F. Cheng, Y. Zhou, H. Zhang, J. Wang, J. Li, Y. Chen, B. Cui, Z.-W. Hu, X. Cao, F. Xiao-Feng Qin and B. Huang, Cancer Cell, 2018, 33, 480–494 CrossRef CAS PubMed.
- C. A. Opitz, U. M. Litzenburger, F. Sahm, M. Ott, I. Tritschler, S. Trump, T. Schumacher, L. Jestaedt, D. Schrenk, M. Weller, M. Jugold, G. J. Guillemin, C. L. Miller, C. Lutz, B. Radlwimmer, I. Lehmann, A. Von Deimling, W. Wick and M. Platten, Nature, 2011, 478, 197–203 CrossRef CAS PubMed.
- J. Yan, D. Chen, Z. Ye, X. Zhu, X. Li, H. Jiao, M. Duan, C. Zhang, J. Cheng, L. Xu, H. Li and D. Yan, Mol. Cancer, 2024, 23, 241 CrossRef PubMed.
- L. Du, Z. Xing, B. Tao, T. Li, D. Yang, W. Li, Y. Zheng, C. Kuang and Q. Yang, Signal Transduct. Targeted Ther., 2020, 5, 10 CrossRef CAS PubMed.
- T. A. Triplett, K. C. Garrison, N. Marshall, M. Donkor, J. Blazeck, C. Lamb, A. Qerqez, J. D. Dekker, Y. Tanno, W.-C. Lu, C. S. Karamitros, K. Ford, B. Tan, X. M. Zhang, K. McGovern, S. Coma, Y. Kumada, M. S. Yamany, E. Sentandreu, G. Fromm, S. Tiziani, T. H. Schreiber, M. Manfredi, L. I. R. Ehrlich, E. Stone and G. Georgiou, Nat. Biotechnol., 2018, 36, 758–764 CrossRef CAS PubMed.
- E. Dolušić, P. Larrieu, L. Moineaux, V. Stroobant, L. Pilotte, D. Colau, L. Pochet, B. Van Den Eynde, B. Masereel, J. Wouters and R. Frédérick, J. Med. Chem., 2011, 54, 5320–5334 CrossRef PubMed.
- S. Hua, F. Chen, G. Xu and S. Gou, Eur. J. Med. Chem., 2019, 169, 29–41 CrossRef CAS PubMed.
- F. Chen, G. Xu, W. Tian and S. Gou, Biochem. Pharmacol., 2021, 193, 114785 CrossRef CAS PubMed.
- D. E. Chapple, P. D. Boyle and J. M. Blacquiere, ChemCatChem, 2021, 13, 3789–3800 CrossRef CAS.
- Z. Li, X.-M. Liu, F. Tan, J.-Q. Wang, X. Qiao, Y.-K. Feng, J.-Y. Xu and J.-H. Hao, J. Med. Chem., 2025, 68, 4352–4372 CrossRef CAS PubMed.
- D. Hoffmann, T. Dvorakova, V. Stroobant, C. Bouzin, A. Daumerie, M. Solvay, S. Klaessens, M.-C. Letellier, J.-C. Renauld, N. Van Baren, J. Lelotte, E. Marbaix and B. J. Van Den Eynde, Cancer Immunol. Res., 2020, 8, 19–31 CrossRef CAS PubMed.
- Y. Ren, R. Wang, S. Weng, H. Xu, Y. Zhang, S. Chen, S. Liu, Y. Ba, Z. Zhou, P. Luo, Q. Cheng, Q. Dang, Z. Liu and X. Han, Mol. Cancer, 2023, 22, 130 CrossRef CAS PubMed.
- Y. Zhang, S. S. Su, S. Zhao, Z. Yang, C.-Q. Zhong, X. Chen, Q. Cai, Z.-H. Yang, D. Huang, R. Wu and J. Han, Nat. Commun., 2017, 8, 14329 CrossRef CAS PubMed.
- K. Rohde, L. Kleinesudeik, S. Roesler, O. Löwe, J. Heidler, K. Schröder, I. Wittig, S. Dröse and S. Fulda, Cell Death Differ., 2017, 24, 83–97 CrossRef CAS PubMed.
- H. K. Matthews, C. Bertoli and R. A. M. De Bruin, Nat. Rev. Mol. Cell Biol., 2022, 23, 74–88 CrossRef CAS PubMed.
- Y. Xie, J. Jiang, Q. Tang, H. Zou, X. Zhao, H. Liu, D. Ma, C. Cai, Y. Zhou, X. Chen, J. Pu and P. Liu, Adv. Sci., 2020, 7, 1903323 CrossRef CAS PubMed.
- Y. Zhou, I. N. Bastian, M. D. Long, M. Dow and S. Shalapour, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025840118 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, F. Guo, S. Qin, N. Zhou, Z. Liu, X. Fan and P. R. Chen, J. Am. Chem. Soc., 2024, 146, 15186–15197 CrossRef CAS PubMed.
- D. Sirnikova, J. Reynisson, A. Brüning-Richardson and C. Kirby, Neuro Oncol., 2023, 25, iii21 CrossRef.
- C. Wu, S. A. Spector, G. Theodoropoulos, D. J. M. Nguyen, E. Y. Kim, A. Garcia, N. Savaraj, D. C. Lim, A. Paul, L. G. Feun, M. Bickerdike and M. Wangpaichitr, Cancer Metab, 2023, 11, 7 CrossRef PubMed.
- G. Zhou, G. Qin, Z. Zhang, H. Zhao and L. Xue, Front. Immunol., 2023, 14, 1283792 CrossRef CAS PubMed.
- F. Exposito, M. Redrado, D. Serrano, L. M. Montuenga, F. Prosper and A. Calvo, Cell Death Dis., 2024, 15, 787 CrossRef CAS PubMed.
- Z. Zhang, X. Kong, M. A. Ligtenberg, B. Baars, E. E. Voest, S. Klarenbeek, M. Altelaar and D. S. Peeper, Cell Rep. Med., 2022, 3, 100655 CrossRef CAS PubMed.
- K. Hänggi, L. Vasilikos, A. F. Valls, R. Yerbes, J. Knop, L. M. Spilgies, K. Rieck, T. Misra, J. Bertin, P. J. Gough, T. Schmidt, C. R. De Almodòvar and W. W.-L. Wong, Cell Death Dis., 2017, 8, e2588 CrossRef PubMed.
- J. Liang, X. Tian, M. Zhou, F. Yan, J. Fan, Y. Qin, B. Chen, X. Huo, Z. Yu, Y. Tian, S. Deng, Y. Peng, Y. Wang, B. Liu and X. Ma, Biomaterials, 2024, 309, 122608 CrossRef CAS PubMed.
- R. L. Van Ineveld, R. Collot, M. B. Román, A. Pagliaro, N. Bessler, H. C. R. Ariese, M. Kleinnijenhuis, M. Kool, M. Alieva, S. M. Chuva De Sousa Lopes, E. J. Wehrens and A. C. Rios, Nat. Protoc., 2022, 17, 3028–3055 CrossRef CAS PubMed.
- F. Zhou, B. Feng, H. Yu, D. Wang, T. Wang, Y. Ma, S. Wang and Y. Li, Adv. Mater., 2019, 31, 1805888 CrossRef PubMed.
- S. Kim, H. Park, E. An, H. Eom, W. Zhang, J. Kim, I. Choi, M. Kwak, P. C. W. Lee and J. Jin, Adv. Funct. Mater., 2023, 33, 2302825 CrossRef CAS.
- Z. Lu, N. McBrearty, J. Chen, V. S. Tomar, H. Zhang, G. De Rosa, A. Tan, A. M. Weljie, D. P. Beiting, Z. Miao, S. S. George, A. Berger, G. Saggu, J. A. Diehl, C. Koumenis and S. Y. Fuchs, Cell Metab., 2022, 34, 1342–1358 CrossRef CAS PubMed.
- S. Tian, H. Xu, X. Wu, Y. Ding, L. Liang, H. Yin, X. Zeng, Y. Liu and W. Zhu, Eur. J. Med. Chem., 2025, 289, 117470 CrossRef CAS PubMed.
- J. Yang, F. Wang, S. Huang, T. Feng, K. Xiong, Y. Chen and H. Chao, Angew. Chem., Int. Ed., 2025, e202505689 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03778f |
| ‡ Zheng-Qi Shen, Binglian Guo and Xiangyu Dai contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |