
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring catalytic activity modulations: photoredox catalysis with substituted copper(I)-dipyridylamine derivatives†
Alondra
Villegas-Menares
 a,
Yannik Sebastian
Hansmann
b,
Max
Bayas
a,
Camilo
Verdugo
a,
Ignacio
Erazo
c,
Cesar
Zuñiga
d,
Iván
Gonzalez
e,
Antonio
Galdámez
f,
Lucrezia
Villa
g,
Mirco
Natali
g and
Alan R.
Cabrera
*a
a,
Yannik Sebastian
Hansmann
b,
Max
Bayas
a,
Camilo
Verdugo
a,
Ignacio
Erazo
c,
Cesar
Zuñiga
d,
Iván
Gonzalez
e,
Antonio
Galdámez
f,
Lucrezia
Villa
g,
Mirco
Natali
g and
Alan R.
Cabrera
*a
aDepartamento de Química Inorgánica, Facultad de Química y de Farmacia, Pontificia Universidad Católica de Chile, Vicuña Mackenna 4860, Macul, Santiago 7820436, Chile. E-mail: arcabrer@uc.cl
bInstitute for Organic Chemistry II and Advanced Materials, Ulm University, Albert-Einstein-Allee 11, Ulm 89081, Germany
cDepartamento de Ingeniería Mecánica y Metalúrgica, Escuela de Ingeniería, Pontificia Universidad Católica de Chile, Vicuña Mackenna 4860, Macul, Santiago 7820436, Chile
dDepartamento de Química de los Materiales, Facultad de Química y Biología, Universidad de Santiago de Chile, Casilla 40, Correo 33, Santiago 9170022, Chile
eDepartamento de Química, Facultad de Ciencias Naturales, Matemática y del Medio Ambiente, Universidad Tecnológica Metropolitana, Las Palmeras 3360, Ñuñoa, Santiago 7800003, Chile
fDepartamento de Química, Facultad de Ciencias, Universidad de Chile, Las Palmeras 3425, Ñuñoa, Santiago 7500008, Chile
gDepartment of Chemical, Pharmaceutical and Agricultural Sciences, University of Ferrara, via L. Borsari 46, Ferrara 44121, Italy
First published on 7th April 2025
Abstract
In this work, we have successfully synthesized five new heteroleptic copper(I) complexes (C1–5), bearing N,N ligands derived from dipyridylamine and S-BINAP as the P,P auxiliary ligand. All complexes were structurally characterized using NMR, FT-IR, and elemental analysis. Furthermore, the molecular structures of C1, C4, and C5 were determined via X-ray diffraction analysis. The photophysical properties of all complexes were assessed using UV-Vis spectroscopy and spectrofluorometric measurements in dichloromethane solution and the solid state. All complexes displayed absorption bands at lower energies, attributed to spin-allowed MLCT transitions. In degassed dichloromethane solution at room temperature, all complexes exhibited broad luminescence in the visible spectrum, mainly assigned to MLCT/LLCT phosphorescence, with excited state lifetimes in the μs time regime. Besides, all complexes were assessed as photoredox catalysts in chlorosulfonylation and bromonitromethylation reactions of styrene, showing remarkable performances, thus highlighting the privileged role of the dpa ligand for the design of Earth-abundant metal photocatalysts.
1. Introduction
One goal of modern organic chemistry is to develop new and sustainable synthetic routes.1 In this context, using light as an eco-friendly energy source is an alternative to thermal energy in substrate activation, aligned with the principles of green chemistry.2 To achieve this, photosensitizers and photoredox catalysts have been designed to absorb light in the visible region and use this energy to initiate the desired chemical transformation.3,4Transition metals like copper have gained interest in this field due to their abundance, low cost, and biocompatibility.5–8 This element is an attractive alternative to precious metals such as ruthenium and iridium, commonly employed in photocatalysis.9–13 In particular, homoleptic copper complexes have been successfully used to form carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds under mild reaction conditions.6,14–16 Among these are atom transfer radical addition (ATRA) reactions, which enable the difunctionalization of double bonds by introducing various functional groups.8,17 One of the most versatile copper photocatalysts in this family has been [Cu(dap)2]Cl (dap = 2,9-(p-anisyl)-1,10-phenanthroline), which absorbs light in the visible spectrum (437 nm), has a strong reduction potential in its excited state (−1.43 V vs. SCE), and a 270 ns lifetime in the excited state in CH2Cl2.14 The research group of Reiser specifically used this complex in various reactions, among the most notable being bromoalkylation, chlorosulfonylation, and, more recently, bromonitromethylation of styrene.8,18,19
On the other hand, heteroleptic copper complexes, which combine different bidentate ligands—mainly commercially available bidentate phosphines—present great versatility, cost-effectiveness, and stability.6,20–22 These complexes absorb in the near-UV-visible region. Still, unlike homoleptic complexes, they exhibit excited state lifetimes on the order of microseconds, allowing them to participate more effectively in single electron transfer (SET) and/or energy transfer (ET) processes.5,8,23,24 Although these complexes exhibit greater stability than their homoleptic counterparts, they remain susceptible to instability under irradiation and electrochemical conditions.25 These complexes are still prone to ligand exchange, photodecomposition, and structural rearrangements, which can significantly affect their performance and durability in practical applications.26,27 Enhancing the performance of these complexes remains a significant challenge, requiring innovative strategies to improve their stability and resilience under operational conditions.19
Several research groups, such as those of Collins, Hu, Evano, and Liu, have reported heteroleptic Cu(I) complexes capable of catalyzing carbon–carbon (C–C), carbon–nitrogen (C–N), carbon–sulfur (C–S) couplings, and other functional group exchanges.6,28–37 A common theme in the vast majority of reported studies is the use of phenanthroline or bipyridine derivatives as N,N-ligands, which have demonstrated their efficacy in a wide range of chemical transformations. However, the functionalization of these ligands, like the renowned dap ligand, is limited, and the synthetic routes for substituted derivatives involve multiple steps, increasing costs and variability.38 This has driven into more accessible alternatives, such as dipyridylamine (dpa) ligand derivatives.39–41 These dpa ligands have been synthesized in a single step and obtained with excellent yields, even for asymmetric derivatives.42
In 2021, we reported the study of heteroleptic Cu(I) complexes with dpa ligands and various commercial phosphines applied in ATRA reactions, decarboxylative coupling, and an Appel-type reaction, demonstrating the wide versatility of these complexes.43 In that work, it was concluded that the most suitable phosphine for all reactions was S-BINAP. Taking this into account, in the present work, the photocatalytic activity of new [Cu(N,N)(S-BINAP)]+ complexes is evaluated. In these complexes, N,N corresponds to dpa-derived ligands, with electron-donating (–OMe) or electron-withdrawing (–CF3) substituents in various positions on the pyridine rings, obtained through a straightforward Buchwald–Hartwig catalyzed reaction. The photocatalysts were structurally, electrochemically, and photophysically characterized. Besides, their catalytic activity in chlorosulfonylation and bromonitromethylation reactions of styrene was evaluated.
2. Experimental section
2.1 General considerations
All commercially available reagents and solvents were used as received unless otherwise specified. Ligands L1, L4, and L5 were prepared as described in the literature.42 NMR spectra were recorded on NMR Bruker AV 400 MHz. Chemical shifts are given in parts per million relative to TMS (1H and 13C, δ(SiMe4) = 0) or an external standard [δ(CFCl3) = 0 for 19F NMR and δ(H3PO4) = 0 for 31P NMR]. Additional 2D experiments supported most NMR assignments. FT-IR spectra were recorded on a Shimadzu IRTracer-100 spectrophotometer using KBr pellets. Elemental analysis was carried out using an Elementar Analysensysteme GmbH, Model Vario EL III. UV-Vis spectra were registered using a Shimadzu spectrophotometer, model UV-1900i. Photoluminescence (PL) spectra of the complexes were measured using an Edinburgh Instrument spectrofluorometer. Excitation was provided near the absorption maximum of the lowest energy transition. The emission quantum yield (Φem) was calculated using the relative method following the literature description. Solutions were degassed using N2 for 20 minutes before analysis. Luminescence measurements at 77 K were performed in 2-MeTHF using a cryostat. Poly(methyl methacrylate) (PMMA) films were prepared from CH2Cl2 solutions of C1–5 and low-molecular-weight PMMA, followed by drop casting onto a glass surface. Emission lifetimes were taken on a laser flash photolysis apparatus comprised of a Continuum Surelite II Nd:YAG laser (excitation at 355 nm, FWHM = 6–8 ns, was provided by THG from the 1064 nm fundamental). Light emitted by the sample was focused onto the entrance slit of a 300 mm focal length Acton SpectraPro 2300i triple grating, flat field, and double exit monochromator equipped with a photomultiplier detector (Hamamatsu R3896). Signals from the photomultiplier were processed employing a TeledyneLeCroy 604Zi (400 MHz, 20 GS per s) digital oscilloscope. Single crystal X-ray diffraction analysis (XRD) for C1 (CCDC 2405276), C4 (CCDC 2405277), and C5 (CCDC 2405278)† was performed at 100 K using a SuperNova four-circle diffractometer in Kappa geometry with a 50 W Cu or Mo (Kα radiation) microfocus tube, an Atlas CCD detector (Rigaku Oxford Diffraction), and a Cryostream 700 Plus cooler (Oxford Cryosystems Ltd). Data collection, cell refinement, data reduction, and absorption correction were done using CrysAlisPro.44 For more details on the experimental part, see the ESI.†2.2 General procedure for the synthesis of ligands
In a nitrogen-flushed 30 mL microwave glass vessel, 2 mmol of the corresponding aminopyridine, 2 mmol of the bromopyridine, 0.06 mmol of bis(dibenzylideneacetone)palladium(0), 0.12 mmol of 1,3-bis(diphenylphosphino)propane and 2.8 mmol of KOtBu were dissolved in 15 mL of anhydrous toluene. The vessel was purged with nitrogen and sealed with a silicone septum. The reaction mixture was stirred at 130 °C in the microwave reactor for 2 h. Then, the solvent was removed under reduced pressure, and the crude product was absorbed into silica gel. The final product was obtained as a solid after flash column chromatography (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA mixture with ratios between 5:2 and 5:3).
:EA mixture with ratios between 5:2 and 5:3).
2.3 General procedure for the synthesis of Cu(I) complexes
In a glass vial, 1 mmol of [Cu(CH3CN)4]BF4, 1 mmol of S-BINAP, and 1 mmol of the corresponding ligand L1–5 were added. Then, the vial was sealed with a septum and was nitrogen flushed for 5 min. Later, 5 mL of anhydrous dichloromethane was added through a purged syringe, and the reaction mixture was stirred at room temperature for 1 hour under a nitrogen atmosphere. Afterward, the volatiles were removed under reduced pressure, and the crude product was purified by crystallization from a mixture of CH2Cl2 and toluene at −20 °C.2.4 Electrochemical analysis
The electrochemical measurements were done with a typical three-electrode electrochemical cell with separate compartments to avoid undesirable parallel reactions. The saturated Calomel electrode (Hg0/Hg2Cl2sat, SCEsat.) was used as a reference electrode within the lugging capillary with a saturated KCl solution. The lugging capillary is used to prevent contamination with the studied organic compound in the porous frit interface of the reference electrode, which can progressively change their interfacial electrochemical potential.45 A Pt0 wire was used as a counter electrode, and glassy carbon was selected as a working electrode (0.196 cm2). The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in anhydrous acetonitrile. As previously reported, the complex concentration was 1 mM in the same electrolyte solution.43,46 The electrochemical measurement was performed with a PalmSens 3 Instrument Potentiostat. The formal potential (E°′) of C1–5 was evaluated through cyclic voltammetry (CV) at 0.1 mV s−1 from −1.5 to 1.3 V vs. SCE (see Fig. S18†). The electrolytic solution was purged with N2 for 15 minutes in each experiment to avoid the presence of O2(gas). To corroborate the acquired formal E°′, squared wave voltammetry (SWV) was performed, as shown in Fig. S19(a–e).† The potential of each complex was determined according to the appearance of cathodic formal potential
was determined according to the appearance of cathodic formal potential  and anodic formal potential peaks
and anodic formal potential peaks  and their relationship using the following equation
and their relationship using the following equation  .47
.47
3. Results and discussion
3.1 Synthesis and structural characterization of copper(I) complexes
Copper(I) complexes C1–5 were prepared by a simple ligand exchange reaction, where one equivalent of the metallic precursor reacted with one equivalent of the respective dipyridylamine N,N ligand (L1–5) and one equivalent of S-BINAP as the auxiliary P,P ligand, in dichloromethane as solvent and under inert conditions (Scheme 1). The complexes with the general formula [Cu(N,N)(S-BINAP)]BF4 were obtained in yields ranging from 89–98% after purification. | ||
| Scheme 1 General synthetic approach for [Cu(N,N)(S-BINAP)]BF4 complexes formation. | ||
These new complexes were structurally characterized by NMR, FT-IR, and EA techniques. The spectroscopic characterization of the compound is consistent with a mononuclear Cu(I) metal center coordinated to an N,N ligand and a P,P phosphine ancillary ligand. The 31P NMR exhibits one broad signal at −1.0 ppm, approximately, with ν1/2 ∼ 75 Hz, attributed to the phosphorous atom coordinated to the metal center. This broad signal shape is typical for the quadrupolar coupling with the copper atom.48 In addition, at −0.6 ppm (11B NMR) and −151 ppm (19F NMR), singlet signals are observed, corresponding to the counterion BF4−. For a detailed spectroscopic characterization of the complexes, see the ESI.†
In addition, the molecular structure of C1, C4, and C5 was determined by XRD, shown in Fig. 1. The crystals suitable for X-ray analysis were obtained by liquid–liquid slow diffusion of n-pentane in a dichloromethane solution containing the complexes. The compound C1 crystallized in tetragonal space group P41212, and compounds C4 and C5 crystallized in orthorhombic space group P212121, all with four molecules per unit cell. All the complexes were identified as heteroleptic mononuclear copper species, consistent with the findings from their spectroscopic characterization. Okuniewski et al. introduced the structural parameter τ4′ to assess the geometry of complexes with coordination numbers of four.49 The τ4′ values range from 0 to 1, where 1 corresponds to tetrahedral geometry (Td), while 0 represents flat quadratic geometry (D4h). Intermediate values indicate distorted geometries. The values obtained for the complexes are 0.82 (C1), 0.84 (C4), and 0.83 (C5). Therefore, all complexes adopt distorted tetrahedral geometries around the copper(I) center. One interesting difference was observed in the shape adopted by the N,N ligand. In the case of C1, the 5,5′ substitution pattern induced a slight deviation from the planarity of the coordination ring (dihedral angle of 12.7(6)° N1–C1–N2–C7). On the contrary, the 4,4′ substitution pattern produces a more significant deviation, observing a boat-shape of the coordination ring, with dihedral angles of −37.7(4)° for C4 (N1–C1–N2–C7) and −41.2(8)° for C5 (N1–C1–N2–C7).
 | ||
| Fig. 1 ORTEP plot of C1, C4, and C5 complexes. Thermal ellipsoids are shown at 30% probability. Hydrogen atoms and counterion were omitted for clarity. | ||
3.2 Electrochemical characterization of complexes C1–5
The electrochemical properties of complexes C1–5 were measured by cyclic voltammetry at 0.1 V s−1 (Fig. S18†) and corroborated through square wave voltammetry (Fig. S19a–e†). The acquired formal potential values are summarized in Table 1. To negative potentials are observed processes corresponding with the ligand reduction L/L−. On the other hand, peaks are detected toward positive applied potentials and assigned to the Cu(I)/(II) oxidation process. These assignments were carefully evaluated and compared to similar molecular structures in the literature.43,46,50,51 In the case of the redox process associated with the metal center, the oxidation faradaic current observed through SWV of
are summarized in Table 1. To negative potentials are observed processes corresponding with the ligand reduction L/L−. On the other hand, peaks are detected toward positive applied potentials and assigned to the Cu(I)/(II) oxidation process. These assignments were carefully evaluated and compared to similar molecular structures in the literature.43,46,50,51 In the case of the redox process associated with the metal center, the oxidation faradaic current observed through SWV of  Cu(I)/(II) and reduction part
Cu(I)/(II) and reduction part 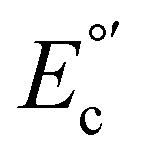 (Cu(II)/(I)) shows a quasi-reversible behavior, which suggests that an adsorption process on the electrode surface stabilizes the oxidized species.50 In addition, a strong non-reversible reducing faradaic peak can be observed through SWV (Fig. S19†) between
(Cu(II)/(I)) shows a quasi-reversible behavior, which suggests that an adsorption process on the electrode surface stabilizes the oxidized species.50 In addition, a strong non-reversible reducing faradaic peak can be observed through SWV (Fig. S19†) between  : 0.25 V and 0.50 V vs. SCE. This peak could be associated with the reduction process from Cu(I) to Cu(0). This observation suggests that the studied molecule structures are unstable within the evaluated range of potentials and supporting electrolytes (TBAPF6 in CH2Cl2), similar to other copper-based complexes reported.52,53
: 0.25 V and 0.50 V vs. SCE. This peak could be associated with the reduction process from Cu(I) to Cu(0). This observation suggests that the studied molecule structures are unstable within the evaluated range of potentials and supporting electrolytes (TBAPF6 in CH2Cl2), similar to other copper-based complexes reported.52,53
| Complex |
a [V vs. SCE] |
|
CH2Cl2 at 298 K | ||
|---|---|---|---|---|---|
| λ/nm | Φ/% | τ/μs | |||
a Cyclic voltammetry recorded in anhydrous acetonitrile solution of C1–5 (1 mM) with 0.1 M TBAPF6 as supporting electrolyte at a scan rate of 0.1 V s−1. Three-electrode cell configuration (glassy carbon working electrode, saturated calomel electrode (Hg0/Hg2Cl2 sat, SCEsat.) reference electrode and Pt wire counter electrode). E1/2 values are referred to SCE. The formal potential  or or  , was obtained as , was obtained as 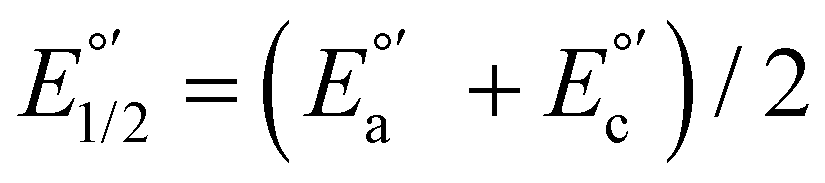 .
b Estimated using either fluorescein in 0.1 M NaOH aqueous solution (Φ = 94%) or [Ir(ppy)3] in 2-MeTHF (Φ = 96%) as a standard.
c Estimated using [Ru(bpy)3]2+ in air-equilibrated H2O as a standard (Φ = 2.8%). .
b Estimated using either fluorescein in 0.1 M NaOH aqueous solution (Φ = 94%) or [Ir(ppy)3] in 2-MeTHF (Φ = 96%) as a standard.
c Estimated using [Ru(bpy)3]2+ in air-equilibrated H2O as a standard (Φ = 2.8%).
|
|||||
| C1 | 1.32 | −0.86 | 503 | 0.40b | 10.7 |
| C2 | 1.10 | −0.80 | 557 | 0.20c | 17.0 |
| C3 | 1.07 | −0.86 | 652 | 0.02c | 0.14 |
| C4 | 1.49 | −0.43 | 499 | 0.30b | 0.17 |
| C5 | 1.24 | −0.78 | 501 | 0.10b | 0.37 |


According to Table 1, the trend observed for complexes substituted in position 5,5′ (C1–C3) is in agreement with their substitution pattern, where C1 is CF3 symmetrically substituted and showed the most positive formal potential ( vs. SCEsat), followed by the asymmetrically substituted with CF3 and OMe (C2:
vs. SCEsat), followed by the asymmetrically substituted with CF3 and OMe (C2:  vs. SCEsat), and then by the OMe symmetrically substituted C3 (
vs. SCEsat), and then by the OMe symmetrically substituted C3 ( vs. SCEsat). These data confirm the modulation of the energy of the HOMO of the complex exerted by the groups in the N,N ligand, where the systematic replacement of an electron-withdrawing group (CF3) by an electro-donating group (OMe) decreases the formal oxidation potential of the copper center. Furthermore, from Table 1 it can be observed that the symmetrical substitution with CF3 in position 4,4′ of the pyridinic ring induced a higher electro-withdrawing effect, exhibiting a formal potential of 1.49 V vs. SCEsat (
vs. SCEsat). These data confirm the modulation of the energy of the HOMO of the complex exerted by the groups in the N,N ligand, where the systematic replacement of an electron-withdrawing group (CF3) by an electro-donating group (OMe) decreases the formal oxidation potential of the copper center. Furthermore, from Table 1 it can be observed that the symmetrical substitution with CF3 in position 4,4′ of the pyridinic ring induced a higher electro-withdrawing effect, exhibiting a formal potential of 1.49 V vs. SCEsat ( of C4), in comparison with its isomer C1 (0.17 V lower). This effect is also observed by comparing C5 and C2, where the 4,4′ pattern has a higher formal potential than the 5,5′ isomer (
of C4), in comparison with its isomer C1 (0.17 V lower). This effect is also observed by comparing C5 and C2, where the 4,4′ pattern has a higher formal potential than the 5,5′ isomer ( vs. SCEsat). Consequently, the descending order in
vs. SCEsat). Consequently, the descending order in  can be described as follows C4 > C1 > C5 > C2 > C3. As to the cathodic side, the potentials of the reduction process fall within a narrow range in the case of C1, C2, and C3, pointing to an almost negligible effect of the substituents. This thus indicates that for these complexes the reduction most likely involves the P,P ligand. On the other hand, the progressive increase of the reduction potential when moving from C5 to C4 strongly suggests that these latter reductions may preferentially occur on the N,N ligand. This result agrees with the more pronounced effects of the substituents in the 4,4′ than the 5,5′ positions as recorded for the Cu(I)/Cu(II) oxidation.
can be described as follows C4 > C1 > C5 > C2 > C3. As to the cathodic side, the potentials of the reduction process fall within a narrow range in the case of C1, C2, and C3, pointing to an almost negligible effect of the substituents. This thus indicates that for these complexes the reduction most likely involves the P,P ligand. On the other hand, the progressive increase of the reduction potential when moving from C5 to C4 strongly suggests that these latter reductions may preferentially occur on the N,N ligand. This result agrees with the more pronounced effects of the substituents in the 4,4′ than the 5,5′ positions as recorded for the Cu(I)/Cu(II) oxidation.
3.3 Photophysical characterization
The absorption spectra of complexes C1–5 measured in CH2Cl2 solution at room temperature are depicted in Fig. 2. They display intense absorptions below 350 nm attributable to spin-allowed π–π* transitions involving the N,N and P,P ligands. Weaker absorption bands are then observed at longer wavelengths and mainly assigned to spin-allowed metal-to-ligand charge transfer (MLCT) transitions. For comparison with the prototype complex [Cu(dpa)(S-BINAP)]BF4,43 and also based upon the electrochemical results, the lowest-energy absorptions most likely result from a combination of Cu,dpa → S-BINAP and Cu → dpa charge transfer transitions (i.e., MLCT/LLCT transitions). Consistently, the energy is expected to be affected by the nature and position of the substituents on the dpa ligand. In this regard, the most red-shifted band is observed for complex C3 (maximum at 378 nm), in agreement with the presence of two electron-donating –OMe groups, the least positive oxidation potential, and the resulting higher-energy HOMO. | ||
| Fig. 2 Absorption spectra of complexes C1–5 in CH2Cl2 at room temperature. | ||
The luminescence properties of complexes C1–5 were then examined in degassed CH2Cl2 at room temperature and in the solid state. The resulting spectra in solution are displayed in Fig. 3, while Table 1 collects the relevant photophysical data. The solid-state luminescence data are instead reported in the ESI.† In CH2Cl2 all complexes exhibit broad, unstructured luminescence in the visible spectrum, independent of the excitation wavelength, mainly assigned to MLCT/LLCT phosphorescence. Conversely, at 77 K (Fig. S20a†), structured emission bands are observed in the range 470–650 nm for all complexes (Table S4†), likely associated with LC phosphorescence.43,54 Similarly, the luminescence spectra measured in a PMMA solid-state matrix (Fig. S20b†) display comparable spectral patterns with two relative maxima at 510–520 and 540–550 nm in the case of complexes C1, C2, C4, and C5 (Table S4†), still consistent with LC phosphorescence. The switch in the excited state nature between room temperature and solid-state matrix likely results from the comparable energy of the triplet MLCT/LLCT and LC excited states and the effective destabilization of the former in the solid-state matrix. A broader emission with a maximum at 569 nm, most likely of MLCT/LLCT character, is instead recorded in PMMA for C3, consistent with its lowest energy band in fluid solution. For this latter, the blue-shift observed in comparison with the emission in CH2Cl2 clearly results from the known rigidochromic effect characteristic of CT states.55
 | ||
| Fig. 3 Luminescence spectra of complexes C1–5 in CH2Cl2 at room temperature. | ||
In CH2Cl2, the emission is generally weak (Φ < 0.4%) akin to the prototype [Cu(dpa)(S-BINAP)]BF4 complex (Φ = 0.09%).43 Interestingly, the emission wavelength and yield are sensitive to the position and type of substituent introduced on the dpa ligand.
For the C1–3 sub-series, i.e., the complexes substituted in position 5,5′, the energy of the emission decreases in the order C1 > C2 > C3, consistent with the similar energy of the LUMO and the progressive destabilization of the HOMO with increasing the number of electron-donating OMe groups, as observed by electrochemical measurements (Table 1). The excited state lifetimes fall within the μs time regime for C1 and C2, and below the μs time range for C3 (Table 1). Estimation of the radiative constants (from the relationship Φ = kR·τ) yields relatively low values for C1–3 (≤103 s−1, see Table S5†), in analogy with the prototype [Cu(dpa)(S-BINAP)]BF4 complex,43 possibly indicating a similar nature of the luminescent excited state in C1–3 and [Cu(dpa)(S-BINAP)]BF4. In this respect, the net drop in emission quantum yield observed in the case of C3 can be mainly associated with faster non-radiative routes (see Table S5†) due to the energy-gap law.
Both complexes C4 and C5 in CH2Cl2 instead display an emission centered at ca. 500 nm, regardless of the presence and type of substituents, with quantum yields of 0.3% and 0.1%, respectively. The similar energy is consistent with the comparable HOMO–LUMO energy gap observed by electrochemistry. For these complexes, the emission lifetimes are in the order of hundred ns, delivering larger radiative constants than those estimated for the C1–3 sub-series (>103 s−1, see Table S5†). These findings suggest a different MLCT character of the luminescent excited state of complexes C4 and C5 in CH2Cl2 at room temperature. According to the electrochemical data above, this can be interpreted assuming a change in the nature of the emitting excited state, i.e., from a dominating Cu,dpa → S-BINAP to a predominant Cu → dpa charge transfer when moving from C1–3 to C4–5. The results shown herein thus indicate that substitution of the dpa ligand has substantial effects on the photophysical behavior of the resulting metal complex when compared to the prototype [Cu(dpa)(S-BINAP)]BF4. In particular, while substitution in position 5,5′ has more pronounced effects on the energy of the excited state, substitution in position 4,4′ has stronger effects on the excited state behavior. Most importantly, besides being poorly luminescent, all complexes display lifetimes in the hundred ns to μs time-scale, essential to effectively partake within light-driven bimolecular processes such as those considered hereafter.
3.4 Photocatalytic evaluation
Continuing our group's research on the photocatalytic application of copper complexes,43 we evaluated the effect of the N,N ligand substituents in C1–5 on the chlorosulfonylation reaction of styrene (Table 2), following the experimental procedure shown in the ESI.† Here, the standard compound used was the complex [Cu(dpa)(S-BINAP)]BF4,43 where dpa stands for the dipyridylamine ligand without any substituent, which showed a 93% yield of the desired product (Table 2, entry 2). Then, we tested compounds C1–5 in the title reaction (Table 2, entries 3–7), which showed that compounds C1 and C4 gave the desired product with excellent yields of 100%. Both complexes possess symmetrical CF3 substitution in the N,N ligand. It is interesting to note that as we change the substitution from symmetric CF3 to symmetric OMe (Table 2, entries 3–5), the catalytic activity of the complex diminished to a 10% yield with the electron-donating groups. Calculation of the reduction potentials of the excited state of the complexes using the Rehm–Weller equation (see Table S6†) suggests that the low activity of C3 most likely arises from the least negative reduction potential within the series which makes activation of the p-toluenesulfonyl chloride substrate via single electron transfer (SET) substantially endergonic (Ered = −0.94 V vs. SCE).18 Regarding the substitution position, by comparing entries 3 with 6 (symmetric CF3, in positions 5,5′ or 4,4′) or entries 4 with 7 (asymmetric CF3/OMe, in positions 5,5′ or 4,4′), there are no significant differences in the yields of the product.
a
which makes activation of the p-toluenesulfonyl chloride substrate via single electron transfer (SET) substantially endergonic (Ered = −0.94 V vs. SCE).18 Regarding the substitution position, by comparing entries 3 with 6 (symmetric CF3, in positions 5,5′ or 4,4′) or entries 4 with 7 (asymmetric CF3/OMe, in positions 5,5′ or 4,4′), there are no significant differences in the yields of the product.
a
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb/% |
|
a Styrene (0.5 mmol, 1.0 equiv.), TsCl (0.5 mmol, 1.0 equiv.), catalyst (1 mol%) in CH3CN (dry, degassed, 2 mL); irradiation at 440 nm (LED) under N2 atmosphere for 24 h at R.T.
b 1H-NMR yield was determined using 1,1,2,2-tetrachloroethane as an internal standard. n.r. = no reaction.
|
||
| 1 | Without | n.r. |
| 2 | [Cu(dpa)(S-BINAP)]BF4 | 93 |
| 3 | C1 | 100 |
| 4 | C2 | 98 |
| 5 | C3 | 10 |
| 6 | C4 | 100 |
| 7 | C5 | 93 |

In 2023, Reiser et al. described a highly efficient Cu(I) photocatalyzed visible light-mediated ATRA reaction involving bromonitromethane derivatives and olefins conducted under environmentally friendly conditions.56 This research highlighted the unique role of copper in photoredox catalysis, demonstrating that it can surpass the highly favorable cyclization of transient to persistent radicals, unlike iridium-based photocatalysts. Consequently, we evaluated our complexes C1–5 in the title reaction (Table 3). All studied complexes, including the prototype [Cu(dpa)(S-BINAP)]BF4, showed catalytic activity towards the bromonitromethylation of styrene in dichloromethane or acetonitrile as solvents (Table 3). However, a strong dependency was observed on the selected solvent (see Table S7†), with acetonitrile being the better solvent for the assay, obtaining higher catalytic activities. Among the evaluated compounds, the symmetric substituted OMe complex C3 exhibited the highest activity (83% yield, Table 3, entry 5), contrasting the observed behavior in the chlorosulfonylation reaction. In this regard, no apparent electronic effect of the substitution pattern on the catalytic performance was observed in the compound series.
a
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb/% |
|
a Styrene (0.5 mmol, 1.0 equiv.), BrCH2NO2 (0.5 mmol, 1.0 equiv.), catalyst (1 mol%) in CH3CN (dry, degassed, 2 mL); irradiation at 440 nm (LED) under N2 atmosphere for 24 h at R.T.
b 1H-NMR yield was determined using 1,3,5-trimethoxybenzene as an internal standard. n.r. = no reaction.
|
||
| 1 | Without | n.r |
| 2 | [Cu(dpa)(S-BINAP)]BF4 | 52 |
| 3 | C1 | 68 |
| 4 | C2 | 66 |
| 5 | C3 | 83 |
| 6 | C4 | 64 |
| 7 | C5 | 46 |

4. Conclusions
In this work, five new copper complexes with dpa ligands substituted with electron-donating and electron-withdrawing groups were obtained. The complexes were structurally characterized, confirming the formation of mononuclear heteroleptic copper species with distorted tetrahedral structures. Additionally, the obtained compounds were characterized electrochemically and optically. The results showed that substitution of the dpa ligand has substantial effects on the behavior of the resulting complex when compared to the prototype [Cu(dpa)(S-BINAP)]BF4. In particular, while substitution in position 5,5′ has more pronounced effects on the energy of the excited state, substitution in position 4,4′ has stronger effects on the excited state behavior.The evaluation of the complexes as photoredox catalysts in the chlorosulfonylation and bromonitromethylation reactions of styrene was achieved. In the chlorosulfonylation reaction, excellent yields were obtained using the complexes with symmetric CF3 substitution on the dpa ligand (C1 and C4). At the same time, the systematic introduction of electron-donating groups (OMe) gradually decreased the observed catalytic activity. Regarding the bromonitromethylation of styrene, although all the complexes were active, especially C3, with an 83% yield, no clear trend was observed between the catalytic activity and the structural or electronic properties of the complexes. These results encourage us to continue our research, especially searching for versatile and easily accessible photoredox catalysts.
Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript. Conceptualization: A. R. C.; methodology: A. V.-M., C. V., I. E., and M. N.; investigation: A. V.-M., Y. S. H., M. B., C. V., I. E., C. Z., and L. V.; resources: C. Z., I. G., M. N., and A. R. C.; formal analysis: A. G.; validation: A. V.-M., and I. E.; writing – original draft: A. V.-M., C. Z., M. N., and A. R. C.; writing – review and editing: A. V.-M., C. Z., M. N., and A. R. C.; project administration: A. R. C.; funding acquisition: A. R. C.; supervision: A. R. C.Data availability
The crystallographic data for complexes C1, C4, and C5 have been deposited in the CCDC under deposition numbers 2405276–2405278†. The spectroscopic datasets supporting this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of Fondo Nacional de Desarrollo Científico y Tecnológico projects 1210661, 1230199, and EQM120021. A. V. M. thanks the Postdoctoral fellowship Fondecyt 3230413, and C. Z. thanks the Postdoctoral fellowship Fondecyt 3220215. M. N. acknowledges financial support from the Italian MUR (PRIN2020, Electrolight4Value, 2020927WY3) and the Università degli Studi di Ferrara (FAR 2024). We thank Dr Dieter Schaarschmidt (Department of Chemistry, University of Hamburg, Germany) for conducting the X-ray diffraction analyses used in this study.References
- J. L. Tyler, F. Katzenburg and F. Glorius, Chem. Sci., 2023, 14, 7408–7410 RSC.
- C. Bihanic, A. Lasbleiz, M. Regnier, E. Petit, P. Le Blainvaux and C. Grison, Molecules, 2021, 26, 7194 CrossRef CAS PubMed.
- N. M. Chauke, R. L. Mohlala, S. Ngqoloda and M. C. Raphulu, Front. Chem. Eng., 2024, 6, 1356021 CrossRef.
- B. H. Jhun, J. Jang, S. Lee, E. J. Cho and Y. You, Nat. Commun., 2024, 15, 6586 CrossRef CAS PubMed.
- Y. J. Zhang, Q. Wang, Z. S. Yan, D. L. Ma and Y. G. Zheng, Beilstein J. Org. Chem., 2021, 17, 2520–2542 CrossRef CAS PubMed.
- J. Beaudelot, S. Oger, S. Perusko, T. A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed.
- A. Reichle and O. Reiser, Chem. Sci., 2023, 14, 4449–4462 RSC.
- S. Engl and O. Reiser, Chem. Soc. Rev., 2022, 51, 5287–5299 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- X. J. Dai, X. L. Xu and X. N. Li, Chin. J. Org. Chem., 2013, 33, 2046–2062 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- W. M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS.
- T. Zhang, J. Rabeah and S. Das, Nat. Commun., 2024, 15, 5208 CrossRef CAS PubMed.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336–7340 CrossRef CAS PubMed.
- T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, ACS Catal., 2018, 8, 3950–3956 CrossRef CAS.
- T. Mandal, N. Katta, H. Paps and O. Reiser, ACS Org. Inorg. Au, 2023, 3, 171–176 CrossRef CAS PubMed.
- J. M. Ahn, T. S. Ratani, K. I. Hannoun, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 12716–12723 CrossRef CAS PubMed.
- A. Hossain, S. Engl, E. Lutsker and O. Reiser, ACS Catal., 2019, 9, 1103–1109 CrossRef CAS.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J. F. Nierengarten and N. Armaroli, Inorg. Chem., 2018, 57, 15537–15549 CrossRef CAS PubMed.
- V. R. Naina, A. K. Singh, F. Krätschmer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Dalton Trans., 2023, 52, 12618–12622 RSC.
- D. Moreno-da Costa, C. Zúñiga-Loyola, F. Droghetti, S. Robles, A. Villegas-Menares, N. Villegas-Escobar, I. Gonzalez-Pavez, E. Molins, M. Natali and A. R. Cabrera, Molecules, 2024, 29, 47 CrossRef CAS PubMed.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J. J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J. F. Nierengarten, Inorg. Chem., 2013, 52, 12140–12151 CrossRef CAS PubMed.
- D. N. Wang, W. H. Hu, C. M. Liu, J. E. Huang and X. Y. Zhang, J. Phys. Chem. Lett., 2023, 14, 10137–10144 CrossRef CAS PubMed.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, in Top. Curr. Chem, ed. V. Balzani and S. Campagna, Springer, Berlin, Heidelberg, 2007, vol. 280, pp. 69–115 Search PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- C. Forster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- C. Minozzi, A. Caron, J. C. Grenier-Petel, J. Santandrea and S. K. Collins, Angew. Chem., Int. Ed., 2018, 57, 5477–5481 CrossRef CAS PubMed.
- J. Sosoe, C. Cruché, M. Morin and S. K. Collins, Can. J. Chem., 2020, 98, 461–465 CrossRef CAS.
- C. Cruché, W. Neiderer and S. K. Collins, ACS Catal., 2021, 11, 8829–8836 CrossRef.
- C. Cruché, S. Gupta, J. Kodanko and S. K. Collins, Molecules, 2022, 27, 3745 CrossRef PubMed.
- M. Alkan-Zambada and X. Hu, J. Org. Chem., 2019, 84, 4525–4533 Search PubMed.
- S. Oger, H. Baguia, T. A. Phan, T. Teunens, J. Beaudelot, C. Moucheron and G. Evano, SynOpen, 2021, 5, 141–144 Search PubMed.
- C. Jacob, H. Baguia, A. Dubart, S. Oger, P. Thilmany, J. Beaudelot, C. Deldaele, S. Perusko, Y. Landrain, B. Michelet, S. Neale, E. Romero, C. Moucheron, V. Van Speybroeck, C. Theunissen and G. Evano, Nat. Commun., 2022, 13, 560 CrossRef CAS PubMed.
- J. Beaudelot, G. Evano and C. Moucheron, Chem. – Eur. J., 2023, 29, e202300758 Search PubMed.
- L. L. Chen, Y. Li, M. F. Han, Y. Peng, X. H. Chen, S. W. Xiang, H. Gao, T. H. Lu, S. P. Luo, B. W. Zhou, H. Y. Wu, Y. F. Yang and Y. K. Liu, J. Org. Chem., 2022, 87, 15571–15581 CrossRef CAS PubMed.
- Y. H. Zhao, H. Y. Li, D. J. Young, X. Q. Cao, D. L. Zhu, Z. G. Ren and H. X. Li, Dalton Trans., 2023, 52, 8142–8154 RSC.
- T. Rawner, PhD in Chemistry, Universitat Regensburg, 2016.
- G. Giobbio, L. M. Cavinato, E. Fresta, A. Montrieul, G. U. Mahoro, J. F. Lohier, J. L. Renaud, M. Linares, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2023, 33, 2304668 Search PubMed.
- G. U. Mahoro, E. Fresta, M. Elie, D. di Nasso, Q. Zhang, J. F. Lohier, J. L. Renaud, M. Linares, R. Wannemacher, J. Cabanillas-Gonzalez, R. D. Costa and S. Gaillard, Dalton Trans., 2021, 50, 11049–11060 Search PubMed.
- M. Elie, M. D. Weber, F. Di Meo, F. Sguerra, J. F. Lohier, R. B. Pansu, J. L. Renaud, M. Hamel, M. Linares, R. D. Costa and S. Gaillard, Chem. – Eur. J., 2017, 23, 16328–16337 CrossRef CAS PubMed.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J. F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J. L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678–14691 Search PubMed.
- M. A. Henriquez, S. Engl, P. Jaque, I. A. Gonzalez, M. Natali, O. Reiser and A. R. Cabrera, Eur. J. Inorg. Chem., 2021, 2021, 4020–4029 CrossRef CAS.
- Y. Agilent Technologies Ltd, Oxfordshire, England, 2014.
- J. B. Hu, A. Stein and P. Bühlmann, TrAC, Trends Anal. Chem., 2016, 76, 102–114 Search PubMed.
- D. Moreno da Costa, M. A. Henriquez, D. Gonzalez-Torres, C. Zuñiga-Loyola, I. Brito, I. González, A. Villegas-Menares, D. MacLeod-Carey, C. Morales-Verdejo and A. R. Cabrera, Inorg. Chim. Acta, 2023, 545, 121249 Search PubMed.
- A. Bard and L. Faulkner, Electrochemical Method. Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2 edn, 2001 Search PubMed.
- F. Asaro, A. Camus, R. Gobetto, A. C. Olivieri and G. Pellizer, Solid State Nucl. Magn. Reson., 1997, 8, 81–88 CrossRef CAS PubMed.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- C. Femoni, S. Muzzioli, A. Palazzi, S. Stagni, S. Zacchini, F. Monti, G. Accorsi, M. Bolognesi, N. Armaroli, M. Massi, G. Valenti and M. Marcaccio, Dalton Trans., 2013, 42, 997–1010 RSC.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- J. Egly, D. Bissessar, T. Achard, B. Heinrich, P. Steffanut, M. Mauro and S. Bellemin-Laponnaz, Inorg. Chim. Acta, 2021, 514, 119971 CrossRef CAS.
- H. Ohara, A. Kobayashi and M. Kato, Dalton Trans., 2014, 43, 17317–17323 RSC.
- T. Kusama and S. Hirata, Front. Chem., 2021, 9, 788577 CrossRef CAS PubMed.
- P. Y. Chen and T. J. Meyer, Chem. Rev., 1998, 98, 1439–1477 CrossRef CAS PubMed.
- A. Reichle, M. Koch, H. Sterzel, L. J. Grosskopf, J. Floss, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2023, 62, e202219086 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2405276–2405278. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03337j |
| This journal is © The Royal Society of Chemistry 2025 |