
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe direct photochemical cross-esterification of alcohols via site-selective C–H bromination
Atsuya
Miyamoto
a,
Hiroyoshi
Takamura
 a,
Isao
Kadota
*a and
Kenta
Tanaka
*b
a,
Isao
Kadota
*a and
Kenta
Tanaka
*b
aGraduate School of Environmental, Life, Natural Science and Technology, Okayama University, 3-1-1 Tsushima-Naka, Kitaku, Okayama 700-8530, Japan. E-mail: kadota-i@okayama-u.ac.jp
bResearch Institute for Interdisciplinary Science, Okayama University, 3-1-1 Tsushima-Naka, Kitaku, Okayama 700-8530, Japan. E-mail: ktanaka@okayama-u.ac.jp
First published on 4th October 2025
Abstract
We have developed a direct photochemical cross-esterification of alcohols that proceeds via the in situ generation of acyl bromides. The C–H bond of a benzyl alcohol is selectively activated by a bromo source under light irradiation, enabling the cross-esterification to afford a variety of functionalized esters.
Esters represent an important structural motif found in a wide variety of bioactive compounds and functional materials. While conventional esterification reactions typically use carboxylic acids or aldehydes as substrates,1 the direct oxidative esterification of alcohols has been developed as an alternative method. The esterification of alcohols typically employs transition-metal catalysts or stoichiometric oxidants (Scheme 1(a)).2 Meanwhile, photochemical transformations have gained significant attention in recent years due to their potential in sustainable energy-conversion systems, and several methods for the photochemical esterification of alcohols have been developed. In 2012, Itoh and co-workers have reported the cross-esterification of benzyl alcohol with aliphatic alcohols such as methanol as the solvent in the presence of CBr4 under an oxygen atmosphere and irradiation from a household lamp (Scheme 1(b)).3 More recently, in 2024, the one-pot, two-step esterification of benzyl alcohol with alcohols using trichloroisocyanuric acid (TCCA), a stoichiometric amount of triethylamine, and a catalytic amount of 4-dimethylaminopyridine (DMAP) under blue-light irradiation has been reported (Scheme 1(c)).4,5 While these reactions represent useful methods that use alcohols as substrates, there are still only a few examples of the photochemical esterification of alcohols. Therefore, the development of more efficient approaches to photochemical esterification reactions remains highly desirable.
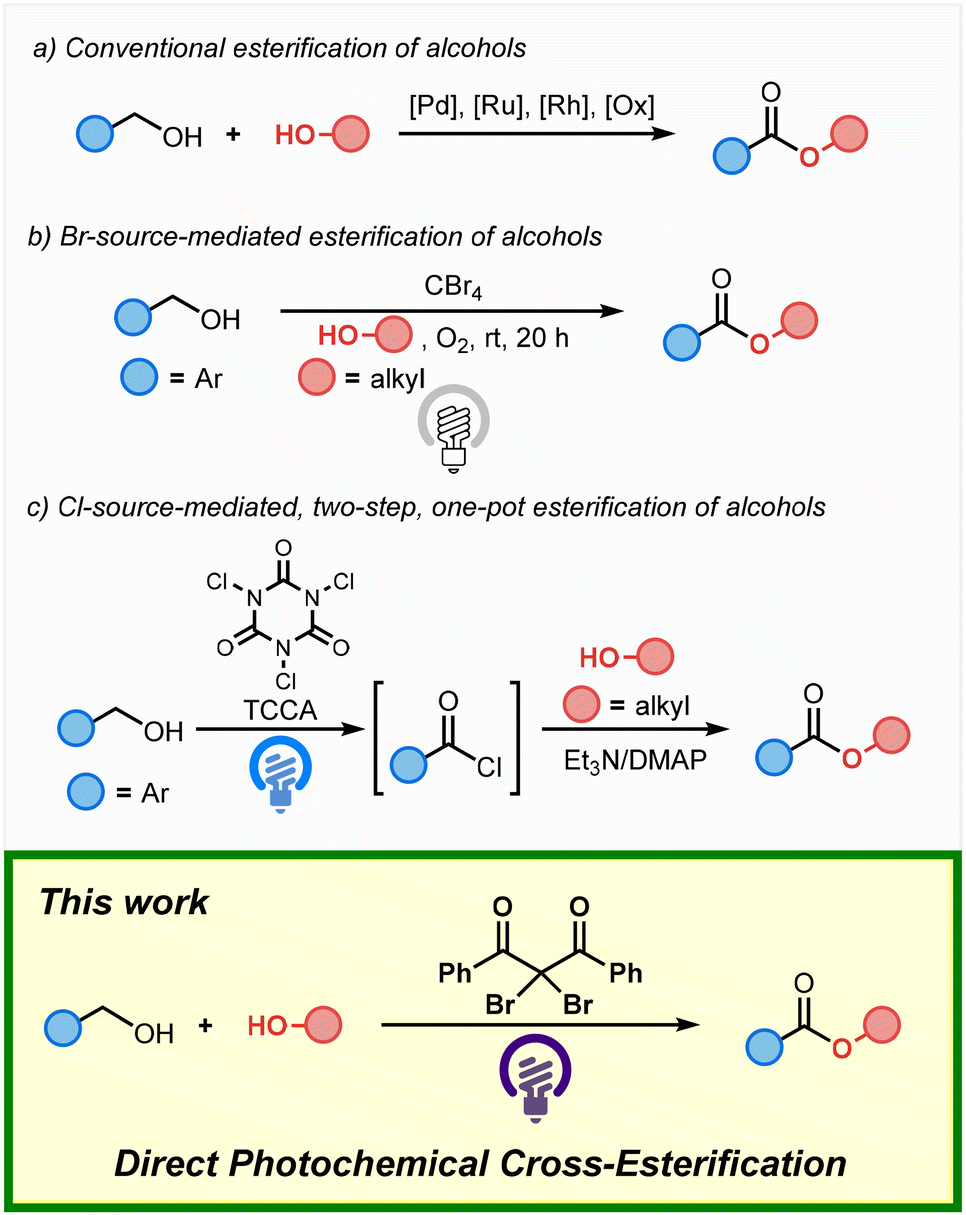 | ||
| Scheme 1 Examples of photochemical cross-esterification reactions of alcohols. | ||
Recently, we have developed the photochemical esterification of aldehydes with alcohols via C–H bromination.6 Given that bromo sources such as bromotrichloromethane (BrCCl3) effectively activate the C–H bonds of aldehydes, we hypothesized that they could also potentially activate the C–H bonds of benzyl alcohols, thus enabling the application of bromo sources in cross-esterification reactions. Here, we report the direct photochemical cross-esterification of alcohols via site-selective C–H bromination (Scheme 1; This work).
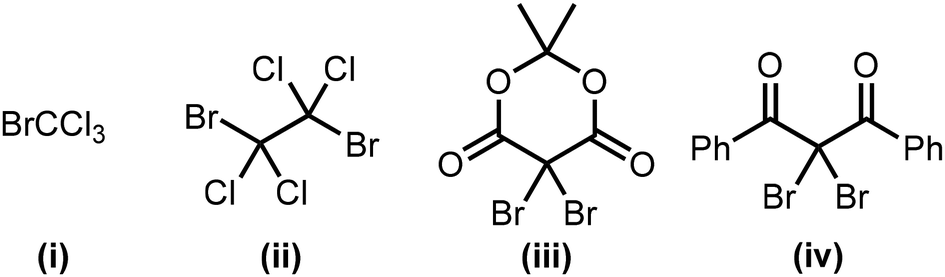
We initially investigated the optimization of the reaction conditions for the photochemical esterification of benzyl alcohol (1a) with 1-butanol (2a) in the presence of BrCCl3 (i) (Table 1). The reaction proceeded in common solvents such as CH3CN, hexane, and toluene, giving the desired product (3a) in low to moderate yield (entries 1–3). Furthermore, halogenated solvents such as 1,2-dichloroethane (DCE) and CH2Cl2 increased the product yield (entries 4 and 5). The esterification proceeded smoothly with various Br sources, including 1,2-dibromo-1,1,2,2-tetrachloroethane (ii), 2,2-dibromo-1,3-diphenyl-1,3-propanedione (iii), and 2,2-dibromo-1,3-diphenyl-1,3-propane-dione (iv) (entries 6–8). Although a lower-energy light source (λex = 405 nm) did not improve the yield of the product, a higher-energy light source (λex = 365 nm) significantly increased the product yield (95%, entries 9 and 10). The molar absorption coefficients of Br source (iv) were determined to be 2.655 × 102 M−1 cm−1 at 405 nm and 4.783 × 102 M−1 cm−1 at 365 nm shown in SI. These results suggest that irradiation at 365 nm excites the Br source more efficiently, promoting cleavage of the C–Br bond and thereby improving the product yield. Reducing the amount of 2a (2.0 equiv.) also afforded the product in high yield (entry 11). Reducing the amount of Br source (iv) (3.0 equiv.) afforded the product in a moderate yield (entry 12). When the reaction was carried out without MS3Å, the product yield decreased to 72% (entry 13). The control experiments, wherein the Br source (iv) or the light source were omitted, did not proceed, and it can therefore be concluded that these two factors effectively promote the reaction (entries 14 and 15). These results also show that the reaction affords the corresponding ester without the requirement for any additives such as bases, and bypasses the need for a sequential one-pot, two-step protocol.4
|
|
||||
|---|---|---|---|---|
| Entry | Br source | Light source (nm) | Solvent | Yield (%) |
| a All reactions were carried out with 1a (0.4 mmol), 2a (1.2 mmol), a Br source (6.0 equiv.) in the specified solvent (4.0 mL) at room temperature for 24 h under LED irradiation. b 0.8 mmol of nBuOH were used. c 3.0 equiv. of the Br source were used. d Without MS3Å. | ||||
| 1 | (i) | 380 | CH3CN | 23 |
| 2 | (i) | 380 | Hexane | 30 |
| 3 | (i) | 380 | Toluene | 52 |
| 4 | (i) | 380 | DCE | 60 |
| 5 | (i) | 380 | CH2Cl2 | 68 |
| 6 | (ii) | 380 | CH2Cl2 | 77 |
| 7 | (iii) | 380 | CH2Cl2 | 78 |
| 8 | (iv) | 380 | CH2Cl2 | 86 |
| 9 | (iv) | 405 | CH2Cl2 | 73 |
| 10 | (iv) | 365 | CH2Cl2 | 95 |
| 11b | (iv) | 365 | CH2Cl2 | 94 |
| 12bc | (iv) | 365 | CH2Cl2 | 68 |
| 13bd | (iv) | 365 | CH2Cl2 | 72 |
| 14b | (iv) | — | CH2Cl2 | 0 |
| 15b | — | 365 | CH2Cl2 | 0 |
|
||||

With the optimal conditions in hand, we next investigated the scope of the alcohols suitable for this reaction (Table 2). Benzyl alcohols with a variety of halogen substituents afforded the corresponding products in high yield (3b–3j). Under the applied reaction conditions, π-conjugated functionalities were also well tolerated (3k–3m). Aromatic alcohols substituted with either electron-donating or withdrawing groups also provided the corresponding products efficiently (3n–3p). Heteroaromatic substrate could be applied to the reaction (3q). The combination of aliphatic alcohols was not suitable for the reaction (3r). Notably, the method was applicable for the esterification of multiples sites in the same molecule, affording multi-substituted esters, which are found in various functional materials (3s–3u).7 As multi-substituted esters are typically prepared via the Fischer esterification,8 the present mild method starting from alcohols offers a valuable alternative for the efficient synthesis of these functional materials. Aliphatic alcohols yielded the desired esters in moderate to high yield, not only for primary alcohols but also in more sterically congested secondary alcohols and cyclic alcohols (3a and 3v–3ac). Owing to the greater steric hindrance of secondary alcohols compared with primary alcohols, extending the reaction time led to a significant increase in product yields (3z, 3aa, 3ac). In contrast, the combination of benzyl alcohols was not suitable for the reaction (3ad).
| a All reactions were carried out using 1 (0.4 mmol), 2 (0.8 mmol), a Br source (iv) (2.4 mmol), and MS3Å (100 mg) in CH2Cl2 (4.0 mL) at room temperature under an argon atmosphere and LED irradiation (λex = 365 nm, 18 W) for 24 h. b The reaction was carried out using 1 (0.4 mmol), 2 (1.6 mmol), a Br source (iv) (2.4 mmol), and MS3Å (100 mg) in CH2Cl2 (4.0 mL) at room temperature under an argon atmosphere and LED irradiation (λex = 365 nm, 18 W) for 72 h. c The reaction was carried out using 1 (0.4 mmol), 2 (0.8 mmol), a Br source (iv) (2.4 mmol), and MS3Å (100 mg) in CH2Cl2 (4.0 mL) at room temperature under an argon atmosphere and LED irradiation (λex = 365 nm, 18 W) for 48 h. |
|---|
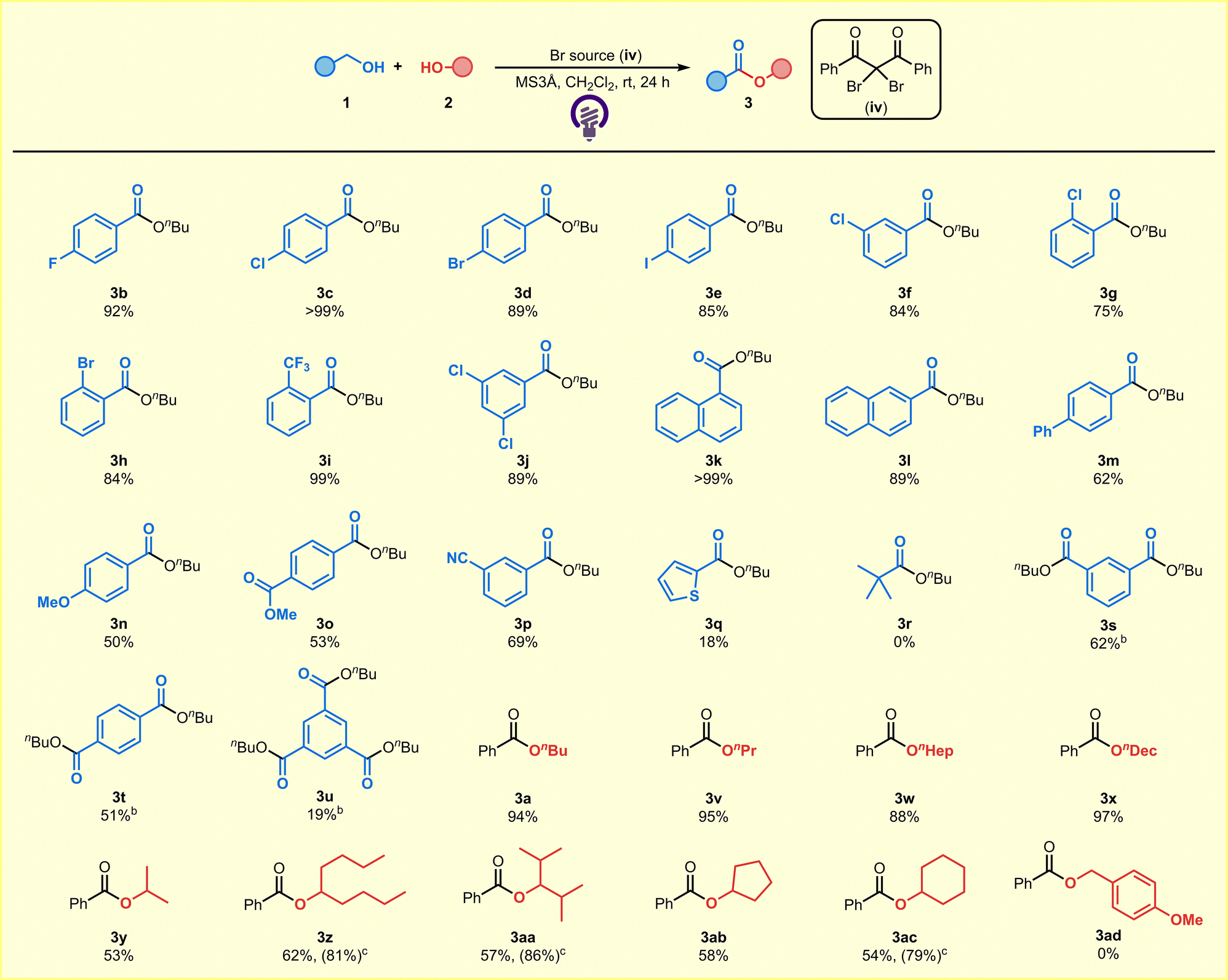
|
To examine the reaction mechanism, a radical scavenger was employed (Scheme 2(1)). When the reaction was carried out in the presence of TEMPO, product formation was not observed. In addition, an acyl radical was trapped by TEMPO and detected as 4a using mass spectrometry, suggesting that the reaction may proceed via a radical pathway. Additionally, when the reaction was conducted in the absence of 2a, the formation of acyl bromide 5 was confirmed by 1H and 13 C NMR spectroscopy (Scheme 2(2)). Therefore, the reaction should involve the generation of an acyl bromide intermediate via C–H bromination. When the reaction was carried out using the aldehyde instead of benzyl alcohol, the desired ester 3j was obtained in 82% yield, suggesting that the reaction would proceed through the aldehyde as an intermediate (Scheme 2(3)).
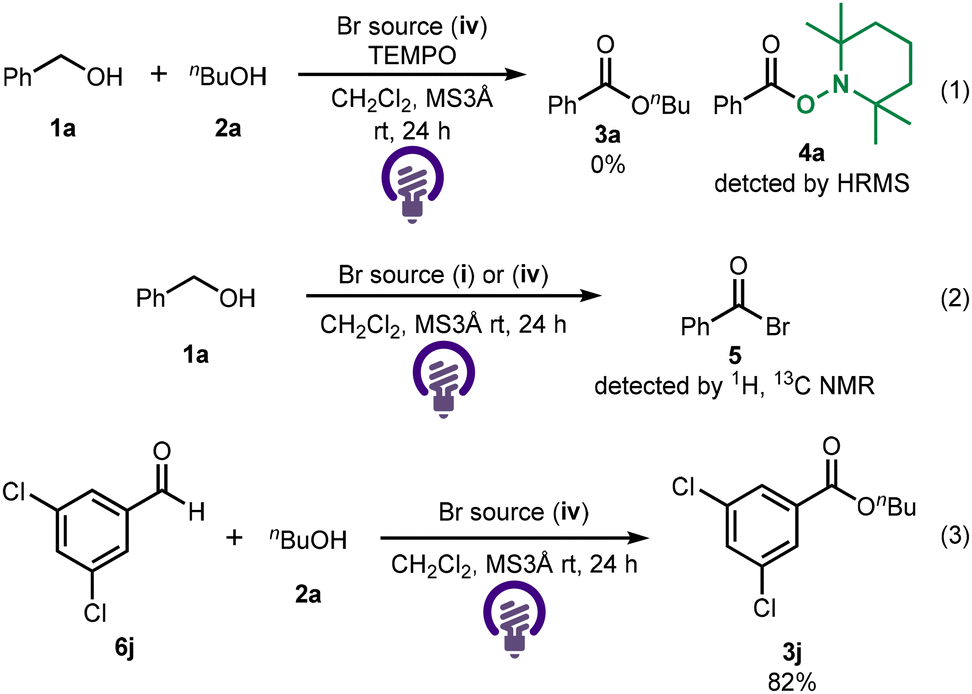 | ||
| Scheme 2 Mechanistic aspects of the photochemical cross-esterification of alcohols. | ||
A feasible reaction mechanism is proposed in Scheme 3. Homolytic cleavage of the Br source (iv) under light irradiation (λex = 365 nm) generates an organic radical (A) and a bromo radical (B).9 Radical A can induce the dissociation of the C–H bond of benzyl alcohol (1a) to form a benzyl radical (C), which then reacts with another equivalent of the Br source (iv) to produce an α-bromo benzyl alcohol (D).10,11 The elimination of HBr from D produces benzaldehyde (E). Radical A dissociates the C–H bond of E to form an acyl radical (F), which then reacts with the Br source (iv) to form acyl bromide 4.11,12 Finally, 1-butanol (2a) reacts with 4 to furnish the desired ester (3a).
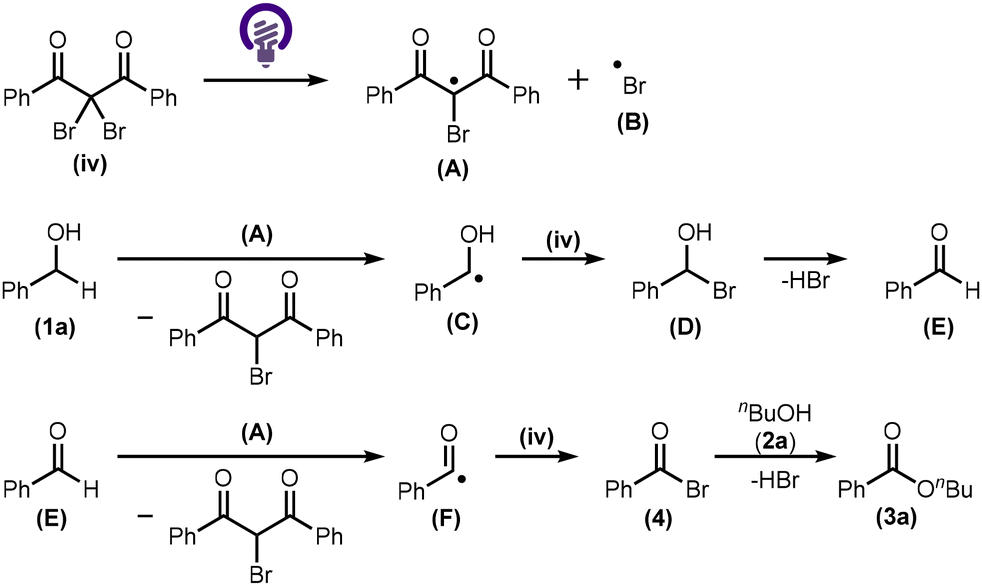 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In summary, we have developed a method for the direct photochemical esterification of alcohols. The reaction affords the corresponding esters without the requirement for any additives such as bases, and bypasses the need for a sequential one-pot, two-step protocol. Various benzyl alcohols bearing electron-donating or electron-withdrawing substituents, as well as π-conjugated functionalities, were well tolerated in the reaction. Notably, this method is applicable to the synthesis of multi-substituted esters, which are commonly found in functional materials and are typically prepared via the Fischer esterification, which often proceeds under much harsher reaction conditions. The present reaction enables the synthesis of a wide range of functionalized esters from alcohols, making it a valuable tool for producing bioactive compounds and functional materials.
This work was supported by Wesco Scientific Promotion Foundation and JSPS KAKENHI Grants JP25K18038. We appreciate the assistance of the Division of Instrumental Analysis at Okayama University with NMR spectroscopy and high-resolution mass spectrometry.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc03371c.Notes and references
- (a) J. Oreta and J. Nishikido, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2nd edn, 2010 Search PubMed; (b) S. Tang, J. Yuan, C. Liu and A. Lei, Dalton Trans., 2014, 43, 13460–13470 RSC; (c) S. Gaspa, A. Porcheddu and L. De Luca, Tetrahedron Lett., 2016, 57, 3433–3440 CrossRef CAS; (d) K. Ekoue-Kovi and C. Wolf, Chem. Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed; (e) L. Ding, P. Su, Y. Shi, H. Huang, L. Zhu, H. Zhang and B. Wang, Ind. Eng. Chem. Res., 2024, 63, 18667–18698 CrossRef CAS; (f) J. Li, D. Zhu, D. J. Young, Y. Wang and H. Li, Eur J Org Chem, 2025, 28, e202401398 CrossRef CAS.
- (a) C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 5144–5148 CrossRef CAS PubMed; (b) D. Srimani, E. Balaraman, B. Gnanaprakasam, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2012, 354, 2403–2406 CrossRef CAS; (c) J. Cheng, M. Zhu, C. Wang, J. Li, X. Jiang, Y. Wei, W. Tang, D. Xue and J. Xiao, Chem. Sci., 2016, 7, 4428–4434 Search PubMed; (d) Y. Yu, J. Lin, A. Qin, H. Wang, J. Wang, W. Wang, G. Wu, Q. Zhang, H. Qian and S. Ma, Org. Lett., 2024, 26, 3469–3474 CrossRef CAS PubMed; (e) R. Ray, R. D. Jana, M. Bhadra, D. Maiti and G. K. Lahiri, Chem. Eur. J., 2014, 20, 15618–15624 CrossRef CAS PubMed; (f) S. Samanta, V. Pappula, M. Dinda and S. Adimurthy, Org. Biomol. Chem., 2014, 12, 9453–9456 RSC; (g) S. Gaspa, A. Porcheddu and L. De Luca, Adv. Synth. Catal., 2016, 358, 154–158 CrossRef CAS.
- T. Nobuta, A. Fujiya, S. Hirashima, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2012, 53, 5306–5308 Search PubMed.
- A. Dellisanti, E. Chessa, A. Porcheddu, M. Carraro, L. Pisano, L. De Luca and S. Gaspa, Molecules, 2024, 29, 570 CrossRef CAS PubMed.
- H. Yi, X. Hu, C. Bian and A. Lei, ChemSusChem, 2017, 10, 79–82 CrossRef CAS PubMed.
- Our recent reports see; (a) H. Ando, S. Kodaki, H. Takamura, I. Kadota and K. Tanaka, Org. Biomol. Chem., 2024, 22, 9032–9035 RSC; (b) H. Ando, H. Takamura, I. Kadota and K. Tanaka, Chem. Commun., 2024, 60, 4765–4768 RSC; (c) S. Nohara, S. Iwai, N. Yamaguchi, Y. Asada, Y. Kamiyama, Y. Tanaka, K. Tanaka and Y. Hoshino, Synlett, 2023, 2525–2529 CAS; (d) K. Tanaka, M. Kishimoto, Y. Tanaka, Y. Kamiyama, Y. Asada, M. Sukekawa, N. Ohtsuka, T. Suzuki, N. Momiyama, K. Honda and Y. Hoshino, J. Org. Chem., 2022, 87, 3319–3328 CrossRef CAS PubMed; (e) M. R. El-kholany, T. Senoo, A. Mizutani, H. Takamura, T. Suzuki, I. Kadota and K. Tanaka, Org. Lett., 2025, 27, 4870–4874 CrossRef CAS PubMed.
- (a) M. Leloire, C. Walshe, P. Devaux, R. Giovine, S. Duval, T. Bousquet, S. Chibani, J. Paul, A. Moissette, H. Vezin, P. Nerisson, L. Cantrel, C. Volkringer and T. Loiseau, Chem. Eur. J., 2022, 28, e202104437 CrossRef CAS PubMed; (b) S. Zhao, J. Zhang, X. Wang, L. Wang, J. Wang and C. Pang, ACS Appl. Polym. Mater., 2023, 5, 2408–2416 CrossRef CAS; (c) O. Plietzsch, A. Schade, A. Hafner, J. Huuskonen, K. Rissanen, M. Nieger, T. Muller and S. Bräse, Eur J Org Chem, 2013, 2013, 283–299 CrossRef CAS.
- (a) S. Huo, X. Deng, N. Yang, M. Qin, X. Zhang, X. Yao, C. An, P. Zhou and X. Lu, Chem. Eng. J., 2024, 481, 148562 CrossRef CAS; (b) B. H. Wilson, H. S. Scott, O. T. Qazvini, S. G. Telfer, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2018, 54, 13391–13394 RSC; (c) D. De, V. Sharma, B. C. Sahu, R. K. Sahu and T. Maharana, Polyhedron, 2023, 230, 116240 CrossRef CAS; (d) G. R. M. O’Donohue, S. Chow, S. D. Houston, T. T. L. Nguyen, G. Paul Savage, H. E. Smyth and C. M. Williams, Chem. Eur. J., 2025, 31, e202404716 CrossRef PubMed.
- (a) P. Bosch, F. Del Monte, J. L. Mateo and R. S. Davidson, J. Photochem. Photobiol., A, 1993, 73, 197–204 CrossRef CAS; (b) I. Saito, T. Sakurai, T. Kurimoto and M. Takayama, Tetrahedron Lett., 1994, 35, 4797–4800 CrossRef CAS; (c) Y.-B. Wang, F. Chen, M. Li, Q. Bu, Z. Du, J. Liu, B. Dai and N. Liu, Green Chem., 2023, 25, 1191–1200 RSC; (d) X. Li, J. Fan, D. Cui, H. Yan, S. Shan, Y. Lu, X. Cheng and T. Loh, Eur. J. Org. Chem., 2022, e202200340 CrossRef CAS.
- C. S. Colley, D. C. Grills, N. A. Besley, S. Jockusch, P. Matousek, A. W. Parker, M. Towrie, N. J. Turro, P. M. W. Gill and M. W. George, J. Am. Chem. Soc., 2002, 124, 14952–14958 CrossRef CAS PubMed.
- (a) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed; (b) E. T. Denisov and V. E. Tumanov, Russ. Chem. Rev., 2005, 74, 825–858 CrossRef CAS.
- (a) N. Gong, Z. Zhao, D. James Young, X. Cao, Z. Ren and H. Li, Chem. Eur. J., 2025, 31, e202404225 CrossRef CAS PubMed; (b) Z. Zhao, W. Li, Q. Shan, D. J. Young, Z.-G. Ren and H.-X. Li, J. Org. Chem., 2025, 90, 1616–1627 CrossRef CAS PubMed; (c) S. Kang, M. T. La and H.-K. Kim, Tetrahedron Lett., 2018, 59, 3541–3546 CrossRef CAS; (d) Y.-D. Kwon, M. T. La and H.-K. Kim, New J. Chem., 2018, 42, 10833–10841 RSC.
| This journal is © The Royal Society of Chemistry 2025 |