
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical impacts of metal cocatalysts on oxidation kinetics and optimal reaction conditions of photocatalytic methane reforming†
Hiromasa
Sato
 a,
Hikaru
Saito
a,
Taisuke
Higashi
a and
Toshiki
Sugimoto
*ab
a,
Hikaru
Saito
a,
Taisuke
Higashi
a and
Toshiki
Sugimoto
*ab
aDepartment of Materials Molecular Science, Institute for Molecular Science, Okazaki, Aichi 444-8585, Japan. E-mail: toshiki-sugimoto@ims.ac.jp
bGraduate Institute for Advanced Studies (SOKENDAI), Okazaki, Aichi 444-8585, Japan
First published on 19th March 2025
Abstract
Metal cocatalysts in photocatalysis are typically regarded as promoting only the reduction reactions. Here, we demonstrate that photocatalytic oxidation kinetics and optimal pressure of methane vary significantly with the loading amount of metal cocatalysts. These variations are well described by kinetic analyses treating molecular-level congestion of oxidation intermediates.
Photocatalysis represents a promising approach for driving redox reactions using light, surpassing thermodynamic limitations.1–6 Most advanced photocatalyst systems rely on noble metal cocatalysts to achieve high photocatalytic activity.7,8 Indeed, numerous studies have demonstrated that the incorporation of metal cocatalysts significantly improves the activity of semiconductor-based photocatalysts.9,10 Conventionally, it has been widely accepted that the metal cocatalysts act mostly as sinks of photogenerated electrons (e−) and serve exclusively as reduction sites.7,8
Some previous studies offered new insights into the role of metal cocatalysts in photocatalysis. For instance, previous density functional theory calculations11 and infrared spectroscopy12 have suggested that holes (h+) generated through band-gap excitation of semiconductor photocatalysts can be trapped by metal cocatalysts. This hypothesis is based on the simple assumption that h+-accumulating metal cocatalysts act as recombination centers for photogenerated e− and h+, without investigating the actual photocatalytic performance. By contrast, our recent study combining operando spectroscopy with real-time mass spectrometry demonstrated that e− and h+ can be separately captured by different metal cocatalysts, and the metal cocatalysts play key roles not only in promoting reduction reactions but also in accelerating oxidation reactions, while minimizing charge recombination.13 Furthermore, metal cocatalysts not only enhance overall photocatalytic activity but also influence the oxidation selectivity.13 These recent findings highlight the need for a deeper understanding of the microscopic roles of metal cocatalysts, beyond the conventional view that they merely promote reduction reactions by accumulating only photogenerated e−.7,8
In this study, we provide further experimental evidence that metal cocatalysts directly impact the photocatalytic oxidation processes. The oxidation process is recognized as the rate-determining step of photocatalysis,14–16 and the oxidation kinetics should be affected by modulation of this step. To examine whether variations in metal cocatalysts alter the dynamics and kinetics of the rate-determining oxidation step, we investigated the effects of loading amounts of metal cocatalysts on photocatalytic methane conversion with water. Methane is the main component of natural gas and a ubiquitous natural resource. Due to its robust C–H bonds, achieving methane conversion under ambient temperatures and pressures poses a significant challenge in advancing sustainable technologies.17 Conventional thermocatalytic methane conversion requires harsh conditions (>1000 K and >20 atm);18 in contrast, photocatalysis offers a more sustainable approach by enabling methane activation and oxidation under ambient conditions (∼300 K and ∼1 atm).17 Herein, we focused on the Pt- or Pd-loaded gallium oxide (Ga2O3) particles as robust model systems for photocatalytic methane reforming.13,15,19 Through controlled experiments under varying methane pressures (PCH4), we observed that the amount of metal cocatalysts significantly influences the PCH4 dependence of the photocatalytic performance, resulting in pronounced modulation of the optimal PCH4 conditions below ambient pressures. This variation suggests that the loading of metal cocatalysts affects the congestion of intermediate species involved in methane oxidation processes. These results highlight the crucial role of metal cocatalysts as active sites also for the oxidation reaction.
β-Ga2O3 is a well-known d10 photocatalyst2 with stable activity and robustness.20 The four β-Ga2O3 samples containing either 0.01 wt% or 1 wt% of metal Pt or Pd cocatalysts (Pt(0.01 wt%)/Ga2O3, Pd(0.01 wt%)/Ga2O3, Pt(1 wt%)/Ga2O3, Pd(1 wt%)/Ga2O3) were prepared by the impregnation method (see Note S1-1, ESI,† for details). As described in detail in Note S1-2 (ESI†), the photocatalytic performance of these metal-loaded Ga2O3 samples was evaluated by irradiating a deep ultraviolet (UV) lamp (∼90 mW cm−2 at 260 ± 15 nm) at various PCH4 under a fixed water vapor pressure (PH2O) of 2 kPa. Notably, the photocatalytic activity under LED light irradiation at 390 nm was low, although loading the Pt and Pd cocatalysts enhanced the light absorption in this wavelength region (Fig. S1-5, ESI†).13 This indicates that e− and h+ species generated by the excitation of the metal cocatalysts21 have little contribution, whereas those generated by the band-gap excitation of Ga2O3 dominantly induce photocatalytic methane conversion.13 Under the irradiation of a deep UV lamp, the sample temperature was ∼318 K and a single layer of adsorbed water molecules covered the sample surfaces at PH2O = 2 kPa (∼20% relative humidity for 318 K samples)22–24 as described in Note S1-3 (ESI†).
Fig. 1 illustrates the PCH4 dependence of the formation rates of H2 (RH2) and CO2 (RCO2) for the four photocatalyst samples. The rate of H2 evolution was approximately four times higher than that of CO2 formation (RCO2 ≈ 0.25RH2), indicating nearly stoichiometric CO2 production in the steam methane reforming reaction (CH4 + 2H2O → CO2 + 4H2). In addition, C2H6 derived from the side reaction (2CH4 → C2H6 + H2)25 was also detected, as will be discussed later (Fig. 3).
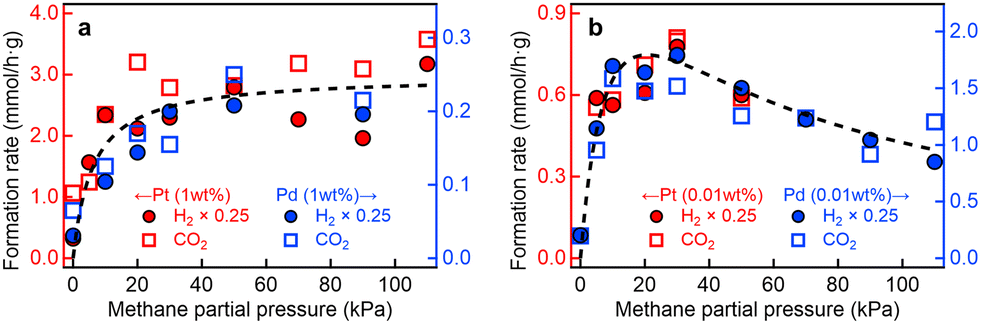 | ||
| Fig. 1 P CH4 profiles of photocatalytic steam methane reforming. Formation rates of H2 and CO2 for (a) Pt(1 wt%)/and Pd(1 wt%)/Ga2O3 photocatalysts, and (b) Pt(0.01 wt%)/and Pd(0.01 wt%)/Ga2O3 photocatalysts under UV irradiation as a function of PCH4 at PH2O = 2 kPa. Curve fitting results based on eqn (3) with U = 40 kJ mol−1 and eqn (4) with U = 37 kJ mol−1 are also shown in (a) and (b), respectively. | ||
As reported previously for the Pt(1 wt%)/Ga2O3 and Pd(1 wt%)/Ga2O3 photocatalysts,13,15RH2 and RCO2 showed a strong dependence on PCH4; the rates increased sharply with PCH4 below ∼30 kPa (0.3 atm) and nearly reached saturation value at ∼100 kPa (1 atm). In contrast, we found that the PCH4 profiles for the Pt(0.01 wt%)/Ga2O3 and Pd(0.01 wt%)/Ga2O3 photocatalysts differ markedly from those for the 1 wt% metal-loaded samples (Fig. 1b); while RH2 and RCO2 increased with PCH4 below ∼20 kPa, they began to decline at PCH4 above ∼20 kPa. Thus, for the 1 wt% metal-loaded samples, the photocatalytic performance for methane conversion is almost maximized at ambient pressure (∼1 atm), whereas the optimal PCH4 is around 20 kPa, well below 1 atm, for the 0.01 wt% metal-loaded samples. The observed variation in the PCH4 profile (Fig. 1) indicates that the microscopic redox reaction kinetics of photocatalytic steam methane reforming is affected by the loading amount of metal cocatalysts. Beyond the conventional assumption that the role of metal cocatalysts is limited solely to reduction reactions in the semiconductor bandgap photoexcitation scheme,7,8 our experimental results clearly show the impact of metal cocatalysts not only on reduction (H2 evolution) but also on oxidation (CO2 formation) processes in this widely used photoexcitation scheme.
To provide microscopic insights on the influence of metal cocatalyst loadings on the reaction kinetics, detailed analysis was carried out on the observed PCH4 dependence of the photocatalytic performance (Fig. 1). In general, the CH4 oxidation process toward CO2 can be divided into three steps (Fig. 2a and b):13,15 (i) dissociative adsorption and desorption of CH4 at vacant active sites, (ii) sequential reactions involving surface intermediate species,26 and (iii) desorption of the final products, primarily CO2 (see also Note S2, ESI†). The C–H cleavage of CH4 in the first step is initiated by hydrogen abstraction from photoactivated interfacial water species,15 resulting in the formation of activated methane species (X1 = ˙CH3) that subsequently chemisorb onto the catalyst surfaces (Fig. 2a and b). This process leads to the PCH4 dependence of the coverage of ˙CH3 (θCH3) similar to the Langmuir adsorption isotherm as follows:13,15
| θCH3 = KPCH4/(1 + KPCH4), | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(U/kBTs). Here, kB and Ts represent the Boltzmann constant and surface temperature, respectively, and U denotes the stabilization energy of the ˙CH3(ad) intermediate (see Note S2-1 for details of the kinetic analysis, ESI†).
exp(U/kBTs). Here, kB and Ts represent the Boltzmann constant and surface temperature, respectively, and U denotes the stabilization energy of the ˙CH3(ad) intermediate (see Note S2-1 for details of the kinetic analysis, ESI†).
 | ||
| Fig. 2 Reaction scheme and kinetic analysis of photocatalytic steam methane reforming. (a) Surface reaction pathway for the conversion of CH4 to CO2, where Xi (i = 1, 2,…,8) represents reaction intermediates; specifically, X1 and X8 denote methyl radical (˙CH3) and adsorbed CO2, respectively. For simplicity, the contribution of OH radical species (˙OH) generated via water oxidation (H2O + h+ → ˙OH + h+)15 is omitted in the diagram. (b) Possible reaction intermediate species from CH4 to CO2 in Fig. 2a through hydrogen abstraction or hydroxylation by photoactivated water species.26 (c) and (d) PCH4 dependence of RCO2 given by (c) eqn (3) and (d) eqn (4) at different values of U (35, 40, and 45 kJ mol−1). | ||
Notably, the product yields linearly increased with irradiation time,13,15 indicating that the reactions proceeded under steady-state conditions. In the case where the reaction of adsorbed ˙CH3 is rate-determining,13,15RCO2 under steady-state conditions is dominated by the reaction of ˙CH3 as follows:
 | (2) |
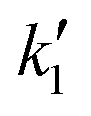 is the rate constant factor for the rate-determining reaction process, and θv ≡ 1 − ∑θXi (θXi is the coverage of the i-th intermediate species (i = 1, 2,…,8, see Fig. 2a and b) that block the reaction sites) represents the fraction of vacant surface sites available for the forward reaction of ˙CH3 species toward CO2 (see also Note S2 for details, ESI†). When the density of surface reaction sites is high enough that few active sites are occupied (θv ≈ 1) in steady reaction conditions, eqn (2) is approximated as
is the rate constant factor for the rate-determining reaction process, and θv ≡ 1 − ∑θXi (θXi is the coverage of the i-th intermediate species (i = 1, 2,…,8, see Fig. 2a and b) that block the reaction sites) represents the fraction of vacant surface sites available for the forward reaction of ˙CH3 species toward CO2 (see also Note S2 for details, ESI†). When the density of surface reaction sites is high enough that few active sites are occupied (θv ≈ 1) in steady reaction conditions, eqn (2) is approximated as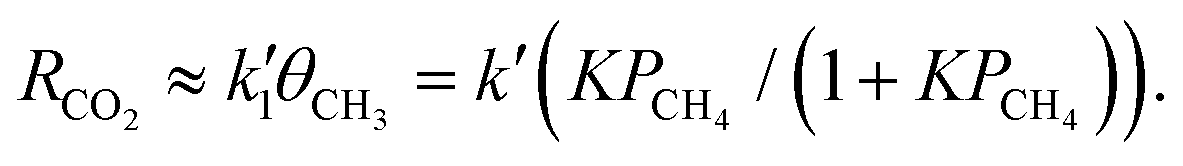 | (3) |
The PCH4 profiles of RCO2 at typical values of U are displayed in Fig. 2c. As U increases, the reaction activity tends to start increasing at the lower PCH4. Then, with increasing PCH4, reaction activity monotonically increases as an upward convex curve and then saturates in response to the abundance of the rate-determining intermediate species.
In contrast, when the density of active sites is not sufficiently high, the coverage of intermediate species competitively affects the fraction of vacant surface sites. Since the majority of the surface intermediates are ˙CH3 (∑θXi ≈ θCH3) in the case where the reaction of adsorbed ˙CH3 intermediates is rate-determining,13,15 the fraction of vacant sites is approximated as θv ≈ 1 – θCH3, yielding the following form of eqn (2):
 | (4) |
In this case, although the reaction activity starts to increase at the lower PCH4 region, it begins to decrease with increasing PCH4 after reaching a maximum value (Fig. 2d). This characteristic PCH4 profile arises from a balance between two competing effects (Fig. S2-3, ESI†): the positive impact of increasing the abundance of the rate-determining intermediate species and the negative impact of molecular-level congestion due to the reduced availability of vacant active sites for further reactions of the rate-determining intermediate species (X1 → X2 in Fig. 2a and b). It should also be noted that, even if the rate-determining step is not the forward reaction of ˙CH3 (X1) but rather another surface reaction involving different intermediate species (Xi → Xi+1 in Fig. 2a and b), the PCH4 dependencies similar to eqn (3) and (4) are derived as discussed in Note S3 (ESI†).
For the Pt(1 wt%)/Ga2O3 and Pd(1 wt%)/Ga2O3 photocatalysts, PCH4 profiles of RCO2 were well-fitted to eqn (3) with a stabilization energy U of 40 kJ mol−1 (Fig. 1a), as reported in our previous studies.13,15 On the other hand, the reaction activity for the Pt(0.01 wt%)/Ga2O3 and Pd(0.01 wt%)/Ga2O3 photocatalysts behaved differently, showing a better fit to eqn (4) compared to eqn (3) with a similar U value of 37 kJ mol−1 (Fig. 1b). These curve-fitting analyses indicate that a sufficient number of active sites are provided for the 1 wt% loading of metal cocatalysts, avoiding competitive occupation of active sites by intermediate species. In contrast, the 0.01 wt% loading provides a limited number of active sites for intermediate species, resulting in the competition of positive and negative effects of active site occupation by intermediate species. Therefore, the observed modulation of reaction kinetics upon loading amount of metal cocatalysts provides compelling evidence that the metal cocatalysts themselves function as reaction fields for the photocatalytic oxidation.13 These results further suggest that the photogenerated e− and h+ migrate separately from Ga2O3 to the metal cocatalyst particles,13 enabling certain metal cocatalysts to act as reduction sites by capturing electrons, while others function as oxidation sites by trapping holes, as discussed in detail in Note S4 (ESI†).
Additional important evidence supporting that metal cocatalysts are deeply involved in molecular-level dynamics and congestion of photocatalytic oxidation processes has been obtained. This is demonstrated by focusing on the simultaneously induced side-reaction of methane into ethane (2CH4 → C2H6 + H2). Fig. 3 illustrates the PCH4 dependence of ethane formation rates (RC2H6) for the four photocatalyst samples. In contrast to RCO2 (Fig. 1), the PCH4 profiles of RC2H6 not only vary depending on the loading amount of metal cocatalysts but also vary depending on the metal element of cocatalysts (Fig. 3). As detailed in our previous study,13 the PCH4 profiles for the Pt(1 wt%)/Ga2O3 and Pd(1 wt%)/Ga2O3 photocatalysts (Fig. 3a) were described by the second-order and first-order reactions of the surface adsorbed ˙CH3 intermediate species, respectively, as follows:
 | (5) |
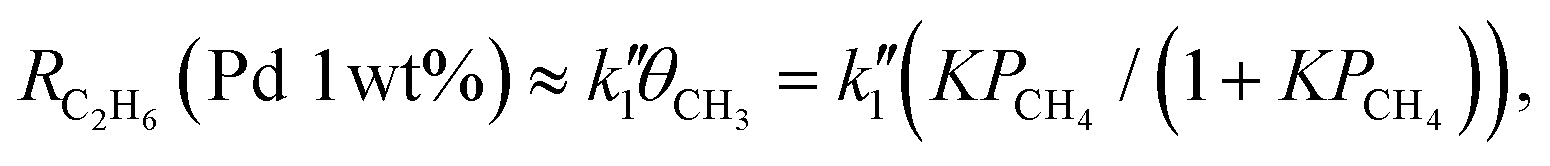 | (6) |
 is a rate constant. The apparent difference of the reaction order indicates that the coupling of ˙CH3 intermediates (2˙CH3 → C2H6) occurs on the catalyst surfaces (eqn (5) and Fig. S5-1a, S5-2, ESI†) for the Pt(1 wt%)/Ga2O3, while it proceeds in the gas phase for the Pd(1 wt%)/Ga2O3 (eqn (6) and Fig. S5-1b, S5-5, ESI†).13
is a rate constant. The apparent difference of the reaction order indicates that the coupling of ˙CH3 intermediates (2˙CH3 → C2H6) occurs on the catalyst surfaces (eqn (5) and Fig. S5-1a, S5-2, ESI†) for the Pt(1 wt%)/Ga2O3, while it proceeds in the gas phase for the Pd(1 wt%)/Ga2O3 (eqn (6) and Fig. S5-1b, S5-5, ESI†).13
 | ||
| Fig. 3 P CH4 profiles of photocatalytic ethane formation from methane. Formation rates of C2H6 for (a) Pt(1 wt%)/and Pd(1 wt%)/Ga2O3 photocatalysts, and (b) Pt(0.01 wt%)/and Pd(0.01 wt%)/Ga2O3 photocatalysts under UV irradiation at PH2O = 2 kPa. Curve fitting results based on eqn (5) and (6) with U = 36 kJ mol−1, eqn (7) with U = 39 kJ mol−1, and eqn (8) with U = 37 kJ mol−1 are also shown in (a) and (b). | ||
As the main reaction (CH4 + 2H2O → CO2 + 4H2), we found that the limited 0.01 wt% loading of metal cocatalysts also has a significant impact on the PCH4 profiles of RC2H6 (Fig. 3b). For the Pt(0.01 wt%)/Ga2O3 photocatalyst, RC2H6 steeply declined at PCH4 > 10 kPa after the initial increase at PCH4 < 10 kPa. A similar trend was observed for the Pd(0.01 wt%)/Ga2O3 photocatalyst, although the decline was more gradual compared to the Pt(0.01 wt%)/Ga2O3 photocatalyst. These non-monotonic PCH4 profiles are derived from the competition between the positive impact of increasing the abundance of the rate-determining ˙CH3 intermediate species and the negative impact of their molecular-level congestion due to the reduced availability of vacant active sites for further reactions. Then, these observed PCH4 profiles were well-fitted with the following equations explicitly incorporating the effect of competing occupation of limited active sites by intermediate species (θv ≈ 1 – θCH3).
 | (7) |
 | (8) |
The decline of RC2H6 is more significant for eqn (7) than eqn (8) (Fig. S5-7b, ESI†). As described in detail in Note S5 (ESI†), the apparent difference of the reaction order for θCH3θv indicates that the homocoupling of methyl radical intermediates (2˙CH3 → C2H6) occurs on the catalyst surfaces (eqn (7) and Fig. S5-1a, S5-3, ESI†) for the Pt(0.01 wt%)/Ga2O3 sample, while it proceeds in the gas phase for the Pd(0.01 wt%)/Ga2O3 sample (eqn (8) and Fig. S5-1b, S5-6, ESI†).13 Even though the homocoupling of ˙CH3 proceeds in the gas phase for the Pd(0.01 wt%)/Ga2O3 sample (Fig. S5-1b, ESI†), manifestation of the negative PCH4 impact in the apparent first order reaction suggests that the desorption of ˙CH3 into the gas phase occurs after it migrates from its initial adsorption site to a nearby vacant site that is more preferable for desorption (see Fig. S5-4, ESI† for more precise picture). Note that, in the situation where the competitive occupation of active sites by intermediate species is negligible (θv ≈ 1), eqn (7) and (8) correspond to eqn (5) and (6), respectively. Thus, our results clearly demonstrate that the competitive occupation of active surface sites by reaction intermediates and the resulting modulation of reaction kinetics emerges also in the side reaction pathway of methane partial oxidation (CH4 → ˙CH3 → C2H6). Owing to this effect, the optimal PCH4 condition for the photocatalytic ethane formation also significantly varied with the amount of metal loading (Fig. 3a and b). Therefore, the observed modulation of ethane formation kinetics also provides compelling evidence that the metal cocatalysts themselves functions as active sites for oxidative photoactivation. Furthermore, the homocoupling pathway for ethane formation is independent of the loading amount of metal cocatalysts (1 wt% or 0.01 wt%) but varies with the elements of metal cocatalysts used (Pt or Pd). This is also a manifestation of the phenomena that the metal cocatalysts fundamentally influence oxidation kinetics and dynamics.
Finally, we emphasize that our concept of metal cocatalysts also provides a rationale for previously reported, yet incomprehensible, results in photocatalytic oxidation. For instance, variations in the activity of photocatalytic oxidation have been reported for semiconductor photocatalysts loaded with metal cocatalysts of different elements and/or loading amount.27–29 Nevertheless, metal cocatalysts have traditionally been regarded solely as the trapping site of e− and the resultant promotors of reduction reactions.7,8 This interpretation arises from the seemingly plausible assumption that more efficient oxidation reactions are simply driven by an increased number of h+ that avoid annihilative charge recombination with e− due to the preferential trapping/consumption of e− facilitated by metal cocatalysts.11,12 Beyond this classical picture, our results provide a series of solid evidence that the metal cocatalyst actively takes part in photocatalytic oxidation and modulates oxidation kinetics/dynamics, shedding new light on the complex interplay between cocatalyst loading and interfacial reaction.
This work was supported by JSPS KAKENHI [JP22H00296, JP24H02205, JP22KJ1427, JP22KJ3098], JST CREST [JPMJCR22L2], JST ACT-X [JPMJAX24D7] and Demonstration Project of Innovative Catalyst Technology for Decarbonization through Regional Resource Recycling, the Ministry of the Environment, Government of Japan.
Data availability
The data and analytical details supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 Search PubMed
.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 Search PubMed
- P. Zhang and X. W. D. Lou, Adv. Mater., 2019, 31, 1900281 Search PubMed
- K. Villa and M. Pumera, Chem. Soc. Rev., 2019, 48, 4966 Search PubMed
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919 CAS
- M. Yamauchi, H. Saito, T. Sugimoto, S. Mori and S. Saito, Coord. Chem. Rev., 2022, 472, 214773 CAS
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CAS
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787 CAS
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490 Search PubMed
- T. Kotani, K. Ogawa, H. Suzuki, K. Kato, O. Tomita, A. Yamakata and R. Abe, EES Catal., 2023, 1, 255 CAS
- C. L. Muhich, Y. Zhou, A. M. Holder, A. W. Weimer and C. B. Musgrave, J. Phys. Chem. C, 2012, 116, 10138 CAS
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218 CAS
- H. Saito, H. Sato, T. Higashi and T. Sugimoto, Angew. Chem., Int. Ed., 2023, 62, e202306058 CAS
- X. Ma, Y. Shi, J. Liu, X. Li, X. Cui, S. Tan, J. Zhao and B. Wang, J. Am. Chem. Soc., 2022, 144, 13565 CAS
- H. Sato, A. Ishikawa, H. Saito, T. Higashi, K. Takeyasu and T. Sugimoto, Commun. Chem., 2023, 6, 8 Search PubMed
- H. Zou, Y. Qi, S. Du, Y. Bao, X. Xin, W. Fan, Y. Xiao, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2024, 146, 28182 CrossRef CAS PubMed
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296 Search PubMed
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965 CrossRef CAS
- H. Sato and T. Sugimoto, J. Am. Chem. Soc., 2024, 146, 24800 CrossRef CAS PubMed
- F. Amano, C. Akamoto, M. Ishimaru, S. Inagaki and H. Yoshida, Chem. Commun., 2020, 56, 6348 RSC
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Chem. Sci., 2021, 12, 5701 RSC
- K. Shirai, T. Sugimoto, K. Watanabe, M. Haruta, H. Kurata and Y. Matsumoto, Nano Lett., 2016, 16, 1323 CrossRef CAS PubMed
- Z. Lin, H. Saito, H. Sato and T. Sugimoto, J. Am. Chem. Soc., 2024, 146, 22276 CrossRef CAS PubMed
- F. Amano, A. Ishikawa, H. Sato, C. Akamoto, S. P. Singh, S. Yamazoe and T. Sugimoto, Catal. Today, 2024, 426, 114375 CrossRef CAS
- G. Wang, X. Mu, J. Li, Q. Zhan, Y. Qian, X. Mu and L. Li, Angew. Chem., Int. Ed., 2021, 60, 20760 CrossRef CAS PubMed
- V. S. Bagotzky, Y. B. Vassiliev and O. A. Khazova, J. Electroanal. Chem., 1977, 81, 229 CrossRef
- H. Yuzawa, S. Yoneyama, A. Yamamoto, M. Aoki, K. Otake, H. Itoh and H. Yoshida, Catal. Sci. Technol., 2013, 3, 1739 Search PubMed
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224 CrossRef CAS
- S. Mori and S. Saito, Green Chem., 2021, 12, 810 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06774f |
| This journal is © The Royal Society of Chemistry 2025 |