
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-induced chemistry for protein functionalisation
Cesare
Berton
 and
Jason P.
Holland
*
and
Jason P.
Holland
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: jason.holland@chem.uzh.ch; Web: https://www.hollandlab.org Tel: +41-44-63-53990
First published on 11th March 2025
Abstract
Derivatising biomolecules like monoclonal antibodies with drugs or imaging agents, whilst preserving their bioactivity, is a challenging task. Protein functionalisation ideally requires methods that operate under mild conditions, are rapid, efficient (high yielding), chemoselective or site-specific, and importantly, non-denaturing. A broad collection of thermally mediated reagents for direct labelling using protein-based reactivity, or bioorthogonal strategies, has been developed, but arguably the most exciting opportunities lie in the application of photochemistry to create new covalent bioconjugate bonds. With current chemical methods for auxochromic tuning of the spectral features of photoactive groups, and with cheap, high-powered light-emitting diodes with precise emission properties, it has never been easier to explore the use of light-induced chemistry for making protein-based bioactive molecules. In biomedicine, the nature of the covalent bond to the protein can have a dramatic impact on the physicochemical properties and performance of the protein-conjugate. Photochemical methods provide access to new types of covalent linkages on protein with the potential to fine-tune biological interactions, leading to improvements in target uptake, binding specificity, metabolic processing, and washout kinetics in vivo. This perspective/review highlights recent advances in the development of photoactive reagents for protein labelling. We also discuss the experimental conditions and critical requirements to implement light-induced synthesis of functionalised protein-conjugates in aqueous media effectively.
 Cesare Berton | Cesare Berton obtained his BSc and MSc degree in Chemistry at the University of Padova, working on the design, synthesis, and characterisation of supramolecular cages under the supervision of Prof. Cristiano Zonta. In 2023, he obtained his PhD at the Swiss Federal Institute of Technology in Lausanne (EPFL), Switzerland, where he focused his attention on dissecting the thermodynamics and kinetics of merocyanine photoacids under the guidance of Prof. Cristian Pezzato. From 2023 onwards, he has been part of the group of Prof. Jason Holland as a postdoc, working on the synthesis of novel radiopharmaceuticals by using supramolecular and photochemical methods. |
 Jason P. Holland | Jason P. Holland is from Yorkshire in the United Kingdom and received a master's degree in Chemistry from the University of York (MChem, 2004) followed by a doctorate from the University of Oxford (DPhil, 2008) with Prof. Jennifer Green and Prof. Jonathan R. Dilworth. He trained as post-doctoral scholar with Prof. Jason S. Lewis in radiochemistry and translational molecular imaging at Memorial Sloan-Kettering Cancer Center in New York (2008–2010). He was then awarded an ETH Fellowship and worked at ETH Zürich and the Paul Scherrer Institute (2010–2012). He was then appointed as Assistant in Chemistry in the Division of Nuclear Medicine and Molecular Imaging at Massachusetts General Hospital, Boston, with a joint faculty appointment as an Instructor in Radiology at Harvard Medical School (2012–2014). In 2015, he worked in the Department of Nuclear Medicine at the University Hospital Freiburg as a Visiting Scientist. In 2022 he was appointed as the Chair of Medicinal Radiochemistry in the Department of Chemistry at the University of Zurich. |
Introduction
The synthesis of functionalised proteins plays a central role in modern biochemical technologies and in new medicines.1–3 For example, antibody-drug conjugates (ADC), and monoclonal antibodies (or related fragments) labelled with fluorescent tags or radioactive payloads underpin state-of-the-art developments in diagnostic imaging and molecularly targeted (radio)therapies.4,5 Bioconjugation chemistry – the ability to form new covalent bonds to protein – lies at the core of these technologies.6–8 Traditional methods for labelling proteins include the use of reagents featuring activated esters like N-hydroxysuccinimide (NHS), or benzyl-iso(thio)cyanates (Bn-NCS) that react with the primary ε-NH2 side chain of lysine residues.9 Other common examples include chemoselective derivatisation of free sulfhydryl groups on cysteine residues by using reagents that present Michael acceptors like maleimides. Several site-specific enzyme–mediate processes, as well as bioorthogonal reaction pairs, have also been developed to increase control over the chemical composition of the functionalised protein.10–12 Others have also explored Ni metal ion13,14 mediated bioconjugation and protein tagging using Au-complexes.15 These thermally mediated bioconjugation strategies work well and have led to over 13 clinically validated ADC (and 3 more expected to be approved by FDA in 2025)16 agents as well as countless examples of fluorescent probes for biochemical analysis in microscopy and related methods, and an increasing number of radiolabelled antibodies for nuclear imaging with positron emission tomography (PET) and radioimmunotherapy (RIT).17When developing new bioconjugation methods to functionalise proteins, a mandatory requirement is to retain the biological integrity of the protein. In practice, this requirement is challenging to fulfil because biomolecules like monoclonal antibodies (mAbs) can be highly sensitive to changes in their physicochemical properties. Fast reaction rates are also ideal for bioconjugate chemistry to ensure efficient and high-yielding reactions when working with protein at low (nano-to-micromolar) concentrations. Many classic bioconjugation methods are relatively slow and incompatible with protein formulation buffers. Employing most classic bioconjugation methods requires pre-processing of the protein sample to alternate conditions that favour the functionalisation step, but this often destabilises the protein leading to denaturation or aggregation. Modern enzymatic methods can address these issues, although they require access to expensive reagents (tailored enzymes with modified active sites that accommodate non-native substrates).1 Bioorthogonal chemistries are also promising but typically rely on pre-functionalisation of the protein with one of the reaction partners through either protein engineering or enzyme reactions, which is then later labelled in a fast, bioorthogonal step.18 Since the nature of the bioconjugate bond can dramatically impact the performance of a protein conjugate – through modification of the target specificity and binding, as well as the overall pharmacokinetic profile in vivo – new methods for creating bioconjugate bonds are essential.19–21
Photochemistry offers several advantages as a means of tagging proteins with bioactive drug molecules or imaging probes.20,22–25 Benefits include: (i) the initiation of the photoreaction under ambient conditions through the application of light at wavelengths that do not inflict photodamage on the protein, (ii) rapid reaction rates using highly reactive intermediates generated from the initial photolysis step, (iii) tolerance of the bioconjugation reactions toward complex biological media, particularly components of the protein formulation buffer employed in the stabilisation of clinical-grade monoclonal antibodies, and (iv) the potential to automate the bioconjugation step using advanced flow-based or reactor based machines. Photochemistry also facilitates the synthesis of new types of reactive handles26,27 or bioconjugate bonds that have yet to be tested in vivo. Accessing different covalent bonds to protein can help fine-tune the performance of protein conjugates with the goals of speeding up the synthesis, improving target uptake, and harnessing new or alternative modes of metabolism to control the pharmacokinetic profile of the agent. Improved chemical control over the adsorption, distribution, metabolism, and excretion (ADME) profile of a protein conjugate can reduce off-target binding alleviating chemotoxicity or radiation dosimetry issues, and thus, improving the therapeutic index of the next generation of drugs. Although highly promising, implementing photochemical methods to derivatise proteins remains challenging because new reagents must be synthesised, and the experimental design is more complex than established conjugation protocols.28
The original use of photochemistry for tagging proteins has a rich history that dates back to the concept of photoaffinity labelling (PAL) introduced by Westheimer and co-workers in 1962 (Fig. 1).29 Subsequent work spanning a timeline up until the 21st Century focused on developing new photoactive handles that can be easily introduced to drug molecules for screening drug-target interactomes.29–41 The basic idea in PAL is that a pre-equilibrium between a biologically active ligand (e.g. drug molecule) and the target protein is established. Subsequently, irradiation with light of a specific wavelength activates the photoreagent, stimulating the formation of highly reactive intermediates such as carbenes, nitrenes, diradicals, and powerful electrophiles. These intermediates react rapidly with protein-based nucleophiles near the binding site of the PAL reagent. In the process, the covalent bioconjugate bond is formed on a timescale ranging from picoseconds to microseconds (Scheme 1A). This mechanism is extremely useful in characterising the binding profile, the protein selectivity, and the mechanism of action for new drug entities.42 However, for the light-induced functionalisation of proteins like immunoglobulins (IgGs) and related fragments, which do not have recognised drug binding sites, a more general bimolecular approach is needed (Scheme 1B).20,22
 | ||
| Fig. 1 Timeline of the progression of photoreagents and their applications in biochemistry or medicinal chemistry. | ||
 | ||
| Scheme 1 Mechanistic differences in the use of light-induced reactions to create covalent bioconjugate bonds on protein via the established concept of (A) photoaffinity labelling with preassociation between the photoreagent and a drug binding site on protein, and (B) direct, bimolecular labelling without preassociation. | ||
Recent reports on the use of light-induced labelling of bioactive proteins have exploited the photochemistry of many different functional groups such as aryl azides, tetrazoles, azirines, diazirines, benzophenones, and photoredox systems (Fig. 1, blue box). The chemistry of these (and other) photoreagents is described in the following sections. The photo-induced processes are stratified into three distinct mechanistic classes. In addition, we begin by presenting a set of idealised requirements for developing new photoreagents in the context of labelling bioactive proteins, as well as a discussion of the experimental aspects that must be considered when using photochemistry in synthesis.
Requirements for light-induced bioconjugation processes
In an ideal photo-induced bioconjugation reaction, several different criteria apply (Fig. 2). First, the reagents should be stable toward thermal degradation, reacting only when exposed to photons of a specific wavelength. Second, to avoid photodegradation of the protein component, photoactivation of the reagents should occur at wavelengths where the protein of interest does not absorb (photo-orthogonality). Contrary to some assertions which suggest that lower energy photons are optimal, we believe that to prolong the shelf-life of the photoreagents, the chromophores should not absorb in the visible region of the electromagnetic spectrum. Activation in the UVA region with photon wavelengths from ∼315–400 nm is an ideal compromise. Light sources that emit in this wavelength range are readily available. This spectral window also has the advantage that the absorption spectrum of many functional groups can be easily tuned through standard application of the Woodward–Fieser–Kuhn rules43,44 for auxochromic shifting of the absorption bands of (poly)aromatic systems using common substituents.20 Third, the photoconjugation process should operate in aqueous conditions and be highly tolerant to oxygen, salts, and standard components found in clinical formulation buffers that are essential to maintain protein stability. For example, high concentrations of amino acids like histidine, polysorbate, sugars, and antioxidants like sodium ascorbate are frequently used in drug formulations.45,46 Fourth, the photoreagents should display high quantum yields for activation, and the bimolecular reaction kinetics with protein should be as rapid as possible to reduce the time required to produce the new bioconjugate bond. This often means optimising reaction conditions to maximise protein labelling whilst simultaneously reducing background quenching reactions including solvent mediated hydrolysis with competing water or hydroxide anions. Fifth, the bioconjugation reactions should be chemoselective and ideally site-specific.47 Sixth, the new bioconjugate bonds should be stable under standard biochemical challenges (such as incubation in human serum at 37 °C) or else offer new options for introducing tissue-specific (typically enzyme-mediated) catabolism to facilitate drug activation and release from ADCs or metabolite excretion for radiotracers.48 Finally, the methods should be amenable to automation, thereby facilitating the reproducible synthesis of protein conjugates under current Good Manufacturing Process (cGMP) conditions with high-throughput. Capturing all these features in one reagent is challenging but we find these design principles a useful starting point when screening different photoreagents for potential applications in antibody-based radiotracer development or ADC technologies.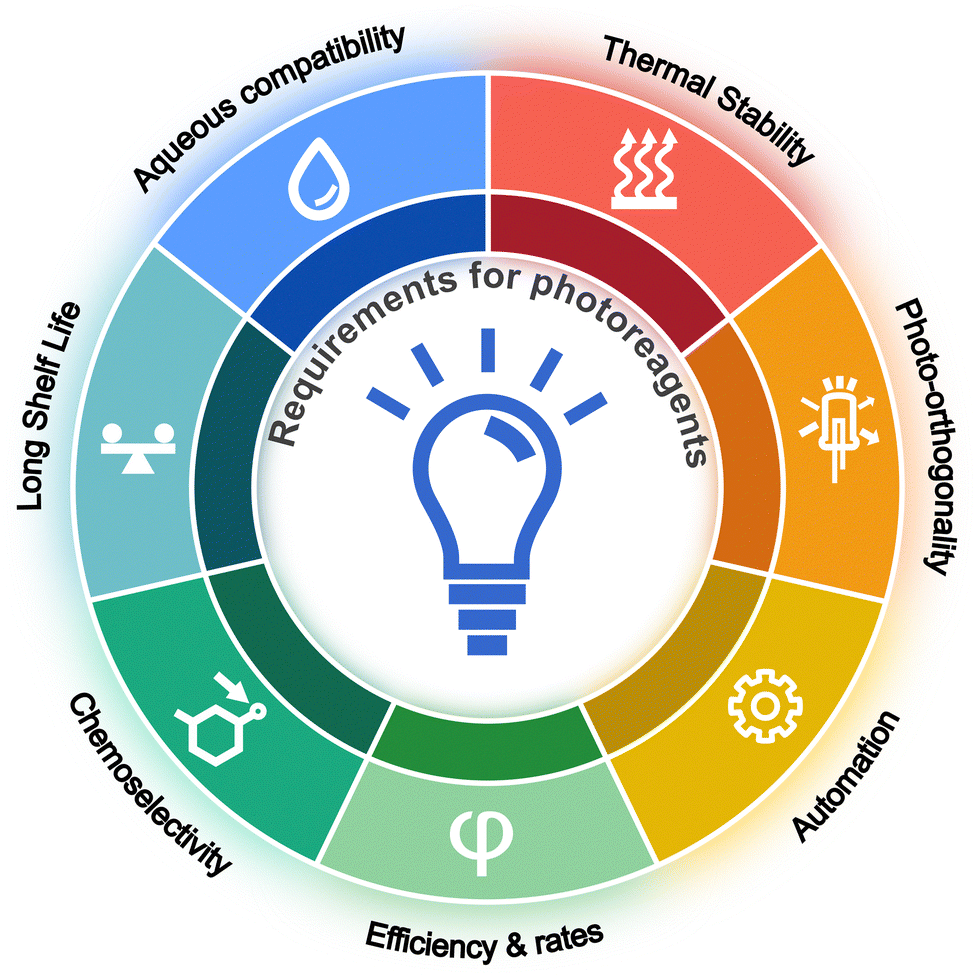 | ||
| Fig. 2 Key requirements of new photoactivable reagents for applications in protein labelling. | ||
Practical considerations for light-induced protein conjugation: the devil is in the detail
All synthetic chemists are skilled in the use of thermally mediated reactions; however, photochemical reactions are not frequently considered as a first option in most synthetic procedures. Successful implementation of photochemical reactions in a robust and reproducible fashion involves additional experimental parameters that are not encountered when reactions are simple stirred and heated. A selection of common pitfalls that occur in synthetic photochemistry, and related practical solutions to overcome problems, is presented in Fig. 3. | ||
| Fig. 3 Illustration of the various experimental design features of light-induced protein conjugation reactions that must be considered during reaction optimisation. (A) Common pitfalls encountered in the setup of photochemical experiments for biomolecular conjugations. Typical errors derive from inconsistent positioning of the light source, variations in reagent concentrations and total reaction volume, inhomogeneity of the sample, temperature fluctuations through heat generation or inefficient heat dissipation, and issues related to the transmission of light through materials (for example, absorption, reflection, or scattering of photons by the reaction vessel which affect photon flux). (B) Potential solutions in experimental design that improve reproducibility and overall efficiency of the photochemical process. Standardised experimental geometries are a critical requirement. In addition, the reaction composition including total volume, light intensities and shape of the light beam delivered into the reaction mixture, reagent homogeneity, temperature, and vessel materials should be tested and standardised to confer experimental reproducibility. | ||
Most pitfalls in photochemical reactions derive from inconsistencies in the experimental setup. Sources of error can be broadly classified into two groups that depend predominantly on either physical effects (e.g. geometry of the apparatus), or the chemistry of the process. The use of a light source is the first major difference between photochemical processes and thermally mediated reactions. Parameters associated with the light source include the wavelength (λ/nm), spectral bandwidth (measured by, for example, the full-width at half-maximum, FWHM/nm), and power of the emission.49 The beam shape, angle, distance and focal point with respect to the reaction mixture also impact the experimental geometry. Other factors include the use of either direct irradiation of the mixture or indirect irradiation through a reaction vessel (e.g. through glass or plastic). Indirect irradiation requires consideration of how reflectance, attenuation, scattering, and fluorescence effects from the reaction vessel impact the photon flux. Single or multi-pass geometries as well as light sources that are used either externally or internally with respect to the reaction mixture also affect the experimental design of photoreactions. On the chemical side, more common issues related to inaccuracies in reagent concentrations, the total volume of the reaction mixture, reaction homogeneity, and control of temperature can influence the success of a photoreaction. Photoreagents may also exhibit wavelength-dependent reactivity and selectivity.49 All these aspects are potential sources of uncertainty, but importantly, each can be controlled and optimised with simple precautions that are easy to implement.
The first practical aspects one must consider are the energetics and the photon wavelengths required for initiating the chemical reaction of interest. Reagents that require impractical (expensive, highly specialised, or custom-made) light sources will struggle to find widespread acceptance compared with other methods that can be initiated by using cheap, commercially available tools. For instance, high-powered light-emitting diodes (LEDs) with narrow bandwidths and peak emission at wavelengths spanning between the UVA (365 nm) and near-infrared (NIR) spectral regions (∼1350 nm) are readily available. These devices have long lifetimes and can produce light with well-defined characteristics. Advantages of using LEDs over common Hg-lamps or lasers include their: (i) simple and low-cost operation, (ii) wide availability around the world, (iii) generation of almost monochromatic (as well as polychromatic) light with high intensity and efficiency, and (iv) modularity and versatility with small apparatus footprints allowing them to fit inside chemical fume-hoods or glove-boxes. A collimator lens can be used for optimal beam focusing to ensure that the LED light source is directed into the reaction mixture with the most effective geometry (Fig. 4A).
 | ||
| Fig. 4 (A) An illustration of LED beam configurations that can be manipulated with a lens to optimise the irradiation of a reaction mixture. (B) Examples of four chemical actinometers that span a wavelength range that is applicable for light-induced functionalisation of protein. The plot shows the spectral range for which accurate quantum yields have been reported including data on temperature and solvent dependencies for the four structures shown.50,51 The image of the light beams in panel A is reproduced with permission from Opsytech Dr Gröbel, Ettlingen, Germany (https://www.opsytec.com/). | ||
Photochemical kinetics of reactions are directly influenced by the light intensity delivered to the reactants. The main contribution to the photon flux is the power output of the light source. Power output should be measured experimentally by electronic spectral radiometers or by chemical actinometry (Fig. 4B). Commercially available devices such as photodiodes, thermal power sensors, and pyroelectric energy sensors give a direct readout of the output power and represent an essential tool for a photochemistry laboratory. Practitioners should note, however, that spectral radiometers only give a threshold value to the experimental light intensity but do not account for the full impact of experimental geometry.
Chemical actinometry can provide a more accurate determination of the light intensity that inherently includes contributions from the experimental setup. However, actinometry can be subject to large experimental errors. Specifically, in chemical actinometry several assumptions including a fixed molar absorption coefficient and quantum yield of the actinometry reagent are required, but these may vary given changes in LED emission profiles, solvent composition, reagent concentration, and temperature. Bolton and co-workers reported extensive data on a collection of chemical actinometers, which are considered to be gold standards for calibration of photon fluxes.51 Examples of chemical actinometers include ferrioxalate, uranyl oxalate, uridine, and the iodide–iodate couple (amongst others) for which detailed temperature, wavelength, and solvent dependency of quantum yields and reaction mechanisms have been reported (Fig. 4B). Another widely available chemical actinometer is o-nitrobenzaldehyde (ONB) which is useful for spectral regions between 280–440 nm.50 The ONB compound undergoes a photoinduced rearrangement to form the corresponding o-nitrosobenzoic acid with well-known quantum yields. The benzoic acid has a characteristic electronic absorption spectrum which is then used to calibrate the photon flux arriving from the light source.50,52
The medium through which the light propagates from the source to the reaction mixture is also important. Direct irradiation of the sample is the simplest design which avoids a host of complicating factors but, in some cases, closed reaction vessels or indirect irradiation through the walls of the reaction vessel are essential. The optical properties of the material used (typically borosilicate glass, quartz glass, or plastics) for the reaction vessel, as well as the vessel shape and other factors like impurities, scratches, or blemishes, influence light transmission and experimental reproducibility. For example, most chemical grade glassware is composed by borosilicate glass which filters wavelengths below 365 nm. Light sources that emit, and photoreactions that proceed, at wavelengths close to the transmission cut-off of the materials used will suffer from low reproducibility if the light is delivered indirectly through the walls of the reaction vessel. An appropriate choice of the light-guiding material and its geometry will ensure a stable and reliable photon flux during the reaction. Typically, quartz represents a good starting point as it is chemically inert and transparent to most wavelengths in the region between 300–1000 nm.
Another aspect to consider is the concentration of the reacting partners in the mixture. Optical density or turbidity can negatively impact light propagation within the sample and must be monitored. Low concentrations of the photoreagent or protein lead to slow bimolecular conjugation kinetics and low yields of the final functionalised protein. In our experience with photo-induced labelling of humanised IgG1 monoclonal antibodies (molecular weight ∼150 kDa) or related fragments, we found that bioconjugation yields decrease rapidly if initial protein concentrations in the reaction drop below ∼10 mg mL−1 (equivalent to a concentration of ∼67 μM [1.5 mg of protein] in a total reaction volume of 0.15 mL).21,53–57 Similarly, the stoichiometric equivalents of the photoreagent should be adjusted to ensure high conjugation yields but to avoid over functionalisation which can impinge on the biophysical properties of the protein. Typically, in our photochemical labelling reactions we adjust the initial of photoreagent-to-protein ratio by a factor associated with the conjugation yield (measured in test reactions) to ensure that on average the final protein conjugate contains a drug-to-antibody (DAR; or equivalently fluorophore or radiometal complex) ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.28
:1.28
Sample homogeneity and thermal stabilisation can be achieved through gentle mixing by using a magnetic stir bar, but slow stirring rates (<100 rpm) and caution should be used to avoid protein foaming, flocculation, and sedimentation. Normally, the temperature of photoreactions should remain stable during the short photolysis step, but it can change if either: (i) high concentrations of chromophores are added that undergo thermal relaxation mechanisms that compete with the desired photochemical processes, (ii) reactions involve strongly exothermic steps, or (iii) the LED source generates heat. Heat sinks, heating mantles or jacketed reaction vessels can be used in situations where tight control over temperature is essential.
Mechanistic classifications of light-induced protein conjugation reactions
In this section, we subdivide the reactivity of photoreagents into three different classes based on their mechanisms of action involving: (i) photodeprotection of masked reagents, (ii) photoactivation to generate highly reactive intermediates, and (iii) photocatalysis (Scheme 2). | ||
| Scheme 2 Bimolecular photoconjugation strategies categorised into three distinct mechanisms depending on the role of the photoactivatable reagent. Mechanism (i) involves the activation of masked functional groups by release of a photolabile protecting group (PG) to give an intermediate that subsequently reacts with protein. Mechanism (ii) involves light-induced activation of a stable ground-state reagent to form an unstable intermediate which is the main protagonist in the bioconjugation reaction. Mechanism (iii) involves photocatalytic reactions in which a photosensitive precatalyst activates by absorption of a suitable photon and promotes reactivity of a substrate in the bioconjugation step. | ||
Photodeprotection reactions are a mainstay of photochemistry and the topic has been reviewed extensively.58,59 Despite appearing to be similar, the photodeprotection and photoactivation mechanisms (Scheme 1, mechanisms i and ii, respectively) are conceptually and empirically distinct. Specifically, photodeprotection reactions generate a stable intermediate which is only reacts in the presence of the corresponding reaction partner. An example is the release of a bioorthogonal reactive strained alkyne upon light-mediated CO release from dibenzocyclopropenones (see below).60,61 These types of reaction are attractive when spatial and temporal control is required over the location of light-induced protein tagging, particularly in labelling components of live cells.62–67
In contrast, photoactivation (Scheme 1, mechanism ii) produces very unstable, highly reactive, and transient intermediates that react on the picosecond-to-microsecond times scale. Examples of photoactivatable reagents include tetrazoles, azirines, sydnones,68 benzophenones, aryl and aliphatic diazirenes, diazo-species, and aryl azides, which undergo photochemical reactions via an electronically excited state. These photochemical reactions transit through highly reactive species including carbenes, nitrenes, biradical species, electrophiles like ketenimines and dipolar species such as nitrile imines and nitrile ylides. In spite of the extreme reactivity of these photoreagents, many compounds have been used in PAL.33,69–84 In the third mechanistic group, (Scheme 1, mechanism iii), the photocatalytic process depends on an initial activation of an additive (a precatalyst) which upon photon absorption, mediates a bioconjugation reaction between a substrate and a protein of interest. The following sections highlight examples of photoreagents which have been employed in the synthesis of functionalised protein conjugates.
Photoreagents and their applications
The following sections summarise the chemistry of eleven of the most prominent photoreagents for direct (bimolecular) functionalisation of proteins. For recent complementary reviews on photochemical reactions and their applications to create bioactive molecules, especially with photo-click reactions, the reader is referred to ref. 58, 85 and 86.Aryl azides
Aryl azides (ArN3) are one of the most well-studied compound classes for photoconjugation chemistry.87 Their development as photoreagents date back to 1969 when Fleet et al.30 used 4-fluoro-3-nitrophenylazide for labelling antibodies, and later this class of compounds was used in PAL.69,70,78–81,88–90 Recently, our group adapted ArN3 chemistry for the photoradiosynthesis of antibody-based radiotracers using a wide range of radioactive nuclides including (amongst others) 89Zr, 68Ga, and 64Cu for positron emission tomography (PET; Scheme 3).21,53,54,56,91 In our hands, the chelating reagents derivatised with the ArN3 functional group display thermal stability up to ∼50 °C, are stable over prolonged periods of time (>2 years) in the freezer at −20 °C. Aryl azides have the advantage that they can be readily installed onto complex drug molecules, fluorophores, or radiometal complexes. Their high chemical stability in buffered aqueous media, and biocompatibility with formulated antibody mixtures, combined with convenient and rapid photochemical activation with photons in the wavelength range 365–450 nm make them close to ideal photoreagents for protein labelling. Upon light-absorption, the ArN3 group is promoted to an electronically excited (singlet) state in which the rapid and almost barrierless extrusion of N2 is favoured, generating highly reactive open-shell singlet nitrenes.87,92 Experimental measurements indicated that the quantum yield of ArN3 activation in water is ∼4%.21 Depending on the electronic nature of the substituents on the aryl ring, the aryl nitrene may react either as a nitrene species or undergo rapid intramolecular bicyclisation then ring opening to form a potent 7-membered ketenimine heterocycle, which acts as a chemoselective electrophile for primary amines.92 Chemoselective coupling of the ketenimine with a lysine side-chain on protein produces a 2-aminoazepine which acts as a stable bioconjugate bond in vivo.62,91 | ||
| Scheme 3 Photoradiosynthesis to label a monoclonal antibody with a positron-emitting 89Zr-desferrioxamine complex via light-induced activation of a ArN3 group. Trastuzumab is linked in a one-pot photoreaction to [89Zr]ZrDFO-ArN3 with high isolated and decay-corrected radiochemical yields and radiochemical purities.28,53,54 | ||
In one example, we used [89Zr]ZrDFO-ArN3 (DFO, desferrioxamine) to photoradiolabel a monovalent engineered antibody onartuzumab with high radiochemical conversions (∼57%) and isolated decay-corrected radiochemical yields (∼41%) of 89Zr-onartuzumab.91 The photoradiolabelled 89Zr-onartuzumab was then tested in cells and in mice bearing human gastric cancer models comprised of subcutaneous human hepatocyte growth-factor receptor (c-MET) overexpressing MKN-45 xenografts. PET imaging, biodistribution, and pharmacokinetic studies revealed superior performance of the photoradiolabelled compound when compared head-to-head against a control 89Zr-onartuzumab radiotracer formed by using the standard DFO-Bn-NCS93 bioconjugation approach that forms a thiourea.91 Notably, the ArN3 photochemistry was found to be compatible with several different clinical formulation buffers. This advance also led to the design of a new radiosynthesiser for automated light-induced synthesis of immunoconjugates (ALISI).54
Tetrazoles
The photochemistry of tetrazoles was originally discovery by Huisgen and co-workers in 1967 where evidence of photoinduced cycloadditions between the heterocycle and ethyl crotonate were reported.94 At that time, and in subsequent studies,31,95,96 it was noted that cross-reactivity between photoactivated tetrazoles with biologically relevant nucleophiles occurs but this fact was subsequently overlooked when tetrazoles and alkene-substituted proteins were combined to develop ‘photo-click’ chemistry in 2008.97,98 Photo-click reactions with tetrazoles were proclaimed to be bioorthogonal but the original report,94 and detailed mechanistic studies in 201699,100 refuted this claim. Indeed, studies have demonstrated photoinduced reactivity of tetrazoles with a wide range of reagents99 with reports of high selectivity towards amino101 carboxyl102,103 thiol104 alkene, alkyne105 and other functionalities,106 depending on the conditions employed. Our own photoradiosynthesis experiments with a novel [89Zr]ZrDFO-tetrazole20 complex for light-initiated labelling of trastuzumab also confirmed that direct protein functionalisation occurs (Scheme 4).20 Similar to the work on ArN3 species, we showed that tetrazole photochemistry is a viable route for synthesising bioactive radiotracers for applications in PET imaging.20 | ||
| Scheme 4 Photo(radio)synthesis of functionalised proteins using light-induced activation of tetrazoles. Formation of the electronic excited state of a tetrazole can lead to intramolecular rearrangements and loss of N2(g) to generate a 1,3-dipolar nitrile imine. This intermediate finds applications in labelling compounds bearing alkene or alkyne reaction partners (photo-click) or in direct reaction with biologically relevant nucleophiles found on protein. | ||
Upon irradiation with UV light, the electronically excited state of tetrazoles undergoes a stepwise rearrangement and photodissociation.92 First, a weak N–N bond breaks to form an electronically excited intermediate which then undergoes spontaneous and rapid loss of N2 to form a nitrile imine. Nitrile imines undergo fast 1,3-dipolar cycloaddition reactions with alkenes and alkynes,92 but also react with a wide variety of biologically relevant nucleophiles. Remarkable examples of their applications are the on-protein synthesis of fluorogenic groups by Qing Lin and co-workers,63,97,107–109 fluorescence microscopy imaging,110,111 and radioimmunoconjugate photosynthesis.20,92
Photolysis of tetrazoles at wavelengths that do not lead to photodegradation of protein requires auxochromic tuning of their absorption spectrum.20 To this end, our group developed naphthyl tetrazole derivatives and performed structure–activity relationship experiments.20 Introduction of a methoxy group onto a naphthyl-tetrazole facilitated rapid and efficient photoactivation at wavelengths between 365 to 395 nm. Detailed computational studies also suggested that the reactivity of tetrazoles with biological nucleophiles might be exploited in future studies by using different experimental conditions to tune the chemoselectivity.92 Overall, tetrazoles are one of the most synthetically versatile classes of photoreagent for labelling proteins.
ortho-Nitrobenzyl alcohols (ONBAs)
The photochemical behaviour of nitroso compounds was first reported in 1901 when the pioneers of photochemistry Ciamician and Silber112 described the photoinduced rearrangement to o-nitrobenzaldehyde to o-nitrosobenzoic acid photolysis.112 Other important works on photochemical reactions of o-nitrobenzyl alcohols (ONBA) and related derivatives were written by Sachs in 1904113 where it was predicted that, “All aromatics which have a hydrogen ortho to a nitro group will be light sensitive.”114 Later in 1970, Woodward and co-workers used ONBAs as photo-removable protecting groups for amino-, carboxy- and ester groups.115 The usefulness of ONBAs and derivatives as photoprotecting groups has been extensively studied by the group of Bochet.59,116–122 Upon photon absorption, ONBAs undergo an intramolecular redox reaction generating the corresponding o-nitrosobenzaldehyde, which until recently, has been exploited mainly for photochemical deprotection of reactive groups. The group of Xiao-Hua Chen repurposed ONBAs for the in situ conjugation of proteins (Scheme 5).123 Here, the photogenerated o-nitrosobenzaldehyde is susceptible to nucleophilic attack from primary amines, condensing to give the corresponding indazolones after intramolecular cyclisation.123,124 This reactivity enables the use of ONBAs as chemoselective photochemical linkers between the ε-NH2 of lysine side-chains and the photogenerated o-nitrosobenzaldehyde. Photochemical conjugation experiments with ONBAs were demonstrated on a wide range of substrates spanning from simple amines and peptides, through to nanobodies. In one study, a nanobody that binds to the human epidermal growth factor receptor type 2 (HER2) was photochemical derivatised by using an alkynyl derivative of ONBAs. Mass spectrometric analyses conducted by MALDI-TOF revealed lysine-specific and near quantitative protein labelling. The ONBA-derived probes were also activated in live cell cultures by UV light (365 nm) and enabled precise temporal and spatial control of bioconjugation, with minimal cellular toxicity. The ONBA strategy enabled kinase profiling in live mammalian cells where a lysine-reactive probe captured over 90 kinases in Jurkat and 76 in K562 cells, outperforming diazirine probes. Structural docking confirmed the selectivity of the probe for conserved lysine residues in kinase ATP-binding sites. The same methodology was also expanded to include mitochondria-specific labelling with a fluorescent rhodamine-ONBA probe. | ||
| Scheme 5 Schematic representation of the light-induced ONBA reaction mechanism labelling a HER2-specific nanobody.123 (Step 1, left) Photoinduced ONBA activation and chemoselective protein derivatisation with an alkynyl group. (Step 2, right) The formation of a covalent bond between the protein and the modified probe via CuI-mediated alkyne–azide click chemistry, illustrating successful bioconjugation towards TAMRA-N3. | ||
Diaryl sydnones
The photoactivity of sydnones was established in 1955 when Tien and Hunsberger reported the synthesis of their pyridyl derivatives which displayed solid-state photochromic behaviour when exposed to sunlight.125 The transparent crystals turned dark blue under light and the transition was reverted by heating or by leaving the crystals in the dark for prolonged times. More recently, Taran and co-workers experimented with the use of sydnones in copper-mediated cycloadditions to alkynes,126 and then further improved strategies were proposed for strained alkynes.127,128The photochemical reactivity of sydnone derivatives on biomolecules came later. In 2018 the group of Zhipeng Yu synthesised a library of diaryl sydnones aiming to tune the electronic structure of these compounds to absorb and react at wavelengths more compatible with applications in biochemistry.129 Their proposed working mechanism resembles the one of tetrazoles.130 Photon absorption by the sydnone group creates an electronically excited state which releases CO2 and forms a nitrile imine. As a consequence, the reactivity patterns of sydnones are anticipated to be comparable to tetrazoles. Initial studies demonstrated that lysozyme, pre-functionalised with strained alkynes, undergoes efficient photo-induced and fluorogenic labelling.131 Similar reactivity was observed with strained alkenes (e.g. trans-cyclooctene, TCO), which undergo fluorogenic cycloadditions with the sydnone-generated nitrile imines forming the corresponding pyrazolines. The pyrazoline products display efficient luminescence properties with high fluorescence quantum yields (0.28 to 0.42) and turn-on enhancement factors (122 to 321-fold).129 The magnitude of these turn-on effects allows for straightforward monitoring and quantification of protein labelling and enables sensitive detection of protein conjugates at sub-micromolar levels.129
Sydnones also undergo cycloaddition reactions via their nitrile imine intermediate with dibenzo-thiadiazepine (DBTD) reagents (Scheme 6).64,132 Notably, dibenzo-thiadiazepines display concomitant photo-isomerisation where the less stable E-isomer was shown to react faster with the nitrile imine. Kinetic studies highlighted that the nitrile imine displayed considerable rate enhancements toward the E-isomer of the DBTD compound which are attributed to increased ring strain (kE = 1.6 (±0.16) × 105 M−1 s−1, kZ = 2.4 (±0.24) × 104 M−1 s−1 for Z, kE/kZ = 6.7). The interaction of sydnones with DBTD is also fluorogenic, thereby facilitating quantification of the protein conjugation step via fluorescence emission coupled to gel electrophoresis methods for protein separation. Photolabelling reactions with a sulfo-Cy3 fluorophore diaryl sydnone derivative exhibited successful bioorthogonal ligation of lysozyme or bovine serum albumin (BSA) where the protein was pre-functionalised with a DBTD reagent.132 Although potentially versatile, current applications of photolabelling chemistry with these DBTB-sydnone reagent pairs remains complex. For instance, the setup requires two light sources for triggering the formation of the nitrile imine at 311 nm and the Z–E photo-isomerisation (quantum yield for isomerisation: ΦZE405 = 0.516 at 298 K) of the DBTD partner at 405 nm using laser light sources. A notable advantage is that the DBTD reagent has excellent stability in the biochemical environment and is resistant to photodegradation (9.4% bleaching after 3500 light-dark cycles). However, rapid thermal reversion from the E-to-Z isomer (3.75 ± 0.23 s−1, 298 K) generates a very narrow reaction window of only a few seconds. This can provide high control over the generation of the reactive intermediate but simultaneously limits the accessible reactive space for bimolecular protein conjugation to only extremely fast bioorthogonal partners.
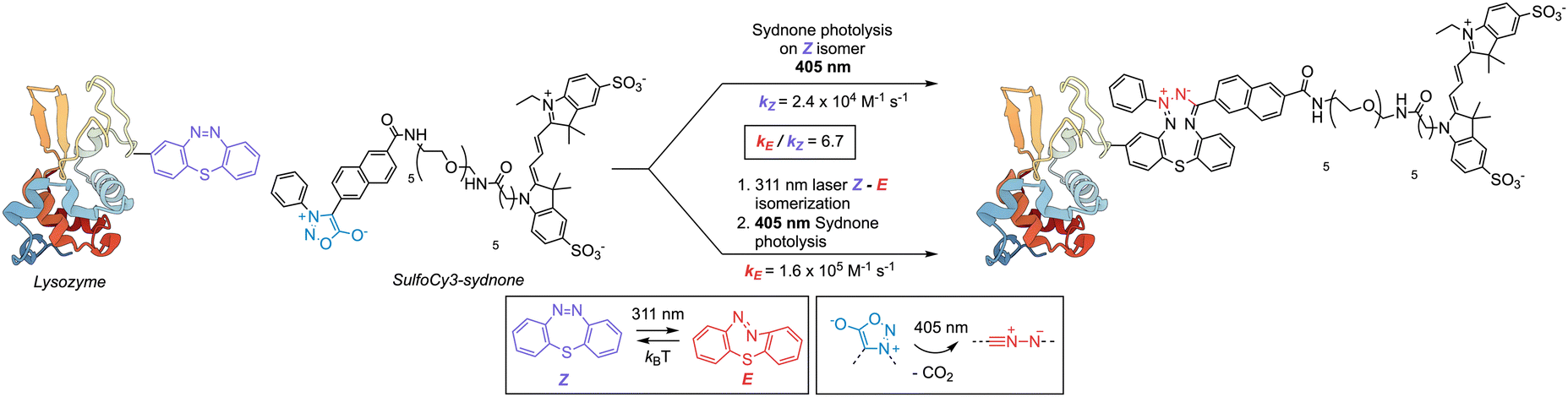 | ||
| Scheme 6 Diaryl sydnones eliminate CO2 under light irradiation forming reactive nitrile imines. In this example a thiadiazepine functionalized lysozyme undergoes photoinduced (405 nm) cycloaddition to the photogenerated nitrile imine with a rate constant kE. The cycloaddition rate constant increases by kE/kZ = 6.7 times following a stepwise procedure in which the diazepine is first converted to the Z geometric isomer. In the subsequent step, the nitrile imine is generated in situ by irradiation with 405 nm light, thus initiating protein conjugation to the Cy3-sydnone derivative.132 | ||
Importantly, the sydnone-DBTD technology has been applied in live cells by in situ labelling of cetuximab131,132 and panitumumab132 derivatives bearing dibenzothiadiazepine. Fluorescence microscopy revealed that irradiating the cell sample with 405 nm resulted in Cy3 accumulating on the cell surface.132 In conclusion, sydnones are promising photoreagents with future prospects for developing both bioorthogonal tools and for potential direct protein conjugation.
Diaryl azirines
The photochemistry of azirines was first reported in 1968 by Arnold133 and later in the 1970s by Padwa31,32 who used these reagents as photochemical building blocks. The main application of diazirines lies in their ability to produce nitrile ylides via photo-induced ring-opening rearrangements.134 Nitrile ylides have been used as cycloadditions reaction partners with dienes or dipolar compounds.134 However, the use of azirines in bioconjugation chemistry remains relatively unexplored. Computational studies have predicted that nitrile ylides display activation parameters which point toward faster reactivity compared with nitrile imines generated from tetrazoles or sydnones.135 This is in line with significantly lower activation free energies for 1,3-dipolar cycloadditions and was corroborated experimentally by their preference to react with unsaturated dicarbonyl compounds bearing electron-withdrawing substituents.135,136Light-induced activation of the 3-membered azirine ring typically requires photons with short wavelengths around 300 nm. Consequently, their application in photoinduced bioconjugation chemistry has been limited. To alleviate this drawback, Barner-Kowollik and co-workers substituted one of the aryl groups with a pyridyl group to redshift the activation wavelength to 405 nm.137
Lin and co-workers prepared azirine-functionalised lysozyme (via classic NHS conjugation methods).136 The bioconjugate then underwent 302 nm photon absorption and a light-induced reaction with unsaturated 1,4-dicarbonyl compounds such as dimethyl fumarate or a PEG-ylated derivative of fumarate (Scheme 7). Successful protein labelling was reported, and the reaction shows characteristics of a bioorthogonal process, but further work is required to establish azirine photochemistry as a reliable method for protein functionalisation. In particular, issues related to the use of high-energy photons, the necessity to introduce the photoactive group via a non-bioorthogonal step, and the slow photoreaction kinetics (photoreaction rate constant of 0.03 s−1 using a 20-fold excess fumarate)136 must be addressed before diaryl azirines can become a more widely use class of photoreagent for protein conjugation.
 | ||
| Scheme 7 Diaryl azirines undergo photoinduced ring-opening reactions to form nitrile ylides, which are established reaction partners for cycloaddition reactions. Here, photolysis of a pre-functionalised lysozyme-diazirine derivative with 302 nm light, stimulates the formation of a new conjugate bond with electron-poor dienes (such as methyl fumarate), forming the cycloaddition product.136 | ||
Photodeprotection of strained alkynes
Strained alkynes have been central to the development of click chemistry for biological applications.138–140 Recently, a new generation of photo-protected strained alkyne reagents has expanded the photochemical reaction palette with cyclopropenones being the most prominent members.67 In 2009, Popik and co-workers reported a novel application involving these compounds and demonstrated their utility for labelling biomolecules in challenging situations such as those involving intact cells.67 The photolysis of the 3-membered cyclic enone results in the rapid generation of the strained alkyne, which serves as an ideal substrate for strain-promoted azide–alkyne cycloadditions (SPAAC).141 Following the formation of the alkyne, the introduction or concurrent presence of an azide derivative initiates the SPAAC reaction, leading to the synthesis of the corresponding triazole with bimolecular rate constants around 0.05 M−1 s−1 (measured in MeOH; for comparison, typical values for SPAAC on unprotected alkynes are ∼0.1 M−1 s−1 in H2O)140,142 Subsequent experiments showed that the photodecaging and SPAAC approach can be employed to label intact Jurkat cells supplied with N-azidoacetyl-sialic acid.67,143 This supplement leads to the selective incorporation of azido groups in glycoproteins and glycolipids synthesised by the cells, thus providing a suitable bioorthogonal substrate for evaluating photodecaging reactivity of alkynes in living systems. For example, azide-presenting cells were subsequently exposed to ultraviolet light in the presence of a cyclopropenone–biotin conjugate, initiating the formation of triazole linkages between biotin fragments and cellular proteins. Incubation of the biotin-functionalised cells with an avidin-Alexa Fluor 488 conjugate showed efficient labelling as detected by using fluorescence microscopy.67The CO-releasing reaction on cyclopropenones has been analysed in detail through a combination of DFT calculations and flash photolysis experiments which probed the nature of the decarbonylation step.60 Electronic transitions responsible for the fragmentation of the molecule have been identified, and mechanistically, the loss of CO is proposed to occur in a stepwise fashion. First, the cyclopropenone undergoes an energy-demanding (∼134 kJ mol−1) ring-opening reaction by forming a ketenylcarbene intermediate (Scheme 8). The ketenylcarbene can be described by two resonance structures, one of which is neutral (carbene form) and the other displays charge separation (zwitterionic form). This intermediate then undergoes fast decarbonylation, highlighting the low activation barrier to cleave the remaining C–CO bond. From the analysis of the computational and experimental results, it was hypothesised that CO release happens in an electronically excited state of the zwitterionic intermediate, as supported by intense solvent polarity effects observed during kinetic experiments.60 An illustration of the photolabelling of proteins with cyclopropenones is presented in Scheme 8.61
 | ||
| Scheme 8 Cyclopropenones are photolabile compounds that undergo decarbonylation reactions upon irradiation. Photochemical CO extrusion leads to the formation of the corresponding alkyne. Specifically, for the dibenzo-derivatives the unmasked alkyne lies within a strained ring system, thereby facilitating classic bioorthogonal click reactions. In this example, an initial SPAAC is performed to connect an N3-derivative of bovine serum albumin (BSA) to the cyclopropenone. This protein conjugate is then photolytically decarbonylated to expose the alkyne, which is used in a second SPAAC reaction to bridge the protein to rhodamine fluorophore.61 | ||
Upon photodeprotection, cyclopropenones also become suitable reagents for inverse electron demand Diels–Alder (IEDDA) reactions.144 In this framework, 1,2,4,5-tetrazine species reacts with a dienophile like a strained alkene or alkyne through a classic [4+2] cycloaddition pathway. The cycloaddition is accompanied by a concomitant release of nitrogen which represents the driving force of the reaction. As discussed above, the alkyne can be generated in situ from the photolysis of a dibenzocyclopropenone, which converts into the corresponding cyclooctyne and then participates in an IEDDA reaction. A remarkable example of this technology has been presented by the Lang group, in which protein engineering was adopted as a biosynthetic strategy to introduce site-specifically tetrazine modified versions of lysine on a variety of proteins including superfolder green fluorescent protein (sfGFP).144 The tetrazine-modified sfGFP was tested for reactivity toward masked alkynes, which yielded nearly quantitative photoreactions upon exposure to light. This system was then further tested on living E. coli samples demonstrating photochemical labelling of intact cells. Protected strained alkynes represent an exemplary case of photoinduced deprotections (Scheme 1, mechanism i), which introduce an additional dimension to synthetic biochemistry.
Photoactivation of tetrazines
Related to the chemistry discussed in the previous section, tetrazines have been used extensively in IEDDA reactions on biological substrates, including for pretargeted in vivo labelling applications in the space of radiotracer imaging and therapy.65,145–149 The tetrazine chemistry and IEDDA reactions have been the subject of numerous excellent reviews, but the majority of the work addresses only thermally-mediated processes.150–152 Light-activated tetrazine chemistry has been reported with activation mechanisms spanning both photodeprotective and photoredox processes (Scheme 1, mechanisms i and iii, respectively).In 2016, Fox and co-workers reported the use of catalytic turn-on photooxidations for converting inactive dihydrotetrazines (DHTZ, Scheme 9) into reactive tetrazines (TZ) under LED irradiation at 600 nm.153 The photooxidation mechanism exploits the sensitising properties of compounds such as methylene blue, rose bengal, and carboxyfluorescein, which act as photocatalysts for the oxidation of DHTZ into TZ. Methylene blue and rose bengal are known as photosensitisers to produce singlet oxygen (1O2), and initial speculations suggested that this species could be the effective oxidant.153 This hypothesis was disproven by conducting control experiments in the presence of NaN3 (a 1O2 quencher)154,155 thus showing that the photocatalytic effect was independent from the formation of reactive oxygen species.
 | ||
| Scheme 9 The photocatalytic oxidation of DHTZ is useful for generating tetrazines on demand, which are potent substrates for bioorthogonal IEDDA-type reactivity in vitro and in vivo. In this example, green fluorescent protein (GFP) was functionalised with a strained trans-cyclooctene (TCO) by using a thiol–maleimide conjugation. Subsequent incubation with a DHTZ reagent, followed by photocatalysed oxidation to unmask the TZ partner led to successful protein functionalisation.153 | ||
These observations led to the conclusion that the photooxidation of DHTZs represents a practical protocol for developing biocompatible functional materials and for the photoinduced formation of protein conjugates. Similar strategies have been employed in other works from the same group in which additional protein derivatives have been photolabelled by using the DHTZ/TZ dyad.66 These preliminary reactions on model proteins served as the foundation for subsequent development of live-cell imaging agents.66 Indeed, the strategy devised by Fox and co-workers facilitates rapid and spatiotemporally resolved labelling of engineered proteins expressed by cells, hence opening new avenues for understanding fundamental biochemical processes.
Forsythe and co-workers applied these concepts in the design of biocompatible and nontoxic materials which can be used in vivo.156 The advantages derived from these methodologies are the short times of photoreaction, red-shifted wavelengths, and the widespread availability of the photocatalysts.
Tetrazine reagents have also been employed in photodeprotection strategies. For instance, Devaraj and colleagues demonstrated tetrazine release from a photocaged dihydrotetrazine, which included a photoprotecting group akin to o-nitrobenzyl carbamates that can be removed by exposure to 425 nm light. The short irradiation times (less than 2 minutes), in conjunction with the high reaction rates and red-shifted absorption, render this methodology a particularly advantageous approach for on-demand biomolecule derivatisation.62
In summary, the development of photooxidation-based protocols for DHTZs and tetrazine derivatives presents a powerful approach for biocompatible material synthesis, live-cell imaging, and on-demand biomolecule modification. These advances, characterised by rapid reaction rates, tunable wavelengths for photoactivation, and the availability of photocatalysts, open new avenues in biomedical research and offer innovative tools for studying and manipulating biochemical processes with high spatial and temporal precision.
Thiol–ene reagents
The distinctive chemistry of sulfhydryl groups (redox chemistry, variable pKa values, photoreactivity, high nucleophilicity and soft Lewis base class B character), coupled with their widespread availability on biomolecules, present an exceptional opportunity of using this functional handle in novel photochemical processes. Most of their thermally mediated chemistry relies on the acid–base properties of the SH group and takes advantage of their innate nucleophilicity. Photochemical reactions between terminal mercaptans and alkenes have been known for more than a century157 but in recent years there has been a ‘rediscovery’ of R-SH chemistry for applications in direct bioconjugation of protein. In their light-induced reactions, thiols become activated and generate thiyl radicals (RS˙). These high-energy intermediates react readily with alkene substrates frequently following anti-Markovnikov patterns,158 although the Markovnikov addition products are observed in rare examples.159 Radical reactivity poses an immense challenge to control, especially on biological substrates under aqueous aerated conditions. Photochemical thiol–ene reactions are not immune from these drawbacks but efforts have been made to mitigate and tame the reactivity of these systems. Numerous research groups have explored the creation of masked thiols including derivatives of coumarin or o-nitrobenzyl which photolytically fragment and release the free thiol.59,160,161 Although the thiol–ene and related thiol–yne reactions display potential in biomolecule functionalisation, the technology needs to mature before it can find broader applications. One issue that must be addressed is that the production of highly chromophoric products after the photodeprotection can impede the efficiency of the thiol–ene reaction. Specifically, these highly absorbing byproducts accumulate within the reaction mixture and progressively function as light filters, consequently impinging the overall efficacy of the reaction.162,163 Controlling the conditions to minimise competing reaction pathways like radical quenching is also essential to improve bioconjugate yields to useful levels.Encouragingly, the group of Sungwoo Hong developed a photochemical system that features mild activation parameters, high selectivity, and biocompatibility. In this work, they prepared a novel generation of quinoline-based photosensitisers poised to activate cysteine side-chain thiols at wavelengths around 440 nm.85 Detailed mechanistic investigation on the thiol–ene reaction confirmed the involvement of radical species, where radical scavengers such as TEMPO (2,2,6,6-tetramethylpiperidine-1-oxide) completely inhibited the formation of the desired products. This reactivity has been exploited for the site-selective formation of thioether bonds on bovine serum albumin (BSA) and ubiquitin K63C (Scheme 10), a version of ubiquitin displaying substitution of lysine 63 for a cysteine residue. Modified variants of these proteins have been synthesised by the thiol–ene method and feature, for instance, a biotin pendant arm or a quinoline derivative reminiscent of the photocatalysts discussed above. In the latter example, the small-molecule reagent served a dual role acting as both the photocatalyst and the alkene source by initiating the thiol–ene reaction and ultimately conjugating with the protein (Scheme 10). Such a strategy provides a convenient instrument for a site-selective, photochemical, and single-step introduction of a fluorophore subunit on proteins that display accessible thiols.
 | ||
| Scheme 10 Thiol–ene reactions proceed through the formation of a photo-generated thiyl radical which is particularly reactive towards unsaturated compounds, such as alkenes or alkynes. In this example, a photosensitised version of the thiol–ene reaction is triggered by 440 nm irradiation to create a new thioether bond bridging a quinoline derivative to ubiquitin-expressing cysteine in position 63.85 Interestingly, the substrate also acts as the photosensitiser, promoting the bioconjugation reaction and producing a fluorescent protein conjugate. | ||
Diazirines
Diazirines quickly found applications as PAL probes75,76,164 in biochemistry in the 1970s when Knowles and co-workers exploited their photochemical reactivity for biomolecule labelling.34,35,165 Their productive photochemistry involves electronic excitation of the diazirine group through absorption of ultraviolet light. Electronic absorption is thought to either promote direct loss of nitrogen to give a carbene or proceed via a two-step mechanism in which photoisomerisation to the diazo species, followed by thermal or secondary photochemical release of N2(g) gives the carbene. Carbenes are highly energetic and display reactivity preferences that depend on their spin state.77 Tang and Lijuan demonstrated the dual reactivity nature of diazirine reagents through an extensive series of labelling and mechanistic experiments (Scheme 11).166 | ||
| Scheme 11 Diazirines photolyse under UV light by forming either a diazo or a carbene intermediate. The diazo species can convert to the carbene via thermal or photochemical pathways but can also react with nucleophiles on proteins. The carbene species can label protein by C–H or X–H bond insertion pathways and are more reactive toward apolar residues. In this example, BSA was first derivatised with a commercially available diazirine N-hydroxysuccinimide derivative (known as SDA) then photolysed to form a new covalent bond, stapling the protein and rigidifying the structure.166 These studies highlight the unique capabilities of diazirines in protein labelling, where their versatile reactivity with both polar and apolar residues under UV irradiation provides a valuable approach for creating covalent linkages to a wide range of biomolecules. | ||
The dual reactivity of diazirines was elucidated through a comprehensive mechanistic analysis of model functional groups, which dissected the behaviour of these photoreagents into two principal modes. The first mode pertains to the interaction of a diazo intermediate with the protein, which preferentially reacts with polar amino acids by liberating N2. Conversely, apolar amino acids predominantly engage with the carbene intermediate, resulting in heterogeneous products typically generated through bond insertion processes. The mechanistic distinction between these two pathways is supported by deconvolution of many elementary steps that were experimentally accessible with the aid of sophisticated photochemical setups. Other examples of the use of diazirine-based photoreagents include direct crosslinkers for biomolecules,72 binding site identification for antibiotics on proteins74 solid-state biomolecule cross-linking167 and mAb radiolabelling.73
Benzophenones
Benzophenone (BP)-based photoreagents were introduced in 1974 when Galardy and co-workers used this class of reagent to map the binding sites of pentagastrin on BSA.33 Their stability, reversible photoactivation in water, and preferential reactivity to otherwise inert C–H bonds (especially methionine residues) crafted the success of this functional group in biomolecule derivatisation.168 The photoactivation mechanism of BPs involves excitation of the carbonyl chromophore by UV light, which promotes an electron from a non-binding orbital based primarily on the oxygen atom to the antibonding π* orbital of the carbonyl group. The diradical formed (either as a singlet or triplet species) is very reactive and tends to interact with C–H bonds resulting hydrogen atom abstraction by the oxygen-centred radical. Ketyl radicals and alkyl radicals on the protein then recombine (potentially after intersystem crossing) to form the final products, which results in covalent C–C bond formation between the BP reagent and the biomolecule of interest.Benzophenone-based compounds have found myriad applications in the radiolabelling of proteins,169–172 lipids,173 protein immobilisation174 and photoconjugation,175,176 and DNA cross-linking.177 Of particular interest, is the development of the methionine proximity assay (MPA) – a method for probing protein structure and ligand-receptor interactions which takes advantage of the intrinsic preference of photoexcited BPs for methionine residues located in the vicinity of the photoprobe.71,178 Seminal work from Escher and colleagues, rigorously investigated a novel Methionine Proximity Assay (MPA) utilising BP photolabelling to elucidate the binding environment of the human angiotensin II type 1 (hAT1) receptor (Scheme 12). By strategically introducing methionine at different positions within transmembrane domains (TMDs) III, VI, and VII, it was possible examine the interaction between these modified sites and photoreactive ligands.178 Upon photoactivation, BP selectively labels methionine through the formation of a charge-transfer complex. Subsequently, cyanogen bromide (CNBr) cleavage generates distinct fragments, thereby revealing specific ligand-receptor contact points. Overall, BP-based photoreagents are valuable tools in biochemical research, particularly in elucidating protein–ligand interactions.
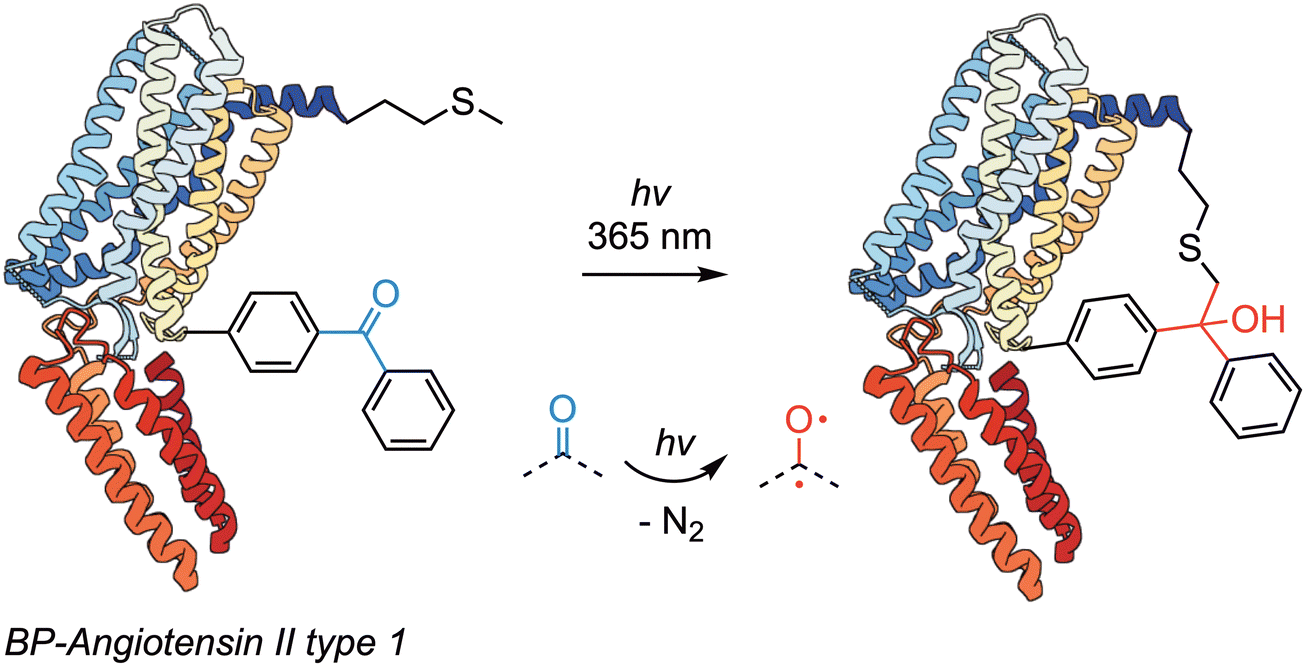 | ||
| Scheme 12 Benzophenones are chromophoric units that undergo reversible photoactivation to give a diradical species in either a singlet or triplet spin state. This diradical can abstract hydrogen atoms from a protein of interest and insert into otherwise unreactive C–H bonds. In this example, BP-derivatised angiotensin II was irradiated with 365 nm light. The electronically excited BP subunit then inserted preferentially into a C–H bond of a proximal methionine residue.178 | ||
Photoredox catalysis for bioconjugation
Photocatalysis is a well-established domain in synthetic chemistry,179 that is also starting to find applications in the functionalisation of biomolecules180–182 and protein labelling in complex cellular environments.183 Within this framework, a photosensitive molecule that remains unreactive in its ground state is added to a reaction mixture containing the biomolecule of interest and an inactive substrate. Photoactivation of the catalyst activates a chemical transformation of the substrate, which then reacts with the protein (Scheme 1, mechanism iii). As an example, McMillan and co-workers utilised the redox properties of lumiflavin in its photoexcited state (E1/2 [3LF/LF˙−] = 1.68 V) to facilitate site-selective N-arylation of phenoxazine on the tyrosine residues of lysozyme (Scheme 13). The site-selectivity of the photoredox system is explained by considering solvent exposure of the amino acid substrates on the protein. Lysozyme has three tyrosine residues (Y20, Y45, and Y54) within its primary structure. Two of them (Y20 and Y54) are buried in hydrophobic pockets or engage in cation–π interactions, making them sterically inaccessible or unreactive. On the other hand, Y45 is presented on the protein surface and is solvent-accessible, thus making it a readily available reactive handle. Upon excitation by visible light, the photocatalyst (PC) lumiflavin transforms into the corresponding nitrogen-based triplet diradical, which can abstract a hydrogen atom from phenoxazine, producing the corresponding N-radical. The phenoxazine radical then combines with Y45 by expelling a proton and concomitantly reducing lumiflavin to the corresponding dihydro form. The dihydrolumiflavin is then oxidized back to the original lumiflavin by oxygen closing the catalytic cycle. The phenoxazine scaffold allows versatile functional group tolerance. For instance, a phenoxazine dialdehyde was employed as a site-selective reagent for introducing reactive aldehyde groups on protein. Subsequent imination of the aldehydes provided a simple synthetic pathway for introducing additional functional fragments such as fluorophores, affinity tags, terminal alkynes and azides. Notably, the reaction is metal–ion free and operates under mild conditions.184 Similar strategies that target methionine residues to install functional groups on aprotinin, lactalbumin, myoglobin, human growth hormone, ribonuclease A, and carbonic anhydrase in a site-selective fashion were also reported.185 | ||
| Scheme 13 Photoredox catalysis proceeds through light-activation of a catalyst which then initiates further chemical transformations on a substrate that can lead to protein bioconjugation. Here, lumiflavin (the photocatalyst, PC) is excited by 405 nm light and undergoes a radical process that abstracts hydrogen from phenoxazine. The phenoxazine radical inserts site-specifically on tyrosine Y45 of lysozyme. By using phenoxazines decorated with additional functional groups (e.g. aldehydes) it is possible to perform further reactions at the heterocycle.185 Although site-selectively was reported in this example, for other proteins like mAbs, which present many solvent exposed tyrosine residues, the method is unlikely to be regioselective. | ||
In other examples, the group of Chien-Wei Chiang reported on the use of triphenylpyrylium tetrafluoroborates as photosensitisers for biocompatible photoredox catalysis that facilitates the introduction of pyrazole derivatives on phenylalanine residues under blue light irradiation.186 Chatterjee and co-workers labelled site-specifically 5-hydroxytryptophan protein derivatives by using methylene blue as the photosensitiser and 455 nm light irradiation.187 Waser and colleagues reported photoredox catalysed decarboxylative alkynylation to produced peptides with alkynes at the C-terminus. This interesting reaction utilised organic dyes to derivatise peptides in the presence of hypervalent iodine compounds, which acted as the alkynyl transfer reagent.188 For a comprehensive discussion of metal complex photocatalysed reactions, we direct the reader to recent review by De Jesus et al.189 Collectively, these advancements underscore the versatility and potential of photoredox catalysis to enable chemo- or potentially site-selective modifications on proteins and peptides, though we note that care must be taken to avoid reaction conditions or substrates that might lead to undesired redox reactions (e.g. disulfide reduction) on protein. This growing body of work demonstrates the innovative approaches being developed to leverage visible light in both metal ion complex and organic dye systems, ultimately broadening the tool-kit available for researchers in chemical biology and organic synthesis.
Conclusions and outlook
This perspective provides a comprehensive overview of the current state of play in the use of photochemical reaction to functionalise biologically active compounds including proteins and peptides. Reaction characteristics and experimental considerations that must be addressed during optimisation procedures are described with common pitfalls and potential solutions highlighted. Notable examples derived from both historical and contemporary literature on photoinduced bioconjugation chemistry are presented. In most cases, the primary biological substrates used are proteins and polypeptides, while other significant biomolecules have been largely neglected. Consequently, there remains unexplored opportunity for adapting photochemical technologies to functionalise carbohydrates, nucleic acids, and lipids through light-induced reactions. These biomolecules exhibit reactivity patterns that differ markedly from those of proteins and amino acids. Also of particular interest in the future will be the development of bioorthogonal and site-selective photochemical reactions. Considering the broad range of photochemical reactions that remain to be tested in a biological context, and the untapped potential of using new types of covalent bond to construct functional molecules like antibody–drug conjguates for imaging and therapy, we believe that that the future looks bright for light-induced biomolecule labelling technologies.Author contributions
C. B. wrote the first draft of the manuscript which was revised by J. P. H.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
No potential conflict of interest relevant to this article was reported.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme/from the European Research Council (under the Grant Agreement No. 101001734, ERC-CoG-2020, PhotoPHARMA), and from the European Innovation Council Pathfinder grant (grant no. 101129886, SMARTdrugs). JPH also thanks the University of Zurich (UZH) for financial support. We thank all members of the Medicinal Radiochemistry group at the UZH for helpful discussions.References
- S. B. Ebrahimi and D. Samanta, Nat. Commun., 2023, 14, 2411 CrossRef CAS PubMed.
- K. Tsuchikama, Y. Anami, S. Y. Y. Ha and C. M. Yamazaki, Nat. Rev. Clin. Oncol., 2024, 21, 203–223 CrossRef CAS PubMed.
- S. B. Ebrahimi and D. Samanta, Nat. Commun., 2023, 14, 2411 CrossRef CAS PubMed.
- A. M. Wu and T. Olafsen, Cancer J., 2008, 14, 191 CrossRef CAS PubMed.
- L. Martiniova, R. J. Zielinski, M. Lin, L. DePalatis and G. C. Ravizzini, Cancer J. Sudbury Mass, 2022, 28, 446–453 CrossRef CAS PubMed.
- G. T. Hermanson and F. L. van Delft, Chemical Linkers in Antibody–Drug Conjugates (ADCs), Royal Society of Chemistry, 2021, pp. 32–70 Search PubMed.
- P. Dennler, E. Fischer and R. Schibli, Antibodies, 2015, 4, 197–224 CrossRef.
- R. Fay and J. P. Holland, J. Nucl. Med., 2019, 60, 587–591 CrossRef CAS PubMed.
- J. Li, J. Chen, Q.-L. Hu, Z. Wang and X.-F. Xiong, Chin. Chem. Lett., 2024, 110126 Search PubMed.
- B. M. Zeglis, C. B. Davis, D. Abdel-Atti, S. D. Carlin, A. Chen, R. Aggeler, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2014, 25, 2123–2128 CrossRef CAS PubMed.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 CrossRef CAS PubMed.
- A. Debon, E. Siirola and R. Snajdrova, JACS Au, 2023, 3, 1267–1283 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- V. Bacauanu, Z. N. Merz, Z. L. Hua and S. B. Lang, J. Am. Chem. Soc., 2023, 145, 25842–25849 CrossRef CAS PubMed.
- S. R. Thomas, R. Bonsignore, J. Sánchez Escudero, S. M. Meier-Menches, C. M. Brown, M. O. Wolf, G. Barone, L. Y. P. Luk and A. Casini, ChemBioChem, 2020, 21, 3071–3076 CrossRef CAS PubMed.
- axispharm, Comprehensive List of FDA-Approved Antibody-Drug Conjugates (ADCs) – Updated for 2024, https://axispharm.com/antibody-drug-conjugatesadcs-list-approved-by-fda2000-2022/.
- FDA Approved ADC Drugs list up to 2022, https://www.bio-itworld.com/pressreleases/2022/11/28/fda-approved-adc-drugs-list-up-to-2022.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- R. Fay and J. P. Holland, Chem. – Eur. J., 2021, 27, 4893–4897 CrossRef CAS PubMed.
- M. Patra, S. Klingler, L. S. Eichenberger and J. P. Holland, iScience, 2019, 13, 416–431 CrossRef CAS PubMed.
- A. Guillou, D. F. Earley and J. P. Holland, Chem. – Eur. J., 2020, 26, 7185–7189 CrossRef CAS PubMed.
- D. Lin, L. M. Lechermann, M. P. Huestis, J. Marik and J. B. I. Sap, Angew. Chem., Int. Ed., 2024, 63, e202317136 CrossRef CAS PubMed.
- K. Kozoriz, O. Shkel, K. T. Hong, D. H. Kim, Y. K. Kim and J.-S. Lee, Acc. Chem. Res., 2023, 56, 25–36 CrossRef CAS PubMed.
- L. Wang, Z. Lv, L. Yang, X. Wu, Y. Zhu, L. Liu, Y. Zhao, Z. Huang, D. A. Nicewicz, Z. Wu, Y. Chen and Z. Li, Bioconjugate Chem., 2024, 35, 1160–1165 CrossRef CAS PubMed.
- B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Nature, 2020, 585, 530–537 CrossRef CAS PubMed.
- P. G. Isenegger, B. Josephson, B. Gaunt, M. J. Davy, V. Gouverneur, A. J. Baldwin and B. G. Davis, Nat. Protoc., 2023, 18, 1543–1562 CrossRef CAS PubMed.
- A. Guillou, D. F. Earley, M. Patra and J. P. Holland, Nat. Protoc., 2020, 15, 3579–3594 CrossRef CAS PubMed.
- A. Singh, E. R. Thornton and F. H. Westheimer, J. Biol. Chem., 1962, 237, PC3006–PC3008 CrossRef CAS.
- G. W. J. Fleet, R. R. Porter and J. R. Knowles, Nature, 1969, 224, 511–512 CrossRef CAS.
- A. Padwa, S. Nahm and E. Sato, J. Org. Chem., 1978, 43, 1664–1671 CrossRef CAS.
- A. Padwa, Acc. Chem. Res., 1976, 9, 371–378 CrossRef CAS.
- R. E. Galardy, L. C. Craig, J. D. Jamieson and M. P. Printz, J. Biol. Chem., 1974, 249, 3510–3518 CrossRef CAS.
- R. A. G. Smith and J. R. Knowles, J. Am. Chem. Soc., 1973, 95, 5072–5073 CrossRef CAS PubMed.
- R. A. G. Smith and J. R. Knowles, J. Chem. Soc., Perkin Trans. 2, 1975, 686–694 Search PubMed.
- M. J. Bouchet, A. Rendon, C. G. Wermuth, M. Goeldner and C. Hirth, J. Med. Chem., 1987, 30, 2222–2227 Search PubMed.
- V. Chowdhry, R. Vaughan and F. H. Westheimer, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1406–1408 CrossRef CAS PubMed.
- H. Scheefers, C. Schreiber and W. Stoffel, Hoppe-Scylers Z Physiol. Chem., 1978, 359, 923–931 CrossRef PubMed.
- M. P. Goeldner, J. E. Hawkinson and J. E. Casida, Tetrahedron Lett., 1989, 30, 823–826 CrossRef CAS.
- J. V. Staros and F. M. Richards, Biochemistry, 1974, 13, 2720–2726 CrossRef CAS PubMed.
- P. E. Nielsen, J. B. Hansen, T. Thomsen and O. Buchardt, Experientia, 1983, 39, 1063–1072 CrossRef CAS.
- Y. Hatanaka and Y. Sadakane, Curr. Top. Med. Chem., 2002, 2, 271–288 CrossRef CAS PubMed.
- R. B. Woodward, J. Am. Chem. Soc., 1941, 63, 1123–1126 CrossRef CAS.
- L. F. Fieser, M. Fieser and S. Rajagopalan, J. Org. Chem., 1948, 13, 800–806 CrossRef CAS PubMed.
- T. J. Zbacnik, R. E. Holcomb, D. S. Katayama, B. M. Murphy, R. W. Payne, R. C. Coccaro, G. J. Evans, J. E. Matsuura, C. S. Henry and M. C. Manning, J. Pharm. Sci., 2017, 106, 713–733 CrossRef CAS PubMed.
- A. L. Daugherty and R. J. Mrsny, Adv. Drug Delivery Rev., 2006, 58, 686–706 CrossRef CAS PubMed.
- D. Schumacher, C. P. R. Hackenberger, H. Leonhardt and J. Helma, J. Clin. Immunol., 2016, 36, 100–107 CrossRef CAS PubMed.
- C. Berton, S. Klingler, S. Prytuliak and J. P. Holland, npj Imaging, 2024, 2, 1–15 CrossRef.
- J. P. Menzel, B. B. Noble, J. P. Blinco and C. Barner-Kowollik, Nat. Commun., 2021, 12, 1691 CrossRef CAS PubMed.
- E. S. Galbavy, K. Ram and C. Anastasio, J. Photochem. Photobiol., C, 2010, 209, 186–192 CrossRef CAS.
- J. Rabani, H. Mamane, D. Pousty and J. R. Bolton, Photochem. Photobiol., 2021, 97, 873–902 CrossRef CAS PubMed.
- K. L. Willett and R. A. Hites, J. Chem. Educ., 2000, 77, 900 CrossRef CAS.
- F. d’Orchymont and J. P. Holland, Angew. Chem., Int. Ed., 2022, 61, e202204072 CrossRef PubMed.
- S. Klingler and J. P. Holland, Sci. Rep., 2022, 12, 668 CrossRef CAS PubMed.
- P. A. Cieslik, S. Klingler, M. Nolff and J. P. Holland, Chemistry, 2024, 30, e202303805 CrossRef CAS PubMed.
- A. Guillou, A. Ouadi and J. P. Holland, Inorg. Chem. Front., 2022, 9, 3071–3081 RSC.
- D. F. Earley, J. Esteban Flores, A. Guillou and J. P. Holland, Dalton Trans., 2022, 51, 5041–5052 RSC.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- A. Poloukhtine and V. V. Popik, J. Phys. Chem. A, 2006, 110, 1749–1757 CrossRef CAS PubMed.
- D. A. Sutton and V. V. Popik, J. Org. Chem., 2016, 81, 8850–8857 CrossRef CAS PubMed.
- L. Liu, D. Zhang, M. Johnson and N. K. Devaraj, Nat. Chem., 2022, 14, 1078–1085 CrossRef CAS PubMed.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766–16769 CrossRef CAS PubMed.
- J. Deng, X. Wu, G. Guo, X. Zhao and Z. Yu, Org. Biomol. Chem., 2020, 18, 5602–5607 RSC.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- A. Jemas, Y. Xie, J. E. Pigga, J. L. Caplan, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2022, 144, 1647–1662 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- K. Porte, M. Riomet, C. Figliola, D. Audisio and F. Taran, Chem. Rev., 2021, 121, 6718–6743 CrossRef CAS PubMed.
- H. Maus, A. Gellert, O. R. Englert, J.-X. Chen, T. Schirmeister and F. Barthels, RSC Med. Chem., 2023, 14, 2365–2379 RSC.
- P. Rousselot, E. Mappus, T. Blachère, M. R. de Ravel, C. Grenot, C. Tonnelle and C. Y. Cuilleron, Biochemistry, 1997, 36, 7860–7868 CrossRef CAS PubMed.
- L. Rihakova, M. Deraët, M. Auger-Messier, J. Pérodin, A. A. Boucard, G. Guillemette, R. Leduc, P. Lavigne and E. Escher, J. Recept. Signal Transduction, 2002, 22, 297–313 CrossRef CAS PubMed.
- A. V. West, G. Muncipinto, H.-Y. Wu, A. C. Huang, M. T. Labenski, L. H. Jones and C. M. Woo, J. Am. Chem. Soc., 2021, 143, 6691–6700 CrossRef CAS PubMed.
- S. Tamamizu, D. H. Butler, J. A. Lasalde and M. G. McNamee, Biochemistry, 1996, 35, 11773–11781 CrossRef CAS PubMed.
- P. Rotsides, P. J. Lee, N. Webber, K. C. Grasty, J. Beld and P. J. Loll, ACS Bio Med Chem Au, 2024, 4, 86–94 CrossRef CAS PubMed.
- A. Walrant and E. Sachon, Mass Spectrom. Rev., 2024, 1–42 Search PubMed.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS PubMed.
- J. Das, Chem. Rev., 2011, 111, 4405–4417 CrossRef CAS PubMed.
- P. R. Kym, K. E. Carlson and J. A. Katzenellenbogen, Bioconjugate Chem., 1995, 6, 115–122 CrossRef CAS PubMed.
- J. Jiang, Y. Liu, S. Yang, H. Peng, J. Liu, Y.-X. Cheng and N. Li, ACS Med. Chem. Lett., 2021, 12, 1905–1911 CrossRef CAS PubMed.
- Y. Hou, H. Lu, J. Li, Z. Guan, J. Zhang, W. Zhang, C. Yin, L. Sun, Y. Zhang and H. Jiang, Cell Chem. Biol., 2022, 29, 133–144.e20 CrossRef CAS PubMed.
- E. Karaj, S. H. Sindi and L. M. Viranga Tillekeratne, Bioorg. Med. Chem., 2022, 62, 116721 CrossRef CAS PubMed.
- S. Chen, C. Liang, H. Li, W. Yu, M. Prothiwa, D. Kopczynski, S. Loroch, M. Fransen and S. H. L. Verhelst, ACS Chem. Biol., 2023, 18, 686–692 CrossRef CAS PubMed.
- S. Zhang, P. Liu, L. Li, Z. Liu, X. Qian, X. Jiang, W. Sun, L. Wang and E. U. Akkaya, ACS Appl. Mater. Interfaces, 2023, 15, 40280–40291 CrossRef CAS PubMed.
- Y. Ma, J. Wang, X. Pan, J. Zhang and Y. Shan, Drug Dev. Res., 2023, 84, 1142–1158 CrossRef CAS PubMed.
- H. Choi, M. Kim, J. Jang and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22514–22522 CrossRef CAS PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed.
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS PubMed.
- C. Bousch, B. Vreulz, K. Kansal, A. El-Husseini and S. Cecioni, Angew. Chem., Int. Ed., 2023, 62, e202314248 CrossRef CAS PubMed.
- M. D. Holborough-Kerkvliet, G. Mucignato, S. J. Moons, V. Psomiadou, R. S. R. Konada, N. J. Pedowitz, M. R. Pratt, T. Kissel, C. A. M. Koeleman, R. T. N. Tjokrodirijo, P. A. van Veelen, T. Huizinga, K. A. J. van Schie, M. Wuhrer, J. J. Kohler, K. M. Bonger, T. J. Boltje and R. E. M. Toes, Glycobiology, 2023, 33, 732–744 CrossRef PubMed.
- Y. Zhang, J. Tan and Y. Chen, Chem. Commun., 2023, 59, 2413–2420 RSC.
- S. Klingler, R. Fay and J. P. Holland, J. Nucl. Med., 2020, 61, 1072–1078 CrossRef CAS PubMed.
- D. F. Earley, A. Guillou, S. Klingler, R. Fay, M. Gut, F. d’Orchymont, S. Behmaneshfar, L. Reichert and J. P. Holland, JACS Au, 2022, 2, 646–664 CrossRef CAS PubMed.
- M. J. W. D. Vosjan, L. R. Perk, G. W. M. Visser, M. Budde, P. Jurek, G. E. Kiefer and G. A. M. S. van Dongen, Nat. Protoc., 2010, 5, 739–743 CrossRef CAS PubMed.
- J. S. Clovis, A. Eckell, R. Huisgen and R. Sustmann, Chem. Ber., 1967, 100, 60–70 CrossRef CAS.
- R. Darkow, M. Yoshikawa, T. Kitao, G. Tomaschewski and J. Schellenberg, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1657–1664 CrossRef CAS.
- H. Meier and H. Heimgartner, Helv. Chim. Acta, 1985, 68, 1283–1300 CrossRef CAS.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J.-S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- S. Zhao, J. Dai, M. Hu, C. Liu, R. Meng, X. Liu, C. Wang and T. Luo, Chem. Commun., 2016, 52, 4702–4705 RSC.
- J. Zhang, J. Liu, X. Li, Y. Ju, Y. Li, G. Zhang and Y. Li, J. Am. Chem. Soc., 2024, 146, 2122–2131 CrossRef CAS PubMed.
- K. Bach, B. L. H. Beerkens, P. R. A. Zanon and S. M. Hacker, ACS Cent. Sci., 2020, 6, 546–554 CrossRef CAS PubMed.
- N. L. Kjærsgaard, T. B. Nielsen and K. V. Gothelf, ChemBioChem, 2022, 23, e202200245 CrossRef PubMed.
- W. Feng, L. Li, C. Yang, A. Welle, O. Trapp and P. A. Levkin, Angew. Chem., Int. Ed., 2015, 54, 8732–8735 CrossRef CAS PubMed.
- S. Jiang, X. Wu, H. Liu, J. Deng, X. Zhang, Z. Yao, Y. Zheng, B. Li and Z. Yu, ChemPhotoChem, 2020, 4, 327–331 CrossRef CAS.
- J. Zhang, J. Liu, X. Li, Y. Ju, Y. Li, G. Zhang and Y. Li, J. Am. Chem. Soc., 2024, 146, 2122–2131 CrossRef CAS PubMed.
- Y. Wang, W. J. Hu, W. Song, R. K. V. Lim and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS PubMed.
- Z. Yu, L. Y. Ho, Z. Wang and Q. Lin, Bioorg. Med. Chem. Lett., 2011, 21, 5033–5036 CrossRef CAS PubMed.
- P. An, Z. Yu and Q. Lin, Org. Lett., 2013, 15, 5496–5499 CrossRef CAS PubMed.
- P. An, Z. Yu and Q. Lin, Chem. Commun., 2013, 49, 9920–9922 RSC.
- Y. Wang, W. Song, W. J. Hu and Q. Lin, Angew. Chem., Int. Ed., 2009, 48, 5330–5333 CrossRef CAS PubMed.
- G. Ciamician and P. Silber, Berichte Dtsch. Chem. Ges., 1901, 34, 2040–2046 CrossRef CAS.
- F. Sachs and S. Hilpert, Berichte Dtsch. Chem. Ges., 1904, 37, 3425–3431 CrossRef CAS.
- B. G. Gowenlock and G. B. Richter-Addo, Chem. Rev., 2004, 104, 3315–3340 CrossRef CAS PubMed.
- A. Patchornik, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- A. Kastrati and C. G. Bochet, J. Org. Chem., 2019, 84, 7776–7785 CrossRef CAS PubMed.
- C. G. Bochet, Pure Appl. Chem., 2006, 78, 241–247 CrossRef CAS.
- T. Šolomek, C. G. Bochet and T. Bally, Chem. – Eur. J., 2014, 20, 8062–8067 CrossRef PubMed.
- T. Šolomek, S. Mercier, T. Bally and C. G. Bochet, Photochem. Photobiol. Sci., 2012, 11, 548–555 CrossRef PubMed.
- A. Blanc and C. G. Bochet, J. Org. Chem., 2002, 67, 5567–5577 CrossRef CAS PubMed.
- C. G. Bochet and A. Blanc, CRC Handbook of Organic Photochemistry and Photobiology, Two Volume Set, CRC Press, 3rd edn, 2012 Search PubMed.
- A.-D. Guo, D. Wei, H.-J. Nie, H. Hu, C. Peng, S.-T. Li, K.-N. Yan, B.-S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X.-H. Chen, Nat. Commun., 2020, 11, 5472 CrossRef CAS PubMed.
- A.-D. Guo, D. Wei, H.-J. Nie, H. Hu, C. Peng, S.-T. Li, K.-N. Yan, B.-S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X.-H. Chen, Nat. Commun., 2020, 11, 5472 CrossRef CAS PubMed.
- J. Lai, Y. Xu, X. Mu, X. Wu, C. Li, J. Zheng, C. Wu, J. Chen and Y. Zhao, Chem. Commun., 2011, 47, 3822–3824 RSC.
- J. M. Tien and I. M. Hunsberger, J. Am. Chem. Soc., 1955, 77, 6604–6607 CrossRef CAS.
- S. Kolodych, E. Rasolofonjatovo, M. Chaumontet, M.-C. Nevers, C. Créminon and F. Taran, Angew. Chem., Int. Ed., 2013, 52, 12056–12060 CrossRef CAS PubMed.
- M. K. Narayanam, Y. Liang, K. N. Houk and J. M. Murphy, Chem. Sci., 2016, 7, 1257–1261 RSC.
- S. Wallace and J. W. Chin, Chem. Sci., 2014, 5, 1742–1744 RSC.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS PubMed.
- V. X. Truong, J. O. Holloway and C. Barner-Kowollik, Chem. Sci., 2022, 13, 13280–13290 RSC.
- X. Zhang, X. Wu, S. Jiang, J. Gao, Z. Yao, J. Deng, L. Zhang and Z. Yu, Chem. Commun., 2019, 55, 7187–7190 RSC.
- J. Gao, Q. Xiong, X. Wu, J. Deng, X. Zhang, X. Zhao, P. Deng and Z. Yu, Commun. Chem., 2020, 3, 1–10 CrossRef PubMed.
- F. P. Woerner, H. Reimlinger and D. R. Arnold, Angew. Chem., Int. Ed. Engl., 1968, 7, 130–131 CrossRef CAS.
- E. Albrecht, J. Mattay and S. Steenken, J. Am. Chem. Soc., 1997, 119, 11605–11610 CrossRef CAS.
- M.-D. Su, H.-Y. Liao, W.-S. Chung and S.-Y. Chu, J. Org. Chem., 1999, 64, 6710–6716 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 7993–7995 RSC.
- J. O. Mueller, F. G. Schmidt, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 10284–10288 CrossRef CAS PubMed.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chem. – Eur. J., 2021, 27, 5057–5073 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- C. D. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M.-K. von Wrisberg, M.-L. Jokisch and K. Lang, Angew. Chem., Int. Ed., 2019, 58, 15876–15882 CrossRef CAS PubMed.
- A. Maggi, E. Ruivo, J. Fissers, C. Vangestel, S. Chatterjee, J. Joossens, F. Sobott, S. Staelens, S. Stroobants, P. V. D. Veken, L. Wyffels and K. Augustyns, Org. Biomol. Chem., 2016, 14, 7544–7551 RSC.
- T. Läppchen, R. Rossin, T. R. van Mourik, G. Gruntz, F. J. M. Hoeben, R. M. Versteegen, H. M. Janssen, J. Lub and M. S. Robillard, Nucl. Med. Biol., 2017, 55, 19–26 CrossRef PubMed.
- U. M. Battisti, K. Bratteby, J. T. Jørgensen, L. Hvass, V. Shalgunov, H. Mikula, A. Kjær and M. M. Herth, J. Med. Chem., 2021, 64, 15297–15312 CrossRef CAS PubMed.
- T. Auchynnikava, A. Äärelä, H. Liljenbäck, J. Järvinen, P. Andriana, L. Kovacs, J. Rautio, J. Rajander, P. Virta, A. Roivainen, X.-G. Li and A. J. Airaksinen, ACS Omega, 2023, 8, 45326–45336 CrossRef CAS PubMed.
- M. Altai, A. Perols, M. Tsourma, B. Mitran, H. Honarvar, M. Robillard, R. Rossin, W. ten Hoeve, M. Lubberink, A. Orlova, A. E. Karlström and V. Tolmachev, J. Nucl. Med., 2016, 57, 431–436 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- M. M. A. Mitry, F. Greco and H. M. I. Osborn, Chem. – Eur. J., 2023, 29, e202203942 CrossRef CAS PubMed.
- K. Kang, J. Park and E. Kim, Proteome Sci., 2017, 15, 15 CrossRef PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- J. R. Harbour and S. L. Issler, J. Am. Chem. Soc., 1982, 104, 903–905 CrossRef CAS.
- M. Y. Li, C. S. Cline, E. B. Koker, H. H. Carmichael, C. F. Chignell and P. Bilski, Photochem. Photobiol., 2001, 74, 760–764 CrossRef CAS PubMed.
- V. X. Truong, K. M. Tsang, F. Ercole and J. S. Forsythe, Chem. Mater., 2017, 29, 3678–3685 CrossRef CAS.
- T. Posner, Berichte Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS.
- Z. Hu, X. Fan, J. Wang, S. Zhao and X. Han, J. Polym. Res., 2010, 17, 815–820 CrossRef CAS.
- N. Kotzur, B. Briand, M. Beyermann and V. Hagen, J. Am. Chem. Soc., 2009, 131, 16927–16931 CrossRef CAS PubMed.
- C. Bao, L. Zhu, Q. Lin and H. Tian, Adv. Mater., 2015, 27, 1647–1662 CrossRef CAS PubMed.
- M. M. Mahmoodi, S. A. Fisher, R. Y. Tam, P. C. Goff, R. B. Anderson, J. E. Wissinger, D. A. Blank, M. S. Shoichet and M. D. Distefano, Org. Biomol. Chem., 2016, 14, 8289–8300 RSC.
- E. B. Brown, J. B. Shear, S. R. Adams, R. Y. Tsien and W. W. Webb, Biophys. J., 1999, 76, 489–499 CrossRef CAS PubMed.
- D. P. Murale, S. C. Hong, Md. M. Haque and J.-S. Lee, Proteome Sci., 2017, 15, 14 CrossRef PubMed.
- H. Bayley and J. R. Knowles, Biochemistry, 1978, 17, 2420–2423 CrossRef CAS PubMed.
- Y. Jiang, X. Zhang, H. Nie, J. Fan, S. Di, H. Fu, X. Zhang, L. Wang and C. Tang, Nat. Commun., 2024, 15, 6060 CrossRef CAS PubMed.
- M. D. Congdon and J. C. Gildersleeve, Bioconjugate Chem., 2021, 32, 133–142 CrossRef CAS PubMed.
- G. Dorman and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef CAS PubMed.
- J. D. Olszewski, G. Dorman, J. T. Elliott, Y. Hong, D. G. Ahern and G. D. Prestwich, Bioconjugate Chem., 1995, 6, 395–400 CrossRef CAS PubMed.
- G. Garcia, D. C. Chiara, S. Nirthanan, A. K. Hamouda, D. S. Stewart and J. B. Cohen, Biochemistry, 2007, 46, 10296–10307 CrossRef CAS PubMed.
- D. Yahalom, M. Rosenblatt and M. Chorev, in Peptides: The Wave of the Future: Proceedings of the Second International and the Seventeenth American Peptide Symposium, June 9–14, 2001, San Diego, California, USA., ed. M. Lebl and R. A. Houghten, Springer Netherlands, Dordrecht, 2001, pp. 308–309 Search PubMed.
- L.-W. Guo, A. R. Hajipour, M. L. Gavala, M. Arbabian, K. A. Martemyanov, V. Y. Arshavsky and A. E. Ruoho, Bioconjugate Chem., 2005, 16, 685–693 CrossRef CAS PubMed.
- P. Wang and T. A. Spencer, J. Labelled Compd. Radiopharm., 2005, 48, 781–788 CrossRef CAS.
- L. Marcon, M. Wang, Y. Coffinier, F. Le Normand, O. Melnyk, R. Boukherroub and S. Szunerits, Langmuir, 2010, 26, 1075–1080 CrossRef CAS PubMed.
- A. Perols and A. E. Karlström, Bioconjugate Chem., 2014, 25, 481–488 CrossRef CAS PubMed.
- S. Delaney, Á. Nagy, A. E. Karlström and B. M. Zeglis, Mol. Imaging Biol., 2023, 25, 1104–1114 CrossRef CAS PubMed.
- J. Jakubovska, D. Tauraitė and R. Meškys, Sci. Rep., 2018, 8, 16484 CrossRef PubMed.
- M. Clément, S. S. Martin, M.-È. Beaulieu, C. Chamberland, P. Lavigne, R. Leduc, G. Guillemette and E. Escher, J. Biol. Chem., 2005, 280, 27121–27129 CrossRef PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. Bottecchia and T. Noël, Chem. – Eur. J., 2019, 25, 26–42 CrossRef CAS PubMed.
- S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed.
- D. K. Kölmel, J. Meng, M.-H. Tsai, J. Que, R. P. Loach, T. Knauber, J. Wan and M. E. Flanagan, ACS Comb. Sci., 2019, 21, 588–597 CrossRef PubMed.
- N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Nat. Chem., 2023, 15, 101–109 CrossRef CAS PubMed.
- B. X. Li, D. K. Kim, S. Bloom, R. Y.-C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed.
- J. Kim, B. X. Li, R. Y.-C. Huang, J. X. Qiao, W. R. Ewing and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 21260–21266 CrossRef CAS PubMed.
- Y. Weng, C.-J. Su, H. Jiang and C.-W. Chiang, Sci. Rep., 2022, 12, 18994 CrossRef CAS PubMed.
- S. J. Singha Roy, C. Loynd, D. Jewel, S. E. Canarelli, E. D. Ficaretta, Q. A. Pham, E. Weerapana and A. Chatterjee, Angew. Chem., Int. Ed., 2023, 62, e202300961 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- I. S. De Jesus, J. A. C. Vélez, E. F. Pissinati, J. T. M. Correia, D. G. Rivera and M. W. Paixao, Chem. Rec., 2024, 24, e202300322 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |