
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple methodology for constructing ferromagnetically coupled Cr(III) compounds†
Hector W. L.
Fraser
 a,
Lucy
Smythe
a,
Sourav
Dey
b,
Gary S.
Nichol
a,
Stergios
Piligkos
c,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
a,
Lucy
Smythe
a,
Sourav
Dey
b,
Gary S.
Nichol
a,
Stergios
Piligkos
c,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ Scotland, UK. E-mail: E.Brechin@ed.ac.uk; Tel: +44 (0)131-650-7545
bDepartment of Chemistry, Indian Institute of Technology Bombay, Mumbai, 400076, India. E-mail: rajaraman@chem.iitb.ac.in
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100, Copenhagen, Denmark
First published on 30th May 2018
Abstract
A large family of chromium(III) dimers has been synthesised and magneto-structurally characterised using a combination of carboxylate and diethanolamine type ligands. The compounds have the general formula [Cr2(R1-deaH)2(O2CR2)Cl2]Cl where R1 = Me and R2 = H (1), Me (2), CMe3 (3), Ph (4), 3,5-(Cl)2Ph (5), (Me)5Ph (6), R1 = Et and R2 = H (7), Ph (8). The compound [Cr2(Me-deaH)2Cl4] (9) was synthesised in order to study the effect of removing/adding the carboxylate bridge on the observed magnetic behaviour. Direct current (DC) magnetic susceptibility measurements showed ferromagnetic (FM) exchange interactions between the Cr(III) centres in the carboxylate bridged family with coupling constants in the range +0.37 < J < +8.02 cm−1. Removal of the carboxylate to produce the dialkoxide-bridged compound 9 resulted in antiferromagnetic (AFM) exchange between the Cr(III) ions. DFT calculations reveal the ferromagnetic exchange is the result of an orbital counter-complementarity effect occuring upon introduction of the bridging carboxylate.
Introduction
Magneto-structural studies of Cr(III) dimers have been reported extensively since the 1970s, with the vast majority revealing antiferromagnetic exchange between the two metal centres.1–3 Indeed, although more than 40 years have passed since the publication of the first ferromagnetically coupled complex in 1976, namely Na4[Cr(mal)2OH]2,4,5 there have been only three further examples: the di-hydroxo bridged compound [(H2O)4Cr(OH)]2[(CH3)3C6H2SO3]4 reported in 1987,6 the di-fluoro bridged [Cr(BTS)F]2 published in 1996,7 and the most recent example a di-hydroxo bridged species of formula (Ph4P)4[Cr(NCS)4OH]2 published in 2014.8 Larger Cr(III) clusters displaying ferromagnetic exchange are even more rare, with only two examples known.9,10 The first is an unusual sulfide-centred tetrahedron externally bridged with acetates, [Cr4S(O2CCH3)8(H2O)4](BF4)2,9 and the second a family of aesthetically pleasing decametallic wheels of general formula [Cr10(OR)20(O2CR′)10] (R = Me or Et, R′ = Me, Et, CMe3).10 In the latter case the bridging unit between pairs of neighbouring Cr(III) ions consists of two alkoxides and one carboxylate. Perhaps of most interest is the observation that the methoxide bridged versions were found to be ferromagnetically coupled to give S = 15 spin ground states, while the ethoxide bridged analogues were antiferromagnetically coupled to afford diamagnetic S = 0 spin ground states. This rather dramatic difference, albeit with just a small difference in the overall magnitude of |J|, was attributed to a change in electron density at the bridging oxygen atom, with the more inductive ethoxide group leading to stronger antiferromagnetic exchange.11 Unfortunately the development of detailed/quantitative magneto-structural correlations for the wheels was hampered by the low symmetry of the molecules, disorder in the ligand framework, and the presence of significant intermolecular exchange interactions at low temperature.11Note that both tetrametallic and decametallic species are heteroleptic, containing two different bridging ligands, including one carboxylate. This perhaps suggests that exploitation of the orbital counter-complementarity effect may be a promising route toward the development of polymetallic Cr(III) species displaying ferromagnetic exchange interactions.12 To this end we recently began a project to examine the magnetic exchange, and uncover the magneto-structural relationship, between the metal ions in [CrIII(OR)2CrIII] and [CrIII(OR)2(O2CR)CrIII] dimers. Our initial study of the di-alkoxo bridged family of dimers showed that the exchange is dependent upon the Cr–O–Cr bridging angle, the orientation of the dihedral angle formed between the bridging Cr2O2 plane and the O–R vector of the bridging group, and the Cr–O–Cr–O dihedral angle.13 The geometric restrictions placed upon dimers of this type essentially means that all such species will be antiferromagnetically coupled, except when a particular, and rather peculiar combination of such values are satisfied, in agreement with the study of Bendix et al.8
Here we extend our study to the [CrIII(OR)2(O2CR)CrIII] family of dimers 1–8 whose structures are similar to the alkoxide bridged species in ref. 13, but with the addition of a single carboxylate bridge. We also include the carboxylate free analogue 9 for direct comparison. The alkoxides employed are diethanolamine ligands, which have been successfully used as flexible bridging supports in the synthesis of a number of interesting 3d,14,15 3d–3d,16 4f,17 and 3d–4f compounds.18 Its range of bridging modes, including a very unusual non-chelating end-on bridging mode,19 have allowed for a plethora of different structural types,20 as recently reviewed by Perlepes and Tasiopoulos.21 In Cr(III) chemistry they have been shown to be successful in the formation of heterometallic cages, with families of Cr23d2 butterflies,22 Cr2Ln2 butterflies,23 and an octanuclear Cr4Dy4 reported.24 Surprisingly however, no homometallic Cr(III) complexes are registered on the CCDC.†
Experimental
Materials and physical measurements‡
All chemicals were procured from commercial suppliers and used as received (reagent grade). Elemental analyses for C, H, N and Cr were performed by Medac Ltd.Synthesis of [Cr2(Me-deaH)2(O2CH)Cl2]Cl (1)§
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaOMe (0.054 g, 1 mmol) in MeCN (15 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was dissolved in MeCN (10 ml) and added to the first solution. The resulting light blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Slow cooling to room temperature yielded blue rod-shaped crystals, which were suitable for X-ray diffraction. Yield 1.8 mg (0.4% by chromium weight). Anal. calcd (%) for C11H25Cl3Cr2N2O6: C 26.87, H 5.13, Cr 21.15, N 5.70; found: C 26.53, H 5.07, Cr 21.21, N 5.68.Synthesis of [Cr2(Me-deaH)2(O2CMe)Cl2]Cl (2)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2CMe (0.082 g, 1 mmol) in MeCN (25 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Slow cooling to room temperature yielded purple block-shaped crystals, which were suitable for X-ray diffraction. Yield 119.8 mg (23.7% by chromium weight). Anal. calcd (%) for C12H27Cl3Cr2N2O6: C 28.50, H 5.38, Cr 20.56, N 5.54; found: C 29.00, H 4.97, Cr 21.22, N 5.46.Synthesis of [Cr2(Me-deaH)2(O2CCMe3)Cl2]Cl (3)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2CCMe3 (0.124 g, 1 mmol) in MeCN (25 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Slow cooling to room temperature yielded purple plate-shaped crystals, which were suitable for X-ray diffraction. Yield 178.6 mg (32.6% by chromium weight). Anal. calcd (%) for C15H33Cl3Cr2N2O6: C 32.89, H 6.07, Cr 18.98, N 5.11; found: C 32.83, H 5.83, Cr 18.99, N 5.11.Synthesis of [Cr2(Me-deaH)2(O2CPh)Cl2]Cl (4)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2CPh (0.144 g, 1 mmol) in MeCN (25 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Slow cooling to room temperature yielded dark purple block-shaped crystals, which were suitable for X-ray diffraction. Yield 193.8 mg (34.1% by chromium weight). Anal. calcd (%) for C17H29Cl3Cr2N2O6: C 35.96, H 5.15, Cr 18.32, N 4.93; found: C 35.85, H 4.82, Cr 18.18, N 4.81.Synthesis of [Cr2(Me-deaH)2(O2C(Cl)2Ph)Cl2]Cl (5)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2C-3,5-Cl2Ph (0.213 g, 1 mmol) in MeCN (25 ml) with continuous stirring for 3 hours. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting green solution was left to stir overnight at room temperature. A 10 ml sample of the green solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. The solution was then filtered. Layering with 2-propanol yielded dark purple plate-shaped crystals, which were suitable for X-ray diffraction. Yield 50.4 mg (7.9% by chromium weight). Anal. calcd (%) for C17H27Cl5Cr2N2O6: C 32.07, H 4.27, Cr 16.33, N 4.40; found: C 31.26, H 4.41, Cr 15.82, N 4.54.Synthesis of [Cr2(Me-deaH)2(O2C(Me)5Ph)Cl2]Cl (6)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2C(Me)5Ph (0.214 g, 1 mmol) in MeCN (25 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. The blue solution was then filtered, layering with 2-propanol yielded purple coloured plate-shaped crystals, which were suitable for X-ray diffraction. Yield 67.6 mg (10.6% by chromium weight). Anal. calcd (%) for C22H39Cl3Cr2N2O6: C 41.42, H 6.16, Cr 16.30, N 4.39; found: C 40.42, H 6.15, Cr 16.06, N 4.55.Synthesis of [Cr2(Et-deaH)2(O2CH)Cl2]Cl (7)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaOMe (0.054 g, 1 mmol) in MeCN (15 ml) with continuous stirring. Et-deaH2 (0.26 ml, 2 mmol) was also dissolved in MeCN (10 ml) with continuous stirring and added to the first solution. The resulting green solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Vapour diffusion of the filtered blue solution with ethyl acetate yielded dark green block-shaped crystals, which were suitable for X-ray diffraction. Yield 5.5 mg (1.1% by chromium weight). Anal. calcd (%) for C13H29Cl3Cr2N2O6: C 30.04, H 5.62, Cr 20.01, N 5.39; found: C 29.87, H 5.50, Cr 20.65, N 5.22.Synthesis of [Cr2(Et-deaH)2(O2CPh)Cl2]Cl·½Et2O (8)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with NaO2CPh (0.144 g, 1 mmol) in MeCN (25 ml) with continuous stirring. Et-deaH2 (0.26 ml, 2 mmol) was added to the green solution and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Vapour diffusion of the filtered solution with diethyl ether yielded dark purple rod-shaped crystals, which were suitable for X-ray diffraction. Yield 1.2 mg (0.2% by chromium weight). Anal. calcd (%) for C19H33Cl3Cr2N2O6: C 38.30, H 5.58, Cr 17.45, N 4.70; found: C 37.82, H 5.52, Cr 18.01, N 4.51.Synthesis of [Cr2(Me-deaH)2Cl4] (9)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved in MeCN (25 ml) with continuous stirring. Me-deaH2 (0.23 ml, 2 mmol) was added and the resulting blue solution was left to stir overnight at room temperature. A 10 ml sample of this solution was heated in a Teflon-lined autoclave at 100 °C for 12 hours. Slow cooling to room temperature yielded dark green rod-shaped crystals, which were suitable for X-ray diffraction. Yield 41.0 mg (8.5% by chromium weight). Anal. calcd (%) for C10H24Cl4Cr2N2O4: C 24.91, H 5.02, Cr 21.57, N 5.81; found: C 23.94, H 4.65, Cr 22.20, N 5.46.X-ray crystallography
Diffraction data for samples 1–9 were collected using a Rigaku Oxford Diffraction SuperNova diffractometer with MoKα (1–4, 6–9) and CuKα (5) radiation, and are given in Tables S1 and S2.† An Oxford Cryosystems Cryostream 700+ low temperature device was used to maintain a crystal temperature of 120 K. The structures were solved using ShelXT or ShelXS by direct (2–7, 9) or intrinsic phasing solution methods (1, 8) and refined with version 2016/6 of ShelXL interfaced with Olex2.25,26 All non-hydrogen atoms were refined using anisotropic displacement parameters. C-bound H atoms were placed in calculated positions geometrically and refined using the riding model. In structures 1, 2, 4, 6 and 9, O-bound H atoms were identified from a difference Fourier map and refined freely. In 3, 5, 7 and 8, O-bound H atoms were identified from a difference Fourier map and refined as riding, with geometric restraints. CCDC 1579642–1579645 & 1579647–1579651.†SQUID magnetometry
Magnetic susceptibility and magnetisation measurements in the temperature range T = 2–300 K were performed on a Quantum Design MPMS XL SQUID magnetometer equipped with a 7 T dc magnet on finely ground samples of 1–9. The observed paramagnetic susceptibilities were corrected for diamagnetic contributions using Pascal's constants. Susceptibility measurements were performed under magnetic fields of 0.1 T, 0.5 T and 1.0 T. Variable-temperature-variable-field dc magnetisation experiments were carried out in the 2–7 K and 0.5–7.0 T temperature and magnetic field ranges, respectively.Computational details
All theoretical calculations were performed using the hybrid B3LYP functional with Ahlrichs triple-ξ basis set as implemented in Gaussian 09.27–30 This methodology has yielded good numerical estimates of the J values in a variety of systems, including several Cr(III) clusters.13,31–38J values were computed from the energy difference between the HS state and BS state. The energy of the HS state was calculated using the single determinant approach, with the energy of the BS state calculated using the broken symmetry approach developed by Noodleman.39 A quadratic convergence method was employed to obtain the most stable wave function. For orbital analysis using the HTH model,40 β orbitals of the HS state were employed as these orbitals have been shown to better represent the interaction between the metal ions.41,42Results and discussion
Structure description
The structures of compounds 1–9 are given in Fig. 1 (representative 2 and 9) and Fig. S1–9,† with the most pertinent structural parameters listed in Table 1. | ||
| Fig. 1 The molecular structures of complexes 2 (A) and 9 (B). Colour code: Cr = dark blue, O = red, N = light blue, C = black, Cl = yellow, H = white. H-atoms are omitted for clarity, except for those on the protonated arm of the Me-deaH− ligands. H-bonds are drawn as dashed black lines. | ||
| Cr–Cr [Å] | r [Å] | Φ [°] | θ [°] | ψ [°] | |
|---|---|---|---|---|---|
| 1 | 2.967 | 1.957–1.971 | 98.41, 97.87 | 38.76, 40.79 | 18.41 |
| 2 | 2.968 | 1.957–1.972 | 98.55, 97.91 | 37.85, 40.85 | 18.74 |
| 3 | 2.957 | 1.946–1.962 | 98.39, 98.34 | 21.80, 41.35 | 18.72 |
| 4 | 2.959 | 1.956–1.965 | 98.20, 97.71 | 39.46, 39.98 | 18.57 |
| 5 | 2.965 | 1.954–1.965 | 98.39, 98.32 | 40.98, 39.53 | 18.11 |
| 2.958 | 1.944–1.964 | 98.38, 98.24 | 40.35, 30.07 | 17.85 | |
| 6 | 2.955 | 1.942–1.956 | 98.60, 98.37 | 39.63, 18.21 | 18.33 |
| 7 | 2.964 | 1.953–1.960 | 98.57, 98.44 | 33.86, 40.65 | 17.44 |
| 8 | 2.971 | 1.954–1.966 | 98.89, 98.39 | 36.12, 40.04 | 19.66 |
| 9 | 3.023 | 1.961–1.964 | 100.73 | 33.77 | 0.00 |
There are two different structure types. The first structure type is that seen in compounds 1–8; the second that exhibited by compound 9 (Fig. 1). Complexes 1–8 consist of two Cr(III) ions bridged by two μ-OR groups belonging to the singly deprotonated Me-deaH ligands, and one μ-O2CR carboxylate bridge. The three remaining coordination sites on the octahedral Cr(III) ions are occupied by the N atom and protonated O atom from a Me-deaH ligand, and a terminally bonded chloride ion. The carboxylate sits above the [Cr2] moiety (as drawn in Fig. 1) causing the two μ-OR bridges to be pushed downwards, distorting the planarity of the [Cr2O2] unit (Table 1). The protonated OH groups from the R-deaH ligands sit below the [Cr2O2] ‘plane’ and are H-bonded to the charge balancing chloride anion (OH⋯Cl, ∼2.1 Å), which sits midway between the two Cr(III) ions. There are numerous inter-dimer interactions in the extended structures of 1–8, with the closest contacts typically being between the terminal Cl ions and methyl H-atoms of R-deaH ligands on neighbouring molecules (∼2.7–2.9 Å). The most relevant interactions are summarized in Table 2 and Fig. S10–S11.†
| Crystal system | Space group | d H(Me)–Cl [Å] | d π–π [Å] | |
|---|---|---|---|---|
| 1 | Monoclinic | P21/c | 2.777 | — |
| 2 | Monoclinic | P21 | 2.860 | — |
| 3 | Orthorhombic | Pbca | 2.875 | — |
| 4 | Monoclinic | P21/c | 2.703 | 3.524 |
| 5 | Triclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2.641 | 3.495 |
| 6 | Orthorhombic | Pbca | 2.771 | 4.487 |
| 7 | Monoclinic | P21/c | 2.593 | — |
| 8 | Monoclinic | P21/c | 2.893 | 3.359 |
| 9 | Monoclinic | P21/n | 2.806 | — |
The second structure type is seen in compound 9 (Fig. 1B and Table 1), and describes a simple di-alkoxo bridged [CrIII(μ-OR)2CrIII] dimer. Complex 9 crystallises in the monoclinic space group P21/n, with the asymmetric unit containing half the molecule. The two deaH ligands are again singly deprotonated with the N atom and protonated O atom terminally bonded to a Cr(III) ion. The remaining coordination sites on the metals are occupied by four terminally bonded Cl ions, two of which H-bond to the protonated arm of the Me-deaH ligands (O–H⋯Cl, ∼2.3 Å). The result is a fully planar [Cr2(OR)2] bridging unit, in contrast to that seen in 1–8. In the extended structure the closest inter-dimer interactions are between the terminally bonded Cl ions and the Me groups of the Me-deaH ligands at a (C–H⋯Cl) distance of ∼2.9 Å (Table 2 and Fig. S10†).
Magnetic measurements
DC magnetic susceptibility measurements were carried out on powdered polycrystalline samples of compounds 1–9 in applied magnetic fields of 0.1 T, 0.5 T and 1.0 T, over the temperature range T = 2–300 K. Fig. 2 shows the experimental results as the χMT product versus T, where χM is the molar magnetic susceptibility.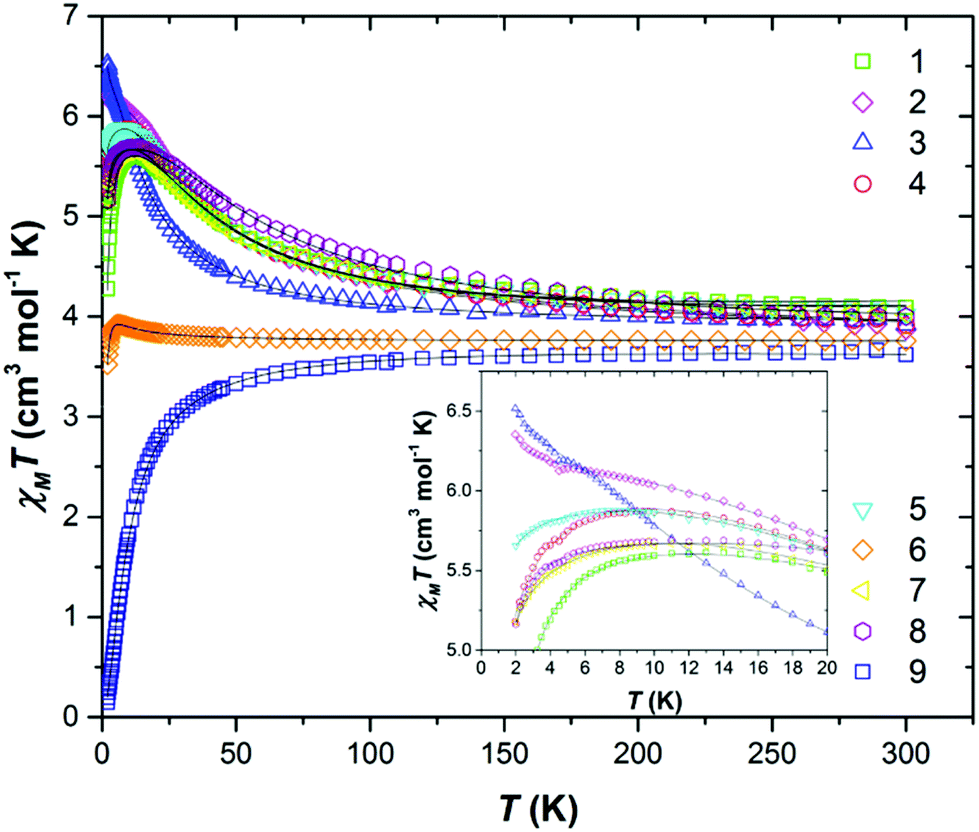 | ||
| Fig. 2 Plot of the χMT product versus T for complexes 1–9 in an applied field of 0.1 T (1–7, 9) and 0.5 T (8). The solid black lines are a fit of the experimental data. See text for details. The inset shows the low temperature region, highlighting the inter-dimer interactions. | ||
At T = 300 K the χMT values were found to be 4.08 (1), 3.87 (2), 3.92 (3), 3.92 (4), 4.02 (5), 3.73 (6), 4.02 (7), 4.00 (8) and 3.62 (9) cm3 K mol−1. The values for compounds 1–8 are slightly higher than that expected for two non-interacting s = 3/2 ions with g = 2.00 (3.75 cm3 K mol−1), while that for compound 9 is somewhat lower. For compound 9 the value of χMT decreases gradually with decreasing temperature to reach a T = 2 K value of 0.15 cm3 K mol−1. This is indicative of a relatively weak antiferromagnetic exchange interaction between the Cr(III) ions, and the stabilisation of a diamagnetic ground state. In contrast, the experimental data for compounds 1–8 show a gradual increase in the value of χMT with decreasing temperature to reach T = 2 K values of 4.28 (peak at 5.61) (1), 6.35 (2), 6.52 (3), 5.18 (peak at 5.87) (4), 5.65 (peak at 5.88) (5), 3.52 (peak at 3.92) (6), 5.17 (peak at 5.69) (7) and 5.16 (peak at 5.69) (8) cm3 K mol−1, indicative of weak ferromagnetic (FM) exchange interactions, leading to S = 3 ground states.
Variable-temperature variable-field (VTVB) magnetisation measurements, performed in the B = 0.5–7 T and T = 2–7 K field and temperature ranges, confirm this assessment (Fig. S12–19†). The χMT vs. T and B vs. H data were fitted simultaneously using isotropic spin-Hamiltonian (1), where the indices i and j refer to each of the Cr(III) ions, μB is the Bohr magneton, B is the applied magnetic field, g is the g-factor of the Cr(III) ions (fixed at g = 2.00), Ŝ is a spin operator and J is the isotropic exchange interaction. Using this model, best-fit parameters were J = +6.04 (1), +4.66 (2), +2.48 (3), +4.86 (4), +4.82 (5), +0.37 (6), +5.41 (7), +8.02 (8) and −1.65 (9) cm−1. These values are listed in Table 3, and compared with values calculated using DFT (vide infra).
 | (1) |
| J exp [cm−1] | J calc [cm−1] | |
|---|---|---|
| 1 | +6.04 | +6.3 |
| 2 | +4.66 | +6.0 |
| 3 | +2.48 | +3.5 |
| 4 | +4.86 | +5.9 |
| 5 | +4.82 | +6.2 |
| 6 | +0.37 | +1.8 |
| 7 | +5.41 | +5.8 |
| 8 | +8.02 | +6.9 |
| 9 | −1.65 | −0.4 |
As can be seen in the inset of Fig. 2, there is some deviation in the very low temperature (T < 10 K) susceptibility data for compounds 1–8. If one assumes that DCr is negligible, then these differences can be attributed to the inter-dimer interactions highlighted above. In the case of compounds 2 and 3 the low temperature χMT value reaches a maximum at ∼11 K and then increases to higher values. In the case of compounds 1 and 4–8, the opposite occurs and a low temperature decrease is observed. This would appear to indicate the presence of FM and AFM intermolecular interactions, respectively – a fit of the data in this region within a mean field approximation affording zJ = −2.742 × 10−3 (1), +5.21 × 10−3 (2), +7.43 × 10−3 (3), −1.06 × 10−2 (4), −9.88 × 10−2 (5), −5.24 × 10−2 (6), −2.44 × 10−1 (7) and −2.15 × 10−2 (8) K. In 2 and 3 the closest intermolecular contacts are mediated via Cl⋯H(Me) interactions. In 4, 5, 6 and 8 these same interactions are present, but occur in combination with numerous π⋯π interactions, the latter most commonly associated with AFM dipolar exchange. The intermolecular nature of the exchange in this temperature regime is confirmed via the field-dependence of the χMT inflection for all three compounds (Fig. S20–27†).
Theoretical studies
To uncover the origin of the ferromagnetic exchange coupling in this class of dimers, DFT calculations have been carried out on complexes 1–9 with the resulting computed J values shown in Table 3. Calculations reproduce both the sign and magnitude of these values extremely well in all cases. Calculations reveal the strongest FM coupling occurs in complexes 1 and 8, and AFM coupling in 9, consistent with the experimental J values. In order to analyse the trend in the observed exchange, one has to analyse all the structural parameters that could control the sign and magnitude of the exchange, namely the Cr–O bond lengths (r = alkoxide; τ = carboxylate), Cr–O–Cr bond angles (Φ), Cr–O–Cr–O dihedral angles (ψ), and the out-of-plane shift between the bridging Cr2O2 plane and the OR vector of the bridging group (θ) (Fig. 3 and Table 1). | ||
| Fig. 3 Diagrammatic representation of the structural parameters influencing the sign and magnitude of magnetic exchange in CrIII dimers. The Cr–Ocarb distances fall in the range 1.967 < τ < 1.989 Å. | ||
For compounds 1–8, the variations in r, τ, Φ and ψ are fairly small, with more variation apparent in θ. Use of the “GHP” model (which considers only r, Φ and θ) previously employed to extract the magneto-structural correlation for the [Cr(OH)]2 core,1 leads to poor reproduction of both the sign and magnitude of J, due to the presence of the additional carboxylate bridge (Table S3†). Indeed, the “GHP” model also failed to reproduce the data for a previously reported family of [Cr(OR)]2 dimers where the hydroxide bridges were replaced with alkoxide bridges.13
The net exchange J has two contributions, JAFM and JFM, denoting the antiferromagnetic and ferromagnetic parts, respectively. Qualitative models based on Hey–Thibeault–Hoffmann (HTH) and Kahn methodologies are widely used to analyse the origin of magnetic exchange in dinuclear complexes.43–47 While the HTH model analyses the differences in energies of symmetric and antisymmetric combinations of the SOMOs, the Kahn model takes into account the overlap between the SOMOs to arrive at a qualitative understanding. Herein, both models are employed in order to probe the origin of the unusual ferromagnetic coupling in 1–8.
Initially a qualitative MO diagram for complex 1 was developed to analyse the energy gap between the symmetric and antisymmetric combinations (Fig. 4). Due to the π-donor Cl ions, the degeneracy of the t2g set of orbitals at the individual Cr(III) centres is lifted, with the dxz and dyz orbitals destabilised compared to the dxy orbital. According to the HTH model, the square of the orbital energies between the symmetric and antisymmetic combinations of dxy, dxz and dyz orbitals is directly proportional to the JAFM component of the exchange. The [(dxy)s–(dxy)as] gap is found to be the largest in all complexes studied (green arrows in Fig. 4), followed by [(dyz)s–(dyz)as] and [(dxz)s–(dxz)as] in all complexes except 2, 3 and 6. For complex 9, the [(dyz)s–(dyz)as] and [(dxz)s–(dxz)as] gaps are nearly the same, suggesting degeneracy. The addition of the bridging carboxylate means that the carboxylate MOs mix with the metal (dyz)s orbital leading to an increase in its energy and a decrease in the [(dyz)s–(dyz)as] energy gap. This is known as an orbital counter-complementarity effect.43,48 This leads to a reduction of the JAFM contribution, and in this instance, to the observation of ferromagnetic exchange. To confirm this, calculations were performed on complexes 1 and 4 where the carboxylate bridges were replaced with water ligands to give [Cr2(Me-deaH)2(H2O)2Cl2]2+ (1a) and [Cr2(Me-deaH)2(H2O)2Cl2]2+ (4a) (Fig. S28†). These models yield J values of +3.2 cm−1 and −0.6 cm−1, respectively. The removal of the carboxylate-bridge clearly enhances the JAFM contribution. This is also reflected in the computed MO energies, where as we move from complex 1 (4) to 1a (4a), the [(dyz)s–(dyz)as] gap increases (vide infra; see Fig. 5 and S29†). This follows for all complexes 1–8 where there is a near linear relationship between the MO energies and the J values.
 | ||
| Fig. 4 Qualitative MO diagram for 1, with corresponding orbital diagrams. The green arrows highlight the difference between symmetric and anti-symmetric orbitals. | ||
 | ||
| Fig. 5 Qualitative MO diagram of 4, 4a and 9 with corresponding orbital diagrams. In 4a the carboxylate-bridge has been replaced by water ligands. | ||
The magnitude of the computed J values for compounds 3 and 6 are found to be comparatively small. This is attributed to the larger energy difference between the symmetric and antisymmetric combinations caused by structural alteration; in particular the small θ values which increase the [(dxy)s–(dxy)as] energy gap. For complex 9, all three orbital energy gaps are relatively large, and the dxy and dyz orbitals are near degenerate due to the removal of the counter-complementarity effect.
We now turn to the analysis of the overlap between SOMOs employing Kahn's methodology. As the AFM part of the exchange is directly proportional to the overlap integral, this can provide a clue to the origin of the FM coupling observed. The computed overlap integrals for complexes 1–9 are given in Table S4.† For complex 1 calculations reveal four dominant overlaps dxy|px|dxy, dxy|px|dyz, dxy|px|dxz and dyz|py|dyz, with dxy|px|dxy contributing to the AFM part of the exchange, JAFM. Spin density plots reveal cubic spin density on the metal ions resulting from the t2g3 configuration of the Cr(III) centres (Fig. 6), with intersection of the directional nodal planes of the three t2g orbitals generating a hole in the face of the ‘cube’. The spin density of the Cr(III) centres is greater than 3.0, implying a dominant spin polarisation mechanism is present. Spin densities on the nitrogen and chlorine atoms show only spin polarisation, with the oxygen atoms exhibiting a mixture of both spin polarisation and spin delocalisation. Strong π-donation of β-electrons from the Cl ions to the empty β-Cr(III) orbitals occurs, leading to residual positive spin densities on the Cl ions in all structures.
 | ||
| Fig. 6 Spin density plots for complex 1 (a) and complex 9 (b). | ||
One potentially pertinent structural difference between compounds 1–8 lies in the nature of the substituent present in the carboxylate group. We therefore investigated the effect of introducing halide atoms to the carboxylates in a model complex of formula [Cr2(Me-deaH)2(O2CX)Cl2]Cl where X = F, Cl, Br and I (Fig. S30†). C–X bond distances were fixed based on literature values. All four substitutions were found to yield J values in the range 6.2–6.4 cm−1, revealing minimal variation in J upon changing the halide. The π* orbital of the carboxylate, which is responsible for the counter-complementarity effect, is unaltered by this substitution resulting in little change in orbital energies.
To further probe the nature of the individual bridges on the magnitude and sign of J, detailed calculations on 1 were undertaken whereby each bridging ligand was removed individually (Fig. 7 and S31†). In model 1b one of the bridging OR groups has been removed leading to super-exchange via the carboxylate and one OR group (Fig. S31a†). This was found to yield very strong antiferromagnetic exchange with J = −98.7 cm−1 due to the very large orbital energy gaps induced. In model 1c both bridging OR groups were removed (Fig. S31b†); this results in a weaker antiferromagnetic exchange, J = −7.7 cm−1. The magnitude of the computed J values correlate nicely to the symmetric and antisymmetric orbital energy gaps shown in Fig. 7. A similar trend is also seen if we compare the orbital energies of complex 4, 4a, and 9 (Fig. 5). Removal of the carboxylate bridge in 4 leads to model 4a, where the energy of the (dyz)as decreases dramatically, falling below (dxy)s and (dyz)s, and becoming the lowest lying energy orbital. The orbital ordering computed for complex 9 is similar to 4a, reiterating that the carboxylate plays a major role in switching the nature of magnetic coupling.
 | ||
| Fig. 7 Evolution of d-orbital energies from complex 1 to model complexes 1a–c. 1a = carboxylate removed; 1b = one bridging –OR group removed; 1c = both bridging –OR groups removed. | ||
To fully comprehend the experimentally observed trend in the J values for this family, a magneto-structural correlation has been developed on a model complex of 1 (Fig. S32†) for four of the structural parameters described earlier: Φ, ψ, θ and τ (there are no significant changes in r in 1–9; Fig. 8).
 | ||
| Fig. 8 Magneto-structural correlations for the four parameters as computed by DFT. (a) Cr–O–Cr angle, Φ (fitting the experimental points gives the relationship J = 9.81 +6.85 exp(−Φ/5.44), R2 = 0.98341). (b) Cr–O–Cr–O dihedral angle, ψ (fitting the experimental points gives the relationship J = 9.65–16.56 exp(−Ψ/10.76), produces R2 = 0.98771). (c) Out of plane displacement of the alkyl group, θ (fitting the experimental points gives the relationship J = −0.23 + 0.18θ, produces R2 = 0.99877) (d) Cr–Ocarb distance, τ. | ||
 | ||
| Fig. 9 Illustration of the splitting of the MOs for the developed magneto-structural correlations for (a) Cr–O–Cr angle (Φ), (b) Cr–O–Cr–O dihedral angle (ψ), (c) out of plane displacement of the alkyl group (θ), and (d) Cr–Ocarb distance (τ). | ||
Conclusions
A family of Cr(III) dimers of general formula [Cr2(R1-deaH)2(O2CR2)Cl2]Cl has been synthesised using a combination of carboxylate and diethanolamine ligands. The compound [Cr2(Me-deaH)2Cl4] was synthesised in order to study the effect of removing/adding the carboxylate bridge. Direct current magnetic susceptibility and magnetisation measurements show ferromagnetic exchange interactions between the Cr(III) centres in the carboxylate bridged family, with coupling constants in the range +0.37 < J < +8.02 cm−1. Removal of the carboxylate to produce the dialkoxide-bridged complex results in antiferromagnetic exchange between the Cr(III) ions. DFT calculations reveal the origin of the ferromagnetic exchange to be an orbital counter-complementarity effect. In particular, the addition of the bridging carboxylate results in the carboxylate MOs mixing with the Cr (dyz)s orbital leading to a decrease in the [(dyz)s–(dyz)as] energy gap.Complexes 1–8 represent only the fifth set of Cr(III) dimers to exhibit ferromagnetic exchange, and the first family of complexes containing multiple members to have a detailed magneto-structural correlation developed. They also provide a clear rationale for the ferromagnetic exchange seen in the polynuclear complexes [Cr4S(O2CCH3)8(H2O)4](BF4)2 and [Cr10(OR)20(O2CR′)10] which hitherto have had no explanation. It also suggests a simple blueprint for the construction of ferromagnetically coupled Cr(III) cages of any nuclearity – exploit the orbital counter-complementarity effect by combining heteroleptic ligand sets, here alkoxides and carboxylates. The [MIII(OR)2(OCO)] bridging unit employed here is an extremely common building block in Mn(III) and Fe(III) chemistry, and we therefore see no reason why similar cages of Cr(III) cannot be made, potentially opening the door to new families of complexes containing multiple Cr(III) ions with controllable exchange interactions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
EKB thanks the EPSRC for funding, and the Velux Foundations for a Villum Visiting Professor programme grant. GR thanks the DST (No. EMR/2014/000247).Notes and references
- J. Glerup, D. J. Hodgson and E. Pedersen, Acta Chem. Scand., Ser. A, 1983, 37, 161–164 CrossRef.
- T. F. Tekut, C. J. O'Connor and R. A. Holwerda, Inorg. Chem., 1993, 32, 324–328 CrossRef.
- (a) H. S. Cheng and L. S. Wang, Phys. Rev. Lett., 1996, 77, 51–54 CrossRef PubMed; (b) S. J. Brudenell, S. J. Crimp, J. K. E. Higgs, B. Moubaraki, K. S. Murray and L. Spiccia, Inorg. Chim. Acta, 1996, 247, 35–41 CrossRef.
- E. D. Estes, R. P. Scaringe, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1976, 15, 1179–1182 CrossRef.
- F. S. Delgado, J. Sanchiz, T. Lopez, F. Lloret, M. Julve and C. Ruiz-Perez, CrystEngComm, 2010, 12, 2711–2721 RSC.
- C. Reber, H. U. Güdel, L. Spiccia and W. Marty, Inorg. Chem., 1987, 26, 3186–3191 CrossRef.
- R. Sanzenbacher, A. Bottcher, H. Elias, M. Huber, W. Haase, J. Glerup, T. B. Jensen, M. Neuburger, M. Zehnder, J. Springborg and C. E. Olsen, Inorg. Chem., 1996, 35, 7493–7499 CrossRef.
- T. J. Morsing, H. Weihe and J. Bendix, Eur. J. Inorg. Chem., 2014, 2014, 5990–5996 CrossRef.
- A. Bino, D. C. Johnston, D. P. Goshorn, T. R. Halbert and E. I. Stiefel, Science, 1988, 241, 1479–1481 Search PubMed.
- E. J. L. McInnes, C. Anson, A. K. Powell, A. J. Thomson, S. Poussereau and R. Sessoli, Chem. Commun., 2001, 89–90 RSC.
- D. M. Low, G. Rajaraman, M. Helliwell, G. Timco, J. van Slageren, R. Sessoli, S. T. Ochsenbein, R. Bircher, C. Dobe, O. Waldmann, H. U. Güdel, M. A. Adams, E. Ruiz, S. Alvarez and E. J. L. McInnes, Chem. – Eur. J., 2006, 12, 1385–1396 CrossRef PubMed.
- Y. Nishida and S. Kida, J. Chem. Soc., Dalton Trans., 1986, 2633–2640 RSC.
- H. W. L. Fraser, G. S. Nichol, G. Velmurugan, G. Rajaraman and E. K. Brechin, Dalton Trans., 2017, 46, 7159–7168 RSC.
- D. Foguet-Albiol, K. A. Abboud and G. Christou, Chem. Commun., 2005, 4282–4284 RSC.
- D. Foguet-Albiol, T. A. O'Brien, W. Wernsdorfer, B. Moulton, M. J. Zaworotko, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2005, 44, 897–901 CrossRef PubMed.
- R. W. Saalfrank, R. Prakash, H. Maid, F. Hampel, F. W. Heinemann, A. X. Trautwein and L. H. Böttger, Chem. – Eur. J., 2006, 12, 2428–2433 CrossRef PubMed.
- G. Abbas, Y. H. Lan, G. E. Kostakis, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2010, 49, 8067–8072 CrossRef PubMed.
- A. M. Ako, V. Mereacre, R. Clérac, I. J. Hewitt, Y. H. Lan, C. E. Anson and A. K. Powell, Dalton Trans., 2007, 5245–5247 RSC.
- O. V. Nesterova, M. V. Kirillova, M. da Silva, R. Boca and A. J. L. Pombeiro, CrystEngComm, 2014, 16, 775–783 RSC.
- J. W. Sharples and D. Collison, Coord. Chem. Rev., 2014, 260, 1–20 CrossRef PubMed.
- A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC.
- V. V. Semenaka, O. V. Nesterova, V. N. Kokozay, R. I. Zybatyuk, O. V. Shishkin, R. Boca, C. J. Gomez-Garcia, J. M. Clemente-Juan and J. Jezierska, Polyhedron, 2010, 29, 1326–1336 CrossRef.
- S. K. Langley, D. P. Wielechowski, N. F. Chilton, B. Moubaraki and K. S. Murray, Inorg. Chem., 2015, 54, 10497–10503 CrossRef PubMed.
- S. K. Langley, C. M. Forsyth, B. Moubaraki and K. S. Murray, Dalton Trans., 2015, 44, 912–915 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. K. Larsen, E. J. L. McInnes, H. El Mkami, J. Overgaard, S. Piligkos, G. Rajaraman, E. Rentschler, A. A. Smith, G. M. Smith, V. Boote, M. Jennings, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 101–105 CrossRef PubMed.
- K. S. Pedersen, G. Lorusso, J. J. Morales, T. Weyhermuller, S. Piligkos, S. K. Singh, D. Larsen, M. Schau-Magnussen, G. Rajaraman, M. Evangelisti and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 2394–2397 CrossRef PubMed.
- M. Atanasov, C. Busche, P. Comba, F. El Hallak, B. Martin, G. Rajaraman, J. van Slageren and H. Wadepohl, Inorg. Chem., 2008, 47, 8112–8125 CrossRef PubMed.
- C. E. Talbot-Eeckelaers, G. Rajaraman, J. Cano, G. Aromí, E. Ruiz and E. K. Brechin, Eur. J. Inorg. Chem., 2006, 3382–3392 CrossRef.
- S. Piligkos, H. Weihe, E. Bill, F. Neese, H. El Mkami, G. M. Smith, D. Collison, G. Rajaraman, G. A. Timco, R. E. P. Winpenny and E. J. L. McInnes, Chem. – Eur. J., 2009, 15, 3152–3167 CrossRef PubMed.
- P. Christian, G. Rajaraman, A. Harrison, J. J. W. McDouall, J. T. Raftery and R. E. P. Winpenny, Dalton Trans., 2004, 1511–1512 RSC.
- S. K. Singh, K. S. Pedersen, M. Sigrist, C. A. Thuesen, M. Schau-Magnussen, H. Mutka, S. Piligkos, H. Weihe, G. Rajaraman and J. Bendix, Chem. Commun., 2013, 49, 5583–5585 RSC.
- T. Gupta and G. Rajaraman, Chem. Commun., 2016, 52, 8972–9008 RSC.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef.
- P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884–4899 CrossRef.
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391–1400 CrossRef.
- E. Ruiz, S. Alvarez, A. Rodríguez-Fortea, P. Alemany, Y. Pouillon and C. Massobrio, in Magnetism: Molecules to Materials II, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001, p. 227 Search PubMed.
- O. Kahn, Molecular Magnetism, Wiley, 1993 Search PubMed.
- C. A. Daul, I. Ciofini and A. Bencini, in Reviews of Modern Quantum Chemistry, World Scientific, 2002, vol. II, pp. 1247–1294 Search PubMed.
- D. Venegas-Yazigi, D. Aravena, E. Spodine, E. Ruiz and S. Alvarez, Coord. Chem. Rev., 2010, 254, 2086–2095 CrossRef.
- O. Kahn and O. Guillou, Research Frontiers in Magnetochemistry, World Scientific, Singapore, 1993 Search PubMed.
- O. Kahn, Angew. Chem., Int. Ed. Engl., 1985, 24, 834–850 CrossRef.
- L. L. Wang, Y. M. Sun, Z. N. Qi and C. B. Liu, J. Phys. Chem. A, 2008, 112, 8418–8422 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details for X-ray crystallography and structure, magnetic measurements and DFT calculations. CCDC 1579642–1579645 and 1579647–1579651. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01963k |
| ‡ All reactions were performed in home-made solvothermal autoclaves. Attempts to make the same complexes under ambient conditions or under reflux were unsuccessful. |
| § The very low yield of 1 can be attributed to the presence of the formate ligand, formed by the in situ oxidation of the MeOH solvent and/or methoxide base. Deliberate addition of formic acid/formate to the reaction mixture did not produce complex 1. |
| This journal is © The Royal Society of Chemistry 2018 |