
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituent effect on the dynamics of the inclusion complex formation between protoberberine alkaloids and cucurbit[7]uril
Zsombor
Miskolczy
 ,
László
Biczók
* and
György
Lendvay
,
László
Biczók
* and
György
Lendvay
Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, P.O. Box 286, 1519 Budapest, Hungary. E-mail: biczok.laszlo@ttk.mta.hu
First published on 14th May 2018
Abstract
The kinetics of entry into and exit from the cavity of cucurbit[7]uril (CB7) was studied by the stopped-flow method in water at various temperatures using pharmaceutically important natural isoquinoline alkaloids as guest compounds. The rate constant of the alkaloid–CB7 complex dissociation was separately determined exploiting the very strong competitive binding of the 1-adamantylammonium cation to CB7. The enthalpy and entropy of activation for the release of berberine from CB7 were significantly lower than those found in the case of the other alkaloids, suggesting different dissociation dynamics. CB7 initially moved from the energetically most stable position encompassing the isoquinoline moiety of berberine to the smaller benzodioxole part, which could leave the macrocycle with less structural distortion. Despite the same thermodynamic parameters of berberine and palmatine inclusion, the latter compound was encapsulated in and set free from CB7 much slower due to the more substantial steric hindrance. The most rapid entry into CB7 and the most exothermic binding were found for epiberberine and coptisine, the alkaloids substituted with the less spacious dioxole ring on their isoquinoline moiety.
Introduction
Cucurbit[n]urils (CBn), pumpkin-shaped molecular containers, have widespread applications in analytical chemistry,1,2 nanotechnology,3–5 catalysis,6,7 drug delivery,8,9 and in the biomedical field10 because they can encapsulate various classes of compounds. The inclusion in their hydrophobic cavity brings about substantial alteration in the acidity,11 solubility,12 fluorescence properties,13 and photostability14,15 of guest molecules. The kinetics of the supramolecular polymerization of two bifunctional monomers possessing cucurbit[7]uril (CB7) and adamantane end groups was controlled using a pH responsive competitive binder compound.16Information on the dissociation rate of CBn complexes is essential for controlled-release applications, in which the guest molecule is expelled from the macrocycle upon external stimuli.17 The knowledge of the kinetics of entry into and exit from the macrocycle is of crucial importance also for the rational design of functional supramolecular systems composed of CBn. A thermodynamically unfavourable state can become stable at room temperature if a substantial activation barrier hinders the structural change. For example, a bistable [2]rotaxane was created in which the movement of cucurbit[6]uril (CB6) along an axle molecule from one site to the other was induced by pH change, but the relocation to the initial state was kinetically impeded.18 A very slow CB6 shuttling was also observed upon protonation within a [3]rotaxane containing a dodecyl spacer, whereas the wheel- and axle-like components of its [3]pseudorotaxane analogue dissociated upon pH enhancement.19 Molecular dynamics simulations contributed to the deeper understanding of the acid-induced movement of CBn in pseudorotaxanes.20 The rate constants of the reversible confinement in CB7 exhibited a substantial solvent effect. The ingression of viologen-containing, dumbbell-shaped guests into CB7 was much faster in aqueous solution than in DMSO.21 A multicomponent system was found in which the kinetics of self-sorting controlled the composition of the products, and the complexes produced immediately after mixing of the reactants differed from those in thermodynamic equilibrium.22
Kinetic measurements provided insight into the mechanistic details of the reactions proceeding in the presence of CBn. Multistep binding of organic ammonium cations in CB6 was inferred from systematic time-resolved NMR studies.23–25 Stopped-flow experiments demonstrated that the colour of an anthocyanin dye was stabilized by CB7 because the decomposition of the flavylium cationic form via hydration could not compete with the rapid host–guest complex formation.26 Bohne and Thomas found two pathways for the formation of the CB7 complex of the 2-aminoanthracenium cation.27 Namely, not only the direct inclusion but also the protonation of the CB7-included 2-aminoanthracene took place. The slower deprotonation in the cavity of CB7 than in water caused the substantial pKa increase upon complexation.
Despite the significance of the rate of the entry and exit in the application of CB7 in the delivery and controlled release of drugs, little information is available on the dynamics of reversible binding in this cavitand,26–31 and only two studies32,33 reported the activation parameters of the formation and dissociation of the inclusion complexes. The substantial fluorescence quantum yield enhancement of berberine34 (B), a biologically active isoquinoline alkaloid, upon embedment in CB7 and CB8 was used to monitor the complexation in real time. The effect of the alteration of the macrocycle size on the stoichiometry of association, the kinetics of the elementary reaction steps, and the thermodynamics of binding was revealed.32,35
Since it is unknown how the modification of the molecular structure affects the dynamics of inclusion in CB7, we performed systematic kinetic studies. The dimensions of the encapsulated protoberberine alkaloids were tuned by changing the methoxy and dioxole substituents in the 2,3 and 9,10 positions, as shown in Scheme 1. The main objective was to unravel how the steric demand of the guest modifies the mechanism and the rate constants of the ingression into and exit from CB7. The revealed substituent effect on the kinetic parameters provided new insights into the dominant inclusion pathway, and the results may contribute to the development of new strategies to control the guest release or capture.
 | ||
| Scheme 1 Structural formula of the guest and host compounds. | ||
Experimental
P (Sigma-Aldrich) was chromatographed on a silica gel (Merck) column eluting with ethanol, EB (ChemFaces), C (CFM Oskar Tropitzsch GmbH) and 1-adamantylamine (A, Sigma-Aldrich) were used without further purification. The latter compound was fully protonated at neutral pH since its conjugated acid had a pKa value36 of 10.55. The alkaloids were chloride salts, and their concentrations were determined spectrophotometrically using the molar absorption coefficient of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 344 nm, 24240 M−1 cm−1 at 355 nm and 22130 M−1 cm−1 at 356 nm in water for P,37 EB, and C,38 respectively. High-purity CB7 was kindly provided by Dr Anthony I. Day (University of New South Wales, Canberra, Australia). Experiments were performed in double distilled water.
000 M−1 cm−1 at 344 nm, 24240 M−1 cm−1 at 355 nm and 22130 M−1 cm−1 at 356 nm in water for P,37 EB, and C,38 respectively. High-purity CB7 was kindly provided by Dr Anthony I. Day (University of New South Wales, Canberra, Australia). Experiments were performed in double distilled water.
The UV-visible absorption spectra were measured with an Agilent Technologies Cary60 spectrophotometer. Corrected fluorescence spectra were recorded with a Jobin-Yvon Fluoromax-P spectrofluorometer. Stopped-flow measurements were carried out with the same instrument using an Applied Photophysics RX2000 rapid mixing accessory and a pneumatic drive. The temperature of the samples was controlled with a Julabo F25-ED thermostat. The results of spectrophotometric and fluorescence titrations as well as stopped-flow measurements were analysed with homemade programs written in MATLAB 7.9. Isothermal calorimetric titrations were performed with a VP-ITC (MicroCal) instrument at 298 K. All solutions were degassed prior to titration. Alkaloid solution (∼90 μM) was injected stepwise (10 μl each) from the computer-controlled microsyringe at an interval of 270 s into ∼9 μM CB7 solutions, while stirring at 300 rpm. The dilution heat, which was obtained by adding alkaloid solution into water under the same conditions as in the titration of CB7, was subtracted from the integrated heat evolved per injection, and the result was divided by the mole number of the injectant. Data were analysed using the one-site binding model and Origin 7 software. The first data point was always removed. The measurements were repeated three times.
Molecular modeling calculations were first carried out with the RM1 semiempirical method using HyperChem 8.0.10 software. Further optimization was performed using density functional theory. The M05-2X functional combination and the 6-31G(d,p) basis set were used for geometry optimization followed by the calculation of vibrational frequencies. From the obtained molecular structures the procedure was repeated with the CAM-B3LYP functional and the same basis set. Rather small geometry changes were seen in the second optimization. The selected functionals are suited for the description of dispersion effects, which are important in host–guest complexes. The GAUSSIAN 09 suite of programs39 was employed for all DFT calculations.
Results
Steady-state fluorescence studies
A previous study demonstrated that the quantum yields of fluorescence and intersystem crossing grow with the diminution of the solvent polarity in the case of B and P due to the deceleration of the internal conversion.40 Similarly, the confinement of alkaloids in the nonpolar and extremely nonpolarizable interior of CB7 significantly slows down the radiationless deactivation from the singlet excited state to the ground state, leading to substantial fluorescence intensity enhancement. Fig. 1 shows that the studied alkaloids weakly emit in water, but an intense fluorescence band with a hypsochromic shift is observed upon inclusion in CB7, analogous to that found for B.34 The gradual increase of the CB7 amount brings about an incipient steep emission intensity rise followed by a levelling off. The lines in the insets to Fig. 1 represent the results of the nonlinear least-squares analysis of the experimental data assuming 1:1 inclusion stoichiometry. The best fits provided K = 2.6 × 107, 5.0 × 108, and 6.2 × 108 M−1 for the equilibrium constants of P, C, and EB confinement in CB7 at 298 K, respectively. The K value of P encapsulation agrees within the limit of experimental errors with that reported for B binding32 (2.4 × 107 M−1). In contrast, a more than an order of magnitude larger K was found for C and EB. When the fluorescence titrations with CB7 were repeated at ∼300-fold larger alkaloid concentrations, the intersection of the two linear regimes in the fluorescence intensity vs. CB7 concentration plots appeared at an equimolar alkaloid:CB7 molar ratio, confirming 1:1 complexation.
 | ||
| Fig. 1 Fluorescence spectra of 0.33 μM P, 0.038 μM EB, and 0.028 μM C in water (red line) and in the presence of a large excess of CB7 (blue line). Insets display the growth of the fluorescence intensity with CB7 concentration. | ||
Structure of the CB7 complexes
The results of the previously published NMR experiments have demonstrated that the isoquinoline moiety of protoberberine alkaloids is encapsulated in the hydrophobic core of CB7, whereas the other end of the guest molecule is located outside the host.34,41–43 To gain further information on the structure of the inclusion complexes, density functional theory (DFT) calculations were performed. The energy-minimized structures obtained at the CAM-B3LYP/6-31G(d,p) level of theory (Fig. 2) demonstrate that the alkaloid orientation in the CB7 cavity remains practically the same irrespective of the substituents. Only partial embedment occurs because the alkaloids are much longer than the height of the host. The substituted isoquinoline moiety is confined and its heterocyclic nitrogen is located in the vicinity of one of the carbonyl-rimmed portals of the macrocycle. The neighbouring two methoxy groups occupy more space than the dioxole ring. Hence, the methoxy at position 10 somewhat protrudes from CB7, whereas the dioxole substituent of the isoquinoline ring is fully embedded. The symmetric round cross section of the host is distorted and becomes oval upon inclusion of alkaloids. | ||
| Fig. 2 Energy-minimized structures for B-CB7 (A), P-CB7 (B), C-CB7 (C), and EB-CB7 (D) complexes calculated by the DFT method. Colour codes: oxygen, red; nitrogen, light blue; carbon, brown; hydrogen, white. | ||
The theoretical calculations allowed us to estimate the relative complexation energies of the four inclusion complexes. As the geometry optimization was performed in vacuum, the results cannot be directly compared with the experimental data obtained in aqueous solution. The difference between the calculated and measured values reflects not only the neat solvation effects but also the enthalpy change due to the release of high-energy water from the CB7 interior upon encapsulation of an alkaloid. Table 1 summarizes the derived binding enthalpies (ΔH) and the energy differences between the molecular structures within the complex and in the free form for alkaloids (Estrain(alkaloid)) and CB7 (Estrain(CB7)) at the CAM-B3LYP/6-31G(d,p) level of theory. The positive values of the latter two quantities indicate that the inclusion complex formation is accompanied by steric distortion of both the host and the guest. The larger Estrain(alkaloid) and (Estrain(CB7)) values upon binding of P and B arise from the more substantial deformation caused by the encapsulation of the more spacious dimethoxyisoquinoline moiety compared with that of the smaller dioxoloisoquinoline unit of C and EB. The smaller enthalpy gain for P and B inclusion in CB7 agrees with the experimental results (vide infra). The considerable difference between the calculated and measured ΔH of host–guest binding implies that the release of water from the CB7 cavity and from the solvate shells significantly alters the binding enthalpy.
| Palmatine | Berberine | Coptisine | Epiberberine | |
|---|---|---|---|---|
| ΔH/kJ mol−1 | −216 | −221 | −235 | −241 |
| E strain(alkaloid)/kJ mol−1 | 27.9 | 28.6 | 20.5 | 20.6 |
| E strain(CB7)/kJ mol−1 | 11.8 | 11.4 | 7.1 | 3.9 |
Stopped-flow experiments at 298 K
The rate constant of the dissociation of the inclusion complex was selectively determined by the accurate method described in our previous paper.33 The 1-Adamantylammonium cation (AH+) was employed as a competitive binder, whose CB7 complex had such a large stability constant44 (K = 1.7 × 1014 M−1) that the dissociation of AH+–CB7 is negligible on the timescale of the measurements. Fig. 3 shows the fluorescence intensity alteration after rapid addition of AH+ to the solution of alkaloid–CB7 complexes. AH+ was used in substantial excess to ensure that the ingression of AH+ is much quicker into CB7 than the entry of the alkaloid. In this case, the release of the alkaloid from CB7 is followed by the fast association of the empty cavitand with AH+, which blocks the reverse formation of the alkaloid–CB7 complex. Hence, the fluorescence intensity (I) decays in Fig. 3 directly reflect the decrease of the concentration of the CB7-bound alkaloid due to dissociation. As this is a first-order process, its rate constant (kout) can be calculated by the nonlinear least-squares fit of an exponential function to the stopped-flow traces.| I = I0exp(−koutt) + I∞ | (1) |
 | ||
| Fig. 3 Diminution of the normalized fluorescence intensity after rapid 1:1 mixing of (1) 0.22 μM P + 0.23 μM CB7 with 4.7 μM Ada (λexcitation = 345 nm, λdetection = 500 nm), (2) 0.16 μM B + 0.16 μM CB7 with 4.0 μM Ada (λexcitation = 340 nm, λdetection = 500 nm), and (3) 0.22 μM C + 0.22 μM CB7 with 4.6 μM Ada (λexcitation = 350 nm, λdetection = 530 nm) aqueous solutions at 298 K. The black lines show the results of the nonlinear least-squares analysis. | ||
The derived kout values are summarized in Table 2 together with other kinetic and thermodynamic parameters. The fluorescence intensity vs. time profiles for EB-CB7 and C-CB7 dissociations were indistinguishable. Therefore, the data only for the latter complex are presented in Fig. 3. The alkaloids possessing methoxy units on their isoquinoline moiety are released much slower from CB7 than EB and C, in which the pair of methoxy groups are replaced by a less spacious dioxole ring. The egression of B is significantly faster than that of P indicating that the introduction of the dioxole substituent instead of the methoxy groups in positions 2 and 3 also considerably accelerates the exit from the host.
| Palmatine | Berberinea | Coptisine | Epiberberine | |
|---|---|---|---|---|
| a Ref. 32. b K = kin/kout. c ΔH = ΔH‡in − ΔH‡out. d ΔS = ΔS‡in − ΔS‡out. e Average of the results of steady-state fluorescence titrations and ITC measurements. | ||||
| k in at 298 K/M−1 s−1 | (5.4 ± 0.4) × 106 | (19 ± 1) × 106 | (71 ± 8) × 106 | (69 ± 8) × 106 |
| ΔH‡in/kJ mol−1 | 37.9 ± 2 | 32.0 ± 2 | 32.7 ± 3 | 32.4 ± 3 |
| ΔS‡in/J mol−1 K−1 | 11 ± 3 | 2.6 ± 2 | 15 ± 4 | 14 ± 4 |
| k out at 298 K/s−1 | 0.20 ± 0.04 | 0.81 ± 0.08 | 0.12 ± 0.02 | 0.10 ± 0.02 |
| ΔH‡out/kJ mol−1 | 76.8 ± 2 | 69.0 ± 2 | 75.8 ± 2 | 75.7 ± 2 |
| ΔS‡out/J mol−1 K−1 | 2 ± 1 | −19 ± 4 | −8 ± 2 | −10 ± 2 |
| K at 298 K/107 M−1 from kinetic datab | 2.7 ± 0.3 | 2.3 ± 0.4 | 59 ± 15 | 69 ± 16 |
| K at 298 K/107 M−1 from fluorescence titration | 2.6 ± 0.3 | 2.4 ± 0.3 | 50 ± 15 | 62 ± 15 |
| ΔH at 298 K/kJ mol−1 ITC measurement | −37 ± 2 | −38 ± 2 | −45 ± 2 | −45 ± 2 |
| ΔH/kJ mol−1 from kinetic datac | −39 ± 3 | −37 ± 3 | −43 ± 5 | −43 ± 5 |
| ΔS/J mol−1 K−1 from kinetic datad | 9 ± 4 | 15 ± 4e | 23 ± 7 | 24 ± 7 |
Fig. 4 displays the rise of the fluorescence intensity after mixing alkaloid and CB7 solutions. Low reactant concentrations were used to slow down the bimolecular encapsulation to the time range accessible to the stopped-flow technique. Because of the substantial fluorescence yield enhancement upon host–guest association, the growth of the fluorescence intensity indicated the rise of the inclusion complex concentration. The signals levelled off when equilibrium was established. The embedment of EB and C in CB7 showed the fastest and practically identical kinetics. In contrast, a marked difference was observed between the rates of B and P confinement, as indicated by the initial slopes of the signals. The kinetics of 1:1 complexation is defined by the following relationship:
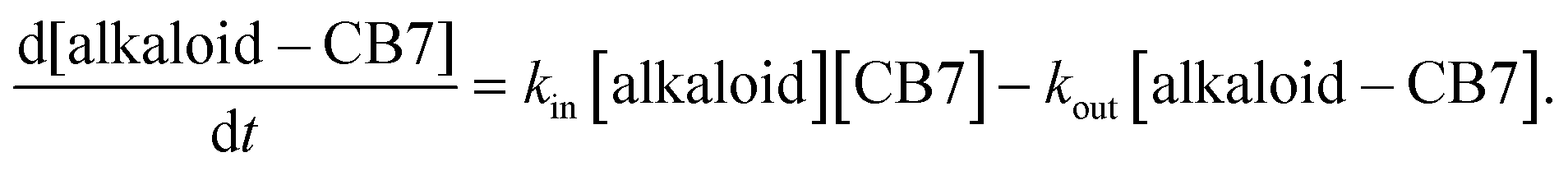 | (2) |
 | ||
| Fig. 4 Rise of the normalized fluorescence intensities after rapid mixing of (1) 0.26 μM P with 0.27 μM CB7 (λexcitation = 345 nm, λdetection = 500 nm), (2) 0.23 μM B with 0.27 μM CB7 (λexcitation = 345 nm, λdetection = 500 nm), and (3) 0.28 μM C with 0.27 μM CB7 (λexcitation = 350 nm, λdetection = 530 nm) aqueous solutions at 298 K. The black lines represent the results of the nonlinear least-squares fit. | ||
To obtain the rate constant of ingression (kin), the numerical solution of this differential equation was fitted to the results of stopped-flow measurements by keeping the previously determined kout constant. The calculated kin values are included in Table 2. The rate constants of the entry into the macrocycle exhibit more than one order of magnitude variation in the series of the structurally very similar alkaloids. The data in Table 2 demonstrate that the binding constants derived from the ratio of the rate constants of ingression and egression (K = kin/kout) agree well with those obtained from steady-state fluorescence titrations.
Effect of temperature change on the rate constants
To gain information on the properties of the transition states and the activation parameters, the aforementioned stopped-flow studies were repeated with the four closely related alkaloids at various temperatures. The Arrhenius plots of the rate constants of entry into and exit from CB7 are presented in Fig. 5 and 6, respectively. Surprisingly, almost parallel linear dependences appear for both the encapsulation and the release of the various alkaloids, suggesting that the activation energies of the formation and dissociation of the inclusion complexes change with the molecular structure of the guests to a small extent. The activation energies (Ea) and A factors were obtained from the nonlinear least-squares fit of the temperature dependence of the rate constants with the Arrhenius equation k = Aexp(−Ea/RT), where R represents the gas constant.
 | ||
| Fig. 5 Logarithm of the rate constants of alkaloid ingression into CB7 in water as a function of reciprocal temperature. Data for berberine were taken from ref. 32. | ||
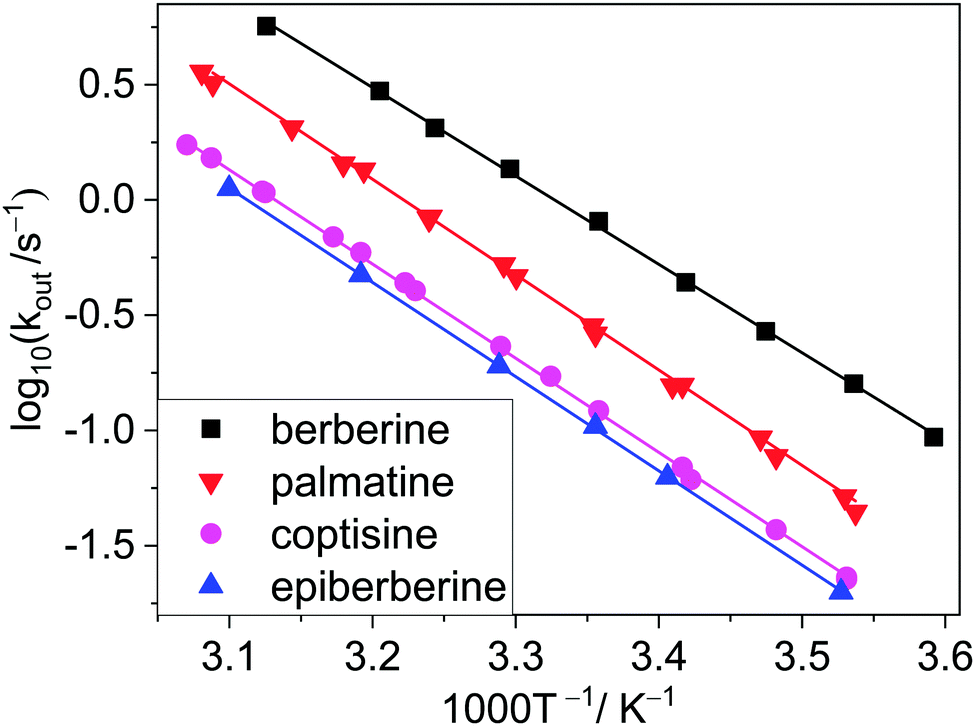 | ||
| Fig. 6 Arrhenius plots of the rate constants of alkaloid egression from CB7 in water. Data for berberine were taken from ref. 32. | ||
On the basis of the transition-state theory, the standard entropy (ΔS‡) and enthalpy (ΔH‡) of activation can be derived as follows:
| ΔH‡ = Ea − RT | (3) |
where e, kB, and h stand for Euler's number, and the Boltzmann and Planck constants, respectively. The transmission coefficient (κ) is usually regarded as unity. The calculated activation parameters are incorporated in Table 2 together with the thermodynamic parameters of host–guest complex formation calculated as a difference of the activation parameters of the entry into and exit from CB7.

Isothermal calorimetric titrations
To corroborate the enthalpy change of complexation derived from kinetic measurements (ΔH = ΔH‡in − ΔH‡out), the evolved reaction heat was directly measured by isothermal titration calorimetry (ITC). The amount of heat developed after each alkaloid injection into ∼9 μM CB7 solution was integrated and corrected with the very small dilution heat. The initial part of the enthalpograms (Fig. 7) indicated practically constant heat generation because of the complete complexation of the added alkaloid. The released heat measured in this domain provided the enthalpy change upon host–guest binding. The ΔH of C and EB inclusion in CB7 did not differ but P embedment was considerably less exothermic. At further successive ligand injections, the heat evolution gradually vanished due to the lessening number of free CB7 macrocycles. The enthalpograms exhibited an inflexion point at a 1:1 component ratio confirming that a CB7 molecule was able to encapsulate only a single alkaloid. To decrease the number of fitted parameters the binding constants determined by fluorescence titrations were kept constant in the non-linear least-squares analysis of the ITC data. The ΔH values obtained from the best fits are in accordance with those determined from activation enthalpies (Table 2).
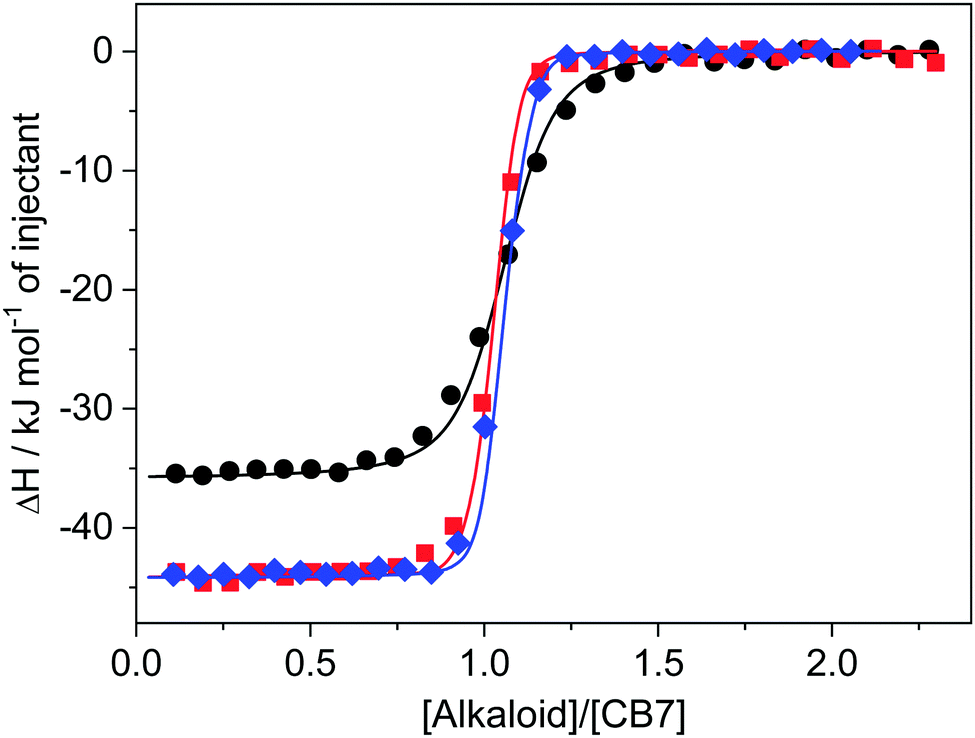 | ||
Fig. 7 Heat release upon addition of ∼90 μM palmatine (●), coptisine ( ), and epiberberine ( ), and epiberberine ( ) solution to ∼9 μM CB7 in water. ) solution to ∼9 μM CB7 in water. | ||
Discussion
The studied alkaloids are encapsulated in CB7 much slower than the diffusion-controlled limit for which a rate constant of 6.5 × 109 M−1 s−1 was reported45 in water at 293 K. The substantial activation enthalpy of ingression (ΔH‡in) implies that the passage of alkaloids through the CB7 portal is sterically hindered. Since the carbonyl-fringed opening of the cavitand is tight compared to the alkaloid dimensions, oval distortion of CB7 is the prerequisite for the penetration into the macrocycle. The energy needed for the partial desolvation of the reactants to reach the transition state also contributes to ΔH‡in. Alkaloid inclusion in CB7 has a positive activation entropy (ΔS‡in) evidencing that the entropy gain upon removal of water from the host interior and the solvate shell of the guest overcompensates the entropy penalty paid upon the ordering of the reactants into the transition state.The kinetic and thermodynamic parameters of C and EB inclusion in CB7 barely differ (Table 2) because the dioxoloisoquinoline moiety of these alkaloids is embedded in the apolar core of the host. The replacement of the dioxole ring with a pair of methoxy groups brings about marked changes in the binding characteristics. The binding affinity becomes much lower due to the diminution of the enthalpy and entropy gains upon complexation, but the ΔH and ΔS of P-CB7 and B-CB7 complex formation exhibit insignificant differences. Hence, we can conclude that only the residues on the encapsulated isoquinoline segment influence the driving force of host–guest interaction. The considerably less exothermic confinement of the 9,10-dimethoxy-substituted alkaloids in CB7 relative to that of the 9,10-dioxole derivatives originates primarily from steric reasons. The bulkier two methoxy groups less easily fit into the CB7 interior than the smaller dioxole ring and may distort slightly more the symmetrical structure of the host. This renders the confinement of P and B enthalpically less favourable. The pair of methoxy substituents have more degrees of conformational freedom to be limited than a dioxole ring. As a consequence, the ΔS of P and B inclusion is below that found for C and EB complexation. The binding of alkaloids into CB7 always leads to a positive ΔS because the entropy benefit in the release of water from the cavitand and from the solvate shell of alkaloids overbalances the entropy loss due to association.
Table 2 demonstrates the lack of correlation between the kinetic and thermodynamic data in the case of the complexation of P and B. The latter compound enters into and exits from CB7 much faster than P despite the practically identical ΔH and ΔS values of P and B binding. This is particularly surprising because the isoquinoline parts of the two alkaloids, which are embedded in CB7, have an identical substitution pattern. The difference in the rate constants arises primarily from the smaller activation enthalpies of B ingression and egression compared with those of P. The lower energy barrier for B insertion suggests different dynamics for complex formation. The penetration of the less bulky benzodioxole part of B is kinetically favourable because less deformation of the reactants is needed (Scheme 2). The entry of the more spacious other end of B is impeded by the narrow portal of CB7 to a larger extent. After the penetration of the benzodioxole moiety into CB7, the macrocycle rapidly slips to the isoquinoline unit producing the thermodynamically most stable structure. As the less spacious benzodioxole segment approaches the transition state, less water is liberated from the core and the hydrophilic portal of CB7 as well as from the alkaloid hydrate shell than in the case of the ingression of the 9,10-dimethoxyisoquinoline part of P. Hence, the activation entropy of B-CB7 formation is lower than that of the association to P-CB7. Since both ends of P are substituted with pairs of methoxy groups, a considerably larger ΔH‡in is observed because the entry into CB7 is hindered irrespective of the dynamics of inclusion. The practically equal activation parameters of EB and C binding in CB7 indicate that the entry of the dioxoloisoquinoline moiety into the host is preferred because this pathway requires the least structural modification.
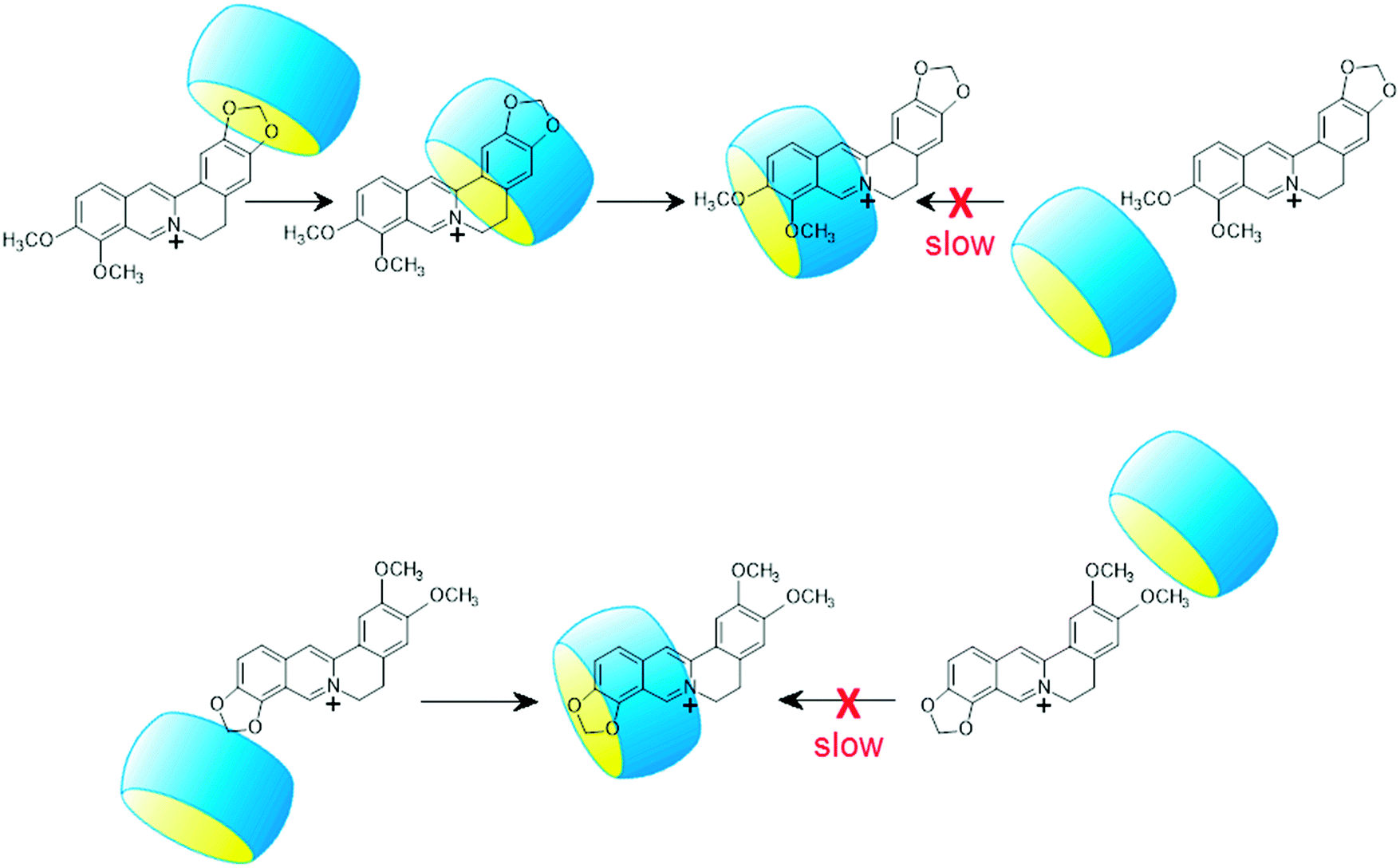 | ||
| Scheme 2 Illustration of the inclusion dynamics for B (upper panel) and EB (lower panel) | ||
The activation enthalpy of alkaloid exit from CB7 (ΔH‡out) is the sum of the activation enthalpy of ingression (ΔH‡in) and the absolute value of the enthalpy change upon host–guest binding (|ΔH|). The ΔH‡out values of EB-CB7 and C-CB7 dissociations correspond to the value found for P-CB7 because the smaller enthalpy gain upon self-assembly to P-CB7 is offset by the larger ΔH‡in of the process. Such a compensation does not occur for B-CB7, and therefore, ΔH‡out is lower by 7 kJ mol−1 than that of the egression of other alkaloids. The 4-fold quicker release of B from CB7 at 298 K compared to the removal of P is attributed to the lower ΔH‡out of B. The dynamics of B egress from CB7 essentially differs from the dissociation of the other alkaloid–CB7 complexes. First, CB7 moves from 9,10-dimethoxyisoquinoline to the benzodioxole part of B, and after that, the smaller benzodioxole moiety leaves the macrocycle because this path needs less structural modification in the transition state due to the smaller size of the benzodioxole moiety. This two-step exit mechanism has no energetic advantage in the case of P-CB7 dissociation because both ends of the guest are substituted by methoxy pairs. The dioxoloisoquinolines of EB and C probably directly egress from CB7 because the steric impediment plays a less important role when the less spacious dioxole is linked to the isoquinoline moiety. The activation entropy of B release from CB7 substantially deviates from that of other alkaloids confirming the different type of transition state for B-CB7 dissociation.
Multistep inclusion mechanisms have been reported for the encapsulation in cucurbit[6]uril (CB6). Nau and coworkers demonstrated that first the cationic moiety of alkylammonium coordinates with the carbonyl rim of CB6, and the hydrophobic moiety of the guest subsequently enters into the host in a ‘‘flip-flop’’ manner.23,24 The formation of both exclusion and inclusion complexes was also observed for 4-methylbenzylammonium binding to CB646 and for 2-aminoanthracenium confinement in CB7.27 Two position of CB6 were found when it slipped through the N1,N6-disubstituted hexane-1,6-diammonium cation.25 In these cases, the two types of complexes are kinetically distinguishable and have their own well-defined energy minimum. In contrast, the two stages of B entry into CB7 probably are not separated by an energy barrier or the energy barrier between them is very low. The ΔH‡in values of alkaloid–CB7 complex formation are considerably lower than those observed by Masson and coworkers25 for the slippage of N1-substituted N6-(3,5-dimethoxybenzyl)hexane-1,6-diammonium cations in CB6 from the initially bound to the thermodynamically stable state (117–88 kJ mol−1). The much larger latter ΔH‡in quantities indicate the more difficult translation of the guests within the confined space of the CB6 interior.
Conclusions
The formation and dissociation of the highly stable protoberberine alkaloid–CB7 1:1 complexes are sterically hindered and possess substantial energy barriers, which depend on the relative size of the guest and the portal of the host macrocycle. Even minor modification in the substitution pattern of alkaloids significantly alters the rate constant of the reversible binding in CB7. In the thermodynamically most favourable complex structure, the isoquinoline part of the studied alkaloids is encompassed by the host. The introduction of bulky substituents into this moiety can essentially alter the dynamics of encapsulation. The sterically less demanding part of the guest passes through the narrow opening of CB7 and afterwards, the most stable position is reached. Kinetic measurements not only provide unique information on the mechanistic details of inclusion complex formation but also represent an appealing alternative to establish the large equilibrium constants of host–guest binding in a simple and reliable way. Large binding constants are usually obtained by multistep sequential competition titrations, in which the proper selection of the series of guests is challenging and the experimental errors accumulate.47 The determination of such association constants is more accurate by simply taking the ratio of the ingression and egression rate constants.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Research, Development and Innovation Office (NKFIH, Grant K123995), the BIONANO GINOP-2.3.2-15-2016-00017 project and the János Bolyai Research Scholarship Program of the Hungarian Academy of Sciences (to ZM). The authors are grateful for the support.References
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef PubMed.
- G. Ghale, A. G. Lanctôt, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762–2765 CrossRef PubMed.
- S. Gurbuz, M. Idris and D. Tuncel, Org. Biomol. Chem., 2015, 13, 330–347 Search PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef PubMed.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106–2115 CrossRef PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178–12190 CrossRef PubMed.
- A. I. Day and J. G. Collins, Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed.
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, Isr. J. Chem., 2011, 51, 616–624 CrossRef.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef PubMed.
- D. H. Macartney, Isr. J. Chem., 2011, 51, 600–615 CrossRef.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef PubMed.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061–1070 Search PubMed.
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2011, 115, 12577–12583 CrossRef PubMed.
- H. Chen, Z. Huang, H. Wu, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16575–16578 CrossRef PubMed.
- L. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2017, 87, 1–12 CrossRef.
- J. Wook Lee, K. Kim and K. Kim, Chem. Commun., 2001, 1042–1043 RSC.
- D. Tuncel and M. Katterle, Chem. – Eur. J., 2008, 14, 4110–4116 CrossRef PubMed.
- L. D. Malhis, K. Bodoor, K. I. Assaf, N. A. Al-Sakhen and M. I. El-Barghouthi, Comput. Theor. Chem., 2015, 1066, 104–112 CrossRef.
- S. Senler, B. Cheng and A. E. Kaifer, Org. Lett., 2014, 16, 5834–5837 CrossRef PubMed.
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef PubMed.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 3155–3160 CrossRef PubMed.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed.
- X. Ling, E. L. Samuel, D. L. Patchell and E. Masson, Org. Lett., 2010, 12, 2730–2733 CrossRef PubMed.
- B. Held, H. Tang, P. Natarajan, C. P. da Silva, V. de Oliveira Silva, C. Bohne and F. H. Quina, Photochem. Photobiol. Sci., 2016, 15, 752–757 Search PubMed.
- S. S. Thomas and C. Bohne, Faraday Discuss., 2015, 185, 381–398 RSC.
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef PubMed.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2007, 13, 7223–7233 CrossRef PubMed.
- Z. Miskolczy, J. G. Harangozó, L. Biczók, V. Wintgens, C. Lorthioir and C. Amiel, Photochem. Photobiol. Sci., 2014, 13, 499–508 Search PubMed.
- M. H. Tootoonchi, S. Yi and A. E. Kaifer, J. Am. Chem. Soc., 2013, 135, 10804–10809 CrossRef PubMed.
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2014, 118, 2499–2505 CrossRef PubMed.
- Z. Miskolczy, L. Biczók and I. Jablonkai, Phys. Chem. Chem. Phys., 2017, 19, 766–773 RSC.
- M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410–3416 Search PubMed.
- Z. Miskolczy and L. Biczók, Phys. Chem. Chem. Phys., 2014, 16, 20147–20156 RSC.
- X. Yao, X. Wang, T. Jiang, X. Ma and H. Tian, Langmuir, 2015, 31, 13647–13654 CrossRef PubMed.
- M. Maiti and G. S. Kumar, J. Nucleic Acids, 2010, 593408 Search PubMed.
- J. Slavik and L. Slavikova, Collect. Czech. Chem. Commun., 1984, 49, 704–711 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. Görner, Z. Miskolczy, M. Megyesi and L. Biczók, Photochem. Photobiol., 2011, 87, 284–291 CrossRef PubMed.
- N. Dong, L.-N. Cheng, X.-L. Wang, Q. Li, C.-Y. Dai and Z. Tao, Talanta, 2011, 84, 684–689 CrossRef PubMed.
- C. Li, J. Li and X. Jia, Org. Biomol. Chem., 2009, 7, 2699–2703 Search PubMed.
- G.-Q. Wang, Y.-F. Qin, L.-M. Du, J.-F. Li, X. Jing, Y.-X. Chang and H. Wu, Spectrochim. Acta, Part A, 2012, 98, 275–281 CrossRef PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, FL, 3rd edn, 2006 Search PubMed.
- R. Hoffmann, W. Knoche, C. Fenn and H.-J. Buschmann, J. Chem. Soc., Faraday Trans., 1994, 90, 1507–1511 RSC.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. M. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. M. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef PubMed.
| This journal is © the Owner Societies 2018 |