
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing p-type dye sensitised solar cell photovoltages using polyoxometalates†
Hani
El Moll‡
a,
Fiona A.
Black
b,
Christopher J.
Wood
b,
Ahmed
Al-Yasari
ac,
Anil
Reddy Marri
a,
Igor V.
Sazanovich
d,
Elizabeth A.
Gibson
 *b and
John
Fielden
*a
*b and
John
Fielden
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: John.Fielden@uea.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle-upon-Tyne, NE1 7RU, UK. E-mail: Elizabeth.Gibson@Newcastle.ac.uk
cCollege of Pharmacy, University of Kerbala, Kerbala, Iraq
dCentral Laser Facility, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK. E-mail: Igor.Sazanovich@stfc.ac.uk
First published on 3rd July 2017
Abstract
Lindqvist polyoxometalate (POM) additives increase VOC in p-type DSSCs by up to 140%, yielding substantial efficiency gains for poorly matched dyes and redox mediators. For better dye/electrolyte combinations, these gains are typically outweighed by losses in JSC. Charge lifetime and transient IR measurements show that this is due to retardation of both recombination and electron transfer to the mediator, and a positive shift in the NiO valence band edge. The POMs also show their own, limited sensitizing effect.
Dye-sensitized photocathodes (p-DSSCs) were first proposed as a way to increase the efficiency of dye-sensitized solar cells over a decade ago.1 Such photocathodes would replace the platinised counter electrode of the Grätzel cell, and by harvesting lower energy photons than the photoanode increase spectral coverage as well as increasing photovoltage. In this way, the p-DSSC could help drive advances from the current DSSC record of 13%,2 towards (or beyond) the 25% attained by crystalline Si:3 the resulting tandem DSSCs would have a maximum theoretical efficiency of 43%, vs. 33% for a single junction device. But to date, no tandem DSSC has exceeded the power conversion efficiency of a state-of-the-art, single-junction TiO2 n-DSSC, because they are limited by the poor performance of the series connected p-DSSC. The only p-DSSC power conversion efficiencies >1% have been achieved using redox mediators whose negative redox potentials would severely reduce the photovoltage produced by the TiO2 side of any tandem cell.4 With the more typical n-DSSC I3−/I− redox couple, the p-DSSC record efficiency is just 0.61%.5
The poor performance of p-DSSCs results from the small energy difference between the valence band (VB) of the p-type semiconductor (usually NiO) and redox mediator, rapid back transfer of photogenerated holes from NiO to the electrolyte, and slow hole diffusion through NiO.6 Consequently, open circuit voltages (VOC) are low – the maximum obtained with I3−/I− is 350 mV using special high crystallinity NiO and a dye with a highly extended conjugated system.7 More typically, VOC is around 100 mV, and rarely exceeds 150 mV. At the same time, fill factors are low (ca. 30%) and photocurrents (JSC) are moderate (up to 8.2 mA cm−2).8 While the improvements achieved through sensitizer design have been impressive,4,6,8 the necessary step change in p-DSSC performance will require exploration of new strategies, and materials.
One simple modification employed in n-DSSCs, but little investigated in p-DSSCs,9a is the use of coadsorbents such as cholic acid derivatives and alkyl phosphonic acids.9b–e These can prevent dye aggregation, and passivate the surface helping to suppress recombination, reduce dark current and increase stability and efficiency. We expect that in p-DSSCs co-adsorbents can play a similar role, while potentially also providing a remote electron acceptor, isolated from the NiO VB that can relay electrons to the mediator and impede recombination. This strategy may have advantages over incorporating the acceptor into the dye – it facilitates a rapid, combinatorial approach to new cells and should reduce communication between electron and hole.
Owing to their fast electrochemistry, tunable potentials and stability to redox cycling,10 polyoxometalates (POMs) appear a good but as yet untested choice of electron acceptor coadsorbent for p-DSSCs. Ultrafast electron transfer (ET) has been observed between POMs and dyes on TiO2,11 and in n-DSSCs, POMs can enhance performance by increasing electron lifetimes.12 They also perform well as replacements or modifiers for Pt counter electrodes,13 and their charge may alter the position of the NiO VB. Here, we demonstrate for the first time that POMs produce dramatic increases in VOC in NiO p-DSSCs. At the same time, they increase JSC for poorly matched dye/redox mediator combinations, and decrease JSC when dye and mediator are better matched. Together, J–V, charge extraction/lifetime and time-resolved IR (TRIR) data indicate a combination of VB shift, POM electron acceptor behaviour and surface screening are responsible, establishing a model for how POMs modify NiO photoelectrochemistry.
We investigated the tetrabutylammonium salt of Lindqvist anion, [NBu4]2[Mo6O19] (1), and its organoimido derivatives 2, 3 and 4 (Fig. 1). As a highly reversible single electron acceptor with a less negative redox potential (−0.12 V vs. NHE), than those of typical photosensitizers (ca. −0.8 V vs. NHE for P1, −0.36 V vs. NHE for CAD3), the Lindqvist hexamolybdate seems an ideal starting point for study of POMs in the p-DSSC. Attaching an organic group to this cluster anion raises the redox level to around −0.3 V vs. NHE (Table S3, ESI†), providing additional driving force for onward ET to the redox mediator (ca. −0.2 V vs., NHE for I3−/I2˙−, +0.3 V vs. NHE for Co3+/2+),14 and enables introduction of binding groups (pyridine, carboxylate) for NiO. Organoimido compounds 2 to 4 were synthesized from 1 and the appropriate amine using established DCC mediated coupling methods (see ESI†). Of these, 2 and 3 are new compounds, and an X-ray crystal structure of 3 is presented in the ESI.†
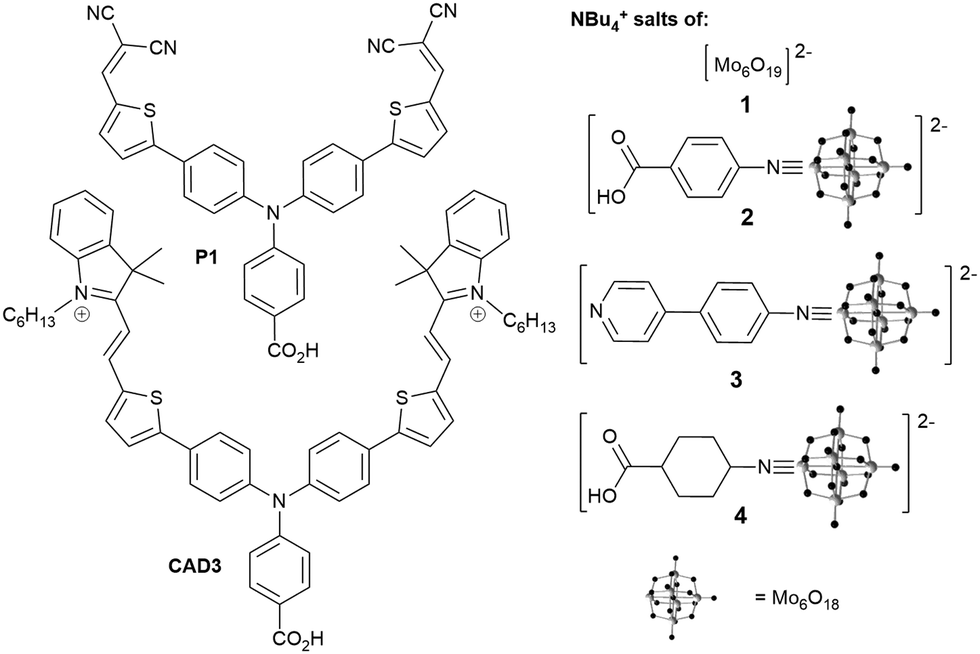 | ||
| Fig. 1 Sensitizer dyes and POM coadsorbents investigated in this study. | ||
As 2 and 3 both show significant visible absorption (between 400 and 450 nm, Fig. S2, ESI†), we first tested the ability of the POM compounds to sensitize NiO with no dye (Table 1 and Fig. S3, S4, ESI†). With I−/I3− electrolyte, 1 to 4 all produce significant VOC (105 to 142 mV, i.e. comparable to or better than typical dyes with this electrolyte) but small JSC (≤0.14 mA cm−1). The highest performing (η = 0.0072%) was 3, likely a result of its more extended visible absorption and the ability of the pyridine group to bind NiO.15 Changing I−/I3− for Co(tBBPY)32+/3+ (tBBPY = 4,4′-di-tert-butyl-2,2′-dipyridyl) further increases VOC, beyond 200 mV for 3 (η = 0.0088%), but generally decreases JSC. This is the first evidence that organoimido-POM charge transfer transitions can sensitize wide-band gap semiconductors, and it is noteworthy that η for NiO-3-Co(tBBPY)32+/3+ is comparable to that of NiO-CAD3-Co(tBBPY)32+/3+ (0.01%) despite far inferior light absorption. This is because ET to Co(tBBPY)32+/3+, for both P1 and CAD3, is slow. Even so, the weak effects observed make it clear that in the coadsorbed dye/POM systems discussed below, all but a small fraction of the observed JSC must result from sensitization by the dye, not the POM.
| POM | Electrolyte | J SC (mA cm−2) | V OC (mV) | FF (%) | η (%) |
|---|---|---|---|---|---|
| a 0.1 M I2, 1 M LiI in MeCN. b 0.1 M CoII(tBBPY)3(ClO4)2, 0.1 M CoIII(tBBPY)3(ClO4)3, 0.1 M LiClO4 in PC. tBBPY = 4,4′-di-tert-butyl-2,2′-dipyridyl. | |||||
| 1 | I−/I3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
0.057 | 109 | 40 | 0.0025 |
| 2 | I−/I3−a |
0.044 | 105 | 36 | 0.0017 |
| 3 | I−/I3−a |
0.14 | 134 | 39 | 0.0072 |
| 4 | I−/I3−a |
0.057 | 142 | 38 | 0.0031 |
| 1 | Co2+/3+b |
0.039 | 23 | 26 | 0.0002 |
| 2 | Co2+/3+b |
0.029 | 130 | 42 | 0.0016 |
| 3 | Co2+/3+b |
0.12 | 208 | 35 | 0.0088 |
| 4 | Co2+/3+b |
0.071 | 164 | 38 | 0.0044 |
To investigate POMs as coadsorbents, we treated NiO films sensitized with either the P1 or CAD3 dyes with 1 to 4 (Table 2). The high point-of-zero charge of NiO favours binding of negatively charged POM species, and evidence of POM binding is seen through Mo peaks in EDX (ESI,† Tables S4 and S5). The films were assembled into dye-sensitized solar cells with I−/I3− or Co(tBBPY)32+/3+ (tBBPY = 4,4′-di-tert-butyl-2,2′-dipyridyl) electrolytes – to provide one system (I−/I3−) where dye and electrolyte are well matched, and another (Co(tBBPY)32+/3+) where they are not. In this way, the influence of the POM's ability to act as an electron accepting, band shifting or screening additive can be probed under different conditions.
| Dye|POM | Electrolyte | J SC (mA cm−2) | V OC (mV) | FF (%) | η (%) |
|---|---|---|---|---|---|
| a 0.1 M I2, 1 M LiI in MeCN. b 0.1 M CoII(tBBPY)3(ClO4)2, 0.1 M CoIII(tBBPY)3(ClO4)3, 0.1 M LiClO4 in PC. tBBPY = 4,4′-di-tert-butyl-2,2′-dipyridyl. | |||||
| CAD3 | I−/I3−a |
4.44 | 93 | 30 | 0.123 |
| CAD3|1 | I−/I3−a |
2.35 | 146 | 37 | 0.127 |
| CAD3|2 | I−/I3−a |
2.14 | 127 | 38 | 0.103 |
| CAD3|3 | I−/I3−a |
1.48 | 117 | 41 | 0.072 |
| CAD3|4 | I−/I3−a |
2.08 | 153 | 39 | 0.123 |
| P1 | I−/I3−a |
2.32 | 110 | 34 | 0.087 |
| P1|1 | I−/I3−a |
1.16 | 138 | 35 | 0.056 |
| P1|2 | I−/I3−a |
1.01 | 130 | 35 | 0.046 |
| P1|4 | I−/I3−a |
0.98 | 160 | 37 | 0.058 |
| CAD3 | Co2+/3+b |
0.40 | 100 | 24 | 0.010 |
| CAD3|1 | Co2+/3+b |
0.58 | 200 | 23 | 0.027 |
| CAD3|2 | Co2+/3+b |
0.75 | 189 | 25 | 0.035 |
| CAD3|4 | Co2+/3+b |
0.74 | 208 | 25 | 0.038 |
| P1 | Co2+/3+b |
0.58 | 146 | 27 | 0.023 |
| P1|1 | Co2+/3+b |
No current | — | — | — |
| P1|2 | Co2+/3+b |
0.67 | 198 | 28 | 0.036 |
| P1|4 | Co2+/3+b |
0.78 | 209 | 27 | 0.044 |
In the presence of the POMs, the VOC of the P1-sensitized cells with the I−/I3− mediator increased (18 to 45%), but JSC fell by at least 50% (J–V curves Fig. S6, ESI†). No clear trends in VOC or JSCvs. redox potential were observed between the different POM coadsorbents. This is surprising as ET from 1 to I3− is thermodynamically uphill, whereas there is a ca. 100–200 mV driving force for ET from 2–4. Similar results were observed with the CAD3|I−/I3− cells, despite ET from CAD3 to 2–4 being almost thermodynamically neutral. All four POMs increased VOC significantly (by 65%, to 153 mV for 4), relative to CAD3 only, but accompanying falls in JSC (43 to 67%) left overall η similar (0.072–0.127%) to the NiO-CAD3 baseline (0.123%) (J–V curves Fig. S5, ESI†). As 3 performed significantly worse with CAD3 than the other POMs it was not investigated further.
Electron transfer from 1–4 to Co(tBBPY)32+/3+ is thermodynamically favourable. Replacing I−/I3− with Co(tBBPY)32+/3+ led to a further ca. 50 mV increase in VOC with CAD3 (up to 108%, to 208 mV) vs. POM free systems (J–V curves Fig. S7 and S8, ESI†), while with P1 the VOC increases were similar to those obtained with I−/I3−. Moreover, with the cobalt electrolyte, the presence of the POM coadsorbent slightly increased JSC in most cases vs. POM free cells. Consequently, η increased, up to near fourfold but from a very low base.
Using the POMs as an electrolyte additive (0.01 or 0.02 M) with both I−/I3− and Co2+/3+ (Table 3 and Fig. S9, S10 in ESI†) produced more pronounced changes in VOC and JSC to the coadsorption experiments. VOC values of up to 293 mV were achieved (P1|1|Co2+/3+) but the highest JSC were only 0.57 mA cm−2. With Co2+/3+, JSC values were also decreased vs. POM free cells (in contrast to adsorbed POM systems), with the exception of 4. This is likely because the total amount of POM present is higher than in the co-adsorption experiments. Electrolytes with only dissolved POM produce cells with zero JSC and low (<100 mV) VOC. This eliminates the possibility that the POM provides an alternative redox mediator, with a more negative redox level (hence increased VOC) but poor electron transfer kinetics.
| POM|electrolyte | J SC (mA cm−2) | V OC (mV) | FF (%) | η (%) |
|---|---|---|---|---|
| a 0.1 M I2, 1 M LiI in PC. b 0.1 M CoII(tBBPY)3(ClO4)2, 0.1 M CoIII(tBBPY)3(ClO4)3, 0.1 M LiClO4, in PC. Added POM 0.02 M (1 to 3) or 0.01 M (4). | ||||
|
1|I−/I3−a |
0.57 | 256 | 30 | 0.043 |
|
2|I−/I3−a |
0.39 | 233 | 27 | 0.025 |
|
3|I−/I3−a |
0.47 | 238 | 33 | 0.037 |
|
4|I−/I3−a |
0.54 | 265 | 31 | 0.045 |
|
1|Co2+/3+b |
0.37 | 293 | 37 | 0.040 |
|
2|Co2+/3+b |
0.32 | 228 | 29 | 0.022 |
|
3|Co2+/3+b |
0.36 | 275 | 24 | 0.033 |
|
4|Co2+/3+b |
0.57 | 230 | 29 | 0.038 |
Two explanations for the increase in VOC with 1–4 are a positive valence band shift due to electrostatic interactions at the NiO|POM interface, and decreased recombination at the NiO|electrolyte interface. It is apparent that a band shift occurs as the charge density vs. photovoltage plot (Fig. 2, top) indicates a shift in the quasi Fermi level for equivalent charge density, and dark current plots (Fig. S5, ESI†) show a >60 mV positive shift in the presence of 1–4. This band shift seems independent of the different electron withdrawing/donating properties of the anchoring group. As it is also independent of any electron transfer or steric/electrostatic screening effects of the POM, it must contribute to the VOC increase in every system studied.
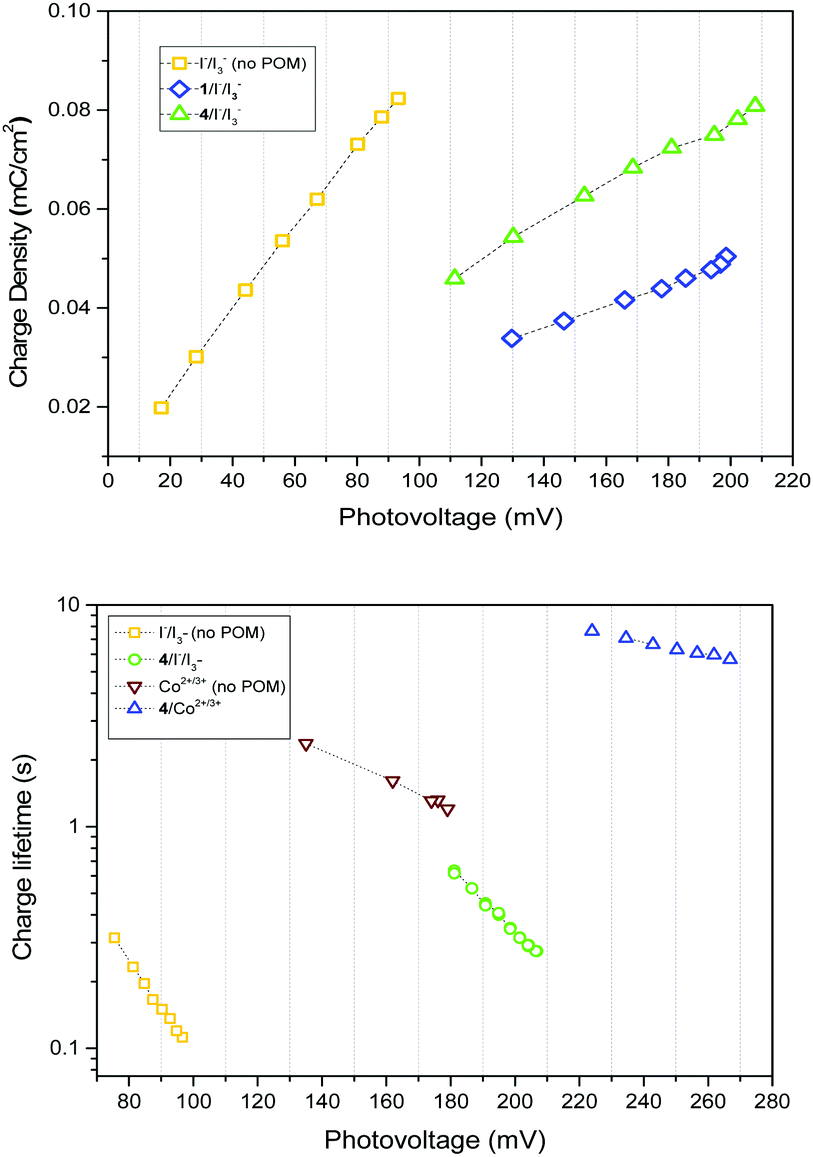 | ||
| Fig. 2 Top: Charge density vs. photovoltage plot for NiO|P1 films in the presence and absence of 1 and 4 in an I−/I3− electrolyte, indicating a positive shift in the Fermi level when 1 and 4 are present. Bottom: Charge lifetime vs. photovoltage of NiO|P1 cells, with and without 4 added to I−/I3− and Co2+/3+ electrolytes. | ||
There is also evidence for the POMs preventing recombination to NiO. Charge-lifetime vs. photovoltage plots (Fig. 2, bottom) reveal a significant increase in charge lifetime for the P1|4 co-adsorbed cells compared to POM free cells, reaching ca. 8 s for 4 with Co2+/3+ and 0.6 s for 4 with I−/I3− (ca. 2 s and 0.3 s respectively without POM). A similar effect is seen in charge lifetime vs. charge density (Fig. S11, ESI†). It is unclear whether this is due to the POM screening the charge in NiO from the electrolyte (electrostatically or sterically), or intercepting the charge and inefficiently releasing it. However, TRIR provides evidence for ET from P1 to 4, by monitoring recombination of the charge-separated state (P1−|NiO+) (Fig. 3). Laser excitation at 540 nm of NiO|P1 adsorbed on NiO (CaF2 window) gave spectra and recombination rates consistent with those previously reported.16 For NiO|P1|4, the spectral features did not change, but transient species generally decayed on a shorter timescale, with an average lifetime of τ = 33 ps. (τ = 65 ps for P1|NiO). This suggests that the POM quenches P1−|NiO+, forming POM−|P1|NiO+.
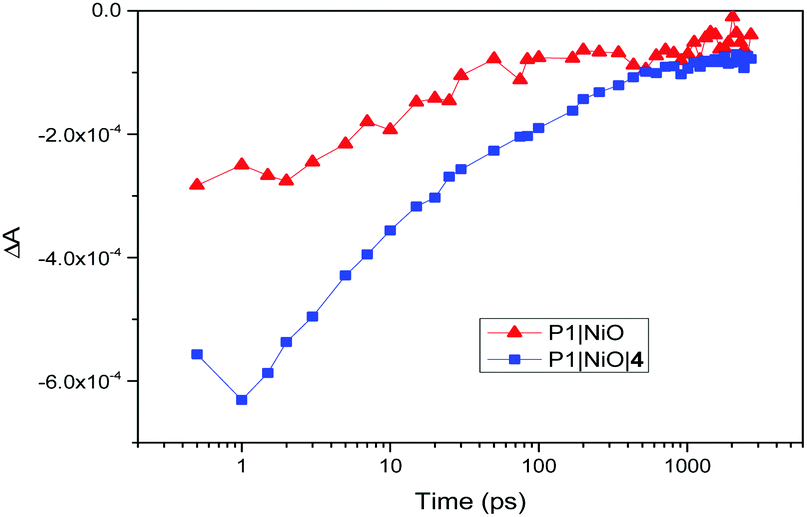 | ||
| Fig. 3 Recovery of the ground state of P1 after laser excitation at 540 nm, taken at 1435 cm−1 (P1|NiO, red triangles) and 1433 cm−1 (P1|NiO|4, blue squares). | ||
TRIR also showed no clear effect on the rate of formation of P1−|NiO+, suggesting that charge-injection is not diminished in the presence of 4 and that lowered photocurrents must be due to other factors. Fully delineating these is beyond the scope of this manuscript, but in real solar cell operating conditions the dye likely transfers electrons to the mediator and POM in parallel. Thus, the POM may increase or decrease JSC depending on how much it increases electron–hole pair lifetimes, and the relative rates of ET to the mediator from dye and POM. Increased JSC for POM coadsorbents with Co2+/3+, that also tend to increase in line with POM redox level, are therefore consistent with an increased electron–hole pair lifetime, and suggest comparable POM-to-mediator and dye-to-mediator ET rates. Decreased JSC for POMs with I/I3− indicate that the increased electron–hole pair lifetime in this system does not compensate for a POM-to-mediator ET that is slow compared to dye-to-mediator ET, and in some cases uphill. A role for screening of the dye from the mediator is also implied by CAD3|4 with I−/I3−: JSC is lowered even though the chance of reducing the POM is minimised by a 40 mV uphill dye-to-POM ET, and 200 mV downhill POM-to-mediator ET.
Previously, films based on Al substituted13a and lacunary silicotungstate Keggin POMs13b showed high efficiency for I−/I3− reduction. It is therefore likely that {Mo6} to mediator electron transfer is less efficient than for these POMs, at least in part due to a weak driving force. The POMs may also adsorb at poorly accessible locations on NiO due to their negative charge (anchoring groups do not seem to influence loading, ESI†). These questions may be addressed in future by expanding the range of POMs studied, and developing deposition methods that ensure location of the POMs close to the electrolyte interface.
In conclusion, using polyoxometalates as coadsorbents in p-DSSCs significantly increases VOC, over 100% in some cases, but with well-matched dye/mediator combinations an accompanying ≥50% decrease in photocurrent eliminates any gain in efficiency. When electron transfer between dye and mediator is poor, however, introduction of POMs at appropriate loadings increases both VOC and JSC. Charge lifetime, charge extraction and TRIR measurements indicate that this behaviour results from a combination of a valence band shift, slowed recombination to NiO, and impeded electron transfer to the mediator due both to electron trapping by the POM, and screening of the dye and NiO from the electrolyte. The results therefore show that charged, electron accepting coadsorbents may provide a simple combinatorial route to suppressing recombination and increasing photovoltage, but materials that enable more efficient transfer of electrons on to the mediator are required. They point towards other applications, e.g. in solar capacitors, photochromic devices or photoelectrochemistry that would benefit from enhanced charge lifetimes.
We thank the UK EPSRC National Mass Spectrometry Facility for MS, and the STFC for access to CLF ULTRA. JF and HEM thank the EPSRC for funding (EP/M00452X/1), EAG the EPSRC and Newcastle University for studentship EPJ5002881 to F. A. B. A. A. Y. thanks the Iraqi government for an HCED PhD scholarship and AMR the EU for Marie Skłodwska Curie Fellowship No. 656658 “NanoCuI”. There are no conflicts of interest to declare.
Notes and references
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2014, 22, 701 Search PubMed.
- (a) I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed., 2015, 54, 3758 CrossRef CAS PubMed; (b) S. Powar, T. Daeneke, M. T. Ma, D. Fu, N. W. Duffy, G. Götz, M. Weidelener, A. Mishra, P. Bäuerle, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 602 CrossRef CAS PubMed.
- X. L. Zhang, Z. Zhang, D. Chen, P. Bauerle, U. Bach and Y.-B. Cheng, Chem. Commun., 2012, 48, 9885 RSC.
- (a) F. Odobel, L. Le Pleux, Y. Pellegrin and E. Blart, Acc. Chem. Res., 2010, 43, 1063 CrossRef CAS PubMed; (b) F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414 CrossRef CAS; (c) F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4, 2551 CrossRef CAS.
- X. L. Zhang, F. Huang, A. Nattestad, K. Wang, D. Fu, A. Mishra, P. Bäuerle, U. Bach and Y.-B. Cheng, Chem. Commun., 2011, 47, 4808 RSC.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915 RSC.
- (a) L. Favereau, Y. Pellegrin, L. Hirsch, A. Renaud, A. Planchat, E. Blart, G. Louarn, L. Cario, S. Jobic, M. Boujtita and F. Odobel, Adv. Energy Mater., 2017, 7, 1601776 CrossRef; (b) K. Hara, Y. Dan-oh, C. Kasad, Y. Ohga, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Langmuir, 2004, 20, 4205 CrossRef CAS PubMed; (c) A. Kay and M. Grätzel, J. Phys. Chem., 1993, 97, 6272 CrossRef CAS; (d) P. Wang, S. M. Zakeeruddin, R. Humphry-Baker, J. E. Moser and M. Grätzel, Adv. Mater., 2003, 15, 2101 CrossRef CAS; (e) P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336 CrossRef CAS.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed.
- (a) X. Xiang, J. Fielden, W. Rodríguez-Córdoba, Z. Huang, N. Zhang, Z. Luo, D. G. Musaev, T. Lian and C. L. Hill, J. Phys. Chem. C, 2013, 117, 918 CrossRef CAS; (b) J. Fielden, J. M. Sumliner, N. Han, Y. V. Geletii, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Chem. Sci., 2015, 6, 5531 RSC.
- (a) S. M. Wang, L. Liu, W.-L. Chen, E.-B. Wang and Z.-M. Su, Dalton Trans., 2013, 42, 2691 RSC; (b) S.-M. Wang, L. Liu, W.-L. Chen, Z.-M. Su, E.-B. Wang and C. Li, Ind. Eng. Chem. Res., 2014, 53, 150 CrossRef CAS; (c) J. Li, X. Sang, W. Chen, C. Qin, S. Wang, Z. Su and E.-B. Wang, Eur. J. Inorg. Chem., 2013, 1951 CrossRef.
- (a) L. C. P. Almeida, A. D. Gonçalves, J. E. Benedetti, P. C. M. L. Miranda, L. C. Passoni and A. F. Nogueira, J. Mater. Sci., 2010, 45, 5054 CrossRef CAS; (b) S.-S. Guo, C. Qin, Y.-G. Li, Y. Lu, Z.-M. Su, W.-L. Chen and E.-B. Wang, Dalton Trans., 2012, 41, 2227 RSC; (c) C. Yuan, S. Guo, S. Wang, L. Liu, W. Chen and E.-B. Wang, Ind. Eng. Chem. Res., 2013, 52, 6694 CrossRef CAS.
- (a) E. A. Gibson, L. LePleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485 CrossRef CAS PubMed; (b) E. A. Gibson, A. L. Smeigh, L. Le Pleux, L. Hammarstrom, F. Odobel, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115, 9772 CrossRef CAS.
- J. Cui, J. Lu, X. Xu, K. Cao, Z. Wang, G. Alemu, H. Yuang, Y. Shen, J. Xu, Y. Cheng and M. Wang, J. Phys. Chem. C, 2014, 118, 16433 CAS.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic, crystallographic and other experimental details, CIF file. CCDC 1537328. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp01558e |
| ‡ Current address: Department of Chemistry, University of Hail, Hail, Saudi Arabia. |
| This journal is © the Owner Societies 2017 |