
Collective formal synthesis of (±)-rhynchophylline and homologues†
Jun Xua,
Li-Dong Shao*a,
Xin Shiab,
Jian Renab,
Chengfeng Xia*a and
Qin-Shi Zhao*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People's Republic of China. E-mail: qinshizhao@mail.kib.ac.cn; xiachengfeng@mail.kib.ac.cn; shaolidong@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 28th June 2016
Abstract
A collective formal synthesis approach to bioactive oxindole alkaloids, including (±)-rhynchophylline, (±)-isorhynchophylline, (±)-mitraphylline, (±)-formosanine, (±)-isomitraphylline, and (±)-isoformosanine, is completed in a protecting-group free manner. Besides multigram-scaled operations, the notable feature of the synthesis is the application of two one-pot, sequential transformations. Namely, two key tetracyclic intermediates pyridinium salt 9 and monoester 14 were prepared by a one-pot N-alkylation/cross-dehydrogenative coupling sequence and a one-pot Michael/Karpocho sequence with minimal purification, respectively.
Introduction
Rhynchophylline (1, Fig. 1) and isorhynchophylline (2) are tetracyclic 3-spirooxindole alkaloids isolated from Uncaria rhynchophylla.1,2 Investigation of the pharmacological effects of rhynchophylline (1) and isorhynchophylline (2) reveal that they exert beneficial effects on cardiovascular disease including hypertension, bradycardia, and arrhythmia. Rhynchophylline (1) and isorhynchophylline (2) both act as Ca2+ channel antagonists, anticoagulants, and vascular smooth muscle cell proliferation inhibitors.3 Moreover, rhynchophylline (1) has been identified as an inhibitor of the receptor tyrosine kinase EphA4, and is implicated in rescuing hippocampal synaptic dysfunctions in AD.4,5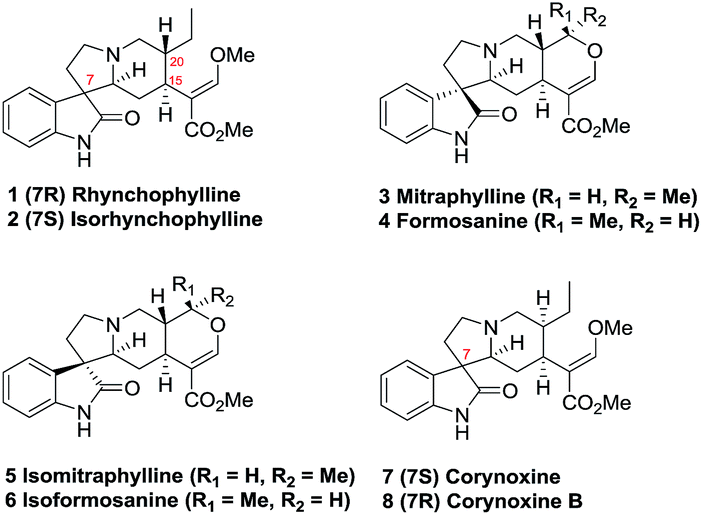 | ||
| Fig. 1 Rhynchophylline and its homologues. | ||
The biological significance and structural complexity of 3-spirooxindole moiety prompted synthetic organic chemists to take up the research project towards the synthesis of rhynchophylline (1) and its homologues (2–8).6,7 Ban et al. achieved the first total synthesis of (±)-1 and (±)-2 via an intramolecular Mannich reaction starting form 2-hydroxytryptamine hydrochloride within 10 steps.6a With the same strategy, Ban et al. completed the synthesis of (±)-mitraphylline (3), (±)-formosanine (4), (±)-isomitraphylline (5), and (±)-isoformosanine (6).6b Martin et al. reported a biomimic formal synthesis of (±)-1 and (±)-2 over 15 steps which 3-spirooxindole tetracyclic fragment was constructed by the oxidation rearrangement of Corynanthe indole intermediate.6c Recently, two enantioselective approaches to 1 and 2 were established by Wang et al.6d and Amat et al.6e in 20 and 18 steps, respectively. Hiemstra et al. reported a collective synthesis of (±)-1, (±)-corynoxine (7), (±)-corynoxine B (8), and (±)-corynoxeine involving an intramolecular Mannich reaction as a key step.6f More recently, we described a cross-dehydrogenative coupling (CDC) approach which provided the direct access to the tetracyclic 3-spirooxindole system and to the application in the facile synthesis of (±)-7 and (±)-8.7 Herein, a collective formal synthesis of six oxindole alkaloids including rhynchophylline (1) and its homologues (2–6) was completed based upon a one-pot N-alkylation/cross-dehydrogenative coupling sequence and a one-pot stereoselective Michael/Karpocho sequence.
Results and discussion
As outlined in Scheme 1, oxindole alkaloids 1–6 could be transformed from Ban's intermediates (16–18)6a,6b and the collective synthesis strategy of them was focused on controlling the stereoselectivity of the functionalization mode of C15 and C20. Unlike corynoxine (7) and corynoxine B (8) which the cis-substituents of C15 and C20 were installed by Johnson–Claisen rearrangement of allylic alcohol 12 with trimethyl orthoacetate in situ.7 The trans-substituents of C15 and C20 of 1–6 could be introduced by a one-pot stereoselective Michael/Krapcho sequence of ketone 13 with methyl malonate. Intermediates 12 and 13 could be formed by dearomatization of pyridinium salt 9. Compound 9 could be prepared by an improved one-pot N-alkylation/CDC sequence of 3-(2-bromoethyl)indolin-2-one (11) which was prepared from tryptophol in 2 steps according to the literature.7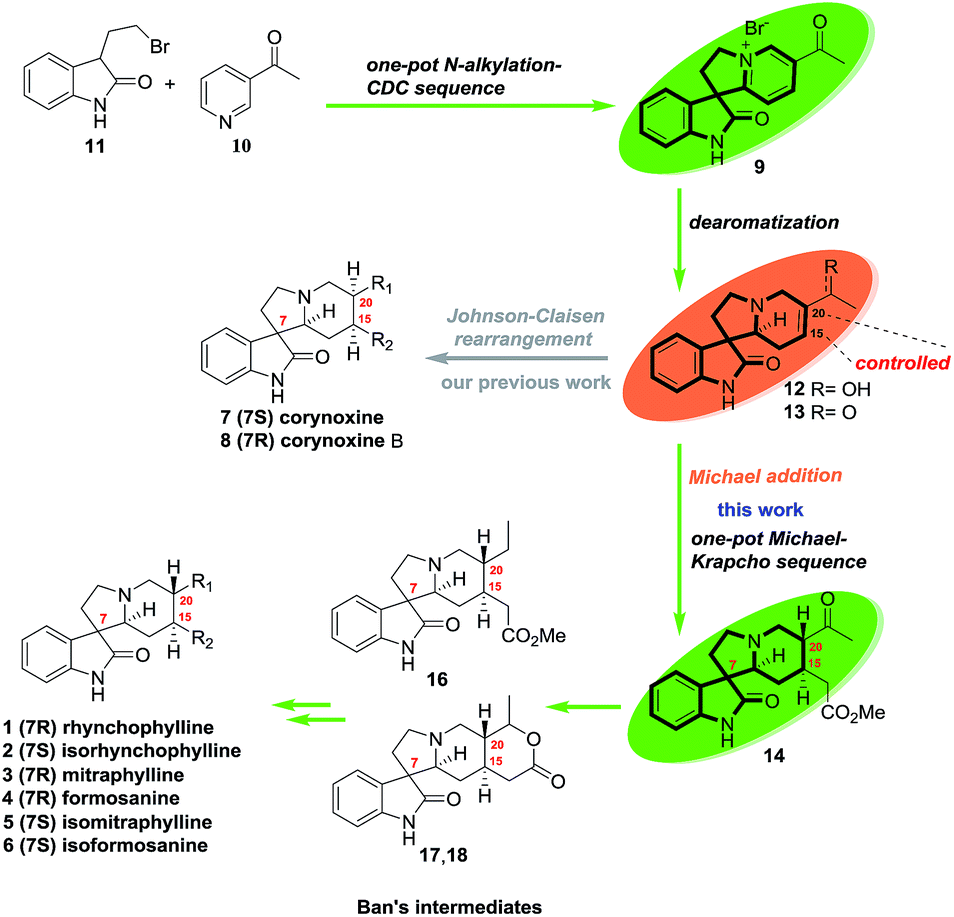 | ||
| Scheme 1 Synthesis strategy of rhynchophylline and its homologues. | ||
The collective synthesis commenced with the multigram-scaled preparation of intermediate 12 and 13 which was shown in Scheme 2. Oxidation of commercially available tryptophol followed by bromination with N-bromosuccinimide (NBS) provided 3-(2-bromoethyl)indolin-2-one (11) in 69.6% yield over 2 steps.7 Then an improved synthesis of 12 and 13 started from a one-pot N-alkylation/CDC protocol which was initiated by heating 11 with 3-acetyl pyridine (10) at 70 °C for 3 h, removing the unreacted 3-acetyl pyridine by adding large amount of ether that contained 10% of MeOH, pouring out of the solvent after stirring vigorously for 30 min. The rest ether was evaporated to give the residue in the flask. Adding Na2CO3 and MeCN into the flask, connecting to an oxygen balloon, the reaction was heated at 50 °C for 12 h to give pyridinium salt 9 in 56% yield over two-steps in one-pot after one purification operation. Subsequently, dearomatization of 9 in dioxane/water with NaBH4 afforded the intermediate 12 in 78% yield as previously reported.7 Interestingly, the reaction was also proceeded smoothly in water without organic solvent to give 12 in 79% yield (Scheme 2).8 Otherwise, dearomatization of 9 by NaBH3CN in THF/AcOH gave ketone 13 in 30% yield.
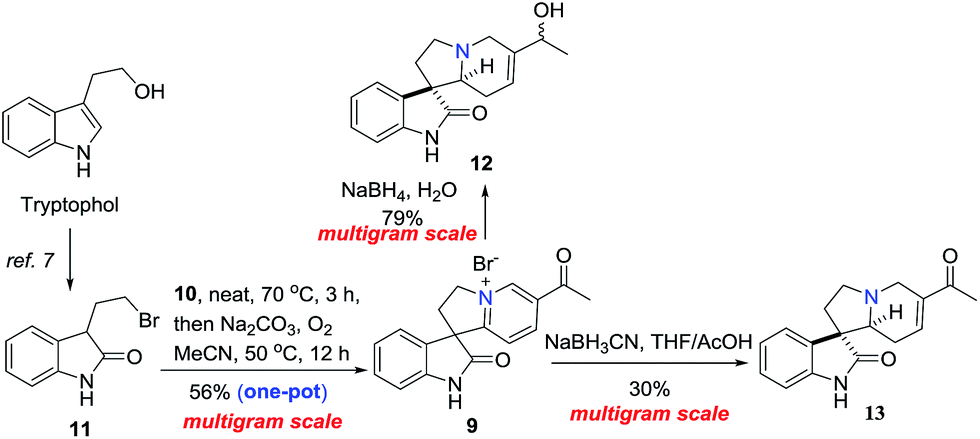 | ||
| Scheme 2 An improved synthesis of intermediate 12 and 13. | ||
Next ketone 13 was converted to Ban's intermediate 16 (ref. 6a) through a series of transformations. As depicted in Scheme 3, ketone 13 could be also prepared by oxidation of allylic alcohol 12 with Dess–Martin periodinane (DMP) in 80% yield.8 Next, a one-pot protocol started from the stereoselective Michael addition reaction of methyl malonate to 13 in the presence of MeONa (1 N).6b,9 After evaporation of MeOH, removing one of the methyl esters under Krapcho's condition by heating the crude diester with LiCl in DMSO at 150 °C gave the key trans-C15,20 substituted keto-monoester 14 in 53% yield over two-reactions in one pot.10,11 Treating 14 with 1,2-ethanedithiol in the presence of BF3·Et2O furnished dithioether 15, which was underwent the desulfuration by RANEY®-Ni to give the desired Ban's intermediate 16 in 81% yield.12–15 Compound 16 could be converted into alkaloids (±)-2 and (±)-1 in 2 and 3 steps, respectively.6a,7 The spectral data of 16 was identical to those reported in the literature by Martin and Wang.6c,6d
 | ||
| Scheme 3 Formal synthesis of (±)-rhynchophylline and (±)-isorhynchophylline. | ||
With compound 14 in hand, both Ban's intermediates 17 and 18 (ref. 6b) could be prepared by diastereoselective hydrogenation of the carbonyl group of 14 (Scheme 4). On one hand, treatment of the monoester 14 with NaBH4 at −40 °C, followed by heating in benzene with TsOH resulted in the desired lactone compound 17 as a single diastereomer in 69% yield over 2 steps, which was further converted into (±)-5 and (±)-3 in 3 and 4 steps, respectively.6b,7 On the other hand, diastereoselective hydrogenation of the monoester 14 with Adam's catalyst in acetic acid resulted in the desired lactone compound 18 as a single diastereomer in 60% yield, which was also further converted into (±)-6 and (±)-4 in 3 and 4 steps, respectively.6b,7 The spectroscopic data of compound 17 and 18 were identical to that reported in the literature.6b
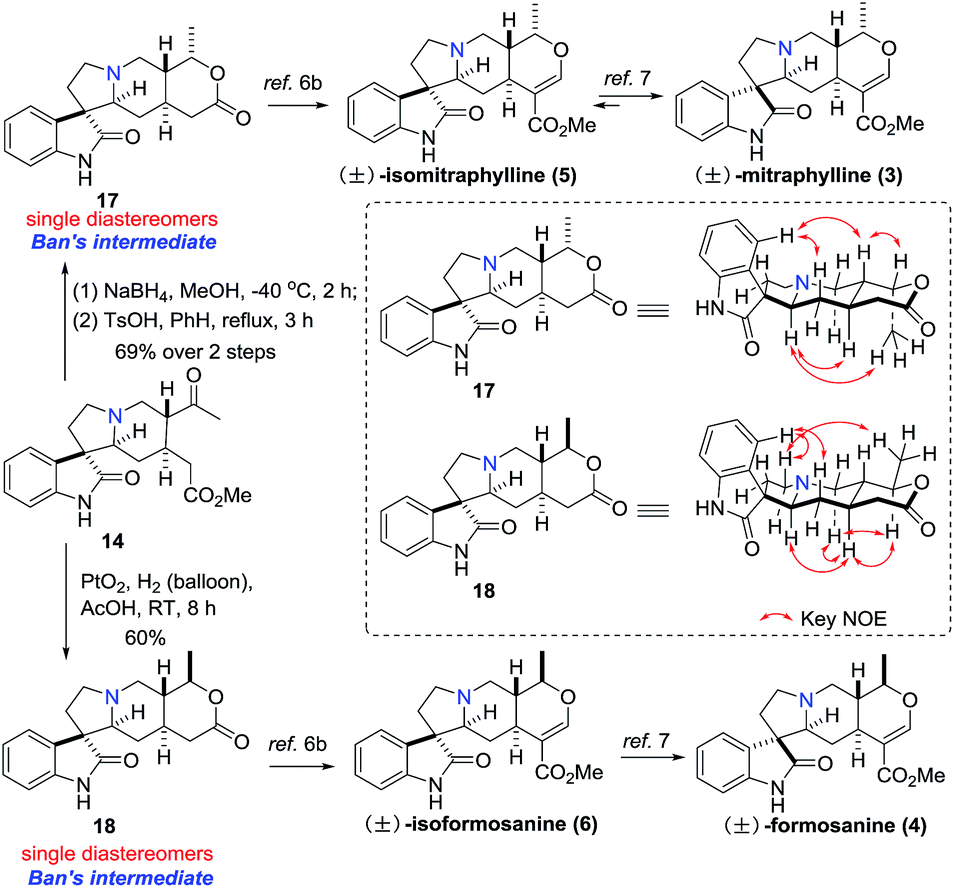 | ||
| Scheme 4 Formal synthesis of (±)-mitraphylline, (±)-isomitraphylline, (±)-formosanine and (±)-isoformosaninec. | ||
Conclusions
In summary, we have accomplished a collective synthesis route to six oxindole alkaloids including (±)-rhynchophylline (1), (±)-isorhynchophylline (2), (±)-mitraphylline (3), (±)-formosanine (4), (±)-isomitraphylline (5), and (±)-isoformosanine (6) within 10 steps from the pyridinium salt 9. The highlighted feature of the synthesis is the application of protecting-group free strategy throughout the synthesis, a one-pot N-alkylation/CDC protocol to furnish key intermediate pyridinium salt 9, and a one-pot Michael/Karpocho protocol to establish the common intermediate 14. Furthermore, many operations could be performed in multigram scale. We anticipate that outlined concise approach can be exploited for the synthesis of other numbers of bioactive tetracyclic 3-spirooxindole alkaloids. Efforts of our laboratory in this field will be reported in due course.Experimental section
General experimental procedures
Anhydrous THF, Et2O, toluene and benzene were distilled over sodium and benzophenone ketyl under N2; Et3N, DCM and CH3CN were refluxed with CaH2 and freshly distilled prior to use; anhydrous MeOH and EtOH were distilled over magnesium under Ar; all other solvents and reagents were used from commercial sources without further purifications. All reactions sensitive to air or moisture were carried out under argon or nitrogen atmosphere in dry and freshly distilled solvents under anhydrous conditions, unless otherwise noted.1H NMR and 13C NMR experiments were performed on Bruker Avance 400, 600 and 800 NMR spectrometer at ambient temperature. The residual solvent protons (1H) or the solvent carbons (13C) were used as internal standards. 1H NMR data were presented as follows: chemical shift in ppm downfield from tetramethylsilane (multiplicity, coupling constant, integration). Chemical shifts (δ) were given in ppm with reference to solvent signals [1H NMR: CDCl3 (7.26), DMSO-d6 (2.50); 13C NMR: CDCl3 (77.16), DMSO-d6 (39.52)]. The following abbreviations are used in reporting NMR data: s, singlet; brs, broad singlet; d, doublet; t, triplet; q, quartet; qt, quartet of triplets; dd, doublet of doublets; dt, doublet of triplets; AB, AB quartet; m, multiplet. HRMS (ESI) was taken on Agilent 6540 Q-TOF spectrometer.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 20:1) to afford product 9 (4.2 g, 56%) as a yellow foam. 1H NMR (400 MHz, CD3OD): δ 9.73 (s, 1H), 8.90 (dd, J = 8.3, 1.0, 1H), 7.65 (d, J = 8.4, 1H), 7.39 (dt, J = 7.9, 5.0, 2H), 7.19–6.95 (m, 2H), 5.39–5.21 (m, 2H), 3.07 (dd, J = 8.5, 6.3, 2H), 2.72 (s, 3H); 13C NMR (100 MHz, CD3OD): δ 192.8, 175.7, 159.6, 145.4, 143.4, 142.6, 135.4, 130.6, 128.3, 124.6, 123.5, 123.5, 110.7, 60.0, 58.2, 48.3, 48.1, 47.8, 47.6, 47.4, 47.2, 47.0, 33.8, 25.9; HRMS (ESI+): calcd for C17H15N2O2 [M]+ 279.1134; found 279.1134.:1) to give allylic alcohol 12 (0.74 g, 79%) as a yellow foam. 1H NMR (400 MHz, CDCl3): δ 8.86 (s, 1H), 7.42–7.32 (m, 1H), 7.18 (t, J = 7.6 Hz, 1H), 7.01 (td, J = 7.4, 2.6 Hz, 1H), 6.90 (d, J = 7.7 Hz, 1H), 5.57 (d, J = 28.9 Hz, 1H), 4.34–4.13 (m, 1H), 3.64 (dd, J = 63.7, 15.5 Hz, 1H), 3.47 (t, J = 8.6 Hz, 1H), 2.97–2.84 (m, 1H), 2.82–2.71 (m, 1H), 2.63 (ddd, J = 17.7, 8.8, 4.1 Hz, 1H), 2.49–2.41 (m, 1H), 2.11 (dt, J = 12.9, 8.5 Hz, 1H), 1.78–1.60 (m, 2H), 1.28 (dd, J = 6.3, 4.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 182.4, 140.5, 140.1, 139.9, 133.4, 127.9, 125.3, 122.6, 119.8, 118.1, 109.8, 70.4, 69.6, 68.1, 68.0, 56.6, 54.5, 53.2, 51.9, 35.4, 35.3, 26.6, 26.5, 22.3, 21.7; HRMS (ESI) m/z calcd for C17H21N2O2 [M + H]+ 285.1598, found 285.1598.:1, v/v, 100 mL) was added NaBH3CN (350 mg, 5.6 mmol) in portions at 0 °C, the temperature was gradually rised to room temperature, and stirred for 12 h. The reaction mixture was poured into saturated NH4Cl aqueous solution, the resulting solution was extracted with DCM (3 × 100 mL). The combined organic layers were washed with brine (3 × 100 mL), dried over Na2SO4, after removal of the solvent, the residue was purified by flash column chromatography on silica gel (DCM/methanol = 80:1) to give ketone 13 (245 mg, 30%) as a colorless oil (spectra data is listed below).:1) to afford ketone 13 (1.62 g, 80%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.45 (s, 1H), 7.35 (d, J = 7.3 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.92 (d, J = 7.7 Hz, 1H), 6.76 (s, 1H), 3.98 (d, J = 16.4 Hz, 1H), 3.52 (dd, J = 8.5, 6.0 Hz, 1H), 2.94 (d, J = 16.0 Hz, 1H), 2.77 (dd, J = 9.0, 4.8 Hz, 1H), 2.73–2.63 (m, 1H), 2.54–2.44 (m, 1H), 2.27 (s, 3H), 2.11 (dt, J = 13.2, 7.8 Hz, 1H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.4, 140.5, 140.1, 139.9, 133.4, 127.9, 125.3, 122.6, 119.8, 118.1, 109.8, 70.4, 69.6, 68.1, 68.0, 56.6, 54.5, 53.2, 51.9, 35.4, 35.3, 26.6, 26.5, 22.3, 21.7; HRMS (ESI) m/z calcd for C17H19N2O2 [M + H]+ 283.1441, found 283.1443.:1) to give product keto-monoester 14 (680 mg, 53%) as a white foam. 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.34 (d, J = 7.4 Hz, 1H), 7.20 (t, J = 7.3 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 3.53 (s, 3H), 3.31 (ddd, J = 10.5, 9.6, 2.8 Hz, 2H), 2.61 (td, J = 11.0, 3.6 Hz, 1H), 2.52 (dd, J = 10.5, 6.8 Hz, 2H), 2.45–2.37 (m, 1H), 2.23 (d, J = 4.2 Hz, 1H), 2.19 (s, 3H), 2.17–2.12 (m, 1H), 2.11–1.95 (m, 3H), 1.39–1.33 (m, 1H), 0.73 (q, J = 11.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 209.8, 181.4, 172.3, 140.4, 133.4, 127.9, 125.1, 122.6, 109.8, 70.9, 56.7, 55.0, 54.9, 53.6, 51.6, 38.5, 35.3, 33.9, 31.1, 30.0; HRMS (ESI) m/z calcd for C20H24N2O4 [M + H]+ 357.1809, found 357.1814.:1) to give dithioketal 15 (41 mg, 86%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.47 (s, 1H), 7.38 (d, J = 7.3 Hz, 1H), 7.18 (t, J = 7.3 Hz, 1H), 7.01 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 3.67–3.60 (m, 1H), 3.55 (s, 3H), 3.44–3.35 (m, 1H), 3.29 (ddt, J = 16.4, 10.7, 5.2 Hz, 3H), 3.20–3.10 (m, 1H), 2.96 (dd, J = 15.5, 2.8 Hz, 1H), 2.57–2.43 (m, 2H), 2.43–2.34 (m, 1H), 2.04 (dddd, J = 32.6, 24.2, 11.9, 6.8 Hz, 5H), 1.69 (s, 3H), 1.24 (dd, J = 9.4, 3.0 Hz, 1H), 0.86 (dd, J = 23.6, 11.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 181.6, 172.8, 140.2, 133.6, 127.8, 125.2, 122.6, 109.7, 71.0, 69.3, 58.0, 56.7, 53.6, 51.9, 51.6, 40.5, 39.5, 37.1, 36.1, 35.6, 32.2, 29.4; HRMS (ESI) m/z calcd for C22H29N2O3S2 [M + H]+ 433.1614, found 433.1618.:1, v/v, 1.5 mL) was added excess RANEY®-Ni (W-2 type, newly activated). After refluxed at 105 °C for 4.5 h, the reaction mixture was filtered through a plug of Celite, the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (DCM/methanol = 80:1) to give 16 (10 mg, 81%) as a colorless oil. 1H NMR (600 MHz, DMSO-d6): δ 10.36 (s, 1H), 7.22 (d, J = 7.3 Hz, 1H), 7.15 (td, J = 7.6, 1.2 Hz, 1H), 6.95 (td, J = 7.5, 0.8 Hz, 1H), 6.81 (d, J = 7.7 Hz, 1H), 3.47 (s, 3H), 3.25–3.17 (m, 2H), 2.47 (dd, J = 15.6, 3.4 Hz, 1H), 2.30 (dd, J = 17.4, 8.7 Hz, 1H), 2.22 (d, J = 10.7 Hz, 1H), 2.17 (t, J = 11.0 Hz, 1H), 1.92–1.77 (m, 2H), 1.69 (t, J = 10.9 Hz, 1H), 1.49 (dd, J = 11.5, 7.2 Hz, 1H), 1.43–1.35 (m, 1H), 1.16 (d, J = 9.4 Hz, 1H), 1.09–0.97 (m, 2H), 0.83 (dd, J = 14.1, 6.7 Hz, 3H), 0.60 (q, J = 11.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6): δ 180.1, 172.8, 141.4, 133.6, 127.5, 124.5, 121.5, 109.2, 71.0, 57.0, 56.0, 53.4, 51.2, 40.6, 37.4, 36.7, 34.7, 31.6, 22.9, 10.9; HRMS (ESI) m/z calcd for C20H27N2O3 [M + H]+ 343.2016, found 343.2022.:1) to afford the reduction product as a colorless oil. This oil was dissolved in benzene (2 mL). To this solution was added TsOH (37 mg, 0.22 mmol) at room temperature. After the reaction was refluxed at 85 °C for 3 h, benzene was removed in vacuo. The residue was diluted with DCM (2 mL) and water (1 mL). The aqueous layer was extracted with DCM (3 × 5 mL), and the combined organic layers were washed with brine, dried over Na2SO4, after removal of the solvent. The crude material was purified by flash chromatography (DCM/methanol = 80:1) to afford 17 (24 mg, 69%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ 7.54 (s, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 7.7 Hz, 1H), 4.67 (m, 1H), 3.33 (t, J = 7.8 Hz, 1H), 3.16 (dd, J = 10.3, 2.8 Hz, 1H), 2.55 (dt, J = 13.1, 6.8 Hz, 2H), 2.48 (d, J = 11.3 Hz, 1H), 2.42 (t, J = 11.2 Hz, 1H), 2.06 (dt, J = 13.1, 8.5 Hz, 1H), 2.02–1.88 (m, 3H), 1.84 (dt, J = 16.1, 5.7 Hz, 1H), 1.37 (d, J = 12.5 Hz, 1H), 1.29 (d, J = 6.7 Hz, 3H), 0.69 (q, J = 11.5 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 180.9, 170.2, 140.0, 133.3, 128.1, 125.2, 122.9, 109.6, 77.6, 70.9, 56.4, 54.1, 54.0, 39.8, 36.6, 35.2, 32.6, 28.7, 17.7; HRMS (ESI) m/z calcd for C19H23N2O3 [M + H]+ 327.1703, found 327.1704. For observation of –NH: 1H NMR (800 MHz, DMSO-d6) δ 10.35 (–NH, s, 1H), 7.17 (d, J = 7.3 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.90 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 7.7 Hz, 1H), 4.60–4.53 (m, 1H), 3.19 (dd, J = 8.4, 6.5 Hz, 1H), 3.06 (dd, J = 10.5, 3.2 Hz, 1H), 2.37 (dd, J = 17.9, 5.4 Hz, 1H), 2.30 (q, J = 8.8 Hz, 1H), 2.25–2.20 (m, 1H), 2.13 (dd, J = 11.4, 8.8 Hz, 1H), 1.87–1.77 (m, 3H), 1.74 (t, J = 10.6 Hz, 2H), 1.13 (d, J = 6.7 Hz, 3H), 1.12–1.08 (m, 1H), 0.50 (q, J = 11.5 Hz, 1H); 13C NMR (200 MHz, DMSO-d6) δ 180.1, 169.7, 141.5, 133.4, 127.7, 124.5, 121.6, 109.3, 76.9, 70.5, 55.7, 53.5, 53.4, 39.0, 36.0, 34.7, 32.00, 28.0, 17.5.:1) to give 18 (6 mg, 60%) as a colorless oil. 1H NMR (800 MHz, CDCl3): δ 7.68 (s, 1H), 7.35 (d, J = 7.4 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 4.16 (m, 1H), 3.34 (td, J = 8.6, 2.2 Hz, 1H), 3.29 (dd, J = 10.8, 3.9 Hz, 1H), 2.60–2.47 (m, 3H), 2.45–2.38 (m, 1H), 2.08–1.98 (m, 2H), 1.86 (t, J = 10.7 Hz, 1H), 1.48 (ddd, J = 21.5, 10.7, 4.1 Hz, 1H), 1.38 (d, J = 6.3 Hz, 3H), 1.31–1.28 (m, 2H), 0.68 (q, J = 11.7 Hz, 1H); 13C NMR (200 MHz, CDCl3): δ 180.9, 170.1, 140.0, 133.3, 128.1, 125.2, 122.8, 109.6, 80.0, 70.7, 56.5, 54.1, 53.8, 43.2, 36.7, 35.05, 34.8, 32.0, 20.0; HRMS (ESI) m/z calcd for C22H29N2O3S2 [M + H]+ 327.1703, found 327.1699. For observation of –NH: 1H NMR (800 MHz, DMSO-d6) δ 10.34 (–NH, s, 1H), 7.15 (d, J = 7.3 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.88 (t, J = 7.4 Hz, 1H), 6.75 (d, J = 7.7 Hz, 1H), 4.10 (m, 1H), 3.20 (d, J = 2.4 Hz, 1H), 3.14 (dd, J = 10.8, 4.0 Hz, 1H), 2.33 (dd, J = 17.8, 5.0 Hz, 1H), 2.29 (d, J = 8.8 Hz, 1H), 2.23 (dd, J = 11.2, 2.0 Hz, 1H), 2.14–2.10 (m, 1H), 1.88 (dd, J = 17.9, 12.5 Hz, 1H), 1.79 (d, J = 12.9 Hz, 1H), 1.69 (t, J = 10.8 Hz, 1H), 1.53 (d, J = 11.6 Hz, 1H), 1.31 (dd, J = 10.7, 4.0 Hz, 1H), 1.18 (d, J = 6.2 Hz, 3H), 1.03 (dd, J = 9.5, 2.9 Hz, 1H), 0.45 (q, J = 11.7 Hz, 1H); 13C NMR (200 MHz, DMSO-d6) δ 180.1, 169.6, 141.5, 133.4, 127.7, 124.5, 121.6, 109.3, 79.0, 70.4, 55.8, 53.5, 53.4, 42.2, 36.1, 34.5, 33.8, 31.3, 19.7.
:methanol = 20:1) to afford product 9 (4.2 g, 56%) as a yellow foam. 1H NMR (400 MHz, CD3OD): δ 9.73 (s, 1H), 8.90 (dd, J = 8.3, 1.0, 1H), 7.65 (d, J = 8.4, 1H), 7.39 (dt, J = 7.9, 5.0, 2H), 7.19–6.95 (m, 2H), 5.39–5.21 (m, 2H), 3.07 (dd, J = 8.5, 6.3, 2H), 2.72 (s, 3H); 13C NMR (100 MHz, CD3OD): δ 192.8, 175.7, 159.6, 145.4, 143.4, 142.6, 135.4, 130.6, 128.3, 124.6, 123.5, 123.5, 110.7, 60.0, 58.2, 48.3, 48.1, 47.8, 47.6, 47.4, 47.2, 47.0, 33.8, 25.9; HRMS (ESI+): calcd for C17H15N2O2 [M]+ 279.1134; found 279.1134.:1) to give allylic alcohol 12 (0.74 g, 79%) as a yellow foam. 1H NMR (400 MHz, CDCl3): δ 8.86 (s, 1H), 7.42–7.32 (m, 1H), 7.18 (t, J = 7.6 Hz, 1H), 7.01 (td, J = 7.4, 2.6 Hz, 1H), 6.90 (d, J = 7.7 Hz, 1H), 5.57 (d, J = 28.9 Hz, 1H), 4.34–4.13 (m, 1H), 3.64 (dd, J = 63.7, 15.5 Hz, 1H), 3.47 (t, J = 8.6 Hz, 1H), 2.97–2.84 (m, 1H), 2.82–2.71 (m, 1H), 2.63 (ddd, J = 17.7, 8.8, 4.1 Hz, 1H), 2.49–2.41 (m, 1H), 2.11 (dt, J = 12.9, 8.5 Hz, 1H), 1.78–1.60 (m, 2H), 1.28 (dd, J = 6.3, 4.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 182.4, 140.5, 140.1, 139.9, 133.4, 127.9, 125.3, 122.6, 119.8, 118.1, 109.8, 70.4, 69.6, 68.1, 68.0, 56.6, 54.5, 53.2, 51.9, 35.4, 35.3, 26.6, 26.5, 22.3, 21.7; HRMS (ESI) m/z calcd for C17H21N2O2 [M + H]+ 285.1598, found 285.1598.:1, v/v, 100 mL) was added NaBH3CN (350 mg, 5.6 mmol) in portions at 0 °C, the temperature was gradually rised to room temperature, and stirred for 12 h. The reaction mixture was poured into saturated NH4Cl aqueous solution, the resulting solution was extracted with DCM (3 × 100 mL). The combined organic layers were washed with brine (3 × 100 mL), dried over Na2SO4, after removal of the solvent, the residue was purified by flash column chromatography on silica gel (DCM/methanol = 80:1) to give ketone 13 (245 mg, 30%) as a colorless oil (spectra data is listed below).:1) to afford ketone 13 (1.62 g, 80%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.45 (s, 1H), 7.35 (d, J = 7.3 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.92 (d, J = 7.7 Hz, 1H), 6.76 (s, 1H), 3.98 (d, J = 16.4 Hz, 1H), 3.52 (dd, J = 8.5, 6.0 Hz, 1H), 2.94 (d, J = 16.0 Hz, 1H), 2.77 (dd, J = 9.0, 4.8 Hz, 1H), 2.73–2.63 (m, 1H), 2.54–2.44 (m, 1H), 2.27 (s, 3H), 2.11 (dt, J = 13.2, 7.8 Hz, 1H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.4, 140.5, 140.1, 139.9, 133.4, 127.9, 125.3, 122.6, 119.8, 118.1, 109.8, 70.4, 69.6, 68.1, 68.0, 56.6, 54.5, 53.2, 51.9, 35.4, 35.3, 26.6, 26.5, 22.3, 21.7; HRMS (ESI) m/z calcd for C17H19N2O2 [M + H]+ 283.1441, found 283.1443.:1) to give product keto-monoester 14 (680 mg, 53%) as a white foam. 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.34 (d, J = 7.4 Hz, 1H), 7.20 (t, J = 7.3 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 3.53 (s, 3H), 3.31 (ddd, J = 10.5, 9.6, 2.8 Hz, 2H), 2.61 (td, J = 11.0, 3.6 Hz, 1H), 2.52 (dd, J = 10.5, 6.8 Hz, 2H), 2.45–2.37 (m, 1H), 2.23 (d, J = 4.2 Hz, 1H), 2.19 (s, 3H), 2.17–2.12 (m, 1H), 2.11–1.95 (m, 3H), 1.39–1.33 (m, 1H), 0.73 (q, J = 11.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 209.8, 181.4, 172.3, 140.4, 133.4, 127.9, 125.1, 122.6, 109.8, 70.9, 56.7, 55.0, 54.9, 53.6, 51.6, 38.5, 35.3, 33.9, 31.1, 30.0; HRMS (ESI) m/z calcd for C20H24N2O4 [M + H]+ 357.1809, found 357.1814.:1) to give dithioketal 15 (41 mg, 86%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.47 (s, 1H), 7.38 (d, J = 7.3 Hz, 1H), 7.18 (t, J = 7.3 Hz, 1H), 7.01 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 3.67–3.60 (m, 1H), 3.55 (s, 3H), 3.44–3.35 (m, 1H), 3.29 (ddt, J = 16.4, 10.7, 5.2 Hz, 3H), 3.20–3.10 (m, 1H), 2.96 (dd, J = 15.5, 2.8 Hz, 1H), 2.57–2.43 (m, 2H), 2.43–2.34 (m, 1H), 2.04 (dddd, J = 32.6, 24.2, 11.9, 6.8 Hz, 5H), 1.69 (s, 3H), 1.24 (dd, J = 9.4, 3.0 Hz, 1H), 0.86 (dd, J = 23.6, 11.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 181.6, 172.8, 140.2, 133.6, 127.8, 125.2, 122.6, 109.7, 71.0, 69.3, 58.0, 56.7, 53.6, 51.9, 51.6, 40.5, 39.5, 37.1, 36.1, 35.6, 32.2, 29.4; HRMS (ESI) m/z calcd for C22H29N2O3S2 [M + H]+ 433.1614, found 433.1618.:1, v/v, 1.5 mL) was added excess RANEY®-Ni (W-2 type, newly activated). After refluxed at 105 °C for 4.5 h, the reaction mixture was filtered through a plug of Celite, the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (DCM/methanol = 80:1) to give 16 (10 mg, 81%) as a colorless oil. 1H NMR (600 MHz, DMSO-d6): δ 10.36 (s, 1H), 7.22 (d, J = 7.3 Hz, 1H), 7.15 (td, J = 7.6, 1.2 Hz, 1H), 6.95 (td, J = 7.5, 0.8 Hz, 1H), 6.81 (d, J = 7.7 Hz, 1H), 3.47 (s, 3H), 3.25–3.17 (m, 2H), 2.47 (dd, J = 15.6, 3.4 Hz, 1H), 2.30 (dd, J = 17.4, 8.7 Hz, 1H), 2.22 (d, J = 10.7 Hz, 1H), 2.17 (t, J = 11.0 Hz, 1H), 1.92–1.77 (m, 2H), 1.69 (t, J = 10.9 Hz, 1H), 1.49 (dd, J = 11.5, 7.2 Hz, 1H), 1.43–1.35 (m, 1H), 1.16 (d, J = 9.4 Hz, 1H), 1.09–0.97 (m, 2H), 0.83 (dd, J = 14.1, 6.7 Hz, 3H), 0.60 (q, J = 11.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6): δ 180.1, 172.8, 141.4, 133.6, 127.5, 124.5, 121.5, 109.2, 71.0, 57.0, 56.0, 53.4, 51.2, 40.6, 37.4, 36.7, 34.7, 31.6, 22.9, 10.9; HRMS (ESI) m/z calcd for C20H27N2O3 [M + H]+ 343.2016, found 343.2022.:1) to afford the reduction product as a colorless oil. This oil was dissolved in benzene (2 mL). To this solution was added TsOH (37 mg, 0.22 mmol) at room temperature. After the reaction was refluxed at 85 °C for 3 h, benzene was removed in vacuo. The residue was diluted with DCM (2 mL) and water (1 mL). The aqueous layer was extracted with DCM (3 × 5 mL), and the combined organic layers were washed with brine, dried over Na2SO4, after removal of the solvent. The crude material was purified by flash chromatography (DCM/methanol = 80:1) to afford 17 (24 mg, 69%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ 7.54 (s, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 7.7 Hz, 1H), 4.67 (m, 1H), 3.33 (t, J = 7.8 Hz, 1H), 3.16 (dd, J = 10.3, 2.8 Hz, 1H), 2.55 (dt, J = 13.1, 6.8 Hz, 2H), 2.48 (d, J = 11.3 Hz, 1H), 2.42 (t, J = 11.2 Hz, 1H), 2.06 (dt, J = 13.1, 8.5 Hz, 1H), 2.02–1.88 (m, 3H), 1.84 (dt, J = 16.1, 5.7 Hz, 1H), 1.37 (d, J = 12.5 Hz, 1H), 1.29 (d, J = 6.7 Hz, 3H), 0.69 (q, J = 11.5 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 180.9, 170.2, 140.0, 133.3, 128.1, 125.2, 122.9, 109.6, 77.6, 70.9, 56.4, 54.1, 54.0, 39.8, 36.6, 35.2, 32.6, 28.7, 17.7; HRMS (ESI) m/z calcd for C19H23N2O3 [M + H]+ 327.1703, found 327.1704. For observation of –NH: 1H NMR (800 MHz, DMSO-d6) δ 10.35 (–NH, s, 1H), 7.17 (d, J = 7.3 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.90 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 7.7 Hz, 1H), 4.60–4.53 (m, 1H), 3.19 (dd, J = 8.4, 6.5 Hz, 1H), 3.06 (dd, J = 10.5, 3.2 Hz, 1H), 2.37 (dd, J = 17.9, 5.4 Hz, 1H), 2.30 (q, J = 8.8 Hz, 1H), 2.25–2.20 (m, 1H), 2.13 (dd, J = 11.4, 8.8 Hz, 1H), 1.87–1.77 (m, 3H), 1.74 (t, J = 10.6 Hz, 2H), 1.13 (d, J = 6.7 Hz, 3H), 1.12–1.08 (m, 1H), 0.50 (q, J = 11.5 Hz, 1H); 13C NMR (200 MHz, DMSO-d6) δ 180.1, 169.7, 141.5, 133.4, 127.7, 124.5, 121.6, 109.3, 76.9, 70.5, 55.7, 53.5, 53.4, 39.0, 36.0, 34.7, 32.00, 28.0, 17.5.:1) to give 18 (6 mg, 60%) as a colorless oil. 1H NMR (800 MHz, CDCl3): δ 7.68 (s, 1H), 7.35 (d, J = 7.4 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 4.16 (m, 1H), 3.34 (td, J = 8.6, 2.2 Hz, 1H), 3.29 (dd, J = 10.8, 3.9 Hz, 1H), 2.60–2.47 (m, 3H), 2.45–2.38 (m, 1H), 2.08–1.98 (m, 2H), 1.86 (t, J = 10.7 Hz, 1H), 1.48 (ddd, J = 21.5, 10.7, 4.1 Hz, 1H), 1.38 (d, J = 6.3 Hz, 3H), 1.31–1.28 (m, 2H), 0.68 (q, J = 11.7 Hz, 1H); 13C NMR (200 MHz, CDCl3): δ 180.9, 170.1, 140.0, 133.3, 128.1, 125.2, 122.8, 109.6, 80.0, 70.7, 56.5, 54.1, 53.8, 43.2, 36.7, 35.05, 34.8, 32.0, 20.0; HRMS (ESI) m/z calcd for C22H29N2O3S2 [M + H]+ 327.1703, found 327.1699. For observation of –NH: 1H NMR (800 MHz, DMSO-d6) δ 10.34 (–NH, s, 1H), 7.15 (d, J = 7.3 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.88 (t, J = 7.4 Hz, 1H), 6.75 (d, J = 7.7 Hz, 1H), 4.10 (m, 1H), 3.20 (d, J = 2.4 Hz, 1H), 3.14 (dd, J = 10.8, 4.0 Hz, 1H), 2.33 (dd, J = 17.8, 5.0 Hz, 1H), 2.29 (d, J = 8.8 Hz, 1H), 2.23 (dd, J = 11.2, 2.0 Hz, 1H), 2.14–2.10 (m, 1H), 1.88 (dd, J = 17.9, 12.5 Hz, 1H), 1.79 (d, J = 12.9 Hz, 1H), 1.69 (t, J = 10.8 Hz, 1H), 1.53 (d, J = 11.6 Hz, 1H), 1.31 (dd, J = 10.7, 4.0 Hz, 1H), 1.18 (d, J = 6.2 Hz, 3H), 1.03 (dd, J = 9.5, 2.9 Hz, 1H), 0.45 (q, J = 11.7 Hz, 1H); 13C NMR (200 MHz, DMSO-d6) δ 180.1, 169.6, 141.5, 133.4, 127.7, 124.5, 121.6, 109.3, 79.0, 70.4, 55.8, 53.5, 53.4, 42.2, 36.1, 34.5, 33.8, 31.3, 19.7.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation (U1502223) and National Basic Research Program of China (2011CB915503).References
- J. S. Shi, J. X. Yu, X. P. Chen and R. X. Xu, Acta Pharmacol. Sin., 2003, 24, 97 CAS.
- G. Laus and K. Wurst, Helv. Chim. Acta, 2003, 86, 181 CrossRef CAS.
- Y. Ng, T. Or and N. Ip, Neurochem. Int., 2015, 89, 260 CrossRef CAS PubMed.
- A. Fu, K. Hung, H. Q. Huang, S. Gu, Y. Shen, E. Cheng, F. Ip, X. H. Huang, W. Fu and N. Ip, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9959 CrossRef CAS PubMed.
- M. Tognolini, M. Incerti and A. Lodola, ACS Chem. Neurosci., 2014, 5, 1146 CrossRef CAS PubMed.
- Total synthesis of rhynchophylline and isorhynchophylline see: (a) Y. Ban, M. Seto and T. Oishi, Chem. Pharm. Bull., 1975, 23, 2605 CrossRef CAS; (b) Y. Ban, N. Taga and T. Oishi, Chem. Pharm. Bull., 1976, 24, 736 CrossRef CAS; (c) A. Deiters, M. Pettersson and S. F. Martin, J. Org. Chem., 2006, 71, 6547 CrossRef CAS PubMed; (d) H. Zhang, X. Ma, H. Kang, L. Hong and R. Wang, Chem.–Asian J., 2013, 8, 542 CrossRef CAS PubMed; (e) M. Amat, C. Ramos, M. Perez, E. Molins, P. Florindo, M. M. M. Santos and J. Bosch, Chem. Commun., 2013, 49, 1954 RSC; (f) M. J. Wanner, S. Ingemann, J. H. van Maarseveen and H. Hiemstra, Eur. J. Org. Chem., 2013, 79, 1100 CrossRef.
- J. Xu, L. D. Shao, D. Li, X. Deng, Y. C. Liu, Q. S. Zhao and C. Xia, J. Am. Chem. Soc., 2014, 136, 17962 CrossRef CAS PubMed.
- R. Oehl, G. Lenzer and P. Rosenmund, Chem. Ber., 1976, 109, 705 CrossRef CAS.
- R. A. Villa, Q. Xu and O. Kwon, Org. Lett., 2012, 14, 4634 CrossRef CAS PubMed.
- D. A. Evans, T. Rovis, M. C. Kozlowski, C. W. Downey and J. S. Tedrow, J. Am. Chem. Soc., 2000, 122, 9134 CrossRef CAS.
- H. L. Zhang, G. Zhao, Y. Ding and B. Wu, J. Org. Chem., 2005, 70, 4954 CrossRef CAS PubMed.
- H. Suginome, S. Yamada and J. B. Wang, J. Org. Chem., 1990, 55, 2170 CrossRef CAS.
- X. Pu and D. Ma, J. Org. Chem., 2003, 68, 4400 CrossRef CAS PubMed.
- C. Kuehm-Caubère, A. Guilmart, S. Adach-Becker, Y. Fort and P. Caubère, Tetrahedron Lett., 1998, 39, 8987 CrossRef.
- A. B. Smith and D. S. Kim, Org. Lett., 2004, 6, 1493 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all isolated compounds. See DOI: 10.1039/c6ra12766e |
| This journal is © The Royal Society of Chemistry 2016 |