Determination of the loading and stability of Pd in an arborescent copolymer in ethanol by microplasma-optical emission spectrometry†
Olivier Nguon,
Mario Gauthier* and
Vassili Karanassios
Waterloo Institute for Nanotechnology, Department of Chemistry, University of Waterloo, Waterloo, ON N2L 3G1, Canada. E-mail: gauthier@uwaterloo.ca; Fax: +1 519 746-0435; Tel: +1 519 888-4567 ext. 35205
First published on 20th January 2014
Abstract
We report, for the first time, the utilization of a microplasma-optical emission spectrometry system for the determination, without sample digestion, of the concentration of Pd loaded in a dendritic graft (arborescent) copolymer dissolved in ethanol. The preparation of polymer-stabilized colloidal Pd particles was achieved by adding palladium acetate to a solution of the copolymer, viz. arborescent polystyrene-graft-poly(2-vinylpyridine), in ethanol. No acid digestion was needed prior to the analysis, and only micro-amounts (μL) of sample were required. Calibration curves obtained for Pd in ethanol were linear in the concentration range of interest, and the percent relative standard deviation (%RSD) ranged from 7.4 to 0.1%. The Pd detection limit was 28 pg (absolute) or 3 ng mL−1 (when using 10 μL samples). The average Pd loading per mole of 2-vinylpyridine unit was determined to be 99.5 mol%. The kinetics of aggregation of the metallic species to Pd black were also determined. The Pd concentration in ethanol without polymer was found to abate to about one third of its initial value after 5 days. In the presence of the copolymer, however, the concentration of Pd in solution remained constant for at least 10 days. The low electric power and gas consumption of the microplasma device, its low operating cost and detection limit, compatibility with organic solvents, and the small sample amount required make this system a greener and cheaper alternative to the inductively coupled plasma (ICP) spectrometry commonly used for Pd quantification.
Introduction
The unique and size-dependent properties of metallic nanoparticles (NPs) present great opportunities for a wide array of applications ranging from sensing to optoelectronics, medicine, and catalysis.1,2 Since the first report by Faraday in 1857,3 various methods have been developed for the preparation of colloidal metallic particles.4One of the most commonly used strategies involves the reduction of a salt precursor in the presence of a stabilizer in solution. Hirai et al. showed that alcohols could serve as reducing agents,5 and that in the presence of a polymeric stabilizer Pd NPs could be prepared.6 Using a similar strategy, poly(2-vinylpyridine) (P2VP) in the form of linear homopolymers,7–10 block copolymers,11–13 and nanospheres14 was shown to complex with palladium species, even at high temperatures and pressures,15 and to lead to the formation of Pd NPs after reduction of the metallic species.
Gauthier et al. rather synthesized arborescent (dendritic graft) copolymers incorporating a branched polystyrene (PS) core and a corona of P2VP chains16,17 to serve as templates for the complexation of Au(III) salts,18 but also for the preparation and the stabilization of Pd NPs in ethanol.19 Polymer-stabilized Pd nanocatalysts were shown to be useful for a wide range of organic reactions such as carbonylation, hydrogenation, oxidation, reduction, and carbon–carbon cross-coupling reactions.20–23
An important way to assess the ability of a polymer to sequester a metal is by determining its loading capacity; that is, by determining the amount of metal loaded per unit amount of stabilizer. This is a key figure of merit for the evaluation of a catalyst system, which makes its precise and accurate determination cardinal. In cross-coupling reactions for instance, the Pd nanocatalyst concentrations used are typically in the low μg mL−1 (part per million, or ppm) range, to even the ng mL−1 (parts per billion, or ppb) range.24–26 However, the determination of such low Pd concentrations in polymers dissolved in organic solvents has been reported to be challenging.26–29
Many analytical techniques have been applied to the determination of Pd concentrations in colloidal systems. This includes cyclic voltammetry,30 UV-visible spectrophotometry,31 neutron activation analysis (NAA),32 X-ray fluorescence (XRF) – such as energy-dispersive (EDX)33,34 or wavelength-dispersive (WDX)35 X-ray spectroscopy, atomic absorption spectroscopy (AAS),13,29,36 and inductively coupled plasma-optical emission spectrometry (ICP-OES).33,34,37–39
Among these, ICP-OES is most widely used due to its desirable analytical performance characteristics, such as limits of detection (LODs) in the low-ppb to sub-ppb range for many elements. Despite their applicability, ICP systems are expensive to operate and have a relatively large carbon footprint. For example, a typical ICP instrument consumes about 20 L min−1 of Ar gas and 1–2 kW of electric power.40 In many cases the high cost-per-analysis prohibits the characterization of a large number of samples, as it would be essential for instance for the systematic “evaluation of catalysts and recycling systems before and after reaction and continuous monitoring of changes during reactions”, as recommended by Molnár.26
Furthermore, when using the most widely employed method to introduce samples into an ICP, viz. a pneumatic nebulizer, the total volume of sample required per analysis ranges from one to a few milliliters. The sample introduction efficiency of a nebulizer is low (1–5%); therefore over 95% of a sample must be collected and disposed of properly. Other issues also arise when using a nebulizer to introduce nanoparticles or polymers directly into an ICP. For instance clogging of the nebulizer by nanoparticles, and sample-to-sample carry-over from polymer adhering to the walls of the spray chamber or on the tubing (memory effects) have been reported.41 To overcome these issues, polymer-stabilized nanocatalysts must be digested (or dissolved), typically with an acid. Although effective, acid digestion increases the risks of both analyte loss during sample processing and contamination from the digestion reagents.42 Furthermore, if organic solvents are introduced into an ICP, the use of a mixed-gas Ar–O2 plasma is required to eliminate plasma instability and to prevent carbon deposits from the solvents.43 Such procedures further increase the complexity, cost-per-analysis, as well as the carbon footprint of the analytical procedure.
Weagant and Karanassios developed a low-cost and greener analytical method (vis-à-vis ICP) using microplasmas (16 × 2 × 9 mm3, length × width × height) that have a low gas flow rate (0.23 L min−1) and a low power consumption (<15 W).44 The same group demonstrated its applicability to solid, liquid, and gaseous samples;45 and so far eleven elements have been characterized by that technique, primarily using dry residues derived from aqueous microsamples.40 The LODs achieved ranged from 5 to 650 picograms (pg).
In this work, we demonstrate for the first time that this microplasma-based analytical method can be used for the direct determination (i.e., without digestion) of Pd loading in arborescent copolymers dissolved in organic solvents. We also apply this method to determine the kinetics of aggregation and the stability of palladium acetate, and of arborescent polymer-stabilized Pd nanoparticles in ethanol.
Experimental section
Materials and methods
ICP-grade Pd standard solutions (Pd-Std) in 10% HCl, 1000 μg mL−1 ± 0.5% (PlasmaCAL, ICP-AES/MS standard, SCP Science, Baie d'Urfé, QC, Canada) were used. The solutions were freshly diluted using either ethanol (undenatured grade, anhydrous, Commercial Alcohols Inc., Brampton, ON, Canada) or Milli-Q water (18.2 MΩ cm, EMD Millipore Systems, Billerica, MA, USA). The ethanol was distilled in a polytetrafluoroethylene (PTFE) apparatus, to eliminate the possibility of metal contamination from glassware. All the samples were stored in acid-washed low density polyethylene (LDPE, Nalgene®) bottles. All the polypropylene micropipette tips (Bevel Point, 1–20 μL, VWR, Mississauga, ON, Canada), LDPE bottles and vials used were acid-washed by soaking for at least 48 h in a 5% (w/v) nitric acid solution, and then rinsed with Milli-Q water. Drying was subsequently performed in a ventilated dust-free enclosure at room temperature for a minimum of 48 h. A Mettler-Toledo XS205 semi-micro balance with a 0.01 mg display was used for sample preparation. Pd-containing microsamples were pipetted with a Corning Lambda™ micropipette (2–20 μL, accuracy ± 5.0%, and a percent relative standard deviation (%RSD) ≤ 1.5%). The carrier gas, also used as the microplasma support gas, was a mixture of 97% Ar–3% H2, v/v, (Praxair Canada Inc., Mississauga, ON, Canada). Palladium(II) acetate (Pd(OAc)2, min. 98%, Strem Chemicals Inc., Newburyport, MA, USA) was the source of Pd.Copolymer synthesis
A first-generation polystyrene-graft-poly(2-vinylpyridine) (G0PS-g-P2VP) arborescent copolymer was synthesized by Gauthier and Munam, by anionic polymerization and grafting techniques, according to a reported procedure.17 The PS core of the copolymer was a comb-branched (or generation 0, G0) polymer prepared by grafting randomly about 17 PS side chains (each with Mn = 5500 g mol−1) onto a linear PS substrate (Mn = 5200 g mol−1). The comb-branched polymer was further grafted with 182 P2VP side chains (Mn = 5500 g mol−1), corresponding to a 2-vinylpyridine (2VP) unit content of 91 mol% in the copolymer obtained. The molar-mass dispersity (Mw/Mn) of the sample was 1.08. The structure of the copolymer obtained, referred to as G1 (overall generation 1), is depicted in Scheme 1.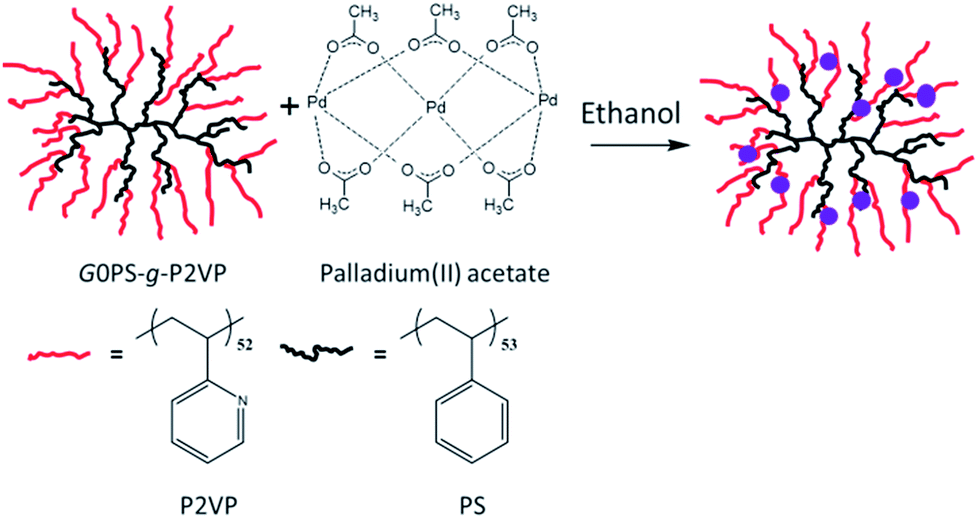 | ||
| Scheme 1 Loading of the G1 arborescent copolymer (G0PS-g-P2VP) with Pd(OAc)2 in ethanol. | ||
Loading of Pd in the copolymer
Loading of the Pd catalyst was achieved by co-dissolution of Pd(OAc)2 with the arborescent copolymer in ethanol solutions as follows: the polymer was dissolved overnight in ethanol (0.2 mg mL−1) in a LDPE vial. Immediately after the dissolution of Pd(OAc)2 in ethanol (0.25 mg mL−1) and sonication for 2 minutes, the desired amount of catalyst solution (either 0.25 or 1.5 molar equiv. of Pd per 2VP unit) was added to the polymer solution to obtain a yellow translucent solution. The separation of free Pd from the polymer-bound Pd was achieved by transferring 15 mL of the polymer–catalyst solution to a dialysis membrane (1000 molecular weight cut-off Spectra/Por® 7 regenerated cellulose), and stirring for 6 h in 200 mL of ethanol while changing the solvent thrice. The polymer–catalyst solution was then recovered from the dialysis membrane and diluted to obtain a 2VP unit concentration of 0.7 μg g−1 (0.7 ppm).Transmission electron microscopy imaging
Imaging by transmission electron microscopy (TEM) was performed in the bright-field mode on a Philips CM10 electron microscope operated at an accelerating voltage of 60 kV. The samples were prepared by depositing two drops of Pd-containing solution (0.07 mg mL−1) onto a 300-mesh Formvar® carbon-coated copper TEM grid (Electron Microscopy Sciences, FCF300-Cu).Instrumentation and operating conditions
A block diagram of the instrumentation used for the quantitative determination of Pd is shown in Fig. 1a. Conceptually, the instrumentation consists of 5 parts: (1) a microsample introduction system; (2) a microplasma device; (3) a scanning monochromator (Heath, 0.35 m Czerny–Turner design equipped with a 1200 grooves per mm grating); (4) a photomultiplier tube (PMT) detector (Hamamatsu model R928 fitted inside a Heath EU-701-30 PMT module); and (5) an amplifier (SRS 570) with a data acquisition sub-system (National Instruments DAQCard 1200) including a computer running a locally developed LabVIEW program for data acquisition. | ||
| Fig. 1 (a) Schematic of the instrumentation used (illustration not to scale) and (b) representative signal. | ||
The electrothermal vaporization microsample introduction system (Fig. 1a) consisted of a vaporization chamber, and a cylindrical ceramic support equipped with a rhenium coiled filament at one end. Cables running through conduits in the ceramic support connected the filament to an external electric power supply. Deposition of the microsamples was performed by retracting the ceramic support from the vaporization chamber, and pipetting a few microliters (3.0–10.0 μL) of solution onto the coil. The ceramic support, along with the sample-carrying coil, was then re-inserted into the vaporization chamber.
Solvent removal from the sample prior to the analysis was found essential to avoid microplasma instability. In the presence of ethanol, the plasma took on a purple color and displayed an erratic background emission. It is well known that plasmas (regardless of their size), when unaided by mixing oxygen gas, do not tolerate organic solvent vapors. The sample drying procedure used for such samples was the following: the electric power applied to the coil was first set to 0 W for 1 min (to allow for the bulk of the volatile solvent to vaporize at room temperature). The electric power was then increased to 0.12 W for 30 s. When the copolymer was present in the sample, an additional step was performed to char the dried polymer remaining on the coil. This was accomplished by applying progressively higher electric power levels; for example, 0.27 W for 15 s, then 0.44 W for 15 s, and subsequently 0.72 W for 30 s. Progressive heating was used to avert possible bursting of sample microdroplets from the rapidly heated coil, which would have resulted in analyte loss. Progressive heating during charring was also used to avoid potential bursting of the polymer matrix. After sample drying (and charring if required) the coil was allowed to cool for 60 s, with the Pd-containing residue remaining on the coil.
The microplasma was subsequently ignited and the visually stable, blue-colored microplasma was allowed to thermally equilibrate for 60 s (so that stable microplasma background emission was obtained). A higher electric power was then applied to the coil (e.g., 44.8 W, corresponding to ca. 2500 °C) to vaporize the Pd-containing residue. This temperature was found sufficient to vaporize the residues from the finely dispersed Pd samples (vide infra), which are expected to display a lower vaporization temperature than their bulk counterparts.46 The gas-phase metal atoms exited the vaporization chamber and were transported to the microplasma by the carrier gas (230 mL min−1, Ar–H2). Argon mixed with hydrogen (3% v/v) was used to prevent oxidation of the Re coil by the low-ppm levels of water and oxygen typically present in commercial compressed gas cylinders.47 Interaction of the vaporized sample residue with the microplasma led to the atomic emission from Pd I at 340.458 nm which was measured by the PMT detector (Fig. 1a). The output of the detector was amplified, digitized, and stored onto a computer system. An example of signal so obtained is shown in Fig. 1b.
Although the emitted signals lasted for only about 0.5 s, data were acquired for 5 s to monitor microplasma background emission during the pre-vaporization and post-vaporization time intervals (Fig. 1b). Furthermore, when a polymer was present, the electric power was applied to the coil for an additional 5 s to remove any carbonaceous material potentially remaining on the coil. At the end of this sequence the electric power was turned off, and the coil was allowed to cool for 60 s before a subsequent run. For each sample or standard solution, 3.0, 6.0 and 10.0 μL injections volumes were used in at least triplicate runs (Tables S1–S3†). There was therefore a minimum of 9 runs per sample or standard solution. It is worth noting that the above steps (drying, charring, and coil-cleaning procedure when applicable) were repeated for more than 1700 vaporization cycles (i.e. analytical runs) without evidence of carbon deposition, fouling of the microplasma electrodes, or coil alteration. The microplasma background emission was also stable, with a %RSD in background variations determined to be less than 0.6% over 10 h of continuous operation. An example of this is shown in Fig. S1.† It is estimated that the operating cost of the microplasma is roughly 100 times lower than for that of a current ICP-OES instrument.
Results and discussion
Pd standard solutions in water and ethanol
Both aqueous and ethanolic solutions, prepared from a Pd standard solution, were analyzed for the first time using the microplasma set-up shown in Fig. 1a. A typical optical emission signal and stable microplasma background emission obtained during the pre- and post-vaporization time intervals are also displayed in Fig. 1b. Calibration curves, obtained using the integrated area of the optical emission peaks, were linear and comparable in the concentration range of interest (with typically R2 = 0.99998 in ethanol) (Fig. S2†).To improve the statistical confidence, three different injection volumes were used (viz. 3.0, 6.0 and 10.0 μL) for each standard solution or sample. In absolute units, the amount of Pd injected ranged between 3.0 and 10.0 ng. The precision, expressed in %RSD, was determined from at least triplicate runs for each of the injection volumes used (Tables S1–S3†). The %RSD obtained for the solution of Pd-Std in water was below 1.7%, and for the Pd-Std solution in ethanol it ranged between 7.4 and 0.7%. For the samples of Pd and polymer dissolved in ethanol, the %RSD was below 3.8%. In general, the %RSD was lower for the aqueous solutions, and for larger injection volumes. Solvent blanks (i.e., without any Pd added) did not show any emission signals.
Copolymer-stabilized Pd solutions in ethanol
The LOD (using the 3σ criterion) for Pd was estimated from the standard deviation of the background before the emission peak when using the same number of data points as for the peak.48 The average LOD estimated using Pd-loaded polymers diluted in ethanol was 28 pg (expressed in absolute amount, Table S1†). When using 10 μL of solution, these LODs correspond to 3 ng mL−1 (or 3 ppb, in relative concentration units). Such a limit of detection is ca. 15 times lower, for instance, than the concentration at which Pd impurities were found to be catalytically active (50 ppb).49 This is also well below some of the “homeopathic” concentrations used (>500 ppb) in a variety of Heck and Suzuki cross-coupling reactions.36,50–52Quantification of Pd loading in the arborescent copolymer
To determine the maximum amount of Pd that can be loaded in the arborescent copolymer described earlier (G0PS-g-P2VP, Scheme 1) an excess of Pd atoms (1.5 molar equiv.) with respect to the 2VP units was added. Dialysis of the polymer–Pd solution in ethanol was then used to remove any unbound metal. The residual Pd content in the polymer was measured and quantified based on a calibration curve constructed from non-dialyzed polymer–Pd solutions diluted in ethanol (R2 = 0.9920). The calibration curve obtained and the amount of Pd measured are shown in Fig. 2. The average Pd content per 2VP unit was found to be 99.5 mol%, with a %RSD below 3.3% (Table S2†). In the remaining of the discussion, this Pd-loaded polymer sample will be referred to as G1-Pd[100 mol%]. | ||
| Fig. 2 Calibration curve for Pd in a solution of G1-Pd(OAc)2 [1.5 equiv.] in ethanol (blue diamonds), and analysis of the dialyzed sample G1-Pd(OAc)2 using the following injection volumes: 3.0 μL (red square), 6.0 μL (green triangle) and 10.0 μL (purple circle). | ||
The Pd concentration was also determined in a polymer-stabilized Pd sample containing 0.25 equiv. of Pd per 2VP unit. The amount of Pd was selected to insure complete loading of the micelles, and has been shown to lead to the formation of stable Pd(II)–polymer hybrid systems.19 After dialysis, the overall Pd content measured using a minimum of 9 runs was 23.7 mol%, with a %RSD ≤ 5.4% (Fig. S3 and Table S3†). This corresponds to 93.3 mol% of the Pd added prior to dialysis. This sample will be referred to as G1-Pd[24 mol%] in the rest of the discussion.
Imaging by TEM of the polymer–Pd solutions confirmed the presence of Pd in the copolymer templates, displaying an overall diameter ca. 18 nm, as shown in Fig. 3. Pd NPs 2–7 nm in diameter, presumably formed through an Ostwald ripening process, are also visible within G1-Pd[100 mol%].
 | ||
| Fig. 3 TEM images obtained from ethanol solutions of G1-Pd[24 mol%] (left), and G1-Pd[100 mol%] (right). The brightness and contrast were adjusted for better visualization (the scale bars represent 50 nm). Inset: magnification over a single micelle, with a dotted circle added to help visualization; the scale bars represent 20 nm. | ||
It was thus concluded that the arborescent copolymer can successfully complex with the Pd(II) species present in solution. It also appears that all the 2VP units in the G1 arborescent copolymer are accessible to the metallic ions and can contribute to forming stable colloidal dispersions. These conclusions are in accordance with earlier work reporting the fast coordination of Pd2+ species,53 and strong interactions of Pd with the lone electron pair of the nitrogen atom in the 2-vinylpyridine units in aqueous media,10,13,14 or even at high pressures and temperatures in organic solvents.15 However the exact nature of the complex formed still requires further investigation. Bekturov et al. suggested a model for the complexation of PdCl2 by P2VP which accounts for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of 2VP units and Pd.7 However, more recently Fernandez et al. reported that up to 1/3 of the Pd in commercial Pd(OAc)2 could be in the form of Pd(0). These reduced species accounted for an excess of Pd measured in polymer brushes decorated with dipyridylamine ligands, when compared to the stoichiometric complexation expected with Pd(II) species.54 The results obtained in the current study are consistent with the formation of a 1:1 complex between the 2VP units and Pd as suggested by Bekturov et al.
:1 molar ratio of 2VP units and Pd.7 However, more recently Fernandez et al. reported that up to 1/3 of the Pd in commercial Pd(OAc)2 could be in the form of Pd(0). These reduced species accounted for an excess of Pd measured in polymer brushes decorated with dipyridylamine ligands, when compared to the stoichiometric complexation expected with Pd(II) species.54 The results obtained in the current study are consistent with the formation of a 1:1 complex between the 2VP units and Pd as suggested by Bekturov et al.
Stability and kinetic studies
From the results shown in Fig. 4, it can be concluded that even for a Pd concentration as low as 2.4 × 10−6 M (0.25 μg mL−1), there is a rapid decrease in the amount of Pd remaining dispersed in solution. After 5 days, for instance, only about one third of the initial Pd content was left in solution. Assuming a first-order rate process, one can write the aggregation rate as: r = −d[Pd]/dt. The apparent rate constant kapp is then obtained from ln([Pd]/[Pd]0) = −kappt, where [Pd]0 and [Pd] represent the initial concentration of Pd in solution and the concentration at time t, respectively. The graphical method seems to confirm the validity of the first order assumption, and the apparent rate constant calculated for Pd aggregation from Pd(OAc)2 in ethanol was (kapp)EtOH = 9.89 × 10−3 h−1 (Fig. S4†).
 | ||
| Fig. 4 Evolution of the mass concentration of Pd in ethanolic solutions of Pd(OAc)2 (red squares), and G1-Pd(OAc)2 [0.25 equiv.] (green triangles) as measured by microplasma-optical emission spectrometry. | ||
In more concentrated solutions and in the absence of a polymeric stabilizer, a deposit of Pd black was observed after only two hours, as shown in Fig. 5b. The reduction of Pd(II) to Pd(0) in ethanol according to Scheme 2 is well-known; in fact this was taken advantage of in the preparation of various polymer-stabilized Pd nanoparticles.4,6,14,34
 | ||
| Fig. 5 Appearance of (a) a Pd standard solution in water, and ethanolic solutions of (b) Pd(OAc)2, (c) G1-Pd[24 mol%], and (d) G1-Pd[100 mol%]. | ||
 | ||
| Scheme 2 Reduction of Pd(II) to Pd(0) in ethanol. | ||
It should be noted that aqueous solutions prepared from a Pd standard solution (by dilution with Milli-Q water to 1 μg mL−1 or 9.40 μM in Pd, and pH 2.7) displayed a similar decrease in concentration, but a plateau was reached after 2 days at about 72 weight% (wt%) of the initial Pd concentration (Fig. S5†). The first-order apparent rate constant of aggregation, determined before reaching the plateau, was (kapp)water = 6.41 × 10−3 h−1 (Fig. S4†). The diluted solutions remained yellow and translucent, as shown in Fig. 5a. The formation of a chlorohydroxypalladium(II) precipitate has indeed been reported for Pd2+ at concentrations above 1 ppm, but this was thought to be less predominant at lower Pd concentrations.56–58 Clearly, the solutions used for calibration purposes should be freshly prepared before use.
Conclusions
We demonstrated the use of a microplasma-based method as an attractive alternative to the more expensive and widely employed ICP technique for Pd quantification. This method fulfills the requirements spelled out by Manning and Grow for a versatile atomic emission source,59 and contributes to the greening of plasma spectrochemistry. The implementation of a charring step alleviated the need for oxygen-containing gas mixtures. The Re coiled filament was used for more than 1700 analytical runs without any noticeable degradation, and the microplasma emission background was stable for more than 10 h of continuous operation. The use of microsamples for the analysis also means that smaller amounts of reagents and catalysts are required. The %RSD achieved ranged from 7.4% to 0.1%, and the average Pd LOD (3σ) was estimated to be 28 pg (in absolute amount) or 3 ng mL−1 (when using 10 μL volumes). Such a LOD was amply sufficient for the determination of Pd loading in microsamples of arborescent copolymers in ethanol. The maximum average Pd content per 2VP unit was determined to be 99.5 mol%.The method developed also enabled kinetic studies of the stability of palladium acetate in ethanol. It was thus found that the apparent rate constant of aggregation in ethanol was 9.89 × 10−3 h−1, with about 70% of the Pd precipitating out of solution after 5 days. A rapid drop in the Pd concentration before reaching a plateau was also observed in aqueous samples of a Pd standard solution. Clearly, Pd calibration standards must be freshly prepared prior to their use if meaningful Pd concentrations are to be obtained. In the presence of the arborescent copolymer, however, the Pd concentration in ethanol remained stable for at least 10 days.
Overall, the microplasma-based approach described above will help address the need for greener and cheaper quantitative analytical methods, and thus facilitate more widespread use of such methods in catalysis, as articulated by Molnár.26 Work is in progress to evaluate other organic solvents (e.g., THF) and aqueous systems, with the aim of making the overall catalytic process greener and cheaper.
Acknowledgements
The authors gratefully thank the Natural Sciences and Engineering Research Council of Canada (NSERC), and GreenCentre Canada for their support of the work.Notes and references
- G. Schmid, in Nanoparticles, Wiley-VCH, Weinheim, 2nd edn, 2010 Search PubMed.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- H. Bönnemann and K. S. Nagabhushana, in Metal Nanoclusters in Catalysis and Materials Science, ed. B. Corain, G. Schmid and N. Toshima, Elsevier Science, Amsterdam, 1st edn, 2008, ch. 2, pp. 21–48 Search PubMed.
- H. Hirai, Y. Nakao, N. Toshima and K. Adachi, Chem. Lett., 1976, 905–910 CrossRef CAS.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594–600 CrossRef CAS.
- E. A. Bekturov, S. E. Kudaibergenov, S. S. Saltybaeva, D. V. Sokolskii, A. K. Zharmagambetova and N. V. Anisimova, React. Polym., 1985, 4, 49–53 CAS.
- E. A. Bekturov, S. E. Kudaibergenov, D. V. Sokolskii, A. K. Zharmagametova, S. G. Mukhamedzhanova and A. S. Kuanyshev, Makromol. Chem., Rapid Commun., 1986, 7, 187–191 CrossRef CAS.
- D. V. Sokolskii, A. K. Zharmagambetova, S. G. Mukhamedzhanova, E. A. Bekturov and S. E. Kudaibergenov, React. Kinet. Catal. Lett., 1987, 33, 387–392 CrossRef CAS.
- W. Hoogsteen and L. G. J. Fokkink, J. Colloid Interface Sci., 1995, 175, 12–26 CrossRef CAS.
- K. Tsutsumi, Y. Funaki, Y. Hirokawa and T. Hashimoto, Langmuir, 1999, 15, 5200–5203 CrossRef CAS.
- T. Hashimoto, M. Harada and N. Sakamoto, Macromolecules, 1999, 32, 6867–6870 CrossRef CAS.
- N. Semagina, E. Joannet, S. Parra, E. Sulman, A. Renken and L. Kiwi-Minsker, Appl. Catal., A, 2005, 280, 141–147 CrossRef CAS PubMed.
- S. Pathak, M. T. Greci, R. C. Kwong, K. Mercado, G. K. S. Prakash, G. A. Olah and M. E. Thompson, Chem. Mater., 2000, 12, 1985–1989 CrossRef CAS.
- M. Harada, M. Ueji and Y. Kimura, Colloids Surf., A, 2008, 315, 304–310 CrossRef CAS PubMed.
- M. Gauthier, J. Li and J. Dockendorff, Macromolecules, 2003, 36, 2642–2648 CrossRef CAS.
- M. Gauthier and A. Munam, RSC Adv., 2012, 2, 3100–3108 RSC.
- J. M. Dockendorff, Ph.D. thesis, University of Waterloo, 2011.
- O. Nguon and M. Gauthier, unpublished work.
- D. Astruc, in Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 1st edn, 2008, ch. 1, pp. 1–48 Search PubMed.
- C.-J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457–2487 RSC.
- Q. Wang and A. E. Ostafin, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific, Valencia, 1st edn, 2004, vol. 5, pp. 475–503 Search PubMed.
- H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3–18 CrossRef CAS.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- W. Kleist, S. S. Pröckl and K. Köhler, Catal. Lett., 2008, 125, 197–200 CrossRef CAS PubMed.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127–14136 CrossRef CAS PubMed.
- D. Astruc, Tetrahedron: Asymmetry, 2010, 21, 1041–1054 CrossRef CAS PubMed.
- K. A. Marvin, J. A. Johnson, S. E. Rodenbusch, L. Gong, D. A. Vanden Bout and K. J. Stevenson, Anal. Chem., 2012, 84, 5154–5158 CrossRef CAS PubMed.
- W. Liao, Y.-C. Chen, J. S. Wang, H. K. Yak and C. M. Wai, Ind. Eng. Chem. Res., 2007, 46, 5089–5093 CrossRef CAS.
- S.-M. Huang and B.-L. He, React. Polym., 1994, 23, 1–9 CrossRef CAS.
- Y. Wu, D. Wang, P. Zhao, Z. Niu, Q. Peng and Y. Li, Inorg. Chem., 2011, 50, 2046–2048 CrossRef CAS PubMed.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef CAS PubMed.
- F. Zhao, M. Shirai and M. Arai, J. Mol. Catal. A: Chem., 2000, 154, 39–44 CrossRef CAS.
- F. Durap, Ö. Metin, M. Aydemir and S. Özkar, Appl. Organomet. Chem., 2009, 23, 498–503 CrossRef CAS.
- A. Ohtaka, T. Teratani, R. Fujii, K. Ikeshita, T. Kawashima, K. Tatsumi, O. Shimomura and R. Nomura, J. Org. Chem., 2011, 76, 4052–4060 CrossRef CAS PubMed.
- Y. He and C. Cai, Catal. Lett., 2010, 140, 153–159 CrossRef CAS.
- S. Weagant, V. Chen and V. Karanassios, Anal. Bioanal. Chem., 2011, 401, 2865–2880 CrossRef CAS PubMed.
- B. Gibson, H. R. Badiei and V. Karanassios, Spectrochim. Acta, Part B, 2006, 61, 753–758 CrossRef PubMed.
- V. Karanassios, F. H. Li, B. Liu and E. D. Salin, J. Anal. At. Spectrom., 1991, 6, 457–463 RSC.
- G. Grindlay, S. Maestre, L. Gras and J. Mora, J. Anal. At. Spectrom., 2006, 21, 1403–1411 RSC.
- S. Weagant and V. Karanassios, Anal. Bioanal. Chem., 2009, 395, 577–589 CrossRef CAS PubMed.
- V. Karanassios, K. Johnson and A. T. Smith, Anal. Bioanal. Chem., 2007, 388, 1595–1604 CrossRef CAS PubMed.
- S. C. Vanithakumari and K. K. Nanda, Phys. Lett. A, 2008, 372, 6930–6934 CrossRef CAS PubMed.
- V. Karanassios, V. Grishko and G. G. Reynolds, J. Anal. At. Spectrom., 1999, 14, 565–570 RSC.
- H. R. Badiei, B. Lai and V. Karanassios, Spectrochim. Acta, Part B, 2012, 77, 19–30 CrossRef CAS PubMed.
- R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. Singer, J. Org. Chem., 2005, 70, 161–168 CrossRef CAS PubMed.
- M. T. Reetz, E. Westermann, R. Lohmer and G. Lohmer, Tetrahedron Lett., 1998, 39, 8449–8452 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed.
- L. M. Bronstein, S. N. Sidorov, P. M. Valetsky, J. Hartmann, H. Cölfen and M. Antonietti, Langmuir, 1999, 15, 6256–6262 CrossRef CAS.
- A. E. Fernandes, A. Dirani, C. d'Haese, G. Deumer, W. Guo, P. Hensenne, F. Nahra, X. Laloyaux, V. Haufroid, B. Nysten, O. Riant and A. M. Jonas, Chem. – Eur. J., 2012, 18, 16226–16233 CrossRef CAS PubMed.
- P. D. Burton, T. J. Boyle and A. K. Datye, J. Catal., 2011, 280, 145–149 CrossRef CAS PubMed.
- C. D. Tait, D. R. Janecky and P. S. Z. Rogers, Geochim. Cosmochim. Acta, 1991, 55, 1253–1264 CrossRef CAS.
- L. I. Elding, Inorg. Chim. Acta, 1972, 6, 647–651 CrossRef CAS.
- A. N. Pankratov, V. B. Borodulin and O. A. Chaplygina, Russ. J. Coord. Chem., 2005, 31, 660–666 CrossRef CAS PubMed.
- T. J. Manning and W. R. Grow, Chem. Educ., 1997, 2, 1–19 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Emission signals, calibration curves, and kinetic plots of the various Pd species. See DOI: 10.1039/c3ra46232c |
| This journal is © The Royal Society of Chemistry 2014 |