 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic carbene transfer allows the direct customization of cyclic purine dinucleotides†
Na
Fei
a,
Daniel
Häussinger
a,
Seraina
Blümli
a,
Benoît-Joseph
Laventie
b,
Lorenzo D.
Bizzini
a,
Kaspar
Zimmermann
a,
Urs
Jenal
b and
Dennis
Gillingham
*a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, 4056, Basel, Switzerland. E-mail: dennis.gillingham@unibas.ch
bBiozentrum, University of Basel, Klingelbergstrasse 50/70, 4056, Basel, Switzerland
First published on 20th June 2014
Abstract
We describe a simple method for the direct modification of nucleobases in cyclic purine dinucleotides, important signalling molecules in both prokaryotes and eukaryotes. The method tolerates all members of the cyclic dinucleotide family and could be used to modulate their function or introduce useful side-chains such as fluorophores and photo-crosslinking groups.
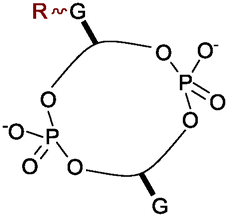
3′,5′- and 2′-5′-linked cyclic dinucleotides (CDNs, see Fig. 1) play diverse and important roles in biology. The cyclic diguanylate derivative (c-di-GMP) is a ubiquitous secondary messenger in prokaryotes.1 Fluctuating levels2 of c-di-GMP regulate a range of bacterial cell functions including motility, adhesion, cell-to-cell communication, exopolysaccharide synthesis, biofilm formation and virulence.3 More recently, c-di-AMP was identified as an important signalling molecule in gram-positive bacteria.4 Moreover recent evidence suggests that humans endogenously produce the unusual CDN c-G(2′,5′)pA(3′,5′)p as part of their innate immune response to cytoplasmic dsDNA and dsRNA.5 Given the diverse roles of CDNs in prokaryotes and eukaryotes, it would be helpful to have probe molecules that could perturb the function of each CDN specifically and independently. Furthermore structural analogues able to evade an immune response or specifically interfere with the mammalian immune system might open the door to CDN derived therapeutics.6
 | ||
| Fig. 1 Structures of the natural CDNs and a phosphorothioate derivative (c-di-GMPS) engineered for phosphodiesterase resistance. | ||
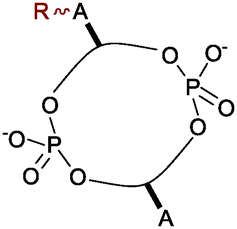
Unravelling and reprogramming the complex biology of CDNs hinges on the ready availability of chemically tailored variants. While total chemical synthesis gives access to any variation, substantial expertise and labour are required.7 Semi-syntheses from the natural CDNs are simpler, but until now these have focussed on changes in the phosphate linkage or 2′ position of the ribose (see Fig. 2).8 We report here that catalytic rhodium-based carbene transfer offers a one-step method to target the exocyclic amine of nucleobases in all types of natural 3′-5′-linked CDNs. Direct nucleobase modification has never been explored, but would provide an important complement to previous approaches.
 | ||
| Fig. 2 CDN sites that can be targeted through direct modification of the natural compounds or with commercial phosphoramidites of the canonical bases. | ||
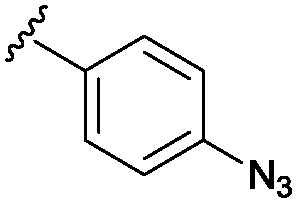
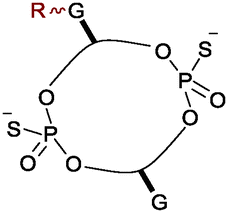
Rhodium catalysed carbene reactions represent an emerging technology in chemical biology.9 We have recently developed Rh-catalysed N–H insertion with diazo compounds as a method for modifying nucleobases in DNA and RNA.10 While the technique is unselective in long single-stranded oligos, CDNs presented the more tractable problem of selecting between only two reactive N–H groups. A handful of diazo compounds bearing common functional tags (amine for water solubility, azide for photo-crosslinking, alkyne for click chemistry) were synthesized and tested with CDNs (see Table 1). The reactions delivered mainly mono-modified CDNs along with some unproductive O–H insertion of the diazo starting material with water (hence 10 equivalents are required); in some cases minor double-modified products were also observed (0–18%). As shown in Table 1 the conversions ranged from 33–80% depending upon the precise substrate and diazo compound. The reactions are fast, requiring at most 2 h to reach completion (entries 1–3). For example, aryl azide modified c-di-GMP (entry 4, Table 1) delivered 58% conversion in just 20 minutes. We also investigated the phosphodiesterase resistant phosphorothioate derivative c-di-GMPS. Sulphur derivatives often hinder carbene transfer reactions, but in this case phosphorothioates were well-tolerated, delivering 80% conversion and 41% isolated yield (entry 6).











One unexpected observation in Table 1 relates to the mixed dinucleotide c-GAMP: in the case of the dimethylamino derived diazo compound a mixture of guanine and adenine modified products were obtained in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture according to HPLC analysis (entry 3), but with the azide containing diazo substrate targeting of the guanine was far more selective (entry 5, >80% selectivity for G alkylation according to integration of HPLC). The structures of the products were gleaned independently from NMR (ROESY, HMQC, and HMBC, see the ESI† for details) and MS–MS fragmentation (Fig. 3). With the sample from entry 3 in Table 1, the major mass peaks matched the guanine-modified structure (see panel A, Fig. 3). In contrast, the minor product from entry 3 in Table 1 delivered the modified adenine fragment (panel b, Fig. 3); while the azide-containing diazo substrate (entry 5) almost exclusively targeted the guanine (panel c, Fig. 3). Alkylation on the phosphate was ruled on the basis that there was a strong HMBC correlation between the α-hydrogen derived from the diazo substrate and the nearest carbon on the nucleobase in each case. We had expected that the unsymmetrical CDN c-GAMP would represent a substantial challenge in chemoselectivity since adenine and guanine display similar functional groups to the catalyst. However, as entries 3 and 5 demonstrate, the substrate can play a role in controlling the site-selectivity. The source of the change could be related to the charge of the dimethylamino group, or the propensity of certain CDNs to form higher-order aggregates in solution.11 Although the product mixture obtained from entry 3 was more complex, we were able to separate each component (the 39% reported yield corresponds to the mixture) and therefore c-GAMP derivatives are available with alterations at either base through one protocol.
:1 mixture according to HPLC analysis (entry 3), but with the azide containing diazo substrate targeting of the guanine was far more selective (entry 5, >80% selectivity for G alkylation according to integration of HPLC). The structures of the products were gleaned independently from NMR (ROESY, HMQC, and HMBC, see the ESI† for details) and MS–MS fragmentation (Fig. 3). With the sample from entry 3 in Table 1, the major mass peaks matched the guanine-modified structure (see panel A, Fig. 3). In contrast, the minor product from entry 3 in Table 1 delivered the modified adenine fragment (panel b, Fig. 3); while the azide-containing diazo substrate (entry 5) almost exclusively targeted the guanine (panel c, Fig. 3). Alkylation on the phosphate was ruled on the basis that there was a strong HMBC correlation between the α-hydrogen derived from the diazo substrate and the nearest carbon on the nucleobase in each case. We had expected that the unsymmetrical CDN c-GAMP would represent a substantial challenge in chemoselectivity since adenine and guanine display similar functional groups to the catalyst. However, as entries 3 and 5 demonstrate, the substrate can play a role in controlling the site-selectivity. The source of the change could be related to the charge of the dimethylamino group, or the propensity of certain CDNs to form higher-order aggregates in solution.11 Although the product mixture obtained from entry 3 was more complex, we were able to separate each component (the 39% reported yield corresponds to the mixture) and therefore c-GAMP derivatives are available with alterations at either base through one protocol.
 | ||
| Fig. 3 MS–MS analysis of alkylation products. Panel A: the major product from entry 3 in Table 1 delivered a daughter ion consistent with guanine alkylation; panel B: the minor product was consistent with adenine alkylation; panel C: the only isolated product from entry 5 in Table 1 delivered a daughter ion consistent with guanine alkylation. | ||
In prokaryotes CDNs are involved in a complicated regulatory network involving a multitude of individual protein components12 and several riboswitches.13 The modified CDN derivatives shown in Table 1 are versatile starting points for exploring the biology of these second messenger molecules. For example, the azide motif can be converted to a fluorescent CDN derivative through a catalytic azide–alkyne cycloaddition (see the ESI,† Fig. S30–S34 for an example). Furthermore, the aryl azide itself is a common photo-crosslinking group and therefore compounds such as those found in Table 1 (entries 4–6) could be used to probe binding sites of c-di-GMP receptors. Although 2′-hydroxyl derived probes are known,8b,14 a family of photo-crosslinkers is important since different receptors will have different binding constraints.
To explore the photo-crosslinking of azide-modified CDNs we selected the known c-di-GMP receptor DgrA – a PilZ homolog that mediates c-di-GMP-dependent control of Caulobacter crescentus cell motility.15 Its high affinity and specificity towards c-di-GMP and the availability of binding mutants make this protein an ideal test bed. The aryl azide modified c-di-GMP (c-di-GMP-N3) was incubated with the protein for 15 h under 366 nm irradiation (see top of Fig. 4) and the mixtures were analysed by high-resolution ESI mass spectrometry, gel electrophoresis, MALDI-TOF, and finally the site of modification was determined by a trypsin digest (see the ESI,† Fig. S35–S39 for details). Even with a single equivalent of c-di-GMP-N3 (lane 3) DgrA was covalently modified in a yield of 17% (according to integration of the gel bands). At 10 equivalents of c-di-GMP-N3 there was complete conversion (lane 8), but the diffuse bands suggested competitive unspecific modification. Previous work has shown that Arg11, Arg12, and Trp75 are important residues for c-di-GMP binding of dgrA: a W75A mutant decreased binding 102–103-fold, while binding was completely abrogated in the R11A/R12A double mutant.15 Consistent with the reported binding studies, reaction of c-di-GMP-N3 with the W75A mutant gave reduced crosslinking (cf. lane 5 versus lane 3) and the R11A/R12A was not detectably modified (lane 7). A trypsin digest of the photo-crosslinking reaction revealed one new peak in the LC-MS whose mass was consistent with modification of the GGR peptide fragment shown in red in Fig. 3. This tripeptide sits directly in the region of the purported c-di-GMP binding site (blue in Fig. 3). Taken together these results demonstrate that c-di-GMP-N3 is a selective cross-linking probe efficient enough to determine binding sites in CDN receptors.
 | ||
| Fig. 4 Modified c-di-GMP-N3 maintains binding to DgrA and can be used for photo-crosslinking. The peak at 14059.2 is likely a post-translational modification which is also cross-linked with the azide compound (14910.7). The baseline impurities in the ESI stem from gradual photolytic degradation of DgrA. | ||
While the cross-linking data provided qualitative validation that N2-modified c-GMP derivatives maintained their ability to bind DgrA, microscale thermophoresis gave a quantitative measure of binding. In panel A of Fig. 5 is a comparison of binding to DgrA of c-di-GMP-N3 and c-di-GMP. The modified CDN still bound with a nanomolar affinity – less than a 3-fold change over the native interaction. Consistent with the cross-linking experiments the W75A DgrA mutant showed substantially attenuated binding (see panel b, Fig. 5), but again the modified CDN bound at a similar level as the natural CDN, further validation that the exocyclic amine is a viable site for modification in the study of CDN biology.
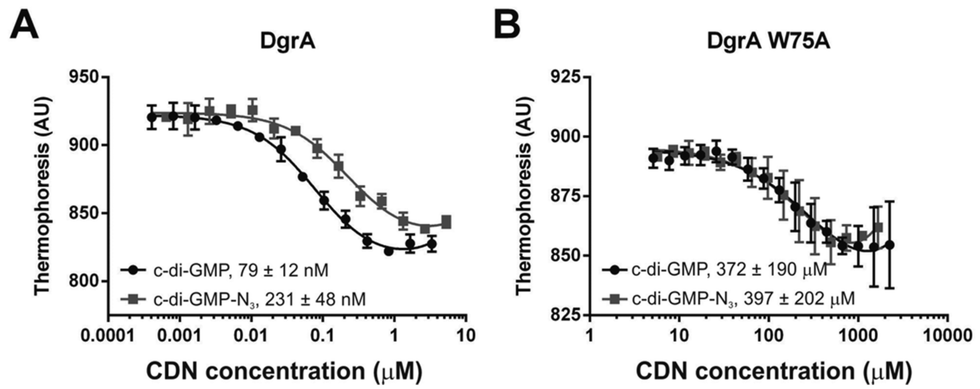 | ||
| Fig. 5 Microscale thermophoresis confirms that c-di-GMP-N3 maintains strong binding to DgrA (panel A). The W75A mutant loses affinity for c-di-GMP and c-di-GMP-N3 to the same extent (panel B). | ||
In summary, we describe a direct method for the synthesis of CDN derivatives modified at the exocyclic amine of the purine bases. The reaction is trivial to execute, making it accessible to non-experts in synthesis and catalysis. The most synthetically challenging aspect of the approach is in the synthesis of the diazo compounds, which typically require 3–5 operations.
New aspects of CDN biology are continually being unveiled. A challenge for chemical biologists is to provide a selective probe for each natural CDN receptor. The process we have described adds a new method for such bespoke probe development. We are currently cataloguing how modifications of the exocyclic amine of CDNs behave with other receptors. A detailed understanding of the different binding requirements of prokaryotic versus eukaryotic receptors will pave the way to creating CDN inspired therapeutics.
Mr Ananth Rao is gratefully acknowledged for assistance with some of the NMR assignments and interpretation.
Notes and references
- (a) H. B. Yan and W. X. Chen, Chem. Soc. Rev., 2010, 39, 2914–2924 RSC; (b) R. Hengge, Nat. Rev. Microbiol., 2009, 7, 263–273 CrossRef CAS PubMed; (c) B. W. Davies, R. W. Bogard, T. S. Young and J. J. Mekalanos, Cell, 2012, 149, 358–370 CrossRef CAS PubMed.
- T. Schirmer and U. Jenal, Nat. Rev. Microbiol., 2009, 7, 724–735 CrossRef CAS PubMed.
- U. Jenal and J. Malone, Annu. Rev. Genet., 2006, 40, 385–407 CrossRef CAS PubMed.
- R. M. Corrigan, J. C. Abbott, H. Burhenne, V. Kaever and A. Grundling, PLoS Pathog., 2011, 7, e1002217 CAS.
- P. Gao, M. Ascano, Y. Wu, W. Barchet, B. L. Gaffney, T. Zillinger, A. A. Serganov, Y. Z. Liu, R. A. Jones, G. Hartmann, T. Tuschl and D. J. Patel, Cell, 2013, 153, 1094–1107 CrossRef CAS PubMed.
- (a) W. X. Chen, R. KuoLee and H. B. Yan, Vaccine, 2010, 28, 3080–3085 CrossRef CAS PubMed; (b) D. K. R. Karaolis, T. K. Means, D. Yang, M. Takahashi, T. Yoshimura, E. Muraille, D. Philpott, J. T. Schroeder, M. Hyodo, Y. Hayakawa, B. G. Talbot, E. Brouillette and F. Malouin, J. Immunol., 2007, 178, 2171–2181 CrossRef CAS; (c) D. X. Gao, J. X. Wu, Y. T. Wu, F. H. Du, C. Aroh, N. Yan, L. J. Sun and Z. J. J. Chen, Science, 2013, 341, 903–906 CrossRef CAS PubMed.
- P. Clivio, S. Coantic-Castex and D. Guillaume, Chem. Rev., 2013, 113, 7354–7401 CrossRef CAS PubMed.
- (a) Y. L. Luo, J. Zhou, S. K. Watt, V. T. Lee, T. K. Dayie and H. O. Sintim, Mol. BioSyst., 2012, 8, 772–778 RSC; (b) I. M. Sharma, T. Dhanaraman, R. Mathew and D. Chatterji, Biochemistry, 2012, 51, 5443–5453 CrossRef CAS PubMed; (c) J. Zhou, D. A. Sayre, J. X. Wang, N. Pahadi and H. O. Sintim, Molecules, 2012, 17, 13376–13389 CrossRef CAS PubMed; (d) C. A. Shanahan, B. L. Gaffney, R. A. Jones and S. A. Strobel, Biochemistry, 2013, 52, 365–377 CrossRef CAS PubMed.
- Z. Chen, F. Vohidov, J. M. Coughlin, L. J. Stagg, S. T. Arold, J. E. Ladbury and Z. T. Ball, J. Am. Chem. Soc., 2012, 134, 10138–10145 CrossRef CAS PubMed.
- K. Tishinov, K. Schmidt, D. Haussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 CrossRef CAS PubMed.
- (a) S. Nakayama, I. Kelsey, J. X. Wang and H. O. Sintim, Chem. Commun., 2011, 47, 4766–4768 RSC; (b) Z. Y. Zhang, S. Kim, B. L. Gaffney and R. A. Jones, J. Am. Chem. Soc., 2006, 128, 7015–7024 CrossRef CAS PubMed.
- C. D. Boyd and G. A. O'Toole, Annu. Rev. Cell Dev. Biol., 2012, 28, 439–462 CrossRef CAS PubMed.
- (a) E. R. Lee, J. L. Baker, Z. Weinberg, N. Sudarsan and R. R. Breaker, Science, 2010, 329, 845–848 CrossRef CAS PubMed; (b) K. D. Smith, S. V. Lipchock, A. L. Livinston, C. A. Shanahan and S. A. Strobel, Biochemistry, 2010, 49, 7351–7359 CrossRef CAS PubMed; (c) J. W. Nelson, N. Sudarsan, K. Furukawa, Z. Weinberg, J. X. Wang and R. R. Breaker, Nat. Chem. Biol., 2013, 9, 834–839 CrossRef CAS PubMed.
- J. Düvel, D. Bertinetti, S. Möller, F. Schwede, M. Morr, J. Wissing, L. Radamm, B. Zimmermann, H.-G. Genieser, L. Jänsch, F. W. Herberg and S. Häussler, J. Microbiol. Methods, 2012, 88, 229–236 CrossRef PubMed.
- M. Christen, B. Christen, M. G. Allan, M. Folcher, P. Jenö, S. Grzesiek and U. Jenal, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4112–4117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc01919a |
| This journal is © The Royal Society of Chemistry 2014 |