
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorine-containing macrocyclic peptides and peptidomimetics
Neshat
Rozatian
 *ab and
Stefan
Roesner
*ac
*ab and
Stefan
Roesner
*ac
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
bCatSci Ltd, CBTC2, Capital Business Park, Wentloog, Cardiff CF3 2PX, UK. E-mail: neshat.rozatian@catsci.com
cSchool of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Byrom Street, Liverpool, L3 3AF, UK. E-mail: s.k.roesner@ljmu.ac.uk
First published on 6th May 2025
Abstract
Fluorination plays a vital role in medicinal chemistry due to the unique properties of the fluorine atom. Macrocyclic peptides offer advantageous properties compared to linear peptides in drug discovery. In recent years, the development of fluorinated macrocyclic peptides and peptidomimetics, such as voxilaprevir, MK-0616 and ulimorelin, has highlighted the growing interest in the combination of fluorination and cyclization for tuning the properties of peptidic drug leads. In this review, the effects of fluorination on biological properties of macrocyclic peptides will be discussed. The use of the fluorine atom as a 19F NMR spectroscopy probe for conformational studies of macrocyclic peptides will be reviewed. Finally, macrocyclic peptides containing the radionucleotide 18F as PET imaging agents will be highlighted.
 Neshat Rozatian | Neshat Rozatian received her PhD from Durham University in 2019, specializing in organofluorine and physical organic chemistries. She then joined the group of Prof. Michael Shipman at Warwick University as a Postdoctoral Research Fellow, working on azetidine-modified macrocyclic peptides. In 2023, she joined Iksuda Therapeutics, developing linker-payloads for incorporation into antibody–drug conjugates. She joined CatSci as a Senior Scientist in 2024, working within the process development team. |
 Stefan Roesner | Stefan Roesner completed his PhD at the University of Bristol, focusing on enantioselective lithiation–borylation reactions. He then moved to the US to undertake postdoctoral research at the Massachusetts Institute of Technology, working on palladium-catalyzed cross-coupling reactions under continuous-flow conditions. Following this, Stefan returned to the UK as a Senior Research Fellow and later Assistant Professor at the University of Warwick, collaborating with Prof. Michael Shipman on peptidomimetics and small nitrogen heterocycles. In April 2024, Stefan became a Senior Lecturer at Liverpool John Moores University, where he continues his research in organometallic and heterocyclic chemistry. |
Introduction
Over the last 30 years, there has been continuously increasing interest in macrocyclic peptides. This is driven by their ability to improve many biophysical properties of peptides, such as binding affinity, specificity and proteolytic stability.1,2 Macrocyclic peptides are predominantly peptidic structures bearing one or more rings containing multiple natural or non-natural amino acid residues. Examples include the antibiotic linopristin and the immunosuppressant cyclosporine. Of the more than 100 peptidic drugs that have reached the market, more than half are macrocyclic.3,4 Cyclic peptides generally display improved pharmacokinetic and pharmacodynamic properties relative to their linear counterparts, making them promising leads in drug discovery.5,6 Macrocyclization of peptides decreases the entropic cost of bonding, which can increase the affinity of peptides to their target.7 A variety of macrocyclization strategies have been developed over the years, including backbone cyclization, side-chain to side-chain cyclization, synthesis of lariat peptides, and many other methods, which have been reviewed elsewhere.1,8–10 Macrocyclic peptides and peptidomimetics occupy a unique chemical space, and work has been done to define these regions according to Principal Component Analysis (PCA).11 Non-peptidic macrocycles derived from amino acids with diverse applications have also been reviewed elsewhere.12Fluorinated small molecules play a vital role in the pharmaceutical and agrochemical industries. 20% of drugs currently on the market contain fluorine and 50% of “blockbuster” drugs are fluorinated.13 The introduction of fluorine into target molecules alters the pKa, metabolic stability, binding affinities and other physicochemical properties, as well as conformational properties.14,15 The unique properties of the C–F bond have enabled the tuning of macromolecule conformations through charge–dipole interactions, dipole–dipole interactions and hyperconjugation.16 The fluorine atom can act as a bioisostere of the hydroxyl moiety, and the difluoromethyl moiety can function as a hydrogen bond donor. The effects of replacing alkoxy or hydroxy groups with a fluorine atom on lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) have been reported, whereby a structure-dependent increase in logP was generally observed.17 The incorporation of fluorine into small molecule drugs can increase hydrophobicity, reduce metabolic degradation and initiate novel interactions with protein targets.18 Synthetic methods of introducing a fluorine atom include electrophilic,19 nucleophilic20 and radical fluorination21,22 as well as trifluoromethylation reagents.23,24 Balz–Schiemann and Swarts halogen exchange processes are widely used for the production of fluoroaromatic and trifluoromethyl aromatic pharmaceuticals, respectively.25 Over the last decade, numerous fluorine-containing building blocks have become commercially available, enabling de novo synthetic approaches.
P) have been reported, whereby a structure-dependent increase in logP was generally observed.17 The incorporation of fluorine into small molecule drugs can increase hydrophobicity, reduce metabolic degradation and initiate novel interactions with protein targets.18 Synthetic methods of introducing a fluorine atom include electrophilic,19 nucleophilic20 and radical fluorination21,22 as well as trifluoromethylation reagents.23,24 Balz–Schiemann and Swarts halogen exchange processes are widely used for the production of fluoroaromatic and trifluoromethyl aromatic pharmaceuticals, respectively.25 Over the last decade, numerous fluorine-containing building blocks have become commercially available, enabling de novo synthetic approaches.
The pharmaceuticals approved by the FDA between 2013 and 2024 included a number of fluorine-containing macrocyclic peptides, such as voxilaprevir, glecaprevir and motixafortide, which will be discussed in detail in this review.26 Fluorine-containing peptides and proteins have increased enzymatic stability.5 The effects of fluorine on conformations of side chains, folding kinetics and activity of linear peptides and proteins have been discussed elsewhere.27–29 Unnatural fluorinated amino acids are useful building blocks for the introduction of fluorine atoms into peptide structures.30 The incorporation of fluoroaromatic linker groups provides an efficient macrocyclization strategy. In this review, we will discuss the strategies employed to alter the biological properties of macrocyclic peptides through fluorination. We will highlight examples where fluorine enabled the analysis of macrocycle conformation as well as the use of fluorine as a 19F NMR reporter. Where relevant, we will include methods of fluorine incorporation into macrocyclic peptides. Ring sizes will be shown in blue at the center of structures. Finally, we will cover examples of macrocyclic peptides labeled with the radionucleotide 18F for positron emission tomography (PET) imaging.
Altering biological properties using fluorine substitution
NS3/4A HCV protease inhibitors
Several fluorine-containing macrocyclic peptidomimetic drugs and lead compounds have been reported that target the hepatitis C virus (HCV) non-structural (NS) protein 3/4A protease. HCV causes chronic liver disease, cirrhosis, and hepatocellular carcinoma. Voxilaprevir (1, GS-9857), developed by Gilead Sciences, was the first fluorinated peptide drug on the market, approved in 2017 (Fig. 1a).31 The key macrocyclic bond forming step involved ring-closing metathesis with Zhan 1b catalyst followed by reduction of the double bond. The difluoromethyl group at the P2 benzopyrazine resulted in improved genotype 3 potency through hydrophobic interactions between the difluoro group and R155, a resistance associated substitution (RAS) (Fig. 1a). A non-natural amino acid at P1 bears a further difluoromethyl group, which contributed toward improved metabolic stability.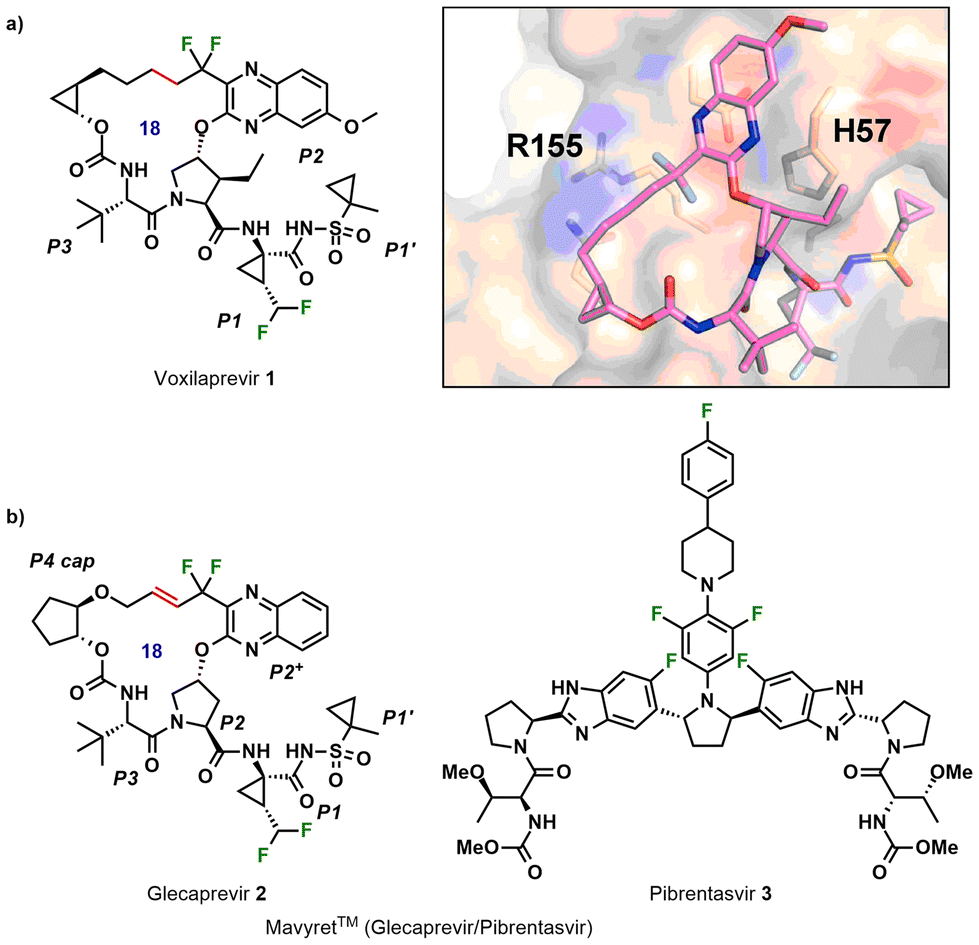 | ||
| Fig. 1 (a) Structure of voxilaprevir 1 and its interaction in crystal structure bound to GT3 surrogate NS3/4A protease. Adapted with permission from ref. 31. Copyright 2019 Elsevier. (b) Structures of glecaprevir 2 and pibrentasvir 3, marketed as Mavyret™, an HCV drug. | ||
Glecaprevir (2, ABT-493) is a next-generation, orally bioavailable drug for the treatment of HCV infections, developed by AbbVie and Enanta.32 Marketed as Mavyret™, it is co-formulated with pibrentasvir (3), an NS5A inhibitor (Fig. 1b).33 Sales of Mavyret™ reached $2.89 billion in 2019.34 Glecaprevir contains four fluorine atoms as well as several amino acid moieties, including tert-Leu, Pro and ACC (1-aminocyclopropanecarboxylic acid). While structurally similar to voxilaprevir, glecaprevir's macrocycle contains an E-alkene, and the quinoxaline is unsubstituted at the C7-position. The medicinal route toward 2 comprised the synthesis of three key building blocks, including the preparation of the difluoromethylene containing quinoxaline derivative 7 (Scheme 1a) and the introduction of fluorine atoms using DAST to construct the difluorocyclopropylene moiety (Scheme 1b).35 Macrocyclization was achieved by a ring-closing metathesis (RCM) reaction using a Ru catalyst (Scheme 1c). A scale up route to glecaprevir has also been reported.36 Glecaprevir has in vitro EC50 values in the low nanomolar range (0.85 to 2.8 nmol L−1) across HCV genotypes 1–6.37 In both voxilaprevir and glecaprevir, the difluoromethylene moiety is responsible for two orthogonal multipolar fluorine-specific interactions.38 The highly electronegative fluorine atom promotes the formation of a caged fluorine-induced hydrogen bond between the hydrogen atom of the difluoromethyl moiety, the backbone carbonyl oxygen of the protease, and the carbonyl oxygen of the P1 group. Thus, the inhibitor is pre-organized for binding. Susceptibility to degradation is decreased through the incorporation of fluorine atoms in benzylic position within the macrocycle backbone. The resultant fluorine-specific interactions improve potency against all proteases genotypes.39
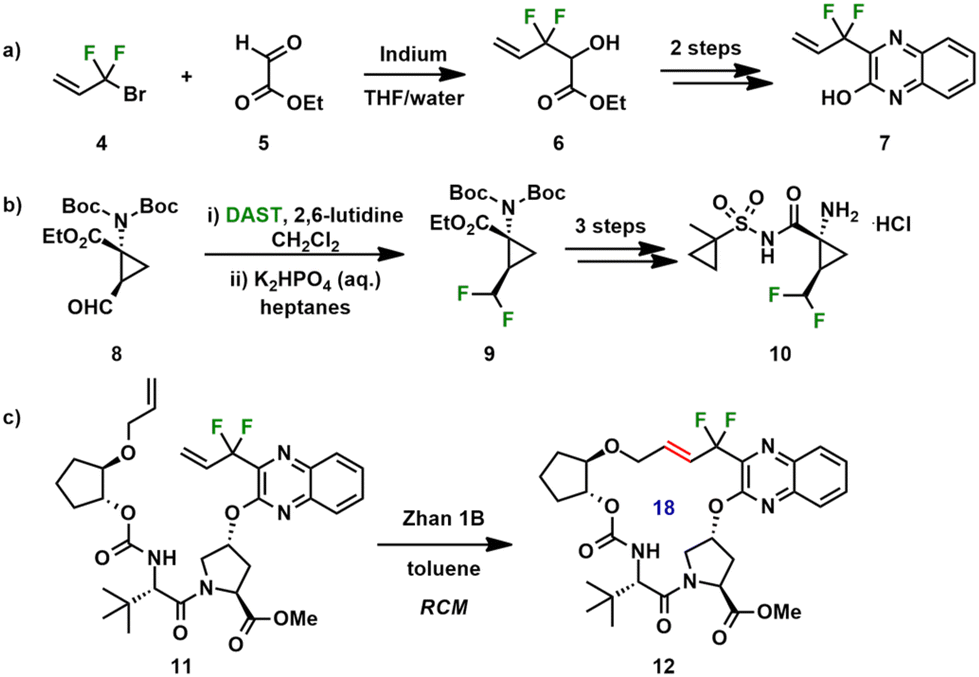 | ||
| Scheme 1 (a) and (b) Synthesis of key fluorinated building blocks toward glecaprevir 2. (c) Macrocyclization via RCM. | ||
Resistance-associated substitutions (RAS) around the NS3/4A protease active site can affect the efficacy of antiviral agents by altering the shape and electrostatic properties of the S4 pocket. In the case of voxilaprevir (1) and glecaprevir (2), it has been shown that the fluorine atoms contribute toward the improved potency and antiviral activity across genotypes and resistant variants compared to the non-fluorinated HCV inhibitor grazoprevir (13, MK-5172, Fig. 2a).31,40 To this end, a series of fluorinated analogs of 13 were synthesized, although the macrocycle was relocated from P2–P4 to P1–P3, to investigate the impact of modifications at the P2+ quinoxaline moiety and the P4 capping group (Fig. 2b). The incorporation of fluorine near sites of RASs was found to improve antiviral activity across several HCV genotypes and resistance variants, due to its unique electronic properties and larger van der Waals radius.41 Against the D168A protease variant, greater potency was retained in analogs fluorinated at P4 caps. Through analysis of co-crystal structures, it was revealed that fluorinated P4 caps could sample alternate binding conformations that enabled adaptation to structural changes induced by the D168A substitution.
 | ||
| Fig. 2 (a) Structure of grazoprevir (13). (b) P1–P3 analogs of grazoprevir with quinoxaline and P4 cap substitutions. | ||
Developed by Array BioPharma and InterMune Inc. and licensed to Roche, danoprevir (32) is a 15-membered macrocyclic peptidomimetic inhibitor of the NS3/4A HCV protease, containing a fluoroisoindoline (Fig. 3a).42,43 This moiety lies in a lipophilic part of the protease's S2 pocket. Crystal structures have shown two equi-energetic conformations of danoprevir, with the fluorine atom either facing the P3 tert-butyl carbamate group or rotated 180° away from it (Fig. 3b). Danoprevir was identified as a clinical candidate due to its favorable potency profile against HCV genotypes 1–6 and key mutants, with IC50 values between 0.2 and 0.4 nM. Clinical trials have recently been conducted to determine the effectiveness of danoprevir, in combination with ritonavir, against COVID-19.44 Patients treated with danoprevir achieved negative nucleic acid test results in fewer days compared with those treated with other antivirals: an average time of 8 days to achieve a negative test result compared with 12.5, and average hospital stay days of 11.4 compared with 16.7, were statistically significant.45
 | ||
| Fig. 3 (a) Structure of danoprevir (32). (b) X-ray crystal structure of 32 bound to NS3 protease active site. Adapted with permission from ref. 42. Copyright 2013 American Chemical Society. (c) Structure of simeprevir (33) and its fluorinated analog 34. | ||
Simeprevir (33, TMC-435), a macrocyclic peptidomimetic, was developed by Janssen Pharmaceuticals Inc. and Medivir AB.46 Milanole et al. reported the synthesis of a fluorinated analog of 33, fluoro-simeprevir (34), a second-generation HCV NS3/4A serine protease inhibitor, containing a fluorocyclopropyl building block (Fig. 3c).47 The synthesis of the unnatural amino acid is outlined in Scheme 2. In the fluorinated analog, the 1-amino-1-carboxyl-2-vinylcyclopropane core is essential for optimal fit in the hydrophobic S1 pocket of the NS3 protease. Installation of a single fluorine to the P1 cyclopropyl moiety in 33, resulted in an 1800-fold increase in EC50, from 8.1 nM to 15 μM. Modelling studies suggested some distortion in the P1–P2 conformation in which repulsion between the fluorine atom and the P2 carbonyl moiety led to a slight rotation that may also disturb the preferred orientation of the acylsulfonamide pharmacophore to the enzyme.
 | ||
| Scheme 2 Synthesis of fluorocyclopropane moiety 42, a key structural component of fluoro-simeprevir 34. | ||
Fluorinated analogs of the peptidomimetic HCV NS3/4A protease inhibitor paritaprevir (43, ABT-450) have been reported (Fig. 4).48,49 Analogs included variants with different heteroaryl substituents at P2+ and the P4 cap, such as compounds 48 and 53. Two general strategies were used to incorporate the difluoromethylene moiety. Direct fluorination of the macrocycle 45 using the fluorinating reagent Morph-DAST (46) selectively converted a ketone into a difluoromethylene moiety (Scheme 3a). Alternatively, fluorine incorporation was achieved using unnatural amino acid 50 (Scheme 3b). Potencies of inhibition were measured against the HCV strains 1a-H77 and 1b-con1 and were reported to show low nanomolar EC50 values.
 | ||
| Fig. 4 Structures of paritaprevir 43 and CF2-containing paritaprevir 44. | ||
 | ||
| Scheme 3 (a) Fluorination of macrocycle 45 using Morph-DAST 46. (b) Incorporation of CF2-moiety into unnatural amino acid 49 and macrocyclization strategy. | ||
BMS-986144 (56) is a pan-genotypic HCV NS3/4A protease inhibitor based on a P1–P3 macrocyclic tripeptide motif, developed as the next generation to the NS3 inhibitor asunaprevir (54) by Bristol Myers Squibb (BMS) (Fig. 5a and b).50 Introduction of the P1–P3 macrocyclic tether to the structure of asunaprevir gave an 8-fold improvement in NS3/4A protease inhibitory potency, and selective functionalization of the tether with alkyl groups optimized both the enzyme inhibitory activity and the metabolic stability. An essential contributor toward higher stability was the trifluorinated Boc group, which addressed the issue of metabolism occurring at this moiety in asunaprevir.51 Thus, the half-life in human liver microsomes (HLM) was increased from 6.4 min for macrocyclic asunaprevir (55) to 33 min for BMS-986144 (56). The isoquinoline moiety bearing a fluorine atom and a deuterated methoxy group (CD3O) further improved the metabolic stability while preserving inhibitory potency.
 | ||
| Fig. 5 (a) Structures of asunaprevir (54), macrocyclic analog 55, and fluorinated macrocycle BMS-986144 (56); HLM = human liver microsomes. (b) Structure of 56 bound to GT-1a NS3/4A protease showing the surface of the enzyme. Adapted with permission from ref. 50. Copyright 2020 American Chemical Society. (c) MK-8831 (58) and its acyclic precursor 57. | ||
MK-8831 (58) is a spiro-proline macrocycle developed by Merck as a pan-genotypic HCV NS3/4A protease inhibitor (Fig. 5c).52 P1–P3 macrocyclization of the acyclic analog 57 improved the potency profile against several mutant variants of genotype 1b, and with the introduction of a trifluoromethyl group to the quinoline, improved genotype 3a activity was also obtained. Following desirable pharmacokinetic profile and safety data, 58 was selected as a preclinical candidate and entered Phase 1 studies.
GS-9256 (59) is a HCV NS3/4A protease inhibitor reported by Gilead Sciences (Fig. 6a).53 The macrocyclic structure features a phosphinic acid pharmacophore and a 2,6-difluorobenzyl group. In structure–activity relationship (SAR) studies, it was found that the 2,6-difluorobenzyl group modulated the low pKa of the phosphinic acid group which was responsible for poor absorption of the non-fluorinated analog. The macrocyclic analogs were seven times more potent NS3 protease inhibitors than non-macrocyclic derivatives. Following modification of the C-8 substituent with a chlorine atom which gave increased bioavailability, the compound with the best potency and pharmaceutical properties, GS-9256 (59), was selected for clinical trials. After a Phase IIb study, 95% of 42 patients treated with GS-9256 in combination with the antivirals tegobuvir, ribavirin and pegylated interferon achieved no detectable levels of HCV twelve weeks post-treatment.54
 | ||
| Fig. 6 Structures of (a) GS-9256 (59), (b) deldeprevir (60) and (c) IDX316 (61) and IDX320 (62). | ||
Deldeprevir (60) (neceprevir, ACH-2684) is a macrocyclic peptidomimetic inhibitor of NS3/4A protease containing a 3,3-difluoropiperidine moiety, developed by Achillion Pharmaceuticals (Fig. 6b).55,56 In Phase Ib clinical trials, it was found to be well-tolerated and reduced the plasma HCV RNA levels in all groups. However, no further developments were reported since 2014.
The development route for the synthesis of macrocyclic trifluoromethylated thiazole structures IDX316 (61) and IDX320 (62) was reported by Idenix Pharmaceuticals Inc., including scale-up to half-kilogram cGMP batches of IDX320 (Fig. 6c).57,58 Single- and multiple-dose studies on IDX320 demonstrated dose-dependent antiviral activity in HCV genotype 1 infected patients.59 However, further development of IDX320 was halted due to the adverse occurrence of increased liver enzymes.
Examples for other therapeutic conditions
MK-0616 (63) is a macrocyclic peptide containing a fluoro-Trp moiety, developed by Merck for the treatment of hypercholesterolemia (Fig. 7).60–6263 is currently in Phase III clinical trials as an oral inhibitor of proprotein convertase subtilisin/kexin type 9 (PCSK9).63 Initially, a series of potent, orally bioavailable tricyclic PCSK9 inhibitors were reported, including compound 64.64 However, this was synthesized by an inefficient linear process and the sulfides were susceptible to oxidation. The redesigned structure 63 was synthesized through a convergent fragment-based strategy, where replacement of the thiol-based linker and central triazole reduced susceptibility to oxidation while maintaining high potency (64Ki = 2 pM vs. 63Ki = 5 pM). 63 was found to inhibit PCSK9 in human plasma (IC50 = 2.5 ± 0.1 nM), had low transcellular permeability, and no off-target activity in screening assays. | ||
| Fig. 7 Structure of MK-0616 (63) and related analog 64. | ||
Fluorine-containing macrocyclic peptides have recently been identified for cancer treatment (Fig. 8). Motixafortide (65), sold under the name Aphexda™, has been approved for the treatment of pancreatic cancer and acute myeloid leukemia.65 LUNA18 (66), reported by Chugai Pharmaceutical, is a highly bioavailable fluorinated macrocyclic peptide that acts as an inhibitor of Kirsten rat sarcoma viral oncogene homolog (KRAS), a common oncogene in numerous cancers.66,67 The 4-CF3-3,5-F2-substituted aryl ring contributed toward improved PPI inhibitory activity, and LUNA18 showed significant efficacy against cancer cell lines with KRAS genetic alterations (IC50 = 0.17–2.9 nM).
 | ||
| Fig. 8 Structures of (a) motixafortide (65) and (b) LUNA18 (66). | ||
Ulimorelin (TZP-101, 67b) is a macrocyclic peptidomimetic ghrelin receptor agonist, developed by Tranzyme Pharma as a first-in-class treatment for postoperative ileus and diabetic gastroparesis (Fig. 9).68 Compound 67a was initially identified as a lead GRLN agonist, and after further development, 67b was selected as the clinical candidate. A key modification was the para-fluoro substituted Phe residue, the choice of which was supported by ligand lipophilicity efficiency (LLE) trends. SAR studies on the phenoxy ring substitution pattern showed a 2-fold improvement in binding potency, Ki, upon fluoro-substitution at the R2 position compared to the unsubstituted derivative 67c. On the other hand, R3 fluoro-substitution in 67e resulted in a 22-fold reduction in potency. PK profiling in rats highlighted desirable properties for 67b and 67d; however, PK profiling in monkeys revealed that the systemic clearance rate (CL) and absolute oral availability (%F) of ulimorelin (67b) were superior to those of 67d, leading to its selection for clinical development.
 | ||
| Fig. 9 Structure of ulimorelin (67b) and its analogs. | ||
MDL-104168 (69) is a macrocyclic human immunodeficiency (HIV) protease inhibitor containing a difluorostatone peptide mimetic, developed by Marion Merrell Dow (Fig. 10).69 The original acyclic HIV-protease inhibitor, MDL-73669 (68), was cyclized via the P1 and P3 side chains, and the resulting macrocyclic peptide 69, still retained good biological activity (Ki = 20 nM, IC50 = 2 nM). Such inhibitors are known to bind via the central hydrated difluorostatone by positioning the fluorine atoms within hydrogen bonding distance from catalytic aspartyl residue carboxylates, thus mimicking a key intermediate formed during the rate limiting step of normal hydrolysis of a substrate.69
 | ||
| Fig. 10 Structures of linear (68) and macrocyclic (69) peptides containing a difluorostatone peptide mimetic. | ||
A series of peptidic macrocycles bearing N-linked peptoid groups were developed targeting CXCR7, a chemokine receptor.70 Building upon previously reported macrocyclic hexapeptide 70 with low nanomolar CXCR7 modulating activity,71 significant structural alterations were made to improve the potency, selectivity and reduced off-target activity (Fig. 11). The potency was increased by addition of a 2,4-difluoro-substituted N-phenylpropyl side chain at the peptoid position. Peptide-peptoid hybrid 71 had the best overall balance between potency (Ki), polarity (EPSA) and passive permeability (Papp); therefore, it was progressed for in vivo oral absorption potential studies. A 3-fold improvement in the activity was obtained for 71 (EC50 = 15 nM) compared to the initial peptide 70 (EC50 = 46 nM).
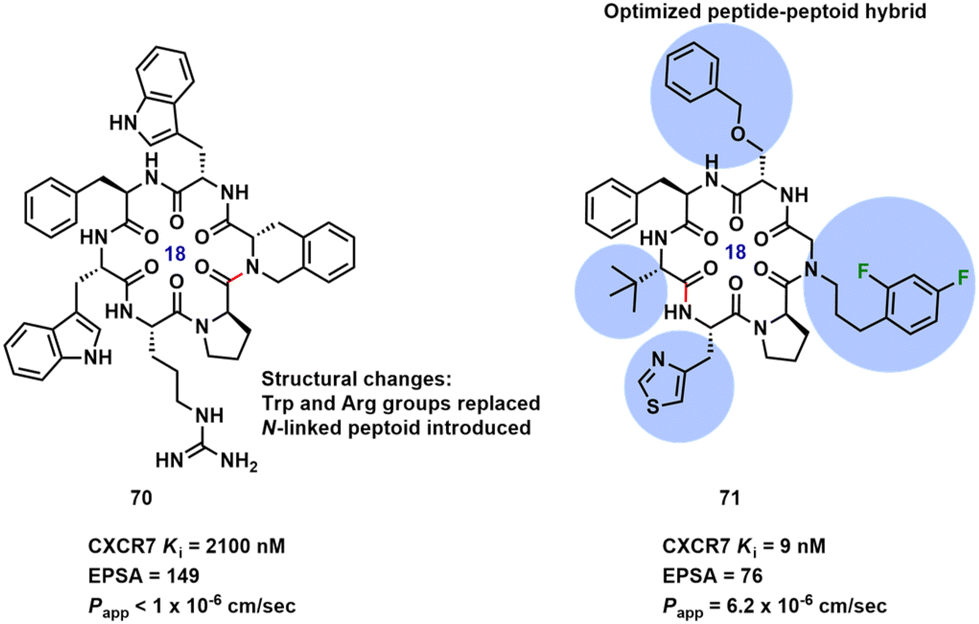 | ||
| Fig. 11 Native macrocyclic hexapeptide structure 70 and structure of optimized peptide-peptoid hybrid 71. | ||
A study reported that the binding affinity of a bicyclic FXIIa inhibitor 72 was enhanced ten-fold by replacing Phe with 4-fluorophenylalanine.72 FXII is a serine protease involved in blood coagulation. A Trp was appended to the C-terminus of bicyclic peptide 72 for quantification by absorption spectrometry to determine Ki values shown in Fig. 12a. Fluorine substitution at the ortho-position reduced the activity approximately 2.5-fold. In contrast, meta- and para-substitution resulted in increased activities by 2- and 11-fold, respectively. The resulting peptide with 4-fluoro-Phe 72i (without C-terminal Trp) had a Ki of 0.84 ± 0.03 nM. Structural modelling of the interactions between the bicyclic peptide and FXIIa showed that the side chain of the 4-fluoro-Phe residue was buried in an aromatic pocket formed by two surface loops (Fig. 12b). It was hypothesized that the short distance between the fluorine atom and both the polarized carbonyl and the α-hydrogen could result in polar interactions. In addition, the fluorine atoms could also perturb the electronic properties of the aromatic ring, thus enhancing the π–π stacking interactions between 4-fluoro-Phe and proximal His393 and Tyr439. Additional replacement of the terminal arginine residues by norarginine was found to increase the target selectivity 27000-fold.
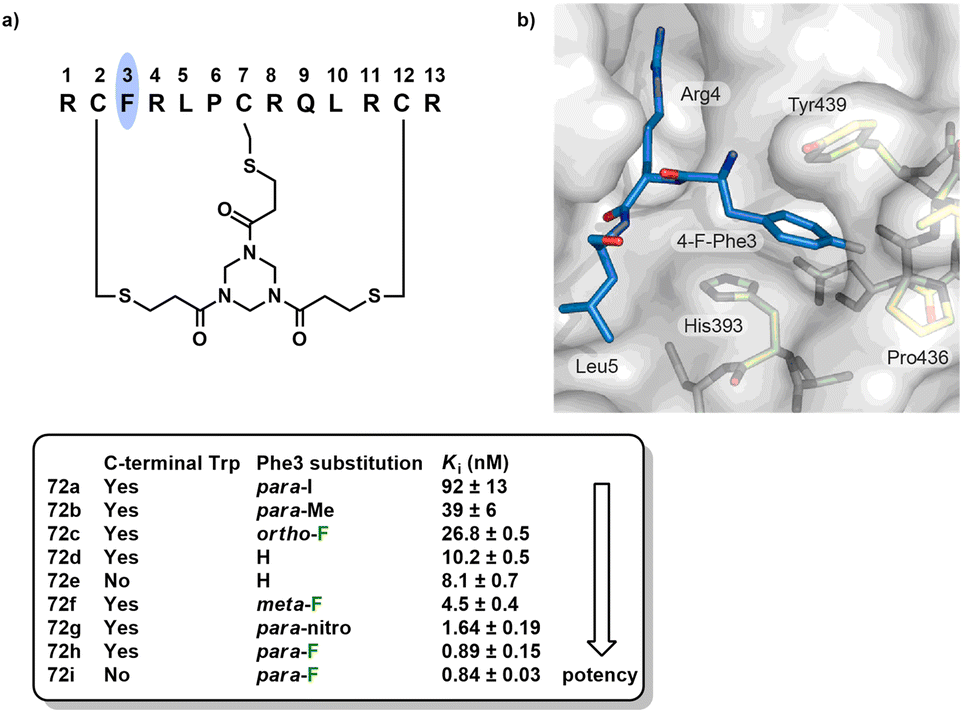 | ||
| Fig. 12 (a) Structures and activities of bicyclic FXIIa inhibitors 72a–i. (b) Structural model for the three key amino acids 4-F-Phe3, Arg4 and Leu5 of bicyclic peptide 72i bound to FXIIa. Adapted with permission from ref. 72. Copyright 2017 American Chemical Society. | ||
Flopristin (73b) is a semi-synthetic antibiotic of the streptogramin class (Fig. 13).73,74 It is a fluorinated derivative of the peptidic macrolactone pristinamycin IIB (73a, also known as virginiamycin M2) and is one of the components of the drug candidate NXL103, along with linopristin. Flopristin exhibited lower minimum inhibitory concentrations (MIC) than pristinamycin against H. influenzae and H. parainfluenzae.73 NXL103 completed Phase II trials but no further progress has been reported. Analogs of flopristin containing the C16-fluoro group were described (73c–e), with the C4-allyl analog 73c displaying up to a 128-fold improvement in MIC compared to the parent compound 73a.75 This compound had moderate activity (32 μg mL−1) against ABC-F-expressing E. faecalis and Gram-negative E. coli (16 μg mL−1), strains highly resistant to steptogramins. 73c also showed greater activity compared to flopristin in multi-drug-resistant S. aureus. Furthermore, flopristin and other fluorinated analogues were found to be more potent than the non-fluorinated lead compounds for inhibition of mitochondrial translation of glioblastoma stem cells. Higher cell membrane permeability was also demonstrated for the fluorinated macrocyclic peptides.76
 | ||
| Fig. 13 Structures of pristinamycin IIB 73a and flopristin 73b and structures of flopristin analogs 73c–e. | ||
Zhou et al. developed the macrocyclic peptidomimetic MCP-1 (74f, R = meta-di-F, Fig. 14), the most potent of a series of fluorinated analogs.77 MCP-1 was found to bind to menin, an essential oncogenic cofactor for mixed lineage leukemia. Fluorine substituents at ortho, meta and para positions were introduced. Due to the acidic residues present along the phenyl binding site in the protein wall, it was proposed that the strong electronegativity of the fluorine atom could enhance polarization of the ring, leading to better binding affinities. meta-F substitution of MCP-1 gave a Ki value of 6.8 nM for binding to menin; hence, the molecule was 4-times more potent than the non-fluorinated derivative. Given the improved binding affinity to menin with F-substitution at the meta position, a new analog with two fluorine substituents at both meta positions was found to bind to menin with a Ki value of 4.7 nM, thus, showing a 6-fold improvement in potency compared to the non-fluorinated derivative. Macrocyclic MCP-1 was determined to be >600 times more potent than the corresponding linear peptide.
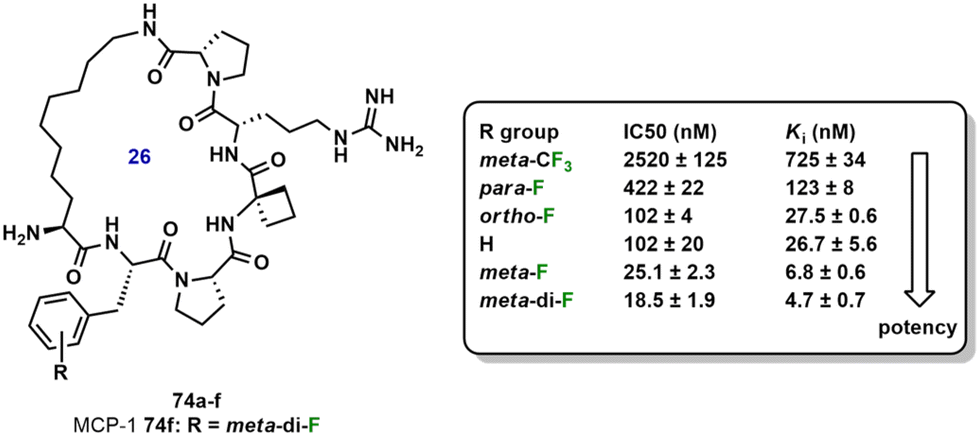 | ||
| Fig. 14 MCP-1 (74f) and its analogs including their potencies. | ||
Tsunemi et al. reported macrocyclization through successive vinylic substitutions via reactions of octafluorocyclopentene (OFCP) with small peptides (Fig. 15) containing combinations of Cys, Tyr, His and Ser residues.78 The macrocyclic peptidomimetics were stable upon storage, except Ser-linked 75a, which underwent ring-opening over several weeks in deuterated DMSO solution. OFCP-linked macrobicyclic peptides such as 75e were synthesized from the linear, unprotected precursors in polysubstitution cascades. Several of the OFCP-derived macrobicycles showed passive permeability in parallel artificial membrane permeability assays (PAMPA).79
 | ||
| Fig. 15 (a) and (b) OFCP-derived macrocyclic peptides 75a–e. | ||
Installation of a trifluoroacetyl (Tfa) moiety on the ε-amino group of lysine (KTfa) was found to be an effective method for the generation of peptide inhibitors of the human sirtuin (SIRT) family.80 Of the three isoforms present, SIRT2 is involved in cell regulation by the deacetylation of ε-N-acetylated lysine residues (KAc) within α-tubulin and histone H4K16.81 The macrocyclic structure of the peptide inhibitors, such as S2iL8 (76) shown in Fig. 16, led to a two-fold increase in the inhibitory effect toward SIRT2 compared to linear analogs. Substitution of KAc with KTfa led to a slower reaction rate between the Tfa group and NAD+, resulting in an unproductive intermediate and thus inhibition of sirtuin activity.
 | ||
| Fig. 16 Structure of the SIRT2-selective CF3-containing macrocyclic peptide S2iL8 (76); the KTfa unit is highlighted. | ||
Fluorinated cyclic pentapeptide analogs of endomorphin-2 (EM-2, 77) have been reported (Fig. 17).82 Endomorphins are endogenous opioid neuropeptides that have attracted attention as pain relief drugs; however, linear EM peptides have suffered from poor receptor selectivity, rapid in vivo degradation, poor blood–brain barrier delivery and toxic side effects. Hence, the structure of EM-2 was modified by both cyclization and fluorination to improve biological activity. Twelve cyclic analogs 78a–l were synthesized by individually replacing Phe residues with fluorinated amino acids (4-fluoro-Phe, 2,4-difluoro-Phe and 4-trifluoromethyl-Phe). Firstly, resistance toward in vivo enzymatic degradation was tested by incubating the cyclic pentapeptides with rat brain homogenate for 90 min and analyzing the amount of remaining peptide by RP-HPLC. 78a–l all displayed less than 7% degradation, while EM-2 was almost completely digested. Next, pharmacological profiles of the analogs were investigated at three opioid receptors: μ-, δ- and κ-opioid peptides (MOP, DOP and KOP). Analogs containing mono- and di-fluorinated Phe residues were full MOP and partial KOP agonists, while showing lower potency and efficacy at the DOP receptor. Analogs containing 4-trifluoromethyl-Phe showed selectivity toward the KOP receptor. Finally, the most potent analogs, 78h and 78j produced a dose-dependent antinociceptive effect after both intracerebroventricular and intraperitoneal injection in mice, indicating that they were able to cross the blood–brain barrier.
 | ||
| Fig. 17 Structures of fluorine-containing cyclic pentapeptides 78a–l based on the linear opioid peptide EM-2 (77). | ||
The influence of a fluorine substituent was explored in a monocyclic analog of the trypsin inhibitor SFTI-1, in which the Lys5 in the substrate-specific P1 position of the wild-type SFTI-1 was replaced by 4-F-Phe to form analog 79 (Fig. 18).83 The addition of the para-fluorine increased the inhibitory activity 15-fold compared to the non-fluorinated Phe analog. It was proposed that additional interactions of the para-substituent with chymotrypsin were responsible for the enhanced inhibitory activity of analog 79. Furthermore, no significant difference in the chymotrypsin hydrolysis pattern was observed between analogs containing 4-F-Phe and Phe. Upon hydrolysis of the P1–P1′ bond, an equilibrium was reached between intact and hydrolyzed forms, where no further proteolysis was observed. This is notable due to the strict structural requirements for inhibitor-enzyme P1–P1′ interactions.
 | ||
| Fig. 18 Structure of 79, a monocyclic analog of SFTI-1, a trypsin inhibitor. | ||
The integration of the flexible in vitro translation (FIT) system into mRNA display technology resulted in the random non-standard peptide integrated discovery (RaPID) system, which has been used to discover de novo fluorinated macrocyclic peptides through ribosomal translation of fluorinated non-canonical amino acids (Fig. 19).84,85 These macrocyclic peptides showed strong binding affinities for human ephrin type-A receptor 2 (EphA2), with KD values of 9.9 nM (Ep-F1, 80a) and 0.25 nM (Ep-F2, 80b).85 The fluorinated macrocyclic peptides were also found to disrupt the outer membrane of E. coli, resulting in lysis, as well as demonstrating broad-spectrum activity against multidrug-resistant Gram-negative bacteria by targeting the β-barrel assembly machinery (BAM) complex.
 | ||
| Fig. 19 Structures of de novo fluorinated macrocyclic peptides selected using the RaPID system targeting EphA2 (Ep-F1 80a and Ep-F2 80b) and Gram-negative pathogens (BAM-f2-R9 80c and BAM-β3-R9 80d). Acp = ε-aminocaproic acid. | ||
Another application of the RaPID and FIT systems was the incorporation of a Cys reactive electrophilic warhead, fluoroamidine, into macrocyclic peptide libraries.86 Fluoroamidine is a small molecule inhibitor of peptidyl arginine deiminase 4 (PADI4), one of the enzymes that catalyze post-translational modification of peptidyl arginine residues to citrulline, which regulates cell signaling processes. Dysregulation of PADI4 is involved in diseases such as rheumatoid arthritis and lupus. In this report, an unnatural amino acid version of fluoroamidine, N-δ-fluoroacetimidoyl ornithine, was ribosomally incorporated into the peptide, such as the highly active cP4_4 (81, Fig. 20). It was found that the fluorine atom forms interactions within the PADI4 active site that are crucial for binding, as well as acting as the leaving group. The macrocyclic peptide itself ensured highly specific target binding and minimal off-target activity.
 | ||
| Fig. 20 Structure of fluoroamidine-containing macrocyclic peptide 81. | ||
mRNA display technology was applied toward the in vivo selection of macrocyclic peptides containing unnatural amino acids, including macrocyclic peptide 82, which bears a 3-fluoro-L-tyrosine and showed low nanomolar affinity for the protease thrombin (Fig. 21).87,88 Following in vitro selection and evolution, the ribosomal synthesis of 82 was achieved via the cysteine residues and dibromoxylene. The macrocyclic nature of the peptides contributed significantly toward their affinity to thrombin, for example, a 25-fold decrease in Kd value was observed for inhibitor 82 compared to its linear counterpart.
 | ||
| Fig. 21 Structure of the thrombin inhibitor 82. | ||
A two-step combinatorial synthesis strategy was developed to synthesize a library of thioether-cyclized peptides, in which “m” random peptides were cyclized by “n” linkers followed by acylation at the peripheral amine with “o” carboxylic acids, to obtain “m × n × o” cyclic peptides (Scheme 4).89 A number of fluorine-containing macrocyclic peptides were included in the library, although the specific effect of the fluorine atom was not investigated. A series of ten dithioether-linker macrocyclic peptides were incubated with liver microsomes and the metabolic stability was monitored by mass spectrometry. No correlation was found between oxidation rate and type of linker. A library of 8448 macrocyclic peptides was synthesized and screened against thrombin and, finally, multiple iterative cycles of library synthesis yielded peptides with oral bioavailability (%F) of up to 18% in rats.
 | ||
| Scheme 4 Combinatory synthesis of a library of thioether-cyclized peptides. | ||
Fluoroaromatic linker for the construction of macrocycles
The use of perfluoroaryl groups as reagents for peptide stapling through cysteine residues has been extensively investigated in recent years.90,91 The field of perfluoroaryl reagents for peptide modification has been reviewed by Brittain and Coxon.92 Perfluoroarylation can increase the lipophilicity of macrocyclic peptides as well as affecting their solubility and cellular penetration, however, the latter is also dependent on the physicochemical properties of the starting peptide.Biocompatible macrocyclizations of peptides have been carried out using decafluoro-diphenylsulfone (DFS, 84, Scheme 5).93 Reaction rates for SNAr at Cys residues with 12 different perfluoroarenes were measured via19F NMR spectroscopy in aqueous buffer, where reactions involving DFS were the fastest. The rates of macrocyclization correlated with the electronegativity of the substituents on the perfluoroarene ring. The rate enhancement was also attributed to the superior water solubility of DFS, compared to other perfluoroarenes such as hexafluorobenzene and perfluoronitrobenzene, which required an organic co-solvent. The reactivity of DFS was validated with model systems 85a–e, with rate constants of up to 180 M−1 s−1. DFS formed complex macrocyclic peptides of varying ring sizes, as demonstrated with six peptide-hormones: melanin-concentrating hormone 88 (MCH, Scheme 5), oxytocin, urotensin II, salmon calcitonin, somatostatin-14, and atrial natriuretic factor (1–28), with ring sizes ranging from 6 to 19 amino acids. The rate of SNAr was sequence dependent, with rate acceleration observed when positively charged Arg was present next to Cys. Conversely, negatively charged Asp reduced SNAr rates. Advantages of the DFS framework over other perfluoroarene nucleophiles included biocompatibility with complex multiprotein complexes in aqueous solutions as demonstrated with bacteriophages, which otherwise would be degraded by organic solvents.
 | ||
| Scheme 5 Rapid biocompatible macrocyclization of peptides with DFS (84) between two cysteine residues. | ||
Independently to the previous report, Lautrette et al. reported the N-arylation of unprotected peptides using fluoroaromatic linker groups to synthesize stapled macrocyclic peptides.94 The electrophilic linkers 84 and 89a–d were ranked according to their relative reactivities with the unprotected linear peptide 90, as determined by HPLC-MS analysis (Scheme 6). Oxidation of the perfluorosulfide to the perfluorosulfone resulted in a linker that was highly reactive, due to the promotion of the SNAr by electron-withdrawing groups.
 | ||
| Scheme 6 N-Arylation of unprotected peptides and scale of reactivity for fluoroaromatic linker groups. Reactive fluorine atoms highlighted in red. | ||
The “scanning approach” allows side-chain to side-chain macrocyclization irrespective of the sequence and position of the two amino acid residues.95 A diversity-oriented synthetic (DOS) approach was taken to “scan” two cysteine residues positioned in sites ranging from i, i + 1 to i, i + 14 within 14 unprotected polypeptides.96 Two complementary strategies were then developed for side-chain macrocyclization of the polypeptides. Cysteine cross-linking was performed either with perfluoroaryl-based linkers, or by first incorporating non-crosslinked perfluoroaryl-based moieties followed by macrocyclization with dithiol reagents. Using seven perfluoroaromatic linkers, 98 macrocyclic peptides were produced without the need for reaction optimization for each transformation.
Macrocyclization by perfluoroarylation has been shown to increase peptide stability and delivery of exon-skipping antisense oligonucleotides to cells.97 Two strategies were reported for the synthesis of arginine-rich bicyclic peptides. The first bicyclization method involved trithiol moiety 92 to link three perfluoroaryl groups on the unprotected peptide chain 93 within 2 h and 62% isolated yield after HPLC purification (Scheme 7a). In the second strategy, kinetically controlled bicyclization was carried out on the purified peptide 95 in DMF in the presence of DIPEA (Scheme 7b). This method took advantage of the slow rates of i, i + 1 cyclization previously observed with decafluorobiphenyl96 and gave complete conversion to the i, i + 7 bicyclic peptide within 5 min. HPLC purification gave the isolated bicycle 96 in 81% yield. The bicyclic peptide was then conjugated to phosphorodiamidate morpholino oligonucleotide (PMO) via a copper-catalyzed “click” reaction (Scheme 7c). PMOs are antisense oligonucleotides, such as eteplirsen, an FDA-approved therapy for the treatment of Duchenne muscular dystrophy (DMD).98 The exon-skipping activity of the PMO-conjugated perfluoroaryl bicyclic peptide was greater than that of the non-fluorinated bicyclic peptide. Furthermore, the proteolytic stability of both bicycles 94 and 96 were greater than the corresponding i, i + 13 monocyclic peptide; after 1 h of incubation with trypsin, 70% of bicycle 94 and 45% of bicycle 96 remained undigested compared to only 5% of the monocyclic peptide.
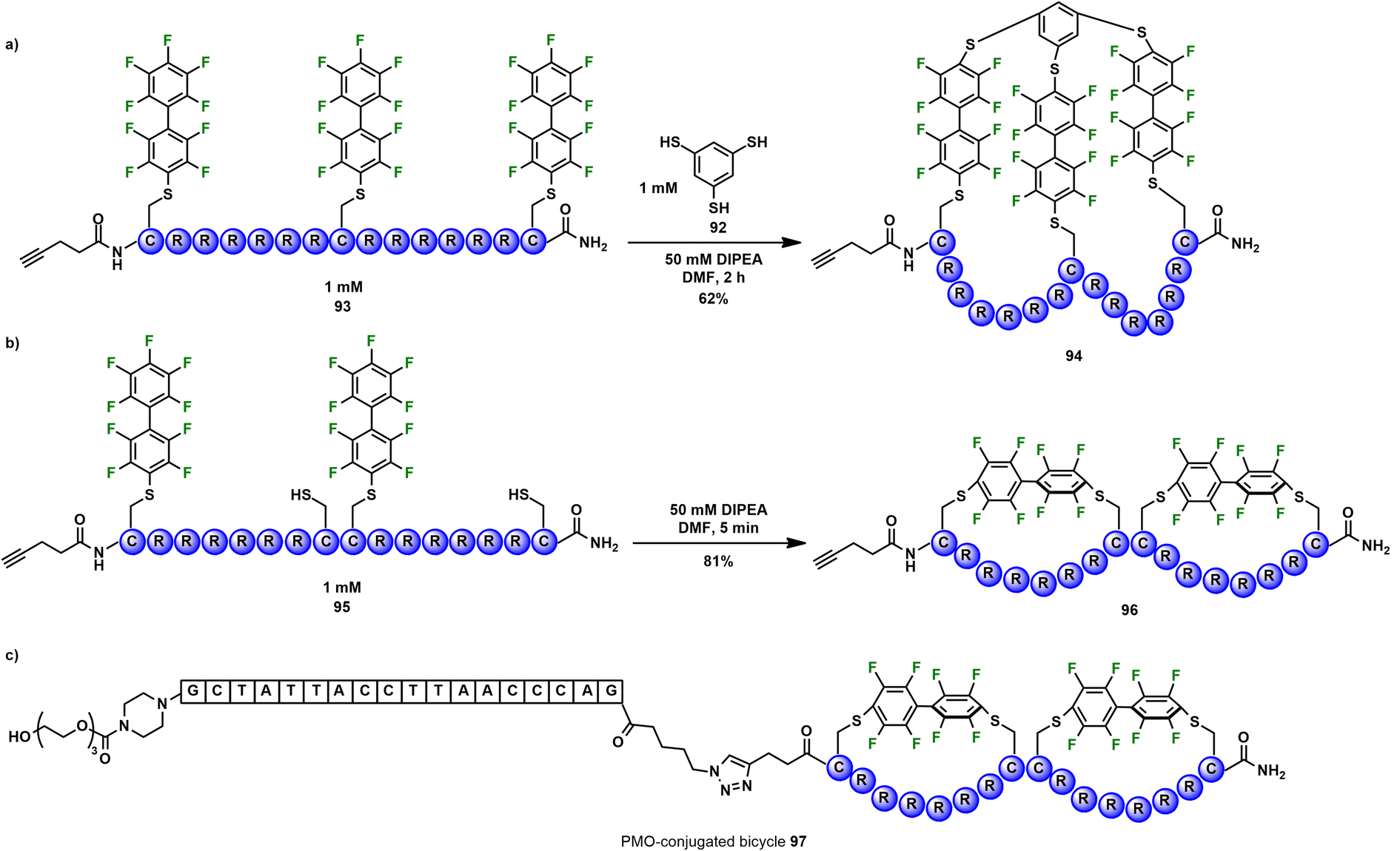 | ||
| Scheme 7 (a) Peptide bicyclization via SNAr reactions of decafluorobiphenyl modified cysteine residues with 1,3,5-benzenetrithiol. (b) High-yielding kinetically controlled bicyclization via intramolecular SNAr at i, i + 7 positions. (c) Bicyclic peptide conjugated to phosphorodiamidate morpholino oligonucleotide (PMO). | ||
Perfluoroaryl macrocyclic peptides were found to have greater penetration into brain endothelial cells, which, coupled with their increased serum stability, improved the delivery of small molecules across the blood–brain barrier.99 This could improve the properties of new drugs for the treatment of brain tumors in diseases such as glioblastoma. Current cancer treatments include Pt(II) complexes, such as cisplatin, carboplatin and oxaliplatin, which have a narrow therapeutic index due to off-target toxicities.100 A possible solution to widen the therapeutic index is administration as the Pt(IV) prodrugs, whereby, following cell internalization, the Pt(IV) prodrug is reduced in situ to the active Pt(II) species. To demonstrate improved delivery of Pt(IV) prodrugs across the blood–brain barrier, a Pt(IV) prodrug-perfluoroaryl macrocyclic peptide conjugate (Pt(IV)–M13, 99) was synthesized (Scheme 8).101 The peptide-prodrug conjugate 98 was prepared by coupling a Pt(IV) prodrug carboxylate to resin-bound peptide TP10 M13. A linear control conjugate, Pt(IV)–L13 conjugate, was also synthesized, in which Cys residues were replaced by Ser. The Pt(IV)–M13 conjugate 99, Pt(IV)–L13 conjugate and cisplatin had IC50 values of 4 μM, 29 μM and 2 μM, respectively.
 | ||
| Scheme 8 Pt(IV)-prodrug peptide 98 cyclized via perfluoroaryl linker 89a. | ||
Fluorine atom as a 19F NMR reporter and a tool for conformational analysis of macrocyclic peptides
Fluorine-containing analogs 100a–f of the cyclic decapeptide gramicidin S (GS) were developed by Wadhwani et al. (Fig. 22).102 The highly NMR sensitive 19F-reporter was attached to the GS backbone, through which the structure and dynamics of GS in its biologically relevant membrane-bound state were studied using solid-state 19F and 15N NMR spectroscopy.103 The antimicrobial properties of GS were attributed to its destabilizing effect on lipid membranes. The alignment of the peptide in the bilayer and its temperature-dependent mobility were determined by analyzing the anisotropic chemical shift of the 19F-labels in macroscopically oriented membranes. A narrowing of the 19F NMR chemical shift dispersion was observed upon raising the temperature to the liquid crystalline state, due to the onset of global rotation of the peptide. It was proposed that the information elucidated from this study could be used to develop an analog more selective toward bacterial membranes. | ||
| Fig. 22 Gramicidin S analogs 100a–f functionalized with 15N and 19F NMR reporter groups. | ||
Iterative conjugate addition/ring expansion (CARE) reactions were reported for the synthesis of macrocyclic peptide mimetics based on β-peptoid linkages.104N-Acylation of δ-valerolactam 101 with acryloyl chloride followed by CARE with p-fluorobenzylamine 102 afforded the 10-membered bis-lactam 103 (Scheme 9). The p-fluorobenzylamine 102 provided a convenient handle for reaction monitoring using 19F NMR spectroscopy. A further N-acylation/CARE sequence was performed on 103 using three different amines (shown in blue) to synthesize 14-membered macrocycles 104a–c. Compounds 104a–c were iteratively expanded using different amines (shown in green) to afford 18-membered β-peptoid-derived tetrapeptide mimetics 105a–c. A limitation of the CARE methodology is that it can only promote 4-atom ring expansions, hence, cyclic peptides with proteinogenic amino acids cannot be targeted.
 | ||
| Scheme 9 Iterative N-acylation and CARE reactions. | ||
To extend the in vivo circulation half-life of peptide therapeutics, a phage-displayed library was reported consisting of albumin-binding macrocyclic peptides modified with decafluoro-diphenylsulfone (DFS, 84) to afford octafluoro-diphenylsulfone-cross-linked macrocycles 106 (Fig. 23).105 Non-specific reactivity of OFS-macrocyclic peptides was observed with thiol nucleophiles, such as glutathione, at basic pH. Hence, the DFS was replaced with the less reactive pentafluorophenyl sulfide, to afford perfluorophenylsulfide (PFS) macrocycles which were unreactive toward free thiol on human serum albumin (HSA). The fluorine handle in the perfluoroaryl macrocyclic peptides allowed the determination of binding constants using 19F NMR spectroscopy, whereby the broadening and disappearance of 19F signals corresponding to fluoroaromatic groups indicated the binding of the macrocyclic peptide to HSA. The measured KD values indicated that both the amino acid sequence and the perfluoroaromatic linker could affect binding. The main function of the perfluoroaromatic linchpin was to constrain the macrocyclic peptide in a productive albumin-binding conformation. The half-lives of PFS-SICRFFCGGG 107 and its analogs were evaluated in mouse plasma, where the concentration of the former was constant after 60 min, while the latter were rapidly cleared. Replacement of the PFS linker with hexafluorobenzene (HFB) and decafluorobiphenyl (DFB) reduced the concentration of the resulting macrocyclic peptides in plasma 10-fold compared to macrocycle 107. This highlights that both the amino acid sequence and the albumin-binding conformational constraints imposed by the PFS linker are crucial for short-term retention in circulation.
 | ||
| Fig. 23 Peptides modified with DFS and PFS linkers. | ||
The C–F bond as a conformational tool within peptides and proteins has been utilized to examine the α-fluoroamide effect and the F–C–C–N gauche effect.106 Stereoselective fluorination has been demonstrated to alter the geometry of cyclic peptides. For their investigations, Hu et al. chose the marine-derived natural product, unguisin A, a cyclic heptapeptide bearing a γ-aminobutyric acid (GABA) residue (109a, Scheme 10).107 The difluorinated amino acids 108a and 108b have been shown to introduce different selectivity patterns in GABA binding assays.108 By replacing the GABA unit with either 108a or 108b or their enantiomers, analogs 110b–e were synthesized in 50–57% yield. Monitoring the macrocyclization by 1H NMR spectroscopy at 0 °C showed completion of the reactions within one minute. To understand the effects of fluorination on the secondary structure, 3JHH, 3JHF and 4JHF values were determined. For isomers 110b–e, the fluorine atoms were gauche but exhibited g+/g− disorder. Furthermore, energy minimizations showed that these fluoro-analogs displayed different geometries from that of 110a. While 110a was flat with a puckered seven-membered H-bonded ring (γ-turn), the syn-difluoro isomer 110b displayed a planar geometry with no H-bonded turns. The other syn-difluoro isomer 110c contained two overlapping H-bonded loops, an equatorial γ-turn and a distorted α-turn (a 13-membered H-bonded ring). 110d and 110e were both highly puckered, with a γ-turn and two β-turns (ten-membered H-bonded rings). Moreover, the role of flexibility in supramolecular interactions of unguisin A was studied using fluorine-containing macrocyclic peptides 110b–e containing the GABA flexible spacer.109 Fluorine substitution in molecules 110b–e rigidified the structure and resulted in a 15–50% lower affinity toward chloride anions compared to unguisin A (110a). Finally, molecular dynamics (MD) simulations found that the difluorinated derivatives of unguisin A were flexible in DMSO solution which, depending on the fluorination pattern, adopted two or more conformations.110
 | ||
| Scheme 10 Structure of unguisin A (109a) and its fluorinated analogs 109b–e. | ||
Extending upon this work, a series of cyclic RGD tetrapeptides containing the fluorine-modified GABA moiety were reported to determine the effect of fluorine pattern on cyclization rates, as well as to assess the ability to tune molecular conformations and biological activities.111 Both enthalpic and entropic contributions of the fluorinated segments were found to be significant for cyclization efficiencies. MD simulations showed substantial variation in conformations of the cyclic tetrapeptides, confirming that fluorination can alter secondary structures. Proof-of-concept studies on cell adhesion and cell spreading were performed, where significant differences were obtained depending on the position of fluorination.
Burade et al. reported the role of fluorine as a reverse turn inducer in acyclic αγα tripeptides containing fluorinated furanoid groups, which have potential applications as anion channels through antiparallel self-assembly of U-shaped monomers.112 This work was extended to the synthesis of fluorinated sugar amino acid derived αγ-cyclic tetra- and hexapeptides. Macrocyclization was achieved using 2-chloro-1-methyl pyridinium iodide (CMPI) and N,N-diisopropylethylamine (DIPEA) in acetonitrile to afford α,γ-cyclic tetrapeptide 112 and α,γ-cyclic hexapeptide 114 (Scheme 11). Observed 4JH,F values of 2.2–3.1 Hz in macrocycles 112 and 114 suggested gauche conformation between C–F and N–H groups, resulting in antiparallel alignments of C–F and adjacent amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The F–C–CO torsion angle of 172.28° and dipole repulsion further supported this alignment, leading to weak intramolecular fluorine-amine hydrogen bonding interactions. These interactions led to the folding of cyclic peptides 112 and 114 into β-strand structures, and further self-assembly into nanotubes in both solution and gas phase. The nanotubes formed by 114 were shown to facilitate anion-selective transport of nitrate ions.
O bonds. The F–C–CO torsion angle of 172.28° and dipole repulsion further supported this alignment, leading to weak intramolecular fluorine-amine hydrogen bonding interactions. These interactions led to the folding of cyclic peptides 112 and 114 into β-strand structures, and further self-assembly into nanotubes in both solution and gas phase. The nanotubes formed by 114 were shown to facilitate anion-selective transport of nitrate ions.
 | ||
| Scheme 11 Synthesis of fluorine-containing α,γ-cyclic peptides 112 and 114. | ||
Cogswell et al. reported the synthesis of a macrocyclic peptide containing an α-fluoroalkoxyaryl group ArOCF2R, which has the potential to function as a tool for studying conformational properties (Scheme 12).113 A difluoroallylated tyrosine served as a building block to synthesize linear peptide 115 in three steps. Following hydrogenation to remove both Cbz and benzyl protecting groups, a macrolactamization reaction was performed to obtain the difluorinated macrocycle 116 in 64% yield. The ArOCF2R group has been shown to reduce in-plane conformational preferences through hyperconjugation effects between low energy σ*C–F antibonding orbitals and oxygen lone pairs.18,114 Although not investigated further in the report by Cogswell et al., it was noted that macrocyclic peptides such as 116 could be used to study the impact of the ArOCF2R group on conformational and physiochemical properties.
 | ||
| Scheme 12 Synthesis of a tyrosine-based difluoromethylene-containing macrocyclic peptide 116. | ||
Numerous tryptophan-derived natural products contain the pyrroloindoline motif. Rose et al. reported a synthetic method where the indole activation step was, itself, the macrocyclization reaction.115 Substrates bearing 5-methyl- and 5-fluoro-L-tryptophan (117–118, Scheme 13a) were synthesized in order to block previously observed pathways of C5 alkylation and, instead, to favor macrocyclization via electrophilic substitution at indole C3.115,116 In an acid-promoted Friedel–Crafts alkylation, 117 and 118 were treated with triflimide in nitromethane to give full conversion to an isomeric product mixture. The electron-withdrawing fluorine substituent led to more effective suppression of reaction at the benzenoid ring than a methyl substituent. Although the Friedel–Crafts macrocyclization approach has the potential to generate libraries of macrocycle isomers that would otherwise be more time consuming to synthesize individually, it was demonstrated that specific isomers can also be prepared convergently (Scheme 13b). Intramolecular Pd0-catalyzed allylation of 5-fluoro-L-tryptophan methyl ester 122 and cinnamyl alcohol 121 gave endo-pyrroloindoline 123. Two further amino acids were then introduced, followed by deprotection of acid labile protecting groups and lactamization with HBTU. Both products 120d and 125 were spectroscopically identical.
 | ||
| Scheme 13 Pyrroloindoline-forming macrocyclization of 5-fluoro-L-tryptophan by (a) acidolysis to promote internal substitution at N1, C2, C3 or C4 and (b) convergent synthesis. | ||
18F labeling of macrocyclic peptides
Positron emission tomography (PET) is a non-invasive medical diagnostic tool for the detection and diagnosis of diseases by visualizing metabolic processes. Receptor-targeting peptide-based imaging agents possess favorable properties such as high affinities and specificities for their targets, improved pharmacokinetic properties, and biosafety.117 Furthermore, the biological half-life of many peptides matches the half-life of radionucleotide 18F (109.77 min). Linear Arg-Gly-Asp (RGD) peptides are often prone to enzymatic degradation, while cyclization has been demonstrated to improve in vivo stabilities. For instance, Davis et al. reported the on-resin head-to-tail cyclization and 18F-radiolabeling of a cyclic peptide, c(RGDyK), a ligand of integrin αvβ3 receptors.118 The acid-sensitive Novasyn® TGA resin was selected to maintain short cleavage times following the on-resin radiolabeling step. Radiolabeling was achieved using a 30 min coupling of [18F]fluorobenzoic acid ([18F]FBA, 126) to the peptidyl-resin 127 (Scheme 14a). This was followed by a 30 min global deprotection and cleavage to obtain the labeled cyclic peptide 128. | ||
| Scheme 14 (a) On-resin 18F-radiolabeling of macrocyclic RGD pentapeptide 127. (b) Direct 18F-radiolabeling of macrocyclic RGD-pentapeptide 130. (c) 18F-radiolabeling of macrocyclic peptides 133a,b using an 18F version of Umemoto's reagent (132). | ||
Chemoselective radio-deoxyfluorination with [18F]fluoride enabled the radiolabeling of peptides containing tyrosine residues bearing a traceless ruthenium activating group (Scheme 14b).119 Metal-mediated 18F-radiofluorination has been reviewed elsewhere.120 A cyclic peptide containing the RGD motif was labeled in 25% decay-corrected radiochemical yield (RCY) after purification by HPLC. The source of [18F]fluoride in typical radiolabeling procedures is an aqueous solution of [18F]fluoride trapped on an anion exchange cartridge, with azeotropic drying of the fluoride. This report developed a strategy involving direct elution with the peptide-ruthenium complex in an ethanol-pivalonitrile solvent system, thereby avoiding the need for aqueous elution and azeodrying. The use of bis(trimethylneopentylammonium) oxalate 129a was vital for successful elution as it increased radiolabeling efficiency 1.7-fold.
18F-trifluoromethylation of peptides bearing cysteine residues was achieved using an 18F-labeled version of Umemoto's reagent (132) and commercial RGD or RAD pentapeptides (Scheme 14c).121 Radiolabeled cyclic peptides cRGDfC(CF218F) 134a and cRADfC(CF218F) 134b were purified and isolated in 19% ± 5% RCY and 33% ± 9% RCY, respectively.
Kee et al. reported the 18F-trifluoromethylation of native aromatic peptide residues using 18F-trifluoromethanesulfinate (CF218FSO2NH4).122 The 18F reagent was prepared in a single step from K18F/K222, a difluorocarbene reagent, and a source of SO2. After initial 18F-labeling of dipeptides, the substrate scope for C–H 18F-trifluoromethylation of Tyr and Trp residues was expanded to biologically active macrocyclic peptides (Scheme 15a). The integrin αvβ3 receptor ligand c(RGDyK) underwent 18F labeling at 33% radiochemical conversion (RCC). Octreotide, an octapeptidic mimic of somatostatin, underwent 18F-trifluoromethylation in 29% RCC. Somatostatin-14, a macrocyclic tetradecapeptide with broad antisecretory activity on endocrine hormones, was Trp-selectively 18F-labeled in 20% RCY.122,123 An automated radiosynthesis of octreotide[Trp(2-CF218F)] 136 was also reported with 133 min total synthesis time from [18F]fluoride, which enabled an in vivo PET imaging experiment on rats.122
 | ||
| Scheme 15 (a) Synthesis of 18F-labeled macrocyclic peptides 135–137. (b) 18F-radiolabeling of a cyclic RGD peptide 139 with organometallic gold(III) reagent 138. (c) Radiolabeling of aminooxy-functionalized octreotate derivative 142 with [18F]FDG 141. (d) Synthesis of 18F-labeled BMS-986229 (147) via CuACC. | ||
18F-labeling of unprotected peptides with an organometallic gold(III) reagent 138 in aqueous media was reported by McDaniel et al. (Scheme 15b).124 A commercially available cyclic RGD peptide 139 was 18F-radiolabeled to demonstrate that S-arylation can be achieved when the Cys residue is in an interchain position. The product 140 was obtained in 94% RCY.
There are challenges associated with the radiolabeling of peptides with the short-lived 18F isotope, due to the large amounts of peptide labeling precursors required as well as extended reaction times. To address these issues, a microfluidic methodology was reported for the radiosynthesis of a clinically relevant octreotate derivative.125 Octreotate is a somatostatin analog, closely related to octreotide which has a terminal Thr reduced to the corresponding amino alcohol. High decay-corrected RCY values of greater than 82% were obtained for the radiolabeling of the aminooxy-functionalized octreotate derivative 142 with [18F]fluorodeoxyglucose ([18F]FDG, 141) (Scheme 15c), compared with 76% RCY obtained via the conventional non-microfluidic method. The reaction time was shortened from 70 to 30 min, which included its HPLC purification. The microfluidic set-up could be tuned to the required radioactivity level for specific patient doses and, in principle, demonstrated the feasibility of this technology toward clinical applications.126
BMS reported the discovery and evaluation of BMS-986229 (147), a novel 18F-labeled macrocyclic peptide radioligand for PET imaging and measurement of PD-L1 expression in tumors (Scheme 15d).127 The programmed death protein (PD-1) is a negative co-stimulatory receptor expressed on activated T- and B-cell surfaces, and PD-L1 is a surface glycoprotein ligand for PD-1.127 Binding of PD-L1 and PD-1 enables immunosuppression on antigen-presenting cells and human cancers by downregulation of T-cell activation and cytokine secretion. The macrocyclic peptide component of 147 was designed using BMS-986189 (highlighted in Scheme 15d), a fully optimized potent PD-L1 antagonist with picomolar affinity.128 A propargyl glycine group was incorporated, which enabled copper-catalyzed azide–alkyne cycloaddition (CuAAC) between peptide 146 and [18F]BMT-187144 (145) to rapidly label the macrocyclic peptide with 18F. A non-decayed corrected RCY of 11% was obtained. In vivo PET imaging in xenograft models showed an increased uptake of BMS-986229 (147) compared to the control. Furthermore, 147 showed a long dissociation off-rate from PD-L1, rapid clearance from the blood and non-PD-L1 tissues, and the largest dynamic range within the non-human primate spleen compared to other PET imaging agents (adnectin and mAb platforms), emphasizing the advantages of using a macrocyclic peptide scaffold.
Conclusions
Fluorine-containing macrocyclic peptides have made a significant impact on medicinal chemistry. The largest category of therapeutic fluorinated macrocyclic peptides are the HCV protease inhibitors. These are 14–18-membered macrocyclic rings, and structures with up to four fluorine atoms have been reported. Other diseases targeted by fluorinated macrocyclic peptides include hypercholesterolemia (MK-0616, 63) and various types of cancer (motixafortide, 65, LUNA18, 66). Fluorine-specific interactions have led to improved enzyme inhibitory activity, increased metabolic stability and decreased susceptibility to degradation.Fluorine-containing non-natural amino acids, such as fluoro-Trp, -Tyr and -Phe, have served as useful building blocks for incorporation into macrocyclic peptides. Structures including 63, 72, 82, 125 and 135–137 have benefited from this synthetic strategy. More complex targets include fluorinated bicyclic peptides such as 72, 75e and 94. A common strategy for bicyclization has involved the reaction of Cys residues with specific linkers. Fluoroaromatic linkers such as decafluoro-diphenylsulfone (DFS) and decafluorobiphenyl (DFB) have enabled the construction of macrocyclic and bicyclic peptides, while the presence of perfluoroaryl has been shown to increase peptide stability and delivery of antisense oligonucleotides to cells, and to improve delivery of Pt(IV) prodrugs across the blood–brain barrier.97,99
mRNA display technology has emerged as a tool for generating large libraries of macrocyclic peptides incorporating non-canonical amino acids.81,88 This technology allows for the rapid selection of high-affinity macrocyclic peptides containing modified backbones, based on target binding. Notable fluorine-containing examples that have been discovered include inhibitors of EphA2, SIRT2 and thrombin.80,85,87
Macrocyclic peptide-based PD-L1 antagonists have been evaluated as PET imaging agents.129 They are advantageous scaffolds for the design of potential PET ligands due to the opportunities for optimization toward high target affinity and rapid tumor uptake. Peptide-based imaging agents also show rapid clearance from blood and background tissues. Additionally, the site-specific modification of peptides enables facile incorporation of radionucleotides.
Despite the numerous benefits of fluorine incorporation into drug leads, it is important to consider the potentially unpredictable effects of fluorine within biological systems. Aromatic fluorination is a well-established strategy to slow metabolism, although aryl fluorides may still undergo oxidative defluorination by enzymes such as cytochrome P450, generating phenolic metabolites and fluoride. In BMS-986144 (56), the trifluorinated Boc group lowered the propensity toward metabolism which had afflicted the non-fluorinated, acyclic precursor asunaprevir.50 Nevertheless, the gain in stability needs to be considered against the toxicity of metabolites from defluorination of macrocyclic peptides.130 Appropriate and precise placement of the fluorine atom will avoid such issues. As noted in previous literature, the challenge within peptide chemistry lies in the ability to balance the specific properties of the fluorine atom with its behavior in its environment and the impact upon uptake and metabolism.28
It is expected that, given their increased drug-likeness, improved proteolytic stability and unique effects on protein–protein interactions, macrocyclic peptides will continue to make an impact on peptide therapeutics. The remarkable and diverse effects of fluorine upon biological molecules have forged a vital role within the pharmaceutical industry. We anticipate that the combination of these two fields will continue to strengthen the medicinal chemists’ toolbox.
Author contributions
N. R. suggested the concept for this review article and designed its figures and schemes. N. R. and S. R. prepared, edited and reviewed the manuscript. All authors have read and approved the final version.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Warwick and Nanna Therapeutics for financial support. We thank Prof. Michael Shipman for support in the writing of this article.References
- A. A. Vinogradov, Y. Yin and H. Suga, Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- A. K. Yudin, Macrocycles: lessons from the distant past, recent developments, and future directions, Chem. Sci., 2015, 6, 30–49 RSC.
- PepTherDia, https://peptherdia.herokuapp.com, (Accessed Dec 2024).
- V. D'Aloisio, P. Dognini, G. A. Hutcheon and C. R. Coxon, PepTherDia: database and structural composition analysis of approved peptide therapeutics and diagnostics, Drug Discovery Today, 2021, 26, 1409–1419 CrossRef PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, The exploration of macrocycles for drug discovery—an underexploited structural class, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- C. Lamers, Overcoming the shortcomings of peptide-based therapeutics, Future Drug Discovery, 2022, 4, FDD75 CrossRef CAS.
- C. Adessi and C. Soto, Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Contemporary strategies for peptide macrocyclization, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Peptide stapling techniques based on different macrocyclisation chemistries, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- J. Wu, J. Tang, H. Chen, Y. He, H. Wang and H. Yao, Recent developments in peptide macrocyclization, Tetrahedron Lett., 2018, 59, 325–333 CrossRef CAS.
- L. A. Viarengo-Baker, L. E. Brown, A. A. Rzepiela and A. Whitty, Defining and navigating macrocycle chemical space, Chem. Sci., 2021, 12, 4309–4328 RSC.
- S. E. Gibson and C. Lecci, Amino Acid Derived Macrocycles—An Area Driven by Synthesis or Application?, Angew. Chem., Int. Ed., 2006, 45, 1364–1377 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, Contribution of Organofluorine Compounds to Pharmaceuticals, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- P. Shah and A. D. Westwell, The role of fluorine in medicinal chemistry: Review Article, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco, D. Kozakov, S. Vajda and A. Whitty, How proteins bind macrocycles, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- R. J. Glyn and G. Pattison, Effects of Replacing Oxygenated Functionality with Fluorine on Lipophilicity, J. Med. Chem., 2021, 64, 10246–10259 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- N. Rozatian and D. R. W. Hodgson, Reactivities of electrophilic N–F fluorinating reagents, Chem. Commun., 2021, 57, 683–712 RSC.
- J. Wu, Review of recent advances in nucleophilic C–F bond-forming reactions at sp3 centers, Tetrahedron Lett., 2014, 55, 4289–4294 CrossRef CAS.
- Z. Zou, W. Zhang, Y. Wang and Y. Pan, Recent advances in electrochemically driven radical fluorination and fluoroalkylation, Org. Chem. Front., 2021, 8, 2786–2798 RSC.
- B. Lantaño and A. Postigo, Radical fluorination reactions by thermal and photoinduced methods, Org. Biomol. Chem., 2017, 15, 9954–9973 RSC.
- S. Barata-Vallejo, B. Lantaño and A. Postigo, Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- A. Harsanyi and G. Sandford, Organofluorine chemistry: applications, sources and sustainability, Green Chem., 2015, 17, 2081–2086 RSC.
- C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, An Update on the Nitrogen Heterocycle Compositions and Properties of U.S. FDA-Approved Pharmaceuticals (2013–2023), J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed.
- M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Fluorinated amino acids: compatibility with native protein structures and effects on protein–protein interactions, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC.
- A. A. Berger, J.-S. Völler, N. Budisa and B. Koksch, Deciphering the Fluorine Code—The Many Hats Fluorine Wears in a Protein Environment, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- S. Huhmann and B. Koksch, Fine-Tuning the Proteolytic Stability of Peptides with Fluorinated Amino Acids: Fine-Tuning the Proteolytic Stability of Peptides with Fluorinated Amino Acids, Eur. J. Org. Chem., 2018, 3667–3679 CrossRef CAS.
- W. D. G. Brittain, C. M. Lloyd and S. L. Cobb, Synthesis of complex unnatural fluorine-containing amino acids, J. Fluor. Chem., 2020, 239, 109630 CrossRef CAS PubMed.
- J. G. Taylor, S. Zipfel, K. Ramey, R. Vivian, A. Schrier, K. K. Karki, A. Katana, D. Kato, T. Kobayashi, R. Martinez, M. Sangi, D. Siegel, C. V. Tran, Z.-Y. Yang, J. Zablocki, C. Y. Yang, Y. Wang, K. Wang, K. Chan, O. Barauskas, G. Cheng, D. Jin, B. E. Schultz, T. Appleby, A. G. Villaseñor and J. O. Link, Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®, Bioorg. Med. Chem. Lett., 2019, 29, 2428–2436 CrossRef CAS PubMed.
- Y. N. Lamb, Glecaprevir/Pibrentasvir: First Global Approval, Drugs, 2017, 77, 1797–1804 CrossRef CAS PubMed.
- P. Lampertico, J. A. Carrión, M. Curry, J. Turnes, M. Cornberg, F. Negro, A. Brown, M. Persico, N. Wick, A. Porcalla, A. Pangerl, E. Crown, L. Larsen, Y. Yu and H. Wedemeyer, Real-world effectiveness and safety of glecaprevir/pibrentasvir for the treatment of patients with chronic HCV infection: A meta-analysis, J. Hepatol., 2020, 72, 1112–1121 CrossRef CAS PubMed.
- J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, Next generation organofluorine containing blockbuster drugs, J. Fluor. Chem., 2020, 239, 109639 CrossRef CAS.
- R. D. Cink, K. A. Lukin, R. D. Bishop, G. Zhao, M. J. Pelc, T. B. Towne, B. D. Gates, M. M. Ravn, D. R. Hill, C. Ding, S. C. Cullen, J. Mei, M. R. Leanna, J. Henle, J. G. Napolitano, N. K. Nere, S. Chen, A. Sheikh and J. M. Kallemeyn, Development of the Enabling Route for Glecaprevir via Ring-Closing Metathesis, Org. Process Res. Dev., 2020, 24, 183–200 CrossRef CAS.
- D. R. Hill, M. J. Abrahamson, K. A. Lukin, T. B. Towne, K. M. Engstrom, R. E. Reddy, A. B. Kielbus, M. J. Pelc, J. Mei, N. K. Nere, S. Chen, R. Henry, S. Chemburkar, C. Ding, H. Zhang and R. D. Cink, Development of a Large-Scale Route to Glecaprevir: Synthesis of the Side Chain and Final Assembly, Org. Process Res. Dev., 2020, 24, 1393–1404 CrossRef CAS.
- S. Zeuzem, G. R. Foster, S. Wang, A. Asatryan, E. Gane, J. J. Feld, T. Asselah, M. Bourlière, P. J. Ruane, H. Wedemeyer, S. Pol, R. Flisiak, F. Poordad, W.-L. Chuang, C. A. Stedman, S. Flamm, P. Kwo, G. J. Dore, G. Sepulveda-Arzola, S. K. Roberts, R. Soto-Malave, K. Kaita, M. Puoti, J. Vierling, E. Tam, H. E. Vargas, R. Bruck, F. Fuster, S.-W. Paik, F. Felizarta, J. Kort, B. Fu, R. Liu, T. I. Ng, T. Pilot-Matias, C.-W. Lin, R. Trinh and F. J. Mensa, Glecaprevir–Pibrentasvir for 8 or 12 Weeks in HCV Genotype 1 or 3 Infection, N. Engl. J. Med., 2018, 378, 354–369 CrossRef CAS PubMed.
- J. Zephyr, N. Kurt Yilmaz and C. A. Schiffer, Viral proteases: Structure, mechanism and inhibition, in The Enzymes, Elsevier, 2021, vol. 50, pp. 301–333 Search PubMed.
- J. Timm, K. Kosovrasti, M. Henes, F. Leidner, S. Hou, A. Ali, N. Kurt Yilmaz and C. A. Schiffer, Molecular and Structural Mechanism of Pan-Genotypic HCV NS3/4A Protease Inhibition by Glecaprevir, ACS Chem. Biol., 2020, 15, 342–352 CrossRef CAS PubMed.
- J. Zephyr, D. Nageswara Rao, S. V. Vo, M. Henes, K. Kosovrasti, A. N. Matthew, A. K. Hedger, J. Timm, E. T. Chan, A. Ali, N. Kurt Yilmaz and C. A. Schiffer, Deciphering the Molecular Mechanism of HCV Protease Inhibitor Fluorination as a General Approach to Avoid Drug Resistance, J. Mol. Biol., 2022, 434, 167503 CrossRef CAS PubMed.
- D. Nageswara Rao, J. Zephyr, M. Henes, E. T. Chan, A. N. Matthew, A. K. Hedger, H. L. Conway, M. Saeed, A. Newton, C. J. Petropoulos, W. Huang, N. Kurt Yilmaz, C. A. Schiffer and A. Ali, Discovery of Quinoxaline-Based P1–P3 Macrocyclic NS3/4A Protease Inhibitors with Potent Activity against Drug-Resistant Hepatitis C Virus Variants, J. Med. Chem., 2021, 64, 11972–11989 CrossRef CAS PubMed.
- Y. Jiang, S. W. Andrews, K. R. Condroski, B. Buckman, V. Serebryany, S. Wenglowsky, A. L. Kennedy, M. R. Madduru, B. Wang, M. Lyon, G. A. Doherty, B. T. Woodard, C. Lemieux, M. G. Do, H. Zhang, J. Ballard, G. Vigers, B. J. Brandhuber, P. Stengel, J. A. Josey, L. Beigelman, L. Blatt and S. D. Seiwert, Discovery of Danoprevir (ITMN-191/R7227), a Highly Selective and Potent Inhibitor of Hepatitis C Virus (HCV) NS3/4A Protease, J. Med. Chem., 2014, 57, 1753–1769 CrossRef CAS PubMed.
- S. D. Seiwert, S. W. Andrews, Y. Jiang, V. Serebryany, H. Tan, K. Kossen, P. T. R. Rajagopalan, S. Misialek, S. K. Stevens, A. Stoycheva, J. Hong, S. R. Lim, X. Qin, R. Rieger, K. R. Condroski, H. Zhang, M. G. Do, C. Lemieux, G. P. Hingorani, D. P. Hartley, J. A. Josey, L. Pan, L. Beigelman and L. M. Blatt, Preclinical Characteristics of the Hepatitis C Virus NS3/4A Protease Inhibitor ITMN-191 (R7227), Antimicrob. Agents Chemother., 2008, 52, 4432–4441 CrossRef CAS PubMed.
- H. Chen, Z. Zhang, L. Wang, Z. Huang, F. Gong, X. Li, Y. Chen and J. J. Wu, First clinical study using HCV protease inhibitor danoprevir to treat COVID-19 patients, Medicine, 2020, 99, e23357 CrossRef CAS PubMed.
- Z. Zhang, S. Wang, X. Tu, X. Peng, Y. Huang, L. Wang, W. Ju, J. Rao, X. Li, D. Zhu, H. Sun and H. Chen, A comparative study on the time to achieve negative nucleic acid testing and hospital stays between danoprevir and lopinavir/ritonavir in the treatment of patients with COVID–19, J. Med. Virol., 2020, 92, 2631–2636 CrossRef CAS PubMed.
- A. Vaidya and C. M. Perry, Simeprevir: First Global Approval, Drugs, 2013, 73, 2093–2106 CrossRef CAS PubMed.
- G. Milanole, F. Andriessen, G. Lemonnier, M. Sebban, G. Coadou, S. Couve-Bonnaire, J.-F. Bonfanti, P. Jubault and X. Pannecoucke, Toward the Synthesis of Fluorinated Analogues of HCV NS3/4A Serine Protease Inhibitors Using Methyl α-Amino-β-fluoro-β-vinylcyclopropanecarboxylate as Key Intermediate, Org. Lett., 2015, 17, 2968–2971 CrossRef CAS PubMed.
- K. F. McDaniel, H. Chen, M. Yeung, T. Middleton, L. Lu and K. Kurtz, Macrocyclic Hepatitis C Serine Protease Inhibitors, WO Pat., WO2012092409A2, 2012 Search PubMed.
- M. Smith and A. Lim, Profile of paritaprevir/ritonavir/ombitasvir plus dasabuvir in the treatment of chronic hepatitis C virus genotype 1 infection, Drug Des., Dev. Ther., 2015, 6083 CrossRef CAS PubMed.
- L.-Q. Sun, E. Mull, S. D'Andrea, B. Zheng, S. Hiebert, E. Gillis, M. Bowsher, S. Kandhasamy, V. R. Baratam, S. Puttaswamy, N. Pulicharla, S. Vishwakrishnan, S. Reddy, R. Trivedi, S. Sinha, S. Sivaprasad, A. Rao, S. Desai, K. Ghosh, R. Anumula, A. Kumar, R. Rajamani, Y.-K. Wang, H. Fang, A. Mathur, R. Rampulla, T. A. Zvyaga, K. Mosure, S. Jenkins, P. Falk, D. M. Tagore, C. Chen, K. Rendunchintala, J. Loy, N. A. Meanwell, F. McPhee and P. M. Scola, Discovery of BMS-986144, a Third-Generation, Pan-Genotype NS3/4A Protease Inhibitor for the Treatment of Hepatitis C Virus Infection, J. Med. Chem., 2020, 63, 14740–14760 CrossRef CAS PubMed.
- L.-Q. Sun, E. Mull, B. Zheng, S. D'Andrea, Q. Zhao, A. X. Wang, N. Sin, B. L. Venables, S.-Y. Sit, Y. Chen, J. Chen, A. Cocuzza, D. M. Bilder, A. Mathur, R. Rampulla, B.-C. Chen, T. Palani, S. Ganesan, P. N. Arunachalam, P. Falk, S. Levine, C. Chen, J. Friborg, F. Yu, D. Hernandez, A. K. Sheaffer, J. O. Knipe, Y.-H. Han, R. Schartman, M. Donoso, K. Mosure, M. W. Sinz, T. Zvyaga, R. Rajamani, K. Kish, J. Tredup, H. E. Klei, Q. Gao, A. Ng, L. Mueller, D. M. Grasela, S. Adams, J. Loy, P. C. Levesque, H. Sun, H. Shi, L. Sun, W. Warner, D. Li, J. Zhu, Y.-K. Wang, H. Fang, M. I. Cockett, N. A. Meanwell, F. McPhee and P. M. Scola, Discovery of a Potent Acyclic, Tripeptidic, Acyl Sulfonamide Inhibitor of Hepatitis C Virus NS3 Protease as a Back-up to Asunaprevir with the Potential for Once-Daily Dosing, J. Med. Chem., 2016, 59, 8042–8060 CrossRef CAS PubMed.
- S. F. Neelamkavil, S. Agrawal, T. Bara, C. Bennett, S. Bhat, D. Biswas, L. Brockunier, N. Buist, D. Burnette, M. Cartwright, S. Chackalamannil, R. Chase, M. Chelliah, A. Chen, M. Clasby, V. J. Colandrea, I. W. Davies, K. Eagen, Z. Guo, Y. Han, J. Howe, C. Jayne, H. Josien, S. Kargman, K. Marcantonio, S. Miao, R. Miller, A. Nolting, P. Pinto, M. Rajagopalan, R. T. Ruck, U. Shah, A. Soriano, D. Sperbeck, F. Velazquez, J. Wu, Y. Xia and S. Venkatraman, Discovery of MK-8831, A Novel Spiro-Proline Macrocycle as a Pan-Genotypic HCV-NS3/4a Protease Inhibitor, ACS Med. Chem. Lett., 2016, 7, 111–116 CrossRef CAS PubMed.
- X. C. Sheng, A. Casarez, R. Cai, M. O. Clarke, X. Chen, A. Cho, W. E. Delaney, E. Doerffler, M. Ji, M. Mertzman, R. Pakdaman, H.-J. Pyun, T. Rowe, Q. Wu, J. Xu and C. U. Kim, Discovery of GS-9256: A novel phosphinic acid derived inhibitor of the hepatitis C virus NS3/4A protease with potent clinical activity, Bioorg. Med. Chem. Lett., 2012, 22, 1394–1396 CrossRef CAS PubMed.
- H. Yang, C. Yang, Y. Wang, G. Rhodes, M. Robinson, G. Cheng, X. Qi, H. Mo, Y. Tian, R. Pakdaman, X. C. Sheng, C. U. Kim and W. E. Delaney, Preclinical Characterization of the Novel HCV NS3 Protease Inhibitor GS-9256, Antiviral Ther., 2017, 22, 413–420 CrossRef CAS PubMed.
- E. De Clercq, Current race in the development of DAAs (direct-acting antivirals) against HCV, Biochem. Pharmacol., 2014, 89, 441–452 CrossRef CAS PubMed.
- J. A. McCauley and M. T. Rudd, Hepatitis C virus NS3/4a protease inhibitors, Curr. Opin. Pharmacol., 2016, 30, 84–92 CrossRef CAS PubMed.
- J. Arumugasamy, K. Arunachalam, D. Bauer, A. Becker, C. A. Caillet, R. Glynn, G. M. Latham, J. Lim, J. Liu, B. A. Mayes, A. Moussa, E. Rosinovsky, A. E. Salanson, A. F. Soret, A. Stewart, J. Wang and X. Wu, Development of Related HCV Protease Inhibitors: Macrocyclization of Two Highly Functionalized Dienyl-ureas via Ring-Closing Metathesis, Org. Process Res. Dev., 2013, 17, 811–828 CrossRef CAS.
- C. C. Parsy, F.-R. Alexandre, V. Bidau, F. Bonnaterre, G. Brandt, C. Caillet, S. Cappelle, D. Chaves, T. Convard, M. Derock, D. Gloux, Y. Griffon, L. B. Lallos, F. Leroy, M. Liuzzi, A.-G. Loi, L. Moulat, M. Chiara, H. Rahali, V. Roques, E. Rosinovsky, S. Savin, M. Seifer, D. Standring and D. Surleraux, Discovery and structural diversity of the hepatitis C virus NS3/4A serine protease inhibitor series leading to clinical candidate IDX320, Bioorg. Med. Chem. Lett., 2015, 25, 5427–5436 CrossRef CAS PubMed.
- J. De Bruijne, A. Van Vliet, C. J. Weegink, W. Mazur, A. Wiercinska-Drapało, K. Simon, G. Cholewińska-Szymańska, J. Kapocsi, I. Várkonyi, X.-J. Zhou, M.-F. Temam, J. Molles, J. Chen, K. Pietropaolo, J. F. McCarville, J. Z. Sullivan-Bólyai, D. Mayers and H. Reesink, Rapid Decline of Viral RNA in Chronic Hepatitis C Patients Treated Once Daily with IDX320: A Novel Macrocyclic HCV Protease Inhibitor, Antiviral Ther., 2012, 17, 633–642 CrossRef CAS PubMed.
- D. G. Johns, L.-C. Campeau, P. Banka, A. Bautmans, T. Bueters, E. Bianchi, D. Branca, P. G. Bulger, I. Crevecoeur, F.-X. Ding, R. M. Garbaccio, E. D. Guetschow, Y. Guo, S. N. Ha, J. M. Johnston, H. Josien, E. A. Kauh, K. A. Koeplinger, J. T. Kuethe, E. Lai, C. L. Lanning, A. Y. H. Lee, L. Li, A. G. Nair, E. A. O'Neill, S. A. Stoch, D. A. Thaisrivongs, T. J. Tucker, P. Vachal, K. Van Dyck, F. P. Vanhoutte, B. Volckaert, D. G. Wolford, A. Xu, T. Zhao, D. Zhou, S. Zhou, X. Zhu, H. J. Zokian, A. M. Walji and H. B. Wood, Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor, Circulation, 2023, 148, 144–158 CrossRef CAS PubMed.
- D. G. Johns, P. Banka, Z. Zhou, A. Klapara, F. Tsay, J. Kong, R. J. Varsolona, R. Desmond, P. E. Maligres, M. Marota, C. Alleyne, G. A. Okoh, J. C. Dinunzio, R. Nofsinger, L. Li, D. J. Smith, M. Mahjour, K. A. Koeplinger, Y. Xiong, P. W. Wuefling and D. J. Higgins, Compounds for treating conditions related to PCSK9 activity, WO Pat., WO2023023245A1, 2023 Search PubMed.
- https://drughunter.com/articles/mk-0616-the-2023-molecule-of-the-year (Accessed Sep 2024).
- C. M. Ballantyne, P. Banka, G. Mendez, R. Garcia, J. Rosenstock, A. Rodgers, G. Mendizabal, Y. Mitchel and A. L. Catapano, Phase 2b Randomized Trial of the Oral PCSK9 Inhibitor MK-0616, J. Am. Coll. Cardiol., 2023, 81, 1553–1564 CrossRef CAS PubMed.
- T. J. Tucker, M. W. Embrey, C. Alleyne, R. P. Amin, A. Bass, B. Bhatt, E. Bianchi, D. Branca, T. Bueters, N. Buist, S. N. Ha, M. Hafey, H. He, J. Higgins, D. G. Johns, A. D. Kerekes, K. A. Koeplinger, J. T. Kuethe, N. Li, B. Murphy, P. Orth, S. Salowe, A. Shahripour, R. Tracy, W. Wang, C. Wu, Y. Xiong, H. J. Zokian, H. B. Wood and A. Walji, A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors, J. Med. Chem., 2021, 64, 16770–16800 CrossRef CAS PubMed.
- S. M. Hoy, Motixafortide: First Approval, Drugs, 2023, 83, 1635–1643 CrossRef CAS PubMed.
- M. Tanada, M. Tamiya, A. Matsuo, A. Chiyoda, K. Takano, T. Ito, M. Irie, T. Kotake, R. Takeyama, H. Kawada, R. Hayashi, S. Ishikawa, K. Nomura, N. Furuichi, Y. Morita, M. Kage, S. Hashimoto, K. Nii, H. Sase, K. Ohara, A. Ohta, S. Kuramoto, Y. Nishimura, H. Iikura and T. Shiraishi, Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor, J. Am. Chem. Soc., 2023, 145, 16610–16620 CrossRef CAS PubMed.
- A. Ohta, M. Tanada, S. Shinohara, Y. Morita, K. Nakano, Y. Yamagishi, R. Takano, S. Kariyuki, T. Iida, A. Matsuo, K. Ozeki, T. Emura, Y. Sakurai, K. Takano, A. Higashida, M. Kojima, T. Muraoka, R. Takeyama, T. Kato, K. Kimura, K. Ogawa, K. Ohara, S. Tanaka, Y. Kikuchi, N. Hisada, R. Hayashi, Y. Nishimura, K. Nomura, T. Tachibana, M. Irie, H. Kawada, T. Torizawa, N. Murao, T. Kotake, M. Tanaka, S. Ishikawa, T. Miyake, M. Tamiya, M. Arai, A. Chiyoda, S. Akai, H. Sase, S. Kuramoto, T. Ito, T. Shiraishi, T. Kojima and H. Iikura, Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets, J. Am. Chem. Soc., 2023, 145, 24035–24051 CrossRef CAS PubMed.
- H. R. Hoveyda, E. Marsault, R. Gagnon, A. P. Mathieu, M. Vézina, A. Landry, Z. Wang, K. Benakli, S. Beaubien, C. Saint-Louis, M. Brassard, J.-F. Pinault, L. Ouellet, S. Bhat, M. Ramaseshan, X. Peng, L. Foucher, S. Beauchemin, P. Bhérer, D. F. Veber, M. L. Peterson and G. L. Fraser, Optimization of the Potency and Pharmacokinetic Properties of a Macrocyclic Ghrelin Receptor Agonist (Part I): Development of Ulimorelin (TZP-101) from Hit to Clinic, J. Med. Chem., 2011, 54, 8305–8320 CrossRef PubMed.
- B. L. Podlogar, R. A. Farr, D. Friedrich, C. Tarnus, E. W. Huber, R. J. Cregge and D. Schirlin, Design, Synthesis, and Conformational Analysis of a Novel Macrocyclic HIV-Protease Inhibitor, J. Med. Chem., 1994, 37, 3684–3692 CrossRef CAS PubMed.
- M. Boehm, K. Beaumont, R. Jones, A. S. Kalgutkar, L. Zhang, K. Atkinson, G. Bai, J. A. Brown, H. Eng, G. H. Goetz, B. R. Holder, B. Khunte, S. Lazzaro, C. Limberakis, S. Ryu, M. J. Shapiro, L. Tylaska, J. Yan, R. Turner, S. S. F. Leung, M. Ramaseshan, D. A. Price, S. Liras, M. P. Jacobson, D. J. Earp, R. S. Lokey, A. M. Mathiowetz and E. Menhaji-Klotz, Discovery of Potent and Orally Bioavailable Macrocyclic Peptide–Peptoid Hybrid CXCR7 Modulators, J. Med. Chem., 2017, 60, 9653–9663 CrossRef CAS PubMed.
- F. O. Gombert, A. Lederer, D. Obrecht, B. Romagnoli, C. Bisang and C. Ludin, Template-Fixed Peptidomimetics with CXCR7 Modulating Activity, WO Pat., WO2011095220, 2011 Search PubMed.
- S. J. Middendorp, J. Wilbs, C. Quarroz, S. Calzavarini, A. Angelillo-Scherrer and C. Heinis, Peptide Macrocycle Inhibitor of Coagulation Factor XII with Subnanomolar Affinity and High Target Selectivity, J. Med. Chem., 2017, 60, 1151–1158 CrossRef CAS PubMed.
- G. A. Pankuch, L. M. Kelly, G. Lin, A. Bryskier, C. Couturier, M. R. Jacobs and P. C. Appelbaum, Activities of a New Oral Streptogramin, XRP 2868, Compared to Those of Other Agents against Streptococcus pneumoniae and Haemophilus Species, Antimicrob. Agents Chemother., 2003, 47, 3270–3274 CrossRef CAS PubMed.
- J. Noeske, J. Huang, N. B. Olivier, R. A. Giacobbe, M. Zambrowski and J. H. D. Cate, Synergy of Streptogramin Antibiotics Occurs Independently of Their Effects on Translation, Antimicrob. Agents Chemother., 2014, 58, 5269–5279 CrossRef PubMed.
- L. Cai, I. B. Seiple and Q. Li, Modular Chemical Synthesis of Streptogramin and Lankacidin Antibiotics, Acc. Chem. Res., 2021, 54, 1891–1908 CrossRef CAS PubMed.
- D. Sighel, G. Battistini, E. F. Rosatti, J. Vigna, M. Pavan, R. Belli, D. Peroni, F. Alessandrini, S. Longhi, M. Pancher, J. Rorbach, S. Moro, A. Quattrone and I. Mancini, Streptogramin A derivatives as mitochondrial translation inhibitors to suppress glioblastoma stem cell growth, Eur. J. Med. Chem., 2023, 246, 114979 CrossRef CAS PubMed.
- H. Zhou, L. Liu, J. Huang, D. Bernard, H. Karatas, A. Navarro, M. Lei and S. Wang, Structure-Based Design of High-Affinity Macrocyclic Peptidomimetics to Block the Menin-Mixed Lineage Leukemia 1 (MLL1) Protein–Protein Interaction, J. Med. Chem., 2013, 56, 1113–1123 CrossRef CAS PubMed.
- T. Tsunemi, S. J. Bernardino, A. Mendoza, C. G. Jones and P. G. Harran, Syntheses of Atypically Fluorinated Peptidyl Macrocycles through Sequential Vinylic Substitutions, Angew. Chem., Int. Ed., 2020, 59, 674–678 CrossRef CAS PubMed.
- A. Mendoza, S. J. Bernardino, M. J. Dweck, I. Valencia, D. Evans, H. Tian, W. Lee, Y. Li, K. N. Houk and P. G. Harran, Cascade Synthesis of Fluorinated Spiroheterocyclic Scaffolding for Peptidic Macrobicycles, J. Am. Chem. Soc., 2023, 145, 15888–15895 CrossRef CAS PubMed.
- J. Morimoto, Y. Hayashi and H. Suga, Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2, Angew. Chem., Int. Ed., 2012, 51, 3423–3427 CrossRef CAS PubMed.
- K. Ito, T. Passioura and H. Suga, Technologies for the Synthesis of mRNA-Encoding Libraries and Discovery of Bioactive Natural Product-Inspired Non-Traditional Macrocyclic Peptides, Molecules, 2013, 18, 3502–3528 CrossRef CAS PubMed.
- J. Piekielna, R. Perlikowska, J. C. do-Rego, J.-L. do-Rego, M. C. Cerlesi, G. Calo, A. Kluczyk, K. Łapiński, C. Tömböly and A. Janecka, Synthesis of Mixed Opioid Affinity Cyclic Endomorphin-2 Analogues with Fluorinated Phenylalanines, ACS Med. Chem. Lett., 2015, 6, 579–583 CrossRef CAS PubMed.
- A. Łęgowska, D. Dębowski, A. Lesner, M. Wysocka and K. Rolka, Introduction of non-natural amino acid residues into the substrate-specific P1 position of trypsin inhibitor SFTI-1 yields potent chymotrypsin and cathepsin G inhibitors, Bioorg. Med. Chem., 2009, 17, 3302–3307 CrossRef PubMed.
- K. Josephson, M. C. T. Hartman and J. W. Szostak, Ribosomal Synthesis of Unnatural Peptides, J. Am. Chem. Soc., 2005, 127, 11727–11735 CrossRef CAS PubMed.
- J. Wu, Y. Wang, W. Cai, D. Chen, X. Peng, H. Dong, J. Li, H. Liu, S. Shi, S. Tang, Z. Li, H. Sui, Y. Wang, C. Wu, Y. Zhang, X. Fu and Y. Yin, Ribosomal translation of fluorinated non-canonical amino acids for de novo biologically active fluorinated macrocyclic peptides, Chem. Sci., 2024, 15, 13889–13898 RSC.
- I. R. Mathiesen, E. D. D. Calder, S. Kunzelmann and L. J. Walport, Discovering covalent cyclic peptide inhibitors of peptidyl arginine deiminase 4 (PADI4) using mRNA-display with a genetically encoded electrophilic warhead, Commun. Chem., 2024, 7, 304 CrossRef CAS PubMed.
- Y. V. Guillen Schlippe, M. C. T. Hartman, K. Josephson and J. W. Szostak, In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef CAS PubMed.
- K. Josephson, A. Ricardo and J. W. Szostak, mRNA display: from basic principles to macrocycle drug discovery, Drug Discovery Today, 2014, 19, 388–399 CrossRef CAS PubMed.
- M. L. Merz, S. Habeshian, B. Li, J.-A. G. L. David, A. L. Nielsen, X. Ji, K. Il Khwildy, M. M. Duany Benitez, P. Phothirath and C. Heinis, De novo development of small cyclic peptides that are orally bioavailable, Nat. Chem. Biol., 2024, 20, 624–633 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- S. J. M. Verhoork, C. E. Jennings, N. Rozatian, J. Reeks, J. Meng, E. K. Corlett, F. Bunglawala, M. E. M. Noble, A. G. Leach and C. R. Coxon, Tuning the Binding Affinity and Selectivity of Perfluoroaryl–Stapled Peptides by Cysteine–Editing, Chem. – Eur. J., 2019, 25, 177–182 CrossRef CAS PubMed.
- W. D. G. Brittain and C. R. Coxon, Perfluoroaryl and Perfluoroheteroaryl Reagents as Emerging New Tools for Peptide Synthesis, Modification and Bioconjugation, Chem. – Eur. J., 2022, 28, e202103305 CrossRef CAS PubMed.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss and R. Derda, Rapid biocompatible macrocyclization of peptides with decafluoro-diphenylsulfone, Chem. Sci., 2016, 7, 3785–3790 RSC.
- G. Lautrette, F. Touti, H. G. Lee, P. Dai and B. L. Pentelute, Nitrogen Arylation for Macrocyclization of Unprotected Peptides, J. Am. Chem. Soc., 2016, 138, 8340–8343 CrossRef CAS PubMed.
- A. G. Jamieson, N. Boutard, D. Sabatino and W. D. Lubell, Peptide Scanning for Studying Structure–Activity Relationships in Drug Discovery, Chem. Biol. Drug Des., 2013, 81, 148–165 CrossRef CAS PubMed.
- Y. Zou, A. M. Spokoyny, C. Zhang, M. D. Simon, H. Yu, Y.-S. Lin and B. L. Pentelute, Convergent diversity-oriented side-chain macrocyclization scan for unprotected polypeptides, Org. Biomol. Chem., 2014, 12, 566–573 RSC.
- J. M. Wolfe, C. M. Fadzen, R. L. Holden, M. Yao, G. J. Hanson and B. L. Pentelute, Perfluoroaryl Bicyclic Cell–Penetrating Peptides for Delivery of Antisense Oligonucleotides, Angew. Chem., Int. Ed., 2018, 57, 4756–4759 CrossRef CAS PubMed.
- K. Siva, G. Covello and M. A. Denti, Exon-Skipping Antisense Oligonucleotides to Correct Missplicing in Neurogenetic Diseases, Nucleic Acid Ther., 2014, 24, 69–86 CrossRef CAS PubMed.
- C. M. Fadzen, J. M. Wolfe, C.-F. Cho, E. A. Chiocca, S. E. Lawler and B. L. Pentelute, Perfluoroarene–Based Peptide Macrocycles to Enhance Penetration Across the Blood–Brain Barrier, J. Am. Chem. Soc., 2017, 139, 15628–15631 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- C. M. Fadzen, J. M. Wolfe, W. Zhou, C.-F. Cho, N. Von Spreckelsen, K. T. Hutchinson, Y.-C. Lee, E. A. Chiocca, S. E. Lawler, O. H. Yilmaz, S. J. Lippard and B. L. Pentelute, A Platinum(IV) Prodrug—Perfluoroaryl Macrocyclic Peptide Conjugate Enhances Platinum Uptake in the Brain, J. Med. Chem., 2020, 63, 6741–6747 CrossRef CAS PubMed.
- P. Wadhwani, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, Optimized Protocol for Synthesis of Cyclic Gramicidin S: Starting Amino Acid Is Key to High Yield, J. Org. Chem., 2006, 71, 55–61 CrossRef CAS PubMed.
- J. Salgado, S. L. Grage, L. H. Kondejewski, R. S. Hodges, R. N. McElhaney and A. S. Ulrich, Membrane-bound structure and alignment of the antimicrobial β-sheet peptide gramicidin S derived from angular and distance constraints by solid state 19F-NMR, J. Biomol. NMR, 2001, 21, 191–208 CrossRef CAS PubMed.
- K. Y. Palate, Z. Yang, A. C. Whitwood and W. P. Unsworth, Synthesis of medium-ring lactams and macrocyclic peptide mimetics via conjugate addition/ring expansion cascade reactions, RSC Chem. Biol., 2022, 3, 334–340 RSC.
- J. Y. K. Wong, A. I. Ekanayake, S. Kharchenko, S. E. Kirberger, R. Qiu, P. Kelich, S. Sarkar, J. Li, K. X. Fernandez, E. R. Alvizo-Paez, J. Miao, S. Kalhor-Monfared, J. D. John, H. Kang, H. Choi, J. M. Nuss, J. C. Vederas, Y.-S. Lin, M. S. Macauley, L. Vukovic, W. C. K. Pomerantz and R. Derda, Genetically encoded discovery of perfluoroaryl macrocycles that bind to albumin and exhibit extended circulation in vivo, Nat. Commun., 2023, 14, 5654 CrossRef CAS PubMed.
- L. Hunter, The C–F bond as a conformational tool in organic and biological chemistry, Beilstein J. Org. Chem., 2010, 6, 38 CrossRef PubMed.
- X.-G. Hu, D. S. Thomas, R. Griffith and L. Hunter, Stereoselective Fluorination Alters the Geometry of a Cyclic Peptide: Exploration of Backbone-Fluorinated Analogues of Unguisin A, Angew. Chem., Int. Ed., 2014, 53, 6176–6179 CrossRef CAS PubMed.
- I. Yamamoto, M. J. T. Jordan, N. Gavande, M. R. Doddareddy, M. Chebib and L. Hunter, The enantiomers of syn-2,3-difluoro-4-aminobutyric acid elicit opposite responses at the GABA C receptor, Chem. Commun., 2012, 48, 829–831 RSC.
- A. D. Ariawan, J. E. A. Webb, E. N. W. Howe, P. A. Gale, P. Thordarson and L. Hunter, Cyclic peptide unguisin A is an anion receptor with high affinity for phosphate and pyrophosphate, Org. Biomol. Chem., 2017, 15, 2962–2967 RSC.
- N. Díaz and D. Suárez, Understanding the Conformational Properties of Fluorinated Polypeptides: Molecular Modelling of Unguisin A, J. Chem. Inf. Model., 2021, 61, 223–237 CrossRef PubMed.
- C. Au, C. Gonzalez, Y. C. Leung, F. Mansour, J. Trinh, Z. Wang, X.-G. Hu, R. Griffith, E. Pasquier and L. Hunter, Tuning the properties of a cyclic RGD-containing tetrapeptide through backbone fluorination, Org. Biomol. Chem., 2019, 17, 664–674 RSC.
- S. S. Burade, T. Saha, N. Bhuma, N. Kumbhar, A. Kotmale, P. R. Rajamohanan, R. G. Gonnade, P. Talukdar and D. D. Dhavale, Self-Assembly of Fluorinated Sugar Amino Acid Derived α,γ-Cyclic Peptides into Transmembrane Anion Transport, Org. Lett., 2017, 19, 5948–5951 CrossRef CAS PubMed.
- T. J. Cogswell, A. Dahlén and L. Knerr, Synthesis of Diverse α–Fluoroalkoxyaryl Derivatives and Their Use for the Generation of Fluorinated Macrocycles, Chem. – Eur. J., 2018, 25, 1184–1187 CrossRef PubMed.
- D. O'Hagan, Understanding organofluorine chemistry. An introduction to the C–F bond, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- T. E. Rose, B. H. Curtin, K. V. Lawson, A. Simon, K. N. Houk and P. G. Harran, On the prevalence of bridged macrocyclic pyrroloindolines formed in regiodivergent alkylations of tryptophan, Chem. Sci., 2016, 7, 4158–4166 RSC.
- K. V. Lawson, T. E. Rose and P. G. Harran, Template-induced macrocycle diversity through large ring-forming alkylations of tryptophan, Tetrahedron, 2013, 69, 7683–7691 CrossRef CAS PubMed.
- X. Sun, Y. Li, T. Liu, Z. Li, X. Zhang and X. Chen, Peptide-based imaging agents for cancer detection, Adv. Drug Delivery Rev., 2017, 110–111, 38–51 CrossRef CAS PubMed.
- R. A. Davis, K. Lau, S. H. Hausner and J. L. Sutcliffe, Solid-phase synthesis and fluorine-18 radiolabeling of cycloRGDyK, Org. Biomol. Chem., 2016, 14, 8659–8663 RSC.
- J. Rickmeier and T. Ritter, Site–Specific Deoxyfluorination of Small Peptides with [18F]Fluoride, Angew. Chem., Int. Ed., 2018, 57, 14207–14211 CrossRef CAS PubMed.
- T. G. Luu and H.-K. Kim, Recent progress on radiofluorination using metals: strategies for generation of C–18 F bonds, Org. Chem. Front., 2023, 10, 5746–5781 RSC.
- S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, 18F-Trifluoromethylation of Unmodified Peptides with 5-18F-(Trifluoromethyl)dibenzothiophenium Trifluoromethanesulfonate, J. Am. Chem. Soc., 2018, 140, 1572–1575 CrossRef CAS PubMed.
- C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, 18F-Trifluoromethanesulfinate Enables Direct C–H 18F-Trifluoromethylation of Native Aromatic Residues in Peptides, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed.
- T. Günther, G. Tulipano, P. Dournaud, C. Bousquet, Z. Csaba, H.-J. Kreienkamp, A. Lupp, M. Korbonits, J. P. Castaño, H.-J. Wester, M. Culler, S. Melmed and S. Schulz, International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature, Pharmacol. Rev., 2018, 70, 763–835 CrossRef PubMed.
- J. W. McDaniel, J. M. Stauber, E. A. Doud, A. M. Spokoyny and J. M. Murphy, An Organometallic Gold(III) Reagent for 18F Labeling of Unprotected Peptides and Sugars in Aqueous Media, Org. Lett., 2022, 24, 5132–5136 CrossRef CAS PubMed.
- V. R. Bouvet and F. Wuest, Application of [18F]FDG in radiolabeling reactions using microfluidic technology, Lab-on-a-Chip, 2013, 13, 4290 RSC.
- S. Richter and F. Wuest, 18F-Labeled Peptides: The Future Is Bright, Molecules, 2014, 19, 20536–20556 CrossRef PubMed.
- D. J. Donnelly, J. Kim, T. Tran, P. M. Scola, D. Tenney, A. Pena, T. Petrone, Y. Zhang, K. M. Boy, M. A. Poss, E. L. Cole, M. G. Soars, B. M. Johnson, D. Cohen, D. Batalla, P. L. Chow, A. O. Shorts, S. Du, N. A. Meanwell and S. J. Bonacorsi, The discovery and evaluation of [18F]BMS-986229, a novel macrocyclic peptide PET radioligand for the measurement of PD-L1 expression and in vivo PD-L1 target engagement, Eur. J. Nucl. Med. Mol. Imaging, 2024, 51, 978–990 CrossRef CAS.
- S. Mukherjee, A. Rogers, G. Creech, C. Hang, A. Ramirez, M. Dummeldinger, S. Brueggemeier, C. Mapelli, S. Zaretsky, M. Huang, R. Black, M. B. Peddicord, N. Cuniere, J. Kempson, J. Pawluczyk, M. Allen, R. Parsons and C. Sfouggatakis, Process Development of a Macrocyclic Peptide Inhibitor of PD-L1, J. Org. Chem., 2024, 89, 6651–6663 CrossRef CAS PubMed.
- M. Badenhorst, A. D. Windhorst and W. Beaino, Navigating the landscape of PD-1/PD-L1 imaging tracers: from challenges to opportunities, Front. Med., 2024, 11, 1401515 CrossRef.
- Y. Pan, The Dark Side of Fluorine, ACS Med. Chem. Lett., 2019, 10, 1016–1019 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2025 |