DOI:
10.1039/D4MD00837E
(Review Article)
RSC Med. Chem., 2025,
16, 1532-1549
Synthetic strategies and medicinal chemistry perspectives of dual acting carbonic anhydrase modulators with monoamine oxidase and cholinesterase inhibitors
Received
26th October 2024
, Accepted 18th January 2025
First published on 6th February 2025
Abstract
Multi-target drug design (MTDD) represents the paradigm shift in pharmaceutical research, moving beyond the conventional one-drug–one-target approach to address the complexity of multifactorial diseases. This strategy aims to develop single therapeutic candidates that can simultaneously modulate multiple biological targets, offering more comprehensive disease management and reducing the likelihood of drug resistance. In this article, we highlighted the design, synthesis, and structure–activity relationships (SARs) of various dual acting inhibitors involved in treatment of neurodegenerative diseases. Dual acting inhibitors targeting carbonic anhydrases (CAs), monoamine oxidases (MAOs), and cholinesterases (ChEs) have emerged as promising therapeutic agents due to their potential in treating complex neurodegenerative and psychiatric disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD). By integrating CA inhibitors with MAO and ChE inhibition, researchers aim to address both the neuroprotective and symptomatic aspects of these disorders. The review also discusses key SAR studies that have guided the optimization of dual inhibitors, focusing on achieving selectivity and potency while minimizing off-target effects. From a medicinal chemistry perspective, the dual inhibition approach offers advantages such as improved efficacy, reduced polypharmacy, and better management of disease progression. However, challenges remain, including maintaining selectivity for target isoforms and overcoming pharmacokinetic limitations. Overall, the development of dual-acting CA–MAO–ChE inhibitors represents a compelling avenue in drug discovery, with the potential to significantly impact the treatment of neurodegenerative diseases.
1. Introduction
Multi-target drugs are therapeutic agents designed to act on multiple molecular targets or pathways simultaneously (Fig. 1). These drugs are becoming increasingly important in the treatment of complex diseases, particularly cancer, metabolic syndrome, neurodegenerative disorders, inflammation and infectious diseases.1 Even if their discovery has long been due to serendipity. Multiple additive or synergistic pharmacodynamic activities may be highly advantageous for treating certain disorders in terms of increased therapeutic efficacy or postponed development of resistance. There are several reasons why multitarget drugs are crucial: 1. complex diseases involve multiple pathways, 2. reduction of drug resistance, 3. synergistic effects, 4. improved patient compliance, and 5. reduced toxicity.2,3
 |
| | Fig. 1 Multi-target drug design. | |
1.1. Complex diseases involve multiple pathways
Many complex diseases like cancer, diabetes, and neurodegenerative diseases, involve a network of signaling pathway. A single-target drug may not be sufficient to manage such diseases effectively. Multitarget medications can achieve more thorough therapeutic effects by targeting multiple pathways. For instance, lapatinib (Fig. 2) is a reversible ATP-competitive inhibitor of the human epidermal growth factor receptor 2 (HER2), and breast cancer is treated by targeting epidermal growth factor receptor (EGFR) tyrosine kinases.4
 |
| | Fig. 2 Structures of clinically used multi-target drugs. | |
1.2. Reduction of drug resistance
One of the main challenges in diseases like cancer and infections is the development of drug resistance. Single-target therapies can lead to the emergence of resistant strains or cells, as the disease can adapt to inhibit one target. Multitarget drugs reduce the likelihood of resistance by simultaneously attacking multiple vulnerabilities within a disease, making it harder for resistant variants to emerge. Antitubercular medication with multi-target characteristics, SQ109 (Fig. 2), for instance, has been shown to inhibit MenA and MenG, which are involved in the manufacture of menaquinones, as well as MmpL3 and MmpL11, which are transporter proteins involved in the formation of cell walls.5
1.3. Synergistic effect
Targeting multiple pathways or molecules can create a synergistic effect, where the combined effect of the drug on different targets is greater than the sum of its individual effects. This can improve the overall efficacy of the treatment and may allow for lower dosages, reducing side effects of drugs, such as caproctamine (Fig. 2), which showed a synergistic cholinergic action against Alzheimer's disease (AD) by antagonizing presynaptic muscarinic acetylcholine M2 autoreceptors and inhibiting acetylcholinesterase (AChE).6
1.4. Improved patient compliance
Multitarget drugs can potentially reduce the number of medications a patient has to take, simplifying treatment regimens. For instance, instead of taking multiple drugs to target different aspects of a disease, a single multitarget drug could achieve the same results. This can lead to better adherence to treatment.7
1.5. Reduced toxicity
By distributing the drug's effects across several targets rather than focusing on a single one at high potency, multitarget drugs may help minimize the risk of off-target effects and toxicity, leading to a better safety profile.8 For example, sorafenib (Fig. 2) was approved by the FDA for the treatment of renal cell carcinoma (RCC) in 2005, unresectable hepatocellular carcinoma (HCC) in 2007 and radioiodine-refractory differentiated thyroid carcinoma in 2013.9–11 Sorafenib inhibits the activity of serine–threonine kinases Raf-1 and B-Raf, vascular endothelial growth factor receptor (VEGFR)-1/2/3, platelet-derived growth factor receptor β (PDGFR-β), c-Kit, RET, and FLT3.12–14
Multi-target drug design for CNS acting molecules focuses on creating compounds that can interact with several biological targets implicated in CNS disorders. Conditions such as Alzheimer's disease, Parkinson's disease, depression, and schizophrenia involve intricate pathological mechanisms. By addressing multiple pathways at once, this approach offers potential therapeutic benefits, tackling the complex and multifactorial nature of these diseases more effectively.15 In the development of multi-target drug design, dual-acting molecules play a crucial role. Dual-acting molecules for CNS disorders are drugs designed to target two distinct biological mechanisms such as monoamine oxidase (MAO) and acetylcholinesterase/butyrylcholinesterase (AChE/BChE), MAO and carbonic anhydrases (CAs)16 or AChE/BChE and CAs, providing broader therapeutic effects. There are several dual-acting drug molecules in the last 5 years for the treatment of CNS diseases, which are discussed below (Fig. 3).
 |
| | Fig. 3 Dual acting drugs used in the treatment of neurological diseases.17–23 | |
Carbonic anhydrases (CAs) are a family of zinc metalloenzymes catalyzing the reversible reaction of CO2 and H2O into bicarbonate ions and protons.24,25 CAs are mainly found in archaea, prokaryotes, and eukaryotes. They are encoded by eight distinct, evolutionarily unrelated genes known as α-CAs (found in vertebrates, eubacteria, algae, and the cytoplasm of green plants), β-CAs (primarily in eubacteria, algae, and the chloroplasts of both monocots and dicots), and γ-CAs (mainly in archaea and some eubacteria), and δ-, η-, θ-, ζ- (found in marine diatom) and ι-CAs (found in diatoms and bacteria).26–30 In mammals, 16 distinct α-CA isozymes or CA-related proteins (CARPs) have been identified, each exhibiting unique subcellular localization and tissue distribution.31–33 Some of these CA isozymes are cytosolic, including CA I, CA II, CA III, CA VII, and CA XIII. Others are membrane-bound, such as CA IV, CA IX, CA XII, CA XIV, and CA XV. CA VA and CA VB are localized in the mitochondria, while CA VI is secreted in saliva and milk. Additionally, there are three known catalytic forms of the CA-related proteins (CARPs), specifically CARP VIII, CARP X, and CARP XI, which also appear to be cytosolic proteins.34 These enzymes catalyze a simple yet vital physiological reaction: the interconversion between carbon dioxide and bicarbonate ions. This process plays a key role in several crucial physiological functions, including respiration and the transport of CO2/bicarbonate between metabolizing tissues and the lungs, maintaining pH and CO2 homeostasis, and facilitating electrolyte secretion in various tissues and organs. They are also involved in biosynthetic pathways like gluconeogenesis, lipogenesis, and ureagenesis, as well as in bone resorption, calcification, tumorigenesis, and many other physiological and pathological processes.35,36 As will be discussed shortly, many of these isozymes are key targets for the design of inhibitors with potential clinical applications. In addition to catalyzing the reversible hydration of CO2 to bicarbonate (reaction (1), Fig. 4), carbonic anhydrases also facilitate several other reactions. These include the hydration of cyanate to carbamic acid and cyanamide to urea (reactions (2) and (3)), the hydration of aldehydes to form gem-diols (reaction (4)), and the hydrolysis of carboxylic acids or sulfonic acids (reactions (5) and (6)). They are also involved in other, less-studied hydrolytic processes, as described in reactions (7)–(9).36,37 The recent advancements in the development of CA inhibitors (CAIs) have led to the creation of novel therapeutic agents targeting a wide range of diseases. The new generations of CAIs are being designed with improved selectivity, potency, and pharmacokinetic properties, including sulfonamides, coumarins, and sulfamates.38,39 CAIs are being explored due to their potential in treating conditions such as glaucoma, epilepsy, obesity, and cancer.40–45 Additionally, CAIs are being investigated for their roles in managing conditions like hypertension, osteoporosis, and neurological disorders. In Alzheimer's disease (AD) and cerebral amyloid angiopathy (CAA), harmful amyloid-beta (Aβ) aggregates build up around and within the walls of cerebral vessels, and form parenchymal plaques in specific brain regions such as the hippocampus and cortex.46 Researchers have shown that cytochrome c is released from the mitochondria of the neurovascular unit (NVU) in a variety of cell types, including endothelial cells (ECs), smooth muscle cells (SMCs), neurons, and glial cells, as a result of Aβ oligomers and protofibrils. Caspases are activated as a result of this release, which causes cell death.47,48 Methazolamide (MTZ) reduced mitochondria-mediated cell death caused by Aβ in all neurovascular cell types. Additionally, MTZ lowered the activation of caspase-3 and caspase-9 in endothelial cells, neurons, and glial cells in vitro, and also reduced caspase-3 activation in vivo.49 Furthermore, in cerebrovascular endothelial cells, acetazolamide (ATZ) and MTZ inhibited Aβ-induced caspase-9 activation and subsequent apoptosis. Sun and Alkon demonstrated that CAAs improve synaptic efficacy, spatial learning, and memory in rats, as shown by the Morris water maze test. This effect occurs because CA activation increases bicarbonate (HCO3−) levels in the hippocampus, which triggers a shift in GABAergic synaptic outputs from inhibitory to excitatory.50 Under these conditions, GABA activates a specific group of hippocampal pyramidal neurons, thereby enhancing associative memory formation. In 2017, research found that CAA (D-phenyl alanine) improves object recognition memory in mice by promoting ERK phosphorylation in the cortex and hippocampus.51 Cytosolic carbonic anhydrases, particularly isoforms II and VII, are thought to be the primary isozymes responsible for this pharmacological effect.52
 |
| | Fig. 4 Reactions catalyzed by α-carbonic anhydrase. | |
Monoamine oxidase (MAO) is a mitochondrial, flavin-dependent enzyme responsible for metabolizing arylalkylamine neurotransmitters such as epinephrine, norepinephrine (NE), dopamine (DA), serotonin (5-HT), and phenylethylamine (PEA), along with various exogenous amines. This process occurs through oxidative deamination, converting these compounds into their corresponding aldehydes and hydrogen peroxide (H2O2) in the central nervous system (CNS) and tissues.53 The Fenton reaction, catalyzed by iron and neuromelanin, transforms hydrogen peroxide (H2O2) into highly dangerous reactive oxygen species (ROS).54 Two isoforms of MAO, known as MAO-A and MAO-B, have been identified based on differences in enzyme inhibition, substrate selectivity, specificity, amino acid sequence, and tissue distribution.55 The enzyme MAO-A preferentially deaminates neurotransmitters such as serotonin, adrenaline, and noradrenaline (e.g., inhibited by clorgyline), while MAO-B primarily oxidizes phenylethylamine and benzylamine (e.g., inhibited by selegiline). Both isoforms, however, are active against tyramine and dopamine.56 The primary differences between MAO-A and MAO-B lie in the specific details of their active sites, which explain their varying substrate and inhibitor selectivities. Despite having a similar overall folding structure, MAO-A and MAO-B share 70% sequence similarity. The availability of structural data has facilitated the development of isoform-selective MAO inhibitors through rational drug design.57 The design strategy for MAO inhibitors requires a careful consideration of selectivity between MAO-A and MAO-B, reversibility, bioavailability, and safety to meet the therapeutic needs for conditions such as depression, Parkinson's disease, and other neuropsychiatric disorders. Recently, the design strategy for the treatment of neurological disease focused on the synthesis of compounds with multitargeting such as MAO and CAs, and MAO and AChE.58
Cholinesterases (ChEs) are essential enzymes involved in several critical fields, including neurobiology, toxicology, and pharmacology. Among these, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are two key groups that play significant roles in the functioning and health of humans and animals.59 AChE, also referred to as true cholinesterase, is predominantly found in the CNS.60 AChE is attached to the cell membranes of excitable tissues and plays a key role in nerve signal transmission. Its primary biological function is to catalyze the breakdown of the neurotransmitter acetylcholine into choline, a process essential for a cholinergic neuron to return to its resting state after being activated. AChE is also present in the membranes of red blood cells, where it is referred to as erythrocyte ChE. BChE, also known as pseudocholinesterase, plasma, or serum ChE, is mainly found in plasma, liver, and muscle tissues. Its exact biological function, however, remains unclear. Although BChE has a molecular structure similar to AChE, it differs in its substrate specificities. AChE primarily hydrolyzes acetyl esters, such as acetylcholine, whereas BChE predominantly hydrolyzes butyrylcholine, although there is some overlap in their substrate preferences.61 The design strategy of ChE inhibitors, which target both AChE and BChE, is crucial for developing therapies for neurodegenerative diseases (like Alzheimer's disease), antidotes for neurotoxins, and insecticides. The design of these inhibitors involves several key aspects, focusing on maximizing potency, selectivity, and bioavailability and minimizing side effects. In recent decades, the primary approach for drug discovery has shifted to designing highly selective drug molecules that target a single drug target.62 However, in many cases, the efficacy of single-target drugs often fails to meet people's expectations. Due to the diversity of the pathogenesis of neurodegenerative diseases, the molecules that regulate the activity of single-target proteins may not significantly change the progression of the disease.63 To improve efficacy, many may consider combining drugs. However, drug combinations can increase the risk of adverse reactions due to contraindications and potential drug–drug interactions.64 In 2005, Morphy et al. introduced a multi-target drug therapy strategy (MTDTS), offering a novel approach to drug discovery.65 As a result, the creation of synergistic multi-target inhibitors has emerged as a more promising strategy for treating diseases, particularly those caused by complex factors like AD.66 AChE is regarded as a primary target, and it can be combined with other targets to design and synthesize multi-target drug ligands (MTDLs), such as AChE with CAs, AChE with BACE-1, AChE with MAO, and AChE with PDE. Therefore, we reviewed studies from the past decade, summarizing the design concepts and structure–activity relationship (SAR) that may offer new pathways for the future development of small molecule candidate drugs used in the treatment of neurodegenerative disease. To the best of our knowledge, this is the first review which covers the chemistry and SAR of dual acting carbonic anhydrase inhibitors with MAO and ChE.
2. Dual acting carbonic anhydrase inhibitors
2.1. Dual acting carbonic anhydrase with monoamine oxidase inhibitors
Giovannuzzi et al. (2024) designed and synthesized various dual acting CAs–MAO-B inhibitors and evaluated their inhibitory activity against an SH-SY5Y human neuroblastoma cell model exposed to toxic Aβ peptides in vitro.67 All the design compounds were synthesized by a linking strategy, where two distinct scaffolds, known for their MAO-B and CAs inhibition, were connected by a linker. They conjugate benzopyrone scaffolds (chalcone or coumarin) as MAO-B inhibitors with a benzenesulfonamide motif (CAs inhibitor) at different positions, using a linker for the connection (Fig. 5).
 |
| | Fig. 5 Drug design strategy of dual acting inhibitors of CAs and MAO-B. | |
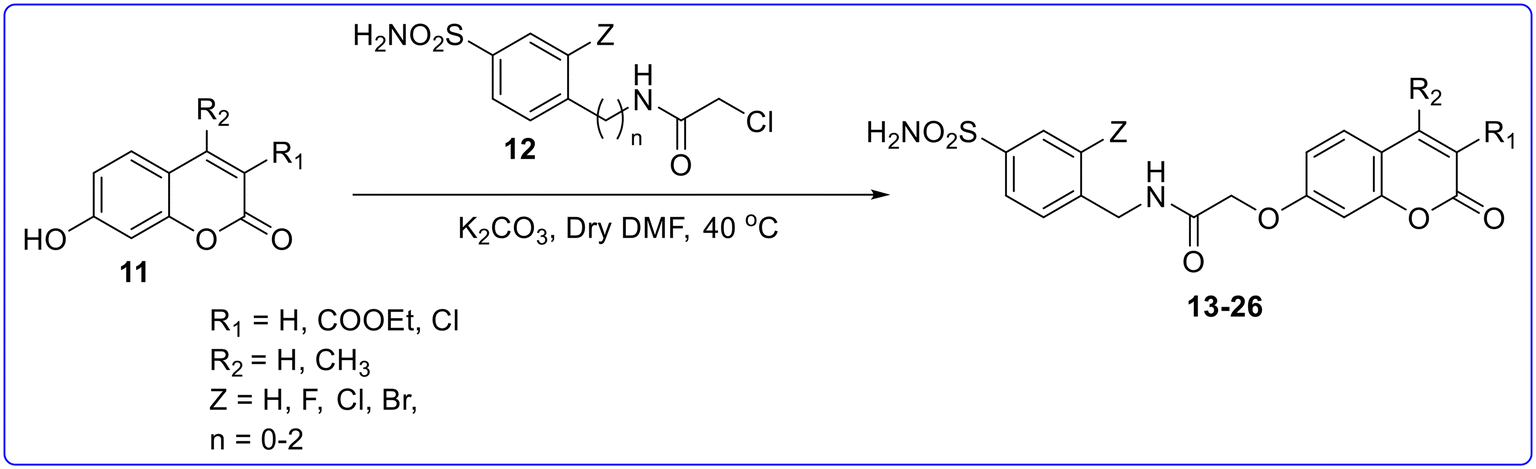
By using this design strategy, they synthesized different sets of coumarin–benzenesulphonamide and chromone–benzenesulphonamide derivatives. The first set of derivatives was synthesized by reacting 7-hydroxycoumarin with benzenesulfonamide bearing a 4-(2-chloroacetamide) moiety in the presence of K2CO3 as a base in anhydrous DMF at 40 °C to obtain hybrid compounds 13–26 (Fig. 6). Among the synthesized derivatives, compound 13 was found to be most potent molecule with a KI value ranging from 0.8 to 9.4 nM against CA II, VA and VII as compared to standard drugs acetazolamide (AAZ) with a KI value = 2.5 to 63 nM and methazolamide (MTZ) with a KI value = 2.1 to 65 nM. Compound 14, featuring a 3-fluoro-sulfanilamide scaffold connected to an unsubstituted coumarin fragment, demonstrated good inhibitory activity against CAs II, VA, VB, and VII, with a KI value ranging from 8.9 to 25.5 nM. The substitution of the fluorine atom with hydrogen or other halogen (compounds 18–20) reduces the CA inhibitory activity. A similar decrease in activity was observed with the elongation of the spacer between the amide linker and the CAI portion (compounds 21 and 22), or with the substitution of the coumarin scaffold with a 3-chloro-4-methyl pattern (compounds 23–26). All the synthesized compounds showed very less or no MAO inhibitory activity as compared to standard drugs clorgyline (CLO) with an IC50 value of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 610 nM for MAO-A and 47.6 nM for MAO-B and selegiline (SEL) with an IC50 value of 3.0 nM for MAO-A and 2501 nM for MAO-B (Fig. 7).
610 nM for MAO-A and 47.6 nM for MAO-B and selegiline (SEL) with an IC50 value of 3.0 nM for MAO-A and 2501 nM for MAO-B (Fig. 7).
 |
| | Fig. 6 Synthesis of compounds 13–26. | |
 |
| | Fig. 7 SARs of coumarin based compounds (13–26). | |
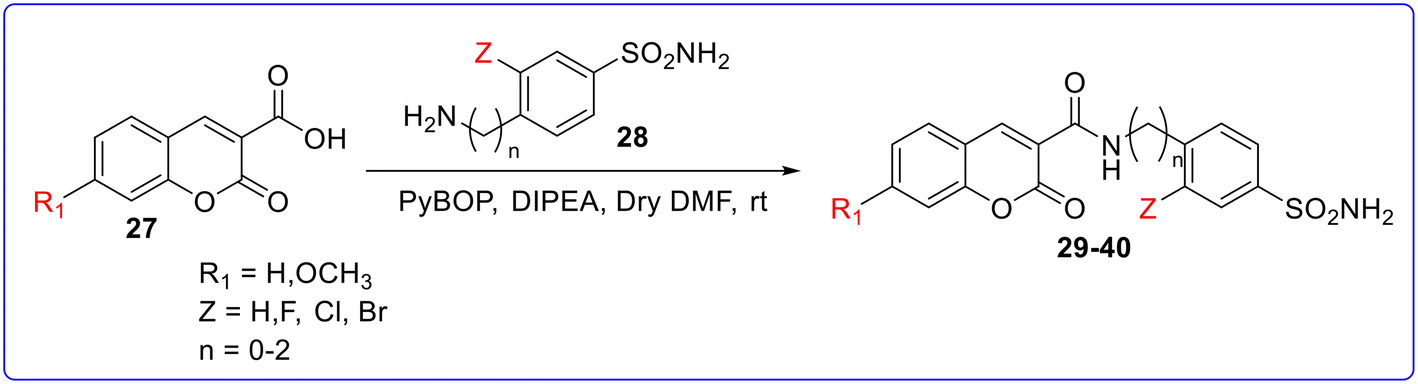
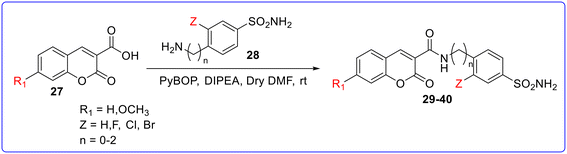
The second set of derivatives was synthesized by a coupling reaction between 3-carboxycoumarin and amine-bearing benzenesulfonamide in the presence of PyBOP as a coupling reagent and DIPEA as a base, in anhydrous DMF to give compounds 29–40 (Fig. 8). Compound 29 with a 3-fluoro-sulfanilamide scaffold connected to an unsubstituted coumarin fragment was the most effective inhibitor of CA II (KI value = 0.4 nM), CA-VA (KI value = 6.9 nM), and CA-VII (KI value = 11 nM) among the produced derivatives. The elongation of the spacer (ethylene group) between the amide linker and CAI portion (30) increases the CA inhibitory activity. The substitution of the fluorine atom with hydrogen or other halogen atom decreases the CA inhibitory activity with respect to standard drugs AZZ (KI value = 2.5 to 63 nM) and MTZ (KI value = 2.1 to 65 nM). Similarly, the elongation of the spacer between the amide linker and CAI portion (34) or substitution at the coumarin scaffold with a 7-OCH3 group (35–40) dramatically decreases the CA inhibitory activity. Compound 40 (KI value 7.0 nM) was found as the most active compound against CA-XII among the synthesized drugs but less than standard drugs AZZ (KI value = 5.7 nM) and MTZ (KI value = 3.4 nM). Some of the synthesized compounds showed increased MAO-B inhibitory activity as compared to the first set of derivatives. Among them, compound 36 was found as the most active compound against MAO-B with an IC50 value of 14.9 nM (Fig. 9).
 |
| | Fig. 8 Synthesis of compounds 29–40. | |
 |
| | Fig. 9 SARs of compounds 29–40. | |
The third set of derivatives (43–45) was synthesized by reacting 7-hydroxycoumarin with 4-sulfamoylbenzyl bromide in the presence of K2CO3 in dry DMF (Fig. 10). Among the synthesized derivatives, compound 43 was found as the most potent CAI against CA-II, VA, VB, VII and XII with KI values of 6.1, 37.1, 15.5, 8.2 and 37.8 nM, respectively. Substitution of chlorine at R1 and the methyl group at the R2 position (compound 46) showed good inhibitory activity against CAs with a KI value ranging from 5.6 to 66.1 nM. Substitution of the COOEt group at R1 and hydrogen at the R2 position decreases the CA inhibitory activity with respect to standard drugs AZZ (KI value = 2.5 to 63 nM) and MTZ (KI value = 2.1 to 65 nM). Notably, compounds 43 and 44 exhibited highly potent MAO-B inhibition, with IC50 values of 9.1 and 6.7 nM, respectively (Fig. 11).
 |
| | Fig. 10 Synthesis of compounds 43–45. | |
 |
| | Fig. 11 SARs of coumarin based compounds (43–45). | |
The fourth set of derivatives (48–53) were synthesized by converting the aldehyde group of compound 46 (chromone derivatives) into acyl bromide through a free radical reaction involving NBS and AIBN in CCl4, followed by a coupling reaction with benzenesulfonamide in the presence of K2CO3 as a base in CCl4 (Fig. 12). All the synthesized chromone derivatives showed less CA inhibition as compared to coumarin derivatives (second set). Among the synthesized derivatives, compound 48 was found as the most potent CAI with a KI value ranging from 8.3 to 58.4 nM. The elongation of the spacer between the amide linker and CAI portion was responsible for increasing CA inhibitory activity. The fluorine substituted compound 50 showed good inhibitory activity with a KI value of 6.4 nM. The substitution of fluorine with hydrogen or other halogen atom dramatically decreases the CA inhibitory activity. The unsubstituted compound 49 showed good MAO-B inhibitory activity with an IC50 value of 32.6 nM (Fig. 13).
 |
| | Fig. 12 Synthesis of compounds 48–53. | |
 |
| | Fig. 13 SARs of chromone based compounds 48–53. | |
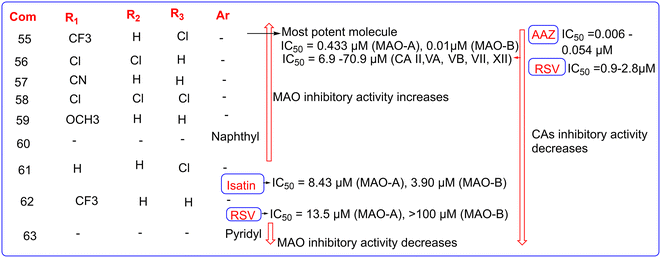
Carradori et al. (2022) synthesized various resveratrol (RSV) analogues (Fig. 14) as dual inhibitors of MAO-B and CAs and evaluated their inhibitory activity by a stopped-flow technique or the esterase activity assay.68–70 All the derivatives were synthesized by a previously reported method.71–74 Among the synthesized derivatives, compound 55 was found as the most potent molecule with IC50 values of 0.433 μM (MAO-A) and 0.01 μM (MAO-B) with respect to the parent compound RSV with IC50 values of 13.5 μM (MAO-A) and >100 μM (MAO-B) and standard drug isatin with IC50 values of 8.43 μM (MAO-A) and 3.90 μM (MAO-B) (Fig. 15). Substitution of the CF3 group at R1 and the chlorine atom at the R3 position is responsible for the inhibitory activity of compound 55. It also showed good inhibitory activity against CAs with IC50 values of 15.6 μM (CA II), 70.9 μM (CA VA), 36.2 μM (CA VB), 0.7 μM (CA VII), and 6.9 μM (CA XII) but less than those of parent molecule RSV and standard drug AAZ with IC50 values ranging from 0.9 to 2.8 μM and 0.006 to 0.054 μM, respectively. The substitution of CF3 with other electron-withdrawing groups (EWGs) like –CN or –Cl at the R1 position, while keeping a halogen (–Cl) or hydrogen at the R2 or R3 positions (compounds 56–58), demonstrated good inhibitory activity against MAO, but resulted in a decrease in CA inhibitory activity. The substitution of CF3 with hydrogen at the R1 position (61) retained the MAO-A and MAO-B inhibitory activity, whereas substituting –Cl with hydrogen at the R3 position (compound 62) reduced the MAO-B inhibitory activity compared to the standard drug isatin. Similarly, substitution of the naphthyl group at the Ar position (60) retained the MAO-B inhibitory activity while the substitution of the pyridyl group at the Ar position (63) decreases the MAO-B inhibitory activity. Substitution of electron withdrawing CF3 with an electron donating group (EDG) such as –OCH3 at the R1 position and hydrogen at R2 and R3 positions (59) retained the MAO-B inhibitory activity. All the synthesized RSV derivatives showed less CA inhibitory activity with respect to parent molecule RSV and standard drug AAZ. The molecular docking study of compound 55 for MAO-A and MAO-B was conducted using Schrödinger Life-Sciences Suite 2021-4 (Maestro v13), with PDB codes of 2Z5X and 2V5Z, respectively. Interestingly, the docking score of compound 55 was found to be similar to the standard drug safinamide (10.336 for MAO-A, 10.375 for MAO-B). The docking study of compound 55 for CAs was performed in GOLD 5.1 program, using the PLP scoring function. The crystal structures of CA I (PDB code 1AZM), CA II (PDB code 4E3H), CA VII (PDB code 3MDZ), CA IX (PDB code 5FL4), and CA XII (PDB code 1JCZ) were obtained from the Protein Data Bank, while previously developed homology models were used for CA VA and CA VB.75,76 The docking study of compound 55 revealed specific and significant interactions within the active sites of both targets.
 |
| | Fig. 14 Structure and synthesized derivatives of RSV. | |
 |
| | Fig. 15 SARs of RSV derivatives. | |
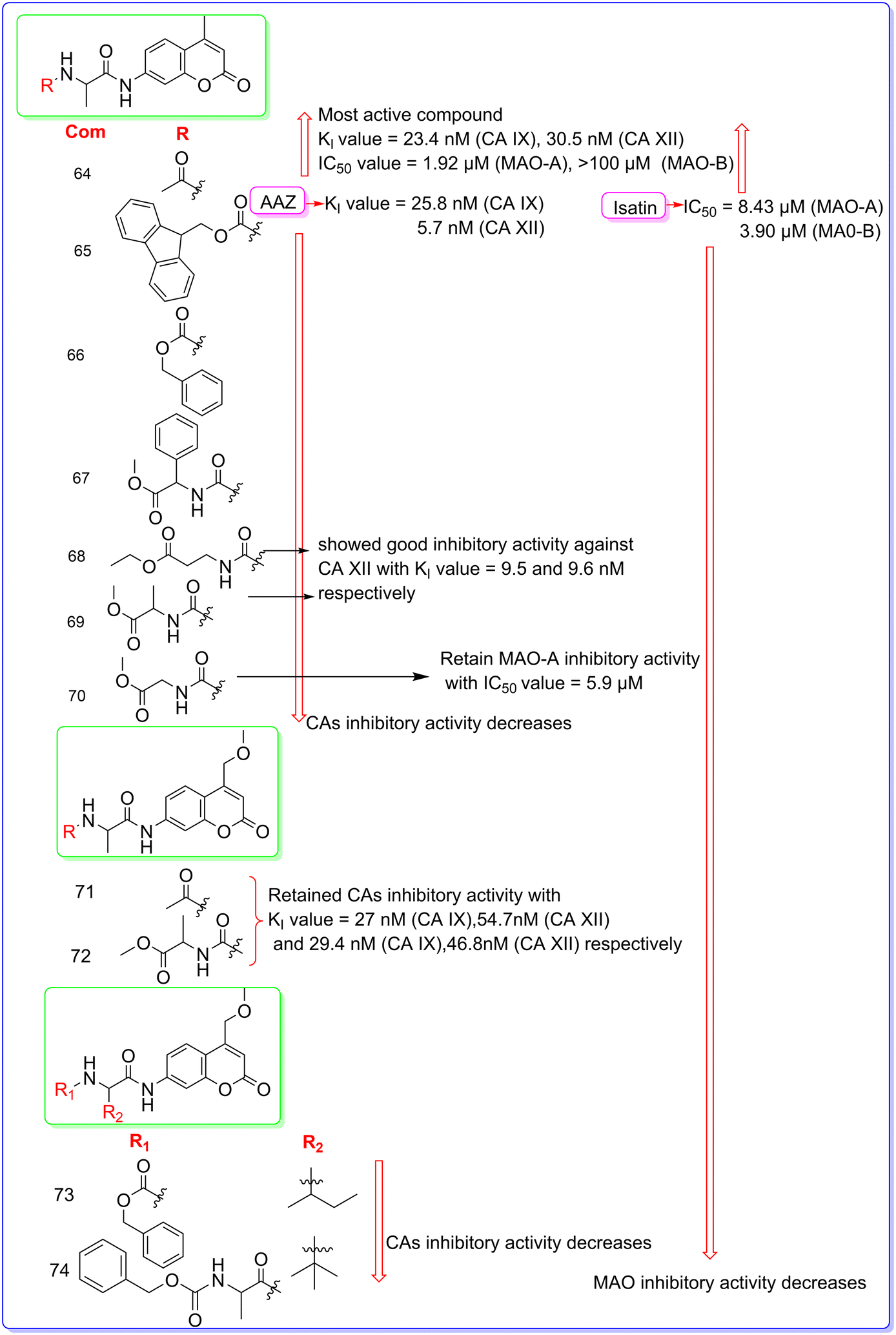
Agamennone et al. (2022) synthesized various coumarin based dual inhibitors of CAs and MAO and evaluated their inhibitory activity against MAO-A, MAO-B, CA I, CA II, CA IX and CA XII.77 A stopped-flow instrument from Applied Photophysics was employed to measure the CO2 hydration activity catalyzed by CA.78 Phenol red, at a concentration of 0.2 mM, served as the indicator, with reactions monitored at a wavelength of 557 nm. A buffer of 20 mM HEPES (pH 7.4) containing 20 mM Na2SO4 was used to maintain constant ionic strength. Amino acyl methyl coumarin (AMC) and amino acyl methoxymethyl coumarin (AMMC) derivatives were synthesized by applying solution phase synthesis (Fig. 16 and 17). Due to the low nucleophilic reactivity of the aromatic amino group, the acylation reactions of coumarins with N-protected amino acids were preferably carried out using the mixed anhydride method. Amino acyl coumarins 65–74 were synthesized by reacting the respective N-protected amino acid with isobutyl chloroformate (IBCF) and triethylamine (TEA) at −10 °C in tetrahydrofuran (THF) under an inert atmosphere. Afterwards, the appropriate coumarin nucleus was added. Once the ice bath was removed, the reaction proceeded for 20–24 hours, yielding the expected conjugate as brown-red slurry. The precipitate was processed, and the compounds were purified through chromatography and recrystallization, resulting in the expected products with satisfactory yields. All the synthesized compounds showed very less or no CA inhibitory activity against CA I and CA II. Among the synthesized derivatives, compound 64 was found as the most active compound with KI values of 23.4 nM (CA IX) and 30.5 nM (CA XII) as compared to standard drug AAZ having KI values of 25.8 nM (CA IX) and 5.7 nM for CA XII. It also demonstrated good MAO inhibition activity with IC50 values of 1.92 μM (MAO-A) and >100 μM (MAO-B) with respect to standard drug isatin having IC50 values of 8.43 μM (MAO-A) and 3.90 μM for MAO-B. The substitution of methyl ketone at position R is responsible for the good inhibitory activity of compound 64. Substitution of methyl ketone with other groups (65–70) dramatically decreased the potency of the molecule except compounds 68 and 69 which showed good inhibitory activity against CA XII with KI values of 9.5 and 9.6 nM, respectively. Substitution of methyl ketone and methyl acetylalaninate at position R of AMMC derivatives (71 and 72) retained the CA inhibitory activity. Further substitution of butane and isobutane at the R2 position decreased the CA inhibitory activity. Substitution of methyl ketone with other groups (65–74) decreased the MAO-A and MAO-B inhibitory activity (Fig. 18). The molecular docking study was performed on Schrödinger Life-Sciences Suite 2021-4 (ref. 79) by using PDB ID 2Z5X (MAO-A), 2V5Z (MAO-B), 6F3B (CA I), 5BNL (CA II), 6G9U (CA IX) and 5LL5 (CA XII).
 |
| | Fig. 16 Synthesis of coumarin derivatives (64–72); reagents and conditions: (a) Z-Cl, 1 N NaOH, 0 °C, 2 h, then r.t., 16 h; (b) Fmoc-OSu, TEA, H2O/THF (2:1), r.t., 4 h; (c) IBCF, TEA, dry THF, −10 °C under N2, 1 h, then r.t., 24 h. | |
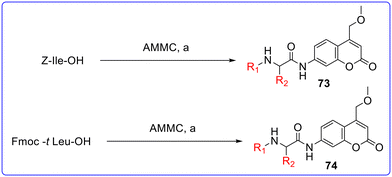 |
| | Fig. 17 Synthesis of coumarin derivatives (73 and 74). Reagents and conditions: (a) IBCF, TEA, dry THF, −10 °C under N2, 1 h, then r.t., 20 h. | |
 |
| | Fig. 18 SARs of coumarin derivatives. | |
2.2. Dual acting carbonic anhydrase with cholinesterase (ChE) inhibitors
Several pyrazole carbamide derivatives are designed and synthesized as selective inhibitors of carbonic anhydrase and cholinesterase (Durgun et al., 2024), and their inhibitory action against acetylcholinesterase (AChE), butyrylcholinesterase (BChE), CA I, and CA II is assessed in vitro.80 All the pyrazole carboxamide derivatives were synthesized by the reaction of diazotization of appropriate sulphonamide derivative 75 to give diazonium salt 76, followed by the reaction with malononitrile in sodium acetate in methanol at 0–10 °C to give 77. On reacting compound 77 with compound 78 in alcohol at 60 °C, pyrazole carboxamide derivatives (79–90) were obtained in desirable yields of 62–85% (Fig. 19). All the synthesized compounds showed good inhibitory activity against ChEs having KI values ranging from 6.60–14.15 nM (AChE) and 54.87–137.20 nM (BChE) and CAs having KI values ranging from 10.69–70.87 nM (CA I) and 20.01–56.6 nM (CA II) as compared to standard drugs tacrine (KI value = 112 nM for AChE, 172 nM for BChE) and AZZ (KI value = 412 nM for CA I, 98.2 nM for CA II). Among the synthesized derivatives, compound 79 was found as the most potent molecule against ChEs with KI values of 6.60 nM for AChE and 68.9 nM for BChE. The substitution of 4,6-dimethylpyrimidin-2-amine at the R1 position and 1,2-dichlorobenzene at the R2 position is responsible for the most potent inhibitory activity of compound 79 against ChEs. Compound 87 was found as the most active compound against CAs with KI values of 10.6 nM (CA I) and 20.01 nM (CA II) as compared standard drug AAZ having KI values of 412 nM for CA I and 98.2 nM for CA II. The substitution of guanidine at the R1 position and p-methyl benzene at the R2 position is responsible for the good inhibitory activity against CAs (Fig. 20). The docking study of the most active compounds (79, 83, 84 and 80) was performed by using Small-Molecule Drug Discovery Suite 2023-2 with PDB ID 7XN1 (AChE) and 4BDS (BChE), and 1AZM (CA I) and 3HS4 (CA II). The docking methodology's reliability was confirmed by the low RMSD values (0.26, 0.12, 0.22, and 0.99 Å for 7XN1, 4BDS, 1AZM, and 3HS4, respectively) and the ability of the docking poses of the co-crystallized ligands to accurately replicate all key interactions. Compounds 79, 83, 84 and 80 formed primary interactions via hydrogen bonding and pi–pi stacking. These inhibitors exhibited docking scores of −9.686, −5.306, −2.958, and −6.376 kcal mol−1 with 7XN1, 4BDS, 1AZM, and 3HS4, respectively. The result of in silico and in vitro study supported to each other (Fig. 21).
 |
| | Fig. 19 Synthesis of pyrazole carboxamide derivatives. | |
 |
| | Fig. 20 SARs of pyrazole carboxamide derivatives. | |
 |
| | Fig. 21 Drug design strategy of dual acting CA activators and AChE inhibitors. | |
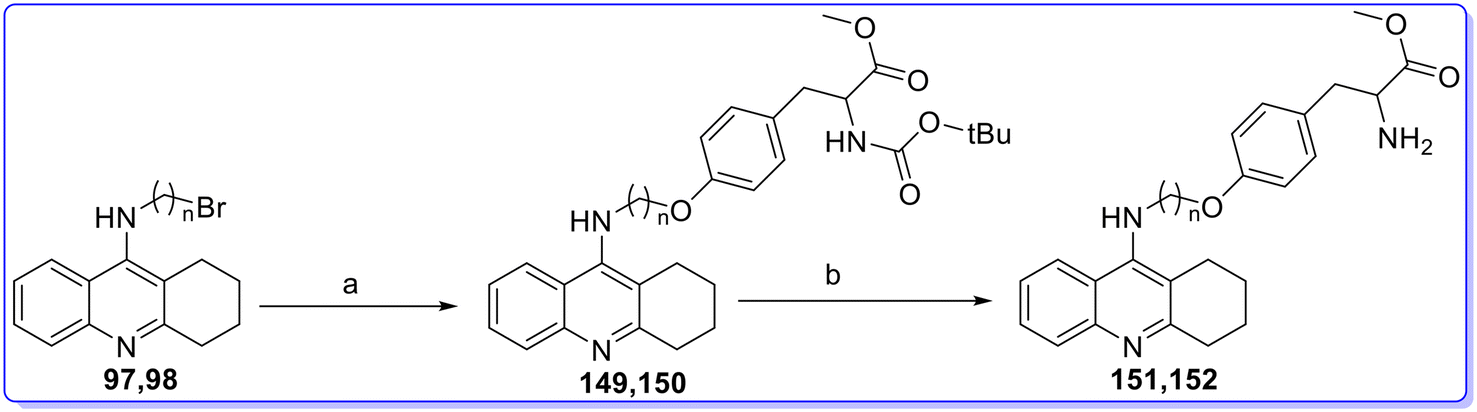
Nocentini et al. (2024) designed and synthesized various dual acting CA activators and AChE inhibitors, and evaluated their biological activity in vitro.81 The intermediate 9-chloro-1,2,3,4-tetrahydroacridine (92) was synthesized through the condensation of anthranilic acid (91) with cyclohexanone in the presence of POCl3 at 115 °C. Subsequently, intermediate 92 was reacted with the appropriate 1,ω-diaminoalkane in the presence of potassium iodide in phenol at 180 °C, yielding the aminoalkyl TAC intermediates 93–96 (Fig. 22). The ω-bromoalkyl intermediates 97 and 98 were prepared via two-step synthesis. First, intermediate 92 was reacted with the appropriate amino alcohol in the presence of KI in phenol at 180 °C. This was followed by the Appel reaction using CBr4 and PPh3 in DMF at room temperature (Fig. 22). Intermediates 93–96 were coupled with commercially available protected L-amino acids using PYBOP as the coupling agent and DIPEA in DMF, resulting in the formation of amide compounds 99–123. Specifically, compounds 99–112 and 121–123 were synthesized using N,N′-diBoc-L-histidine and N-Boc-N′-Cbz-L-lysine, respectively, to prevent undesired side reactions. The N-Boc groups of compounds 99–123 were subsequently removed by treatment with 1.25 M methanolic HCl at room temperature, yielding the corresponding amine compounds 124–138 (Fig. 23). Intermediates 97 and 98 underwent an SN2 reaction with methyl ester L-amino acids in the presence of K2CO3 and KI, resulting in the formation of amine-linked hybrids 139–146 (Fig. 24). Similarly, the TAC hybrids 147 and 148 were synthesized using histamine dihydrochloride (Fig. 25). Finally, the tyrosine derivatives 149 and 150 were synthesized by reacting intermediates 97 and 98 with N-Boc-L-tyrosine methyl ester. The N-Boc groups of compounds 149 and 150 were subsequently removed by treatment with 1.25 M HCl in methanol at room temperature, yielding hybrids 151 and 152. All the synthesized compounds showed less CA activator activity against CA I and CA II as compared to standard drug L-phenylalanine having KA values of 70 nM and 13 nM, respectively. Among the synthesized derivatives, compound 148 was found as the most active compound against CA I with a KA value of 236 nM but less than standard drug L-phenylalanine. Similarly, compound 126 was found as the most active compound against CA II having a KA value of 65.1 nM but less than standard drug L-phenylalanine. Among the synthesised derivatives, compounds 135 and 124 were found as the most potent CA activators against CA IV with KA values of 541 nM and 619 nM, respectively, as compared to standard drug L-Phe having a KA value of 36000 nM. Similarly, compounds 135 and 127 were found as the most potent CA activators against CA VB with KA values of 353 nM and 544 nM, respectively, as compared to standard drug L-phenylalanine having a KA value of 10000 nM. Among the synthesized derivatives, compounds 127 and 126 were found as the most potent CA activators against CA VII with KA values of 82.8 nM and 96.4 nM, respectively, as compared to standard drug L-phenylalanine having a KA value of 10000 nM. All derivatives, except for the histamine-derived compound 148, demonstrate selective inhibition of BChE over AChE, resembling the lead compound tacrine. Among the synthesized compounds, compounds 148 and 135 showed the most potent inhibitory activity against AChE with IC50 values of 2.2 μM and 2.4 μM, respectively, as compared to standard drug tacrine having an IC50 value of 33.9 μM. Similarly, compounds 151 and 100 showed the most potent inhibitory activity against BChE with IC50 values of 0.048 μM and 0.064 μM, respectively, as compared to standard drug tacrine having an IC50 value of 3.9 μM (Fig. 27). Docking study was performed in the Maestro Schrödinger suite against hCA II (PDB: 2ABE),82 hCA VII (PDB: 6H38),83 hAChE (PDB: 4BDT)84 and hBChE (PDB: 6I0B) (Fig. 26).85
 |
| | Fig. 22 Synthesis of intermediate compounds (91–98). Reagents and conditions: (a) cyclohexanone, POCl3, 0 to 115 °C, (b) 1-ω-diaminoalkane, KI, phenol, 180 °C, (c) ω-aminoalkan-1-ol, KI, phenol, 180 °C, and (d) CBr4, PPh3, DMF, rt. | |
 |
| | Fig. 23 Synthesis of dual acting CA activators and AChE inhibitors (124–138). Reagents and conditions: (a) protected L-amino acid (N,N′-diBoc-L-histidine, N-Boc-L-phenylalanine, N-Boc-L-tryptophan, N-Boc-N′-Cbz-L-lysine), PYBOP, DIPEA, DMF, rt and (b) 1.25 M methanolic HCl, 0 °C to rt. | |
 |
| | Fig. 24 Synthesis of dual acting CA activators and AChE inhibitors (139–146). Reagents and conditions: (a) L-amino acid methyl ester (L-histidine, L-phenylalanine, L-tryptophan, and N′-Cbz-L-lysine), K2CO3, KI, DMF, rt. | |
 |
| | Fig. 25 Synthesis of dual acting CA activators and AChE inhibitors (147 and 148). Reagents and conditions: (a) histamine dihydrochloride, K2CO3, KI, DMF, rt. | |
 |
| | Fig. 26 Synthesis of dual acting CA activators and AChE inhibitors (149, 152). Reagents and conditions: (a) N-Boc-L-tyrosine methyl ester, K2CO3, KI, DMF, rt and (b) 1.25 M methanolic HCl, MeOH, 0 °C to rt. | |
 |
| | Fig. 27 SARs of dual acting CA activators and AChE inhibitors. | |
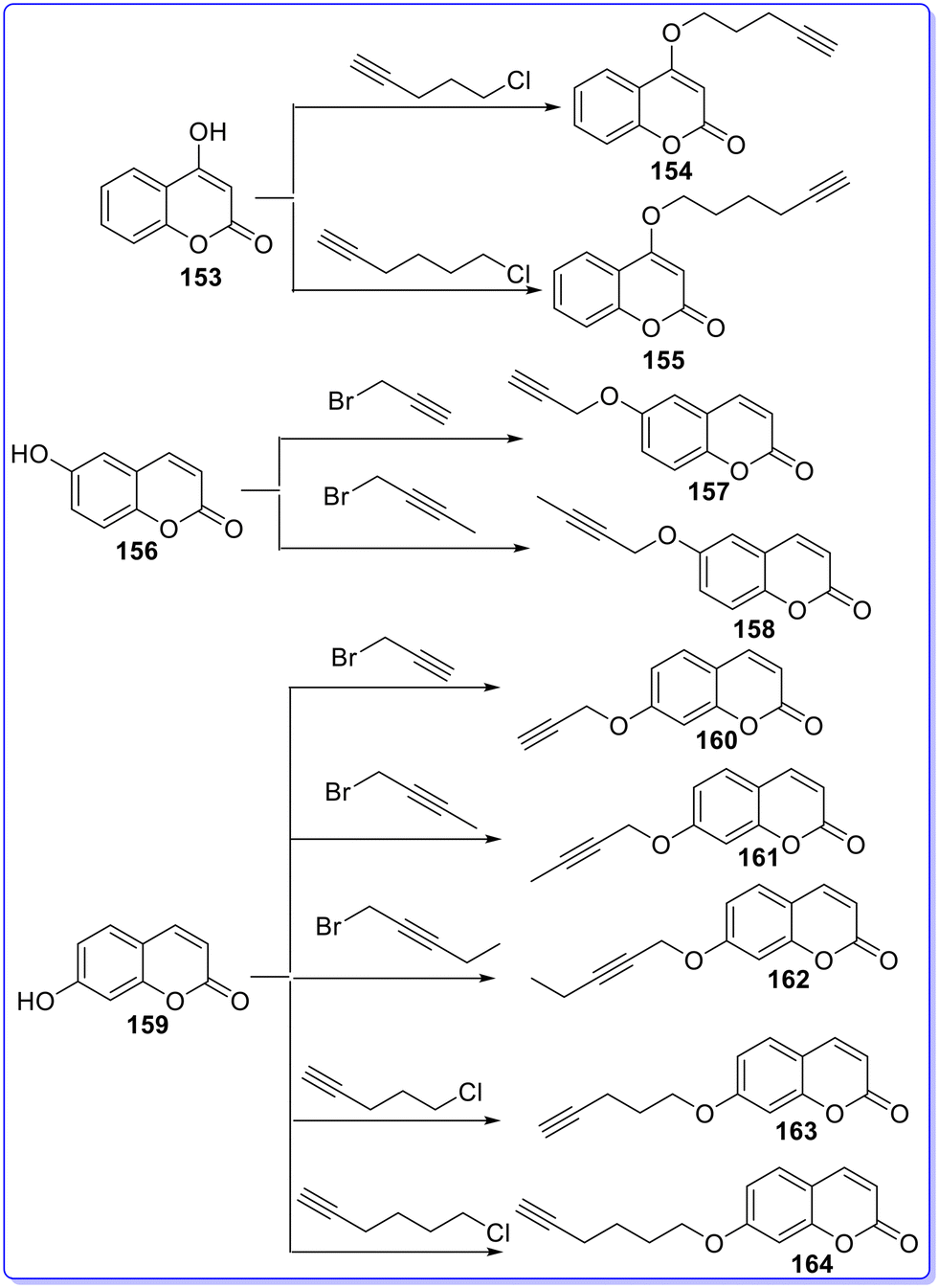
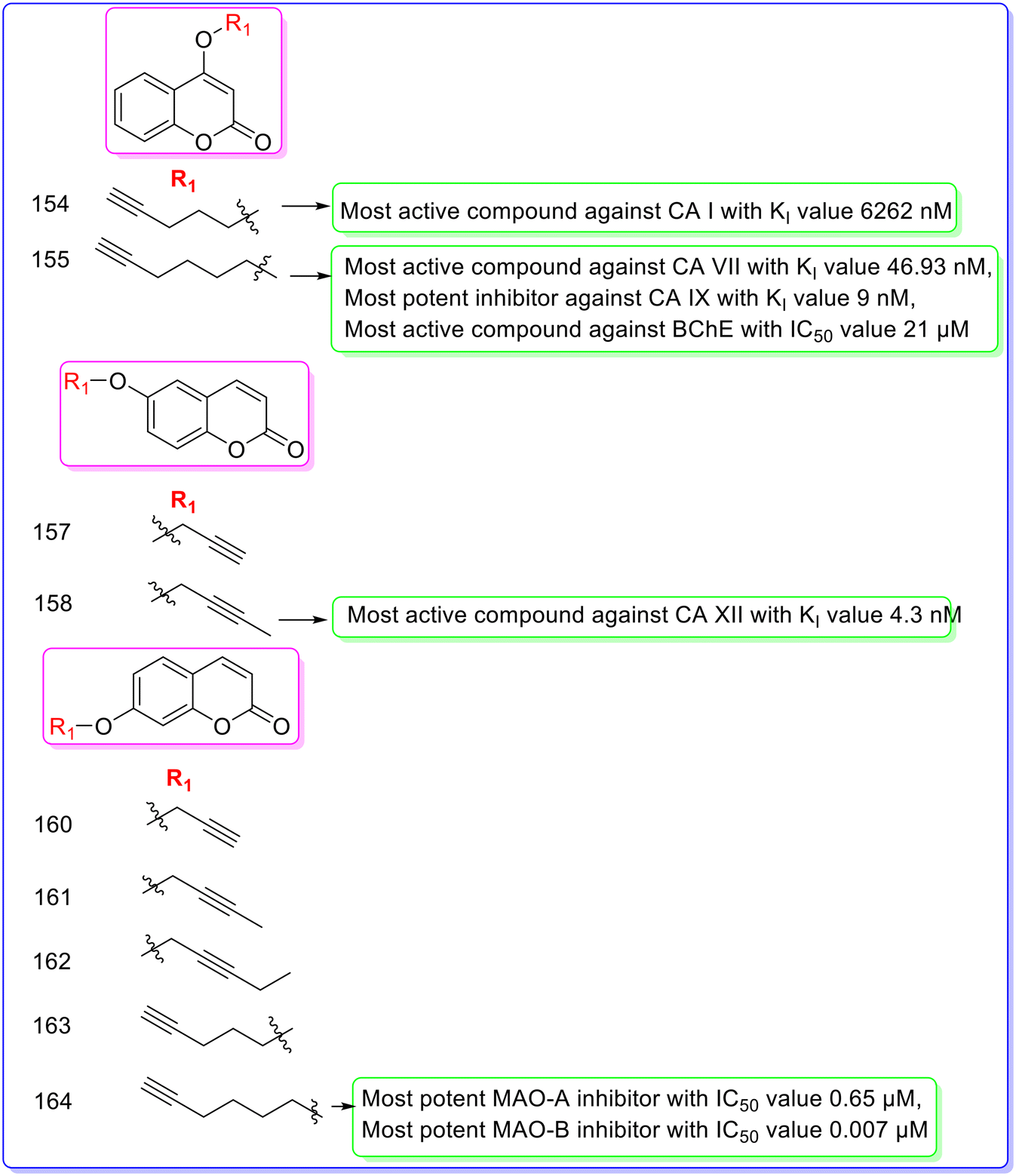
Berrino et al. (2023) designed and synthesized various alkyl substituted coumarins and evaluated their biological activity in vitro against CAs, MAO-A, MAO-B, AChE and BChE.86 The target compounds 154 and 155 were synthesized by reacting starting material 153 with 5-chloropent-1-yne and 6-chlorohex-1-yne, respectively. The compound 156 was reacted with 3-bromopropane-1-yne and 1-bromobut-2-yne in dry DMF at 150 °C to give compounds 157 and 158, respectively. Compounds 160–164 were obtained by a similar reaction between compound 159 and 3-bromoprop-1-yne, 1-bromobut-2-yne, 1-bromopent-2-yne, 5-chloropent-1-yne, and 6-chlorohex-1-yne in dry DMF at 150 °C (Fig. 28). All the synthesized derivatives showed less CA I, CA II, CA VII inhibitory activity as compared to standard drug AZZ having KI values of 250 nM, 12.1 nM, and 2.5 nM, respectively. Among the synthesized derivatives, compound 154 was found as the most active CA I inhibitor with a KI value of 6262 nM, but less active than standard drug AZZ. All the synthesized compounds showed less CA II inhibitory activity with KI values of >10000 nM as compared to standard drug AZZ. Among the synthesized derivatives, compound 155 was found as the most active CA VII inhibitor with a KI value of 46.93 nM, but less active than standard drug AZZ. Compound 155 also showed the most potent inhibitory activity against CA IX with a KI value of 9 nM as compared to standard drug AZZ having a KI value of 25.8 nM. The substitution of the hex-1-yne group at the R1 position is responsible for the most potent inhibitory activity of compound 155. Among the synthesized derivatives, compound 158 showed the most potent inhibitory activity against CA XII with a KI value of 4.3 nM as compared to standard drug AZZ having a KI value of 5.7 nM. Substitution of the but-2-yne group at the R1 position is responsible for the most potent inhibitory activity of compound 158. All the synthesized derivatives showed less AChE and BChE inhibitory activity as compared to standard drug galantamine having IC50 values of 1.29 μM (AChE) and 5.4 μM (BChE). All the synthesized compounds showed less AChE inhibitory activity with IC50 values of >100 μM as compared to standard drug galantamine. Among the synthesized derivatives, compound 155 was found as the most active compound with an IC50 value of 21 μM, but less active than the standard drug. Among the synthesized derivatives, compound 164 was found as the most potent inhibitor against MAO-A and MAO-B with IC50 values of 0.65 μM and 0.007 μM, respectively, as compared to standard drug curcumin having IC50 values of 5.54 μM for MAO-A and 4 μM for MAO-B. The substitution of the hex-1-yne group at the R1 position is responsible for the most potent inhibitory activity of compound 164 (Fig. 29).
 |
| | Fig. 28 Synthesis of alkyl substituted coumarins. | |
 |
| | Fig. 29 SARs of alkyl substituted coumarins. | |
Kurban et al. (2024) designed and synthesized a new class of sulfonamide derivatives and evaluated their inhibitory activity in vitro against CA I, CA II, AChE and BChE.87 All the derivatives were synthesized by reacting compound 165 with phenolic aldehyde in TEA and DCM under reflux conditions to give compounds 166–170, which were reacted with compound 171 in ethanol under reflux conditions to give target compounds 172–176. Among the synthesized derivatives, compound 172 was found as the most potent CAI and CA II inhibitor with KI values of 8.98 nM and 7.55 nM, respectively, as compared to standard drug AZZ having KI values of 250 nM for CA I and 12 nM for CA II. Among the synthesized derivatives, compound 176 was found as the least active CAI and CA II inhibitor with KI values of 14.56 nM and 13.85 nM, respectively. All the synthesized derivatives showed more AChE and BChE inhibitory activity as compared to standard drug neostigmine having KI values of 55.5 nM for AChE and 33.3 nM for BChE. Among the synthesized derivatives, compound 174 was found as the most potent inhibitor against AChE and BChE with KI values of 12.90 and 6.21 nM, respectively, as compared to standard drug neostigmine. Among the synthesized derivatives, compound 172 showed the least potent inhibitory activity against AChE and BChE with KI values of 16.38 and 14.52 nM, respectively. The docking study of the synthesized compound was performed by using PDB ID 1AZM for CA I, 4Q6E for CA II, 4EY7 for AChE and 4BDS for BChE. The docking scores of the most active compound 172 against CAs were found to be −5.209 (CA I) and −5.863 (CA II), and those of 174 against ChEs were found to be −10.500 (AChE) and −6.825 (BChE) (Fig. 30 and 31).
 |
| | Fig. 30 Synthesis of a new class of sulfonamide derivatives. | |
 |
| | Fig. 31 SARs of a new class of sulfonamide derivatives. | |
Conclusion
Multi-target drug design (MTDD) offers a transformative approach to drug discovery, particularly for the treatment of complex and multifactorial diseases such as neurodegenerative disorders. By simultaneously modulating multiple biological targets, MTDD addresses the limitations of traditional single-target therapies, offering potential for improved therapeutic efficacy, reduced drug resistance, and more comprehensive disease management. In this article, we provided a summary of dual-acting drugs used in the treatment of neurodegenerative diseases over the past five years. In this article, we emphasize the design, synthesis, structure–activity relationships (SARs), docking study and biological activity of various dual-acting inhibitors targeting CAs, MAO, and ChE involved in the treatment of neurodegenerative diseases. The review also discusses key SAR studies that have guided the optimization of dual inhibitors, focusing on achieving selectivity and potency while minimizing off-target effects. SAR analysis of dual-acting drugs has proven essential in the development of effective therapeutic agents for neurodegenerative diseases. By understanding how structural modifications influence the biological activity and selectivity of dual inhibitors targeting multiple pathways, researchers can optimize drug candidates for enhanced efficacy and reduced side effects. The insights gained from SAR studies facilitate the fine-tuning of molecular properties, such as potency, selectivity, and pharmacokinetics, thereby addressing the complex nature of neurodegenerative conditions. As the landscape of drug discovery continues to evolve, integrating SAR findings with advanced computational methods and high-throughput screening will further enhance the design of dual-acting agents. While challenges such as maintaining selectivity for specific isoforms and managing potential off-target effects remain, ongoing research in SAR will be pivotal in overcoming these hurdles. Ultimately, the strategic application of SAR in the development of dual-acting drugs holds great promise for improving treatment outcomes for patients with neurodegenerative diseases, paving the way for more effective and personalized therapeutic strategies.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant no. (DGSSR-2024-01-02246).
References
- H. Zheng, M. Fridkin and M. Youdim, Pharmaceuticals, 2014, 7, 113–135 CrossRef PubMed.
- E. Proschak, H. Stark and D. Merk, J. Med. Chem., 2019, 62, 420–444 CrossRef CAS PubMed.
- B. Mathew, D. G. T. Parambi, G. E. Mathew, Md. S. Uddin, S. T. Inasu, H. Kim, A. Marathakam, M. K. Unnikrishnan and S. Carradori, Arch. Pharm., 2019, 352(11), e1900177 CrossRef PubMed.
- G. E. Konecny, M. D. Pegram, N. Venkatesan, R. Finn, G. Yang, M. Rahmeh, M. Untch, D. W. Rusnak, G. Spehar, R. J. Mullin, B. R. Keith, T. M. Gilmer, M. Berger, K. C. Podratz and D. J. Slamon, Cancer Res., 2006, 66, 1630–1639 CrossRef CAS PubMed.
- K. Li, L. A. Schurig-Briccio, X. Feng, A. Upadhyay, V. Pujari, B. Lechartier, F. L. Fontes, H. Yang, G. Rao, W. Zhu, A. Gulati, J. H. No, G. Cintra, S. Bogue, Y.-L. Liu, K. Molohon, P. Orlean, D. A. Mitchell, L. Freitas-Junior, F. Ren, H. Sun, T. Jiang, Y. Li, R.-T. Guo, S. T. Cole, R. B. Gennis, D. C. Crick and E. Oldfield, J. Med. Chem., 2014, 57, 3126–3139 CrossRef CAS PubMed.
- O. Benek, J. Korabecny and O. Soukup, Trends Pharmacol. Sci., 2020, 41, 434–445 CrossRef CAS PubMed.
- W. Löscher, Front. Pharmacol., 2021, 27(12), 730257 CrossRef PubMed.
- X. H. Makhoba, C. Viegas Jr., R. A. Mosa, F. P. Viegas and O. J. Pooe, Drug Des., Dev. Ther., 2020, 14, 3235–3249 CrossRef CAS PubMed.
- M. S. Brose, C. M. Nutting, B. Jarzab, R. Elisei, S. Siena, L. Bastholt, C. de la Fouchardiere, F. Pacini, R. Paschke, Y. K. Shong, S. I. Sherman, J. W. A. Smit, J. Chung, C. Kappeler, C. Peña, I. Molnár and M. J. Schlumberger, Lancet, 2014, 384, 319–328 CrossRef CAS PubMed.
- A.-L. Cheng, Y.-K. Kang, Z. Chen, C.-J. Tsao, S. Qin, J. S. Kim, R. Luo, J. Feng, S. Ye, T.-S. Yang, J. Xu, Y. Sun, H. Liang, J. Liu, J. Wang, W. Y. Tak, H. Pan, K. Burock, J. Zou, D. Voliotis and Z. Guan, Lancet Oncol., 2009, 10, 25–34 CrossRef CAS PubMed.
- S. Wilhelm, C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz, R. Simantov and S. Kelley, Nat. Rev. Drug Discovery, 2006, 5, 835–844 CrossRef CAS PubMed.
- S. M. Wilhelm, C. Carter, L. Tang, D. Wilkie, A. McNabola, H. Rong, C. Chen, X. Zhang, P. Vincent, M. McHugh, Y. Cao, J. Shujath, S. Gawlak, D. Eveleigh, B. Rowley, L. Liu, L. Adnane, M. Lynch, D. Auclair, I. Taylor, R. Gedrich, A. Voznesensky, B. Riedl, L. E. Post, G. Bollag and P. A. Trail, Cancer Res., 2004, 64, 7099–7109 CrossRef CAS PubMed.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed.
- Y. S. Chang, J. Adnane, P. A. Trail, J. Levy, A. Henderson, D. Xue, E. Bortolon, M. Ichetovkin, C. Chen, A. McNabola, D. Wilkie, C. A. Carter, I. C. A. Taylor, M. Lynch and S. Wilhelm, Cancer Chemother. Pharmacol., 2007, 59, 561–574 CrossRef CAS PubMed.
- H. Geerts and L. Kennis, Future Med. Chem., 2014, 6, 1757–1769 CrossRef CAS PubMed.
- M. Agamennone, M. Fantacuzzi, S. Carradori, A. Petzer, J. P. Petzer, A. Angeli, C. T. Supuran and G. Luisi, Molecules, 2022, 27, 7884 CrossRef CAS PubMed.
- G. E. Mathew, J. M. Oh, K. Mohan, M. V. Kumudhavalli, S. Jayanthi, H. Kim and B. Mathew, Process Biochem., 2020, 99, 246–253 CrossRef CAS.
- B. Mathew, S. C. Baek, D. G. Thomas Parambi, J. P. Lee, G. E. Mathew, S. Jayanthi, D. Vinod, C. Rapheal, V. Devikrishna, S. S. Kondarath, Md. S. Uddin and H. Kim, Arch. Pharm., 2019, 352(4), e1800309 CrossRef PubMed.
- R. Sasidharan, B. H. Eom, J. H. Heo, J. E. Park, M. A. Abdelgawad, A. Musa, N. Gambacorta, O. Nicolotti, S. L. Manju, B. Mathew and H. Kim, J. Enzyme Inhib. Med. Chem., 2021, 36, 188–197 CrossRef CAS PubMed.
- J. M. Oh, T. M. Rangarajan, R. Chaudhary, R. P. Singh, M. Singh, R. P. Singh, A. R. Tondo, N. Gambacorta, O. Nicolotti, B. Mathew and H. Kim, Molecules, 2020, 25, 2356 CrossRef CAS PubMed.
- A. Venkidath, J. M. Oh, S. Dev, E. Amin, S. P. Rasheed, A. Vengamthodi, N. Gambacorta, A. Khames, M. A. Abdelgawad, G. George, O. Nicolotti, H. Kim and B. Mathew, Molecules, 2021, 26, 6004 CrossRef CAS PubMed.
- V. P. Vishal, J. M. Oh, A. Khames, M. A. Abdelgawad, A. S. Nair, L. R. Nath, N. Gambacorta, F. Ciriaco, O. Nicolotti, H. Kim and B. Mathew, Pharmaceutics, 2021, 13, 850 CrossRef PubMed.
- G. S. Jeong, S. Kaipakasseri, S. R. Lee, N. Marraiki, G. E. Batiha, S. Dev, A. Palakkathondi, F. S. Kavully, N. Gambacorta, O. Nicolotti, B. Mathew and H. Kim, ChemMedChem, 2020, 15, 2257–2263 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146–189 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2010, 20, 3467–3474 CrossRef CAS PubMed.
-
Carbonic Anhydrase, ed. C. T. Supuran, A. Scozzafava and J. Conway, CRC Press, 2004 Search PubMed.
- A. Bertucci, A. Moya, S. Tambutté, D. Allemand, C. T. Supuran and D. Zoccola, Bioorg. Med. Chem., 2013, 21, 1437–1450 CrossRef CAS PubMed.
- C. T. Supuran, Future Med. Chem., 2011, 3, 1165–1180 CrossRef CAS PubMed.
- S. Fossati, P. Giannoni, M. E. Solesio, S. L. Cocklin, E. Cabrera, J. Ghiso and A. Rostagno, Neurobiol. Dis., 2016, 86, 29–40 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146–189 CrossRef CAS PubMed.
- A. Innocenti, S. Beyza Öztürk Sarıkaya, İ. Gülçin and C. T. Supuran, Bioorg. Med. Chem., 2010, 18, 2159–2164 CrossRef CAS PubMed.
- C. Supuran, Curr. Top. Med. Chem., 2007, 7, 825–833 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- A. Guerri, F. Briganti, A. Scozzafava, C. T. Supuran and S. Mangani, Biochemistry, 2000, 39, 12391–12397 CrossRef CAS PubMed.
- J. Lehtonen, B. Shen, M. Vihinen, A. Casini, A. Scozzafava, C. T. Supuran, A.-K. Parkkila, J. Saarnio, A. J. Kivelä, A. Waheed, W. S. Sly and S. Parkkila, J. Biol. Chem., 2004, 279, 2719–2727 CrossRef CAS PubMed.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS PubMed.
- A. Maresca, C. Temperini, H. Vu, N. B. Pham, S.-A. Poulsen, A. Scozzafava, R. J. Quinn and C. T. Supuran, J. Am. Chem. Soc., 2009, 131, 3057–3062 CrossRef CAS PubMed.
- A. Scozzafava, T. Owa, A. Mastrolorenzo and C. Supuran, Curr. Med. Chem., 2003, 10, 925–953 CrossRef CAS PubMed.
- S. M. Monti, C. T. Supuran and G. De Simone, Expert Opin. Ther. Pat., 2013, 23, 737–749 CrossRef CAS PubMed.
- A. Bertucci, A. Moya, S. Tambutté, D. Allemand, C. T. Supuran and D. Zoccola, Bioorg. Med. Chem., 2013, 21, 1437–1450 CrossRef CAS PubMed.
- A. Thiry, J.-M. Dogne, C. Supuran and B. Masereel, Curr. Top. Med. Chem., 2007, 7, 855–864 CrossRef CAS PubMed.
- E. Berrino, B. Michelet, A. Martin-Mingot, F. Carta, C. T. Supuran and S. Thibaudeau, Angew. Chem., Int. Ed., 2021, 60, 23068–23082 CrossRef CAS PubMed.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2023, 93, 129411 CrossRef CAS PubMed.
- A. Nocentini and C. T. Supuran, Expert Opin. Ther. Pat., 2018, 28, 729–740 CrossRef CAS PubMed.
- J. Ghiso, S. Fossati and A. Rostagno, J. Alzheimer's Dis., 2014, 42, S167–S176 Search PubMed.
- S. Fossati, J. Cam, J. Meyerson, E. Mezhericher, I. A. Romero, P. O. Couraud, B. B. Weksler, J. Ghiso and A. Rostagno, FASEB J., 2010, 24, 229–241 CrossRef CAS PubMed.
- R. Parodi-Rullán, J. Ghiso, E. Cabrera, A. Rostagno and S. Fossati, Aging Cell, 2020, 19(11), e13258 CrossRef PubMed.
- S. Fossati, P. Giannoni, M. E. Solesio, S. L. Cocklin, E. Cabrera, J. Ghiso and A. Rostagno, Neurobiol. Dis., 2016, 86, 29–40 CrossRef CAS PubMed.
- M. K. Sun and D. L. Alkon, J. Pharmacol. Exp. Ther., 2001, 297, 961–967 CrossRef CAS PubMed.
- L. Canto de Souza, G. Provensi, D. Vullo, F. Carta, A. Scozzafava, A. Costa, S. D. Schmidt, M. B. Passani, C. T. Supuran and P. Blandina, Neuropharmacology, 2017, 118, 148–156 CrossRef CAS PubMed.
- P. Blandina, G. Provensi, M. B. Passani, C. Capasso and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2020, 35, 1206–1214 CrossRef CAS PubMed.
- R. Ipe, J. M. Oh, S. Kumar, I. Ahmad, L. R. Nath, S. Bindra, H. Patel, K. Y. Kolachi, P. Prabhakaran, P. Gahtori, A. Syed, A. M. Elgorbanh, H. Kim and B. Mathew, Mol. Diversity, 2024, 1–17 Search PubMed.
-
B. Mathew, J. M. Oh, D. G. T. Parambi, S. T. Sudevan, S. Kumar and H. Kim, Enzyme inhibition assays for monoamine oxidase, in Neuroprotection, ed. S. K. Ray, Methods in Molecular Biology, Humana, New York, 2024, vol. 2761, pp. 329–336 Search PubMed.
- B. Mathew, D. G. T. Parambi, G. E. Mathew, Md. S. Uddin, S. T. Inasu, H. Kim, A. Marathakam, M. K. Unnikrishnan and S. Carradori, Arch. Pharm., 2019, 352(11), e1900177 CrossRef PubMed.
- M. B. H. Youdim, D. Edmondson and K. F. Tipton, Nat. Rev. Neurosci., 2006, 7, 295–309 CrossRef CAS PubMed.
- P. Riederer, L. Lachenmayer and G. Laux, Curr. Med. Chem., 2004, 11, 2033–2043 CrossRef CAS PubMed.
- N. Dinh Thanh, D. Son Hai, V. Ngoc Toan, H. Thi Kim Van, N. Thi Kim Giang and N. Minh Tri, Arch. Pharm., 2024, 357(5), e2300557 CrossRef PubMed.
- N. A. Rehuman, A. G. Al-Sehemi, D. G. T. Parambi, T. M. Rangarajan, O. Nicolotti, H. Kim and B. Mathew, ChemistrySelect, 2021, 6, 7162–7182 CrossRef CAS.
- G. Evtugyn, Talanta, 1998, 46, 465–484 CrossRef CAS PubMed.
- Y. Miao, N. He and J.-J. Zhu, Chem. Rev., 2010, 110, 5216–5234 CrossRef CAS PubMed.
- S. Tian, X. Wang, L. Li, X. Zhang, Y. Li, F. Zhu, T. Hou and X. Zhen, J. Chem. Inf. Model., 2017, 57, 1474–1487 CrossRef CAS PubMed.
- Y.-G. Chen, China Med. J., 2018, 131, 1618–1624 CrossRef CAS PubMed.
- S. Kummar, H. X. Chen, J. Wright, S. Holbeck, M. D. Millin, J. Tomaszewski, J. Zweibel, J. Collins and J. H. Doroshow, Nat. Rev. Drug Discovery, 2010, 9, 843–856 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48(21), 6523–6543 CrossRef CAS PubMed.
- X. Zhao, Q. Hu, X. Wang, C. Li, X. Chen, D. Zhao, Y. Qiu, H. Xu, J. Wang, L. Ren, N. Zhang, S. Li, P. Gong and Y. Hou, Eur. J. Med. Chem., 2024, 279, 116810 CrossRef CAS PubMed.
- S. Giovannuzzi, D. Chavarria, G. Provensi, M. Leri, M. Bucciantini, S. Carradori, A. Bonardi, P. Gratteri, F. Borges, A. Nocentini and C. T. Supuran, J. Med. Chem., 2024, 67, 4170–4193 CrossRef CAS PubMed.
- S. Carradori, M. Fantacuzzi, A. Ammazzalorso, A. Angeli, B. De Filippis, S. Galati, A. Petzer, J. P. Petzer, G. Poli, T. Tuccinardi, M. Agamennone and C. T. Supuran, Molecules, 2022, 27, 7816 CrossRef CAS PubMed.
- M. Şentürk, İ. Gülçin, Ş. Beydemir, Ö. İ. Küfrevioğlu and C. T. Supuran, Chem. Biol. Drug Des., 2011, 77, 494–499 CrossRef PubMed.
- A. Innocenti, I. Gülçin, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2010, 20, 5050–5053 CrossRef CAS PubMed.
- R. Florio, B. De Filippis, S. Veschi, V. di Giacomo, P. Lanuti, G. Catitti, D. Brocco, A. di Rienzo, A. Cataldi, I. Cacciatore, R. Amoroso, A. Cama and L. De Lellis, Int. J. Mol. Sci., 2023, 24, 1977 CrossRef CAS PubMed.
- M. Fantacuzzi, M. Gallorini, N. Gambacorta, A. Ammazzalorso, Z. Aturki, M. Balaha, S. Carradori, L. Giampietro, C. Maccallini, A. Cataldi, O. Nicolotti, R. Amoroso and B. De Filippis, Pharmaceuticals, 2021, 14, 984 CrossRef CAS PubMed.
- P. Di Fermo, S. Di Lodovico, R. Amoroso, B. De Filippis, S. D'Ercole, E. Di Campli, L. Cellini and M. Di Giulio, Antibiotics, 2020, 9, 891 CrossRef CAS PubMed.
- E. S. Di Filippo, L. Giampietro, B. De Filippis, M. Balaha, V. Ferrone, M. Locatelli, T. Pietrangelo, A. Tartaglia, R. Amoroso and S. Fulle, Molecules, 2020, 25, 5770 CrossRef PubMed.
- I. Nishimori, D. Vullo, A. Innocenti, A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, J. Med. Chem., 2005, 48, 7860–7866 CrossRef CAS PubMed.
- S. Carradori, M. Fantacuzzi, A. Ammazzalorso, A. Angeli, B. De Filippis, S. Galati, A. Petzer, J. P. Petzer, G. Poli, T. Tuccinardi, M. Agamennone and C. T. Supuran, Molecules, 2022, 27, 7816 CrossRef CAS PubMed.
- M. Agamennone, M. Fantacuzzi, S. Carradori, A. Petzer, J. P. Petzer, A. Angeli, C. T. Supuran and G. Luisi, Molecules, 2022, 27, 7884 CrossRef CAS PubMed.
- R. G. Khalifah, J. Biol. Chem., 1971, 246, 2561–2573 CrossRef CAS PubMed.
- D. Pirolli, B. Righino, C. Camponeschi, F. Ria, G. Di Sante and M. C. De Rosa, Sci. Rep., 2023, 13, 1494 CrossRef CAS PubMed.
- M. Durgun, S. Akocak, N. Lolak, F. Topal, Ü. M. Koçyiğit, C. Türkeş, M. Işık and Ş. Beydemir, Chem. Biodiversity, 2024, 21(2), e202301824 CrossRef CAS PubMed.
- A. Nocentini, A. Costa, A. Bonardi, A. Ammara, S. Giovannuzzi, A. Petreni, G. Bartolucci, B. Rani, M. Leri, M. Bucciantini, J. G. Fernández-Bolaños, Ó. López, M. B. Passani, G. Provensi, P. Gratteri and C. T. Supuran, J. Med. Chem., 2024, 67, 16873–16898 CrossRef CAS PubMed.
- C. Temperini, A. Scozzafava, D. Vullo and C. T. Supuran, J. Med. Chem., 2006, 49, 3019–3027 CrossRef CAS PubMed.
- M. R. Buemi, A. Di Fiore, L. De Luca, A. Angeli, F. Mancuso, S. Ferro, S. M. Monti, M. Buonanno, E. Russo, G. De Sarro, G. De Simone, C. T. Supuran and R. Gitto, Eur. J. Med. Chem., 2019, 163, 443–452 CrossRef CAS PubMed.
- A. Saxena, A. M. G. Redman, X. Jiang, O. Lockridge and B. P. Doctor, Chem.-Biol. Interact., 1999, 119–120, 61–69 CrossRef CAS PubMed.
- F. Nachon, E. Carletti, C. Ronco, M. Trovaslet, Y. Nicolet, L. Jean and P.-Y. Renard, Biochem. J., 2013, 453, 393–399 CrossRef CAS PubMed.
- E. Berrino, S. Carradori, F. Carta, F. Melfi, M. Gallorini, G. Poli, T. Tuccinardi, J. G. Fernández-Bolaños, Ó. López, J. P. Petzer, A. Petzer, P. Guglielmi, D. Secci and C. T. Supuran, Antioxidants, 2023, 12, 2044 CrossRef CAS PubMed.
- M. G. Kurban, R. Çakmak, E. Başaran, B. Türkmenoğlu and M. Şentürk, J. Mol. Struct., 2024, 1314, 138798 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c,
Mohamed A.
Abdelgawad
d,
Samy
Selim
e,
Sunil
Kumar
b and
Bijo
Mathew
*c,
Mohamed A.
Abdelgawad
d,
Samy
Selim
e,
Sunil
Kumar
b and
Bijo
Mathew