Mechanism study on enhancing the combustion performance of aluminum with polyvinylidene fluoride
Received
10th July 2025
, Accepted 18th September 2025
First published on 13th October 2025
Abstract
As a key metal fuel in high-energy solid propellants, the improvement of the combustion efficiency of aluminum (Al) is the focus of current research. In order to optimize the combustion performance of Al, polyvinylidene fluoride (PVDF) was used as a surface modifier in this study to modulate the combustion process of Al at the atomic scale by taking advantage of its unique high exothermicity and controllable fluorine-release properties. This study elucidates the molecular-level synergistic combustion mechanism in Al/PVDF systems using multiscale simulation approaches. By integrating quantum chemical calculations with reactive molecular dynamics, the interfacial reaction network between the PVDF thermal decomposition products and the Al oxide layer is systematically resolved. Specifically, Gaussian 16W calculations at the B3LYP/6-311+G(3d,2p) level demonstrate that PVDF-derived hydrogen fluoride (HF) molecules selectively capture the bridging oxygen atoms in Al2O3via a dual-step synergistic mechanism, resulting in a three-fold increase in the porosity of the oxide layer. Transition state theory analysis identified the rate-determining steps for the HF/AlO and HF/Al2O3 reactions, while bond order analysis quantitatively revealed the fluorine-induced Al–O bond cleavage mechanism. These insights facilitate in-depth analysis of the fluorine-catalyzed oxide stripping mechanism through thermodynamic parameter optimization. These findings not only establish a constitutive model for PVDF-modified Al combustion but also promote a research paradigm for tailoring metal combustion performance via molecular interfacial engineering.
1. Introduction
In the field of materials science and technology, research and development of high-performance and multifunctional composites have been pivotal in driving scientific and technological advancements. In recent years, fluoropolymer-based composites (such as fluorine rubber (Viton),1 polytetrafluoroethylene (PTFE),2 polyvinylidene fluoride (PVDF)3 and tetrafluoroethylene-hexafluoropropylene-vinylidene fluoride (THV)4) terpolymers combined with aluminum (Al), have garnered significant attention for their potential applications in solid propellants. Fluoropolymers are renowned for their low surface energy and excellent chemical inertness.5 Alternatively, Al is widely regarded as a critical additive for energetic fuels and propellants due to its high volumetric energy density (31 kJ cm−3),6 low oxygen equilibrium, and rapid exothermic oxidation properties.7 However, Al2O3 passivation is produced by forming a thin shell of Al oxide on the metal surface, which lowers energy release.8,9 Thus, to fix this problem, researchers have mixed fluoropolymers with Al. Wang et al.10 found that the introduction of PTFE produces an additional exothermic peak (up to 697 J g−1) at 540–580 °C, increasing the underwater explosion impulse by 23.3%. Koul et al.11 compared the modification effects of PTFE and Viton on hydroxyl-terminated polybutadiene (HTPB)-based solid fuels loaded with nano-aluminum (nAl). The study showed that nAl-PTFE improves fuel recovery by 178%, outperforming the 122% improvement observed with Viton. Research has found that a PVDF coating prevents Al particles from forming a passive oxide layer while releasing fluorine radicals at high temperatures, which helps Al burn more completely. Chen et al.12 confirmed that PVDF forms a continuous coating layer on the surface of Al, and thermal analysis results showed that PVDF coatings significantly increased the heat of combustion for Al particles. Yang et al.13 demonstrated that a PVDF coating promotes the reaction between hydrogen fluoride (HF) and Al2O3, which occurs at 400–500 °C and effectively destroys the oxide layer.
PVDF is a type of fluoropolymer that works well to improve how Al spreads and reacts.14 This is due to the significant thermal decomposition of PVDF at about 400 °C, which releases reactive fluorides that form stable chemical bonds with the Al surface.15,16 PVDF, as an important fluorinated polymer material, has a molecular structure composed of –CH2–CF2– repeating units formed by the polymerization of vinylidene fluoride (VFD) monomers.17 The extremely high electronegativity of fluorine atoms results in a C–F bond energy of 485 kJ mol−1, markedly exceeding that of 413 kJ mol−1 of C–H bonds. This difference in bond strength explains the superior corrosion resistance of PVDF. As the second most widely produced fluoropolymer after PTFE, PVDF has significantly better solubility in common polar solvents than PTFE. It contains 59.4 wt% fluorine (F) and 3 wt% hydrogen (H), which collectively contribute to its outstanding physicochemical properties.18 Ji et al.19 made Al/PVDF with a core–shell structure using a simple solvent method. These particles showed better pressure output and burned well even with little oxygen. Li et al.20 used electrospraying to make nAl/PVDF composites, which exhibited a 50% improvement in combustion heat and a significant increase in combustion rate. Kim et al.21 made Al/PVDF composites with a core–shell structure. The PVDF coating on the Al particles helped reduce oxide formation and made them burn faster than pure Al particles. Also, DeLisio et al.22 used electrospray deposition to create Al/PVDF films and carefully studied the preheating reaction and the underlying mechanisms between Al and PVDF. Sun et al.23 used transmission electron microscopy (TEM) and scanning electron microscopy (SEM) to characterize the findings, revealing that a PVDF coating layer effectively reduces the porosity of the Al oxide layer on the surface of nAl, promoting the formation of a more compact core–shell structure. Chen et al.24 confirmed through SEM observations that compared to uncoated samples, PVDF-coated nAl particles exhibited a marked reduction in surface cracks, resulting in a more complete and continuous core–shell structure.
The above-mentioned research clearly confirms the significant role of PVDF in the Al-oxide membrane removal process and provides reliable experimental evidence. In addition, existing research indicates that the reaction mechanism of fluoropolymer-modified Al powder has been preliminarily revealed through theoretical simulation methods. Zhang et al.25 used DFT calculations to confirm that C2F4 generated from the decomposition of PTFE can react with AlO and Al2O, revealing the microscopic mechanism by which fluorinated organic compounds (FOCs) inhibit the formation of Al2O3. Some scholars have also used molecular simulation methods to study the mechanism of the Al/PVDF system. For example, Padhye et al.26 demonstrated via DFT calculations that HF derived from PVDF preferentially attacks the terminal hydroxyl sites on Al2O3, releasing energy and accelerating the degradation of the passivation layer. Bello et al.27 employed DFT simulations to demonstrate that the energy of dehydrated tetracoordinated Al sites and PVDF increases from −37 kJ mol−1 to −110.54 kJ mol−1, thereby accelerating the fluorination reaction kinetics. These investigations not only deepened our understanding of the reaction mechanism between PVDF and the Al oxide layer interface but also laid a solid theoretical foundation for this study and provided key experimental evidence. However, currently, there is a lack of research on the atomic-scale interaction mechanisms between the PVDF decomposition products and Al oxides. Although existing studies have advanced Al/PVDF composites, their key molecular mechanisms remain unclear. It remains unclear which bonds break first in energetic compound molecules and whether reactions happen alone or between molecules. Therefore, the atomic-level analysis of fluoropolymer-assisted combustion (especially PVDF) is urgently needed.
This study employs DFT to investigate the underlying mechanisms governing the superior combustion performance of PVDF-coated Al-based composites. From a molecular perspective, the decomposition process of PVDF is systematically elucidated, which releases reactive fluorine species, with particular emphasis on the temperature-dependent pathways for HF generation. Furthermore, the dynamic interfacial reactions between these decomposition products and Al oxides are examined, including the transient AlO intermediate and stable Al2O3 surfaces. A key focus lies in uncovering how fluorine-containing intermediates disrupt the native oxide layer on metallic Al, thereby participating in and accelerating its oxidation. The insights gained not only advance fundamental combustion science but also provide critical guidance for designing next-generation energetic materials with precisely tunable reaction properties. By establishing atomic-scale structure–performance relationships, this study lays a solid foundation for optimizing the design of high-performance fluoropolymer–metal composites.
2. Theoretical calculation methods
This study used Gaussian 16 to model the molecular reactions.28 The B3LYP/6-311+G(3d,2p) approach was applied to explore the reaction mechanisms of HF/AlO and HF/Al2O3 molecules, respectively.29 This method achieves a good balance between computational efficiency and accuracy in accurately describing the hydrogen bonding interaction mechanism and electron transfer process between HF molecules and the Al oxide surface. In contrast, smaller basis sets such as 6-31G* underestimate the adsorption energy and bond length accuracy, while larger basis sets, such as those combined with the M06-2X functional, although more precise, substantially increase the computational costs. The geometric configurations of the reactant (Re), intermediates (IM), transition state (TS) and production (Pr) involved in the reaction process were systematically calculated at the B3LYP/6-311+G(3d,2p) level. These calculations reveal how the molecules interact, while also confirming that these structures remain stable during vibration, as shown by the frequency analysis at the same level of theory. GaussView 6.0 was used to visualize all key molecular structures, giving clear pictures of how the reactions progress.30 The IRC method was used to verify that each transition state was truly the highest energy point along the reaction path.31 These calculations pointed out the easiest route molecules take from the starting materials to the final products, showing the correctness and rationality of the reaction mechanism obtained from the calculations.
Recent progress in computer modeling and quantum chemistry has created strong tools for studying how reactions work at the molecular level. At the heart of this is transition state theory (TST), which explains how starting materials change into products by first forming a short-lived transition state.32 This theory explains the detailed mechanisms of chemical reactions at the molecular level. Catalysts significantly enhance the reaction kinetic efficiency by modulating the transition state configuration and reducing the reaction activation energy. To better understand how HF reacts with both AlO and Al2O3, the reaction rates at various temperatures were calculated using TST. To ensure reliability, all calculations were conducted with the GPOP program.33 The temperature-dependent reaction rate (k) was calculated using the Arrhenius equation.34
The reaction rate constant k, which is related to the temperature T, is obtained using the Arrhenius equation, as follows:35,36
where
k is the reaction rate constant at temperature
T;
A is a preexponential factor, with the same dimension as
k;
Ea is the apparent activation energy, which can generally be regarded as a constant independent of temperature, with the units of J mol
−1 or kJ mol
−1;
T is the thermodynamic temperature, in units of K; and
R is the molar gas constant, with units of J mol
−1 K
−1.
Performing an indefinite integral on the differential form of the Arrhenius equation produces the following solution:
| | 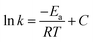 | (2) |
According to the above-mentioned equation, the plot of reaction rate constant (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k) against the reciprocal of temperature (1/T) should be a straight line, which has a slope of −Ea/R. This pattern is commonly observed in the context of many chemical reactions. In cases where temperatures T1 and T2 are involved, the corresponding reaction rate constants are denoted as k1 and k2, respectively.
k) against the reciprocal of temperature (1/T) should be a straight line, which has a slope of −Ea/R. This pattern is commonly observed in the context of many chemical reactions. In cases where temperatures T1 and T2 are involved, the corresponding reaction rate constants are denoted as k1 and k2, respectively.
This showed exactly how temperature affects the reactions of HF with both AlO and Al2O3, revealing important differences in their reaction speeds at various temperatures. Multiwfn 3.4.1 was used to study the reaction sites in detail, focusing on their surface analysis feature to examine molecular properties.37–39 This method maps molecular surface charges to reveal key interaction patterns and reactive sites. To enhance the intuition and dynamic visualization of the analysis results, the electrostatic potential distribution data were integrated with the molecular dynamics simulation visualization software visual molecular dynamics (VMD) in this study. VMD simulations create dynamic visualizations of these electrostatic potential distributions, directly showing where and how reactions occur.
3. Results and discussion
3.1 Single-molecule gas-phase reaction mechanism of PVDF
At high temperatures, PVDF decomposes via a multi-step process involving the gradual fragmentation of its molecular chains and subsequent free radical reactions. Although PVDF normally starts decomposing above 300 °C (572 °F), this temperature can vary depending on the conditions.40 The actual decomposition temperature of PVDF can vary its molecular structure and added chemicals (such as flame boosters and retardants) may lower or alter its breakdown behavior. When PVDF heats above its breakdown point, its molecular chains become unstable. As the temperature increases, the weak spots in these chains break apart, releasing large amounts of energy and free radicals.
These highly reactive free radicals initiate chain reactions with surrounding molecules. As this happens, the structure of PVDF breaks down step by step, producing a variety of decomposition products including oxides, fluorides, and carbon compounds. PVDF undergoes thermal decomposition via a sequential process involving chain scission followed by free radical reactions. The breakdown of PVDF was studied using computational simulations with C4F4H4, the basic building block of PVDF, as a simplified model. All calculations were performed using the B3LYP/6-311+G(d,p) basis set.41 Stable and accurate results for HF were obtained for subsequent analysis through the simulations.
3.1.1 Resolution mechanism of the PVDF structure.
The thermal decomposition of the C4F4H4 structure was successfully simulated using the B3LYP/6-311+G(d,p) basis set. The reaction process involves one Re structure, one IM structure, two TS structures and one Pr structure, as shown in Fig. 1. Fig. 2 illustrates the reaction energy profile of the C4F4H4 system, providing a clear visualization of the energy variations along the reaction pathway. The gradual decomposition of these structures to generate the key product HF can be clearly observed by trajectory analysis. This result indicates the sequential breaking of the chemical bonds in PVDF and elucidates the generation mechanism of HF, a key reaction product of PVDF pyrolysis.
 |
| | Fig. 1 Structures of the C4F4H4 system calculated at the B3LYP/6-311+G(3d,2p) level, where all bond angles and lengths are given in degrees (°) and angstroms (Å), respectively. | |
 |
| | Fig. 2 Potential energy diagram for the C4F4H4 structure reaction calculated at the B3LYP/6-311+G(d,p) level, where the energy is represented in kcal mol−1. | |
The decomposition mechanism of the C4F4H4 structures under high-temperature conditions was investigated in detail. During the reaction of the C4F4H4 structure, the precise pathway for the generation of HF was successfully observed. The key mechanistic feature of this reaction involves H atom transfer (HAT, which represents a fundamental class of radical reactions)42 of the H1 atom. Due to the axisymmetric distribution of the H2 and H3 atoms around the C1 atom, their electron arrangements remain relatively stable, resulting in low reactivity during the reaction. In contrast, the H1 atom, owing to its unique chemical environment, becomes the primary participant in the reaction. Initially, the C1–H1 (1.094 Å) bond breaks, releasing free H1 atoms. Then, the H1 atom migrates toward the C2 atom, forming the transition-state structure TSp1. As the reaction progresses, the H1 atoms gradually approach the C3 atom, ultimately forming a stable C3–H1 (1.091 Å) bond. This process can be described as follows:
| | | Re → TSp1 (C4F4H3⋯H) → IMp1 | (R1) |
Then, IM2 continues to react. The two reaction channels that it can follow are described below:
| | | IMp1 → TSp2 → Pr (C4F3H3⋯HF) | (R2) |
Due to the unique attraction between F1 and H1, the C4–F4 (1.350 Å) bond and the C3–H1 (1.093 Å) bond break sequentially, regenerating free H1 and F1 atoms. Finally, H1 combines with F1 to form a stable H1–F1 (0.927 Å) bond, indicating the production of HF. This pathway overcomes a total reaction energy barrier of 68.21 kcal mol−1. The reaction process involves multiple steps, including the migration of H1 atoms and the breaking and reorganization of bonds, ultimately transforming the initial structure into a stable final product. This detailed reaction pathway not only elucidates the dynamic mechanism of the structural changes in C4F4H4 but also provides a significant theoretical foundation for understanding the chemical behavior of similar systems. Table S1 shows the rotation constants (GHz) and vibration frequencies (cm−1) of the stationary points, products, and transition states in the C4F4H4 system calculated using the B3LYP/6-311+G(3d,2p) method. The reaction occurs through one main pathway with two distinct channels (R1) and (R2).
In this study, the Gibbs free energy change and relative energy of each structure in the C4F4H4 system were systematically calculated at 298.15 K using the B3LYP/6-311+G(d,p) level of theory, with the detailed results presented in Table S2. Through these calculations, the energy evolution during the thermal decomposition of PVDF can be elucidated. The reaction pathway is well-defined and proceeds as follows: Re → TSp1 → IMp1 → TSp2 → Pr (Fig. 2).
3.1.2 Reaction kinetics.
One of the most critical parameters in kinetic studies is the accurate determination of the rate constants for elementary reactions, which is essential for improving the precision of models used in experimental research. Herein, the temperature-dependent reaction rate coefficients of the (R3) and (R4) channels were calculated using TST theory. The calculations were conducted over the temperature range of 300–3000 K, with a sampling interval of 100 K. The rate coefficient, kpi, for each reaction channel (i = 1–2) was calculated as follows:| | | Re → TSp1 → IMp1 (kp1) | (R3) |
| | | IMp1 → TSp2 → Pr (kp2) | (R4) |
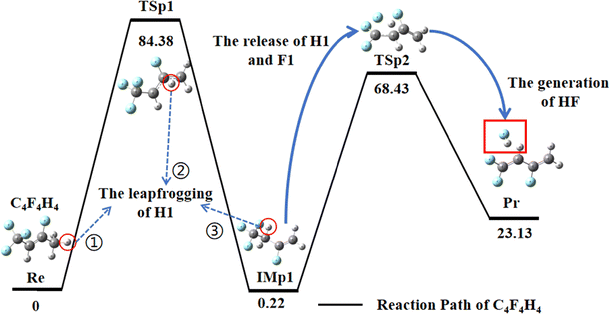
The rate constants for each reaction channel within the temperature range of 300 to 3000 K are presented in Table S3. At the same level of theory, the parameters required to fit the Arrhenius expression, including the Ea and A for each reaction channel, were calculated and are summarized in Table S4. The decomposition of C4F4H4 proceeds through channels RP1 and RP2, ultimately yielding C4F3H3 and stabilized HF. The reaction rate constants for these pathways, fitted to the Arrhenius expression, are as follows: kp1(T) = 1.30 × 1011exp(−81.91 kcal mol−1/RT), kp2(T) = 3.87 × 1011exp(−65.57 kcal mol−1/RT).
In the temperature range of 300 < T < 800 K, the rate constants kp1 and kp2 exhibit the maximum temperature dependence, suggesting that a large portion of the active ingredient HF is released from PVDF. The kinetic analysis (Fig. 3) shows that the rate constants kp1 and kp2 exhibit a significant temperature dependence within the temperature region of 300–800 K, which corresponds to the primary reaction stage of the PVDF main chain C–F bond breaking to release the main HF product. When the temperature rises to 800 K, the thermodynamic analysis confirms that PVDF undergoes violent decomposition, corresponding to the maximum production of HF.43 In the high-temperature interval of 800–3000 K, the reaction rate increases exponentially with an increase in temperature, which is in line with the trend predicted by the transition state theory.
 |
| | Fig. 3 Reaction rate constants of different channels (s−1) calculated at the B3LYP/6-311+G(d,p) level for the C4F4H4 decomposition system. | |
3.1.3 Electrostatic potential (ESP) analysis.
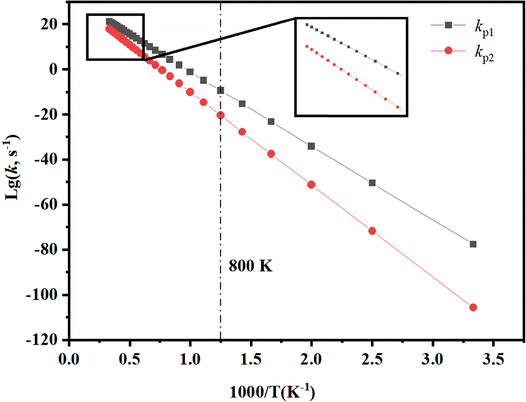
The ESP analysis of the Prp1 structure of the C4F4H4 system (Fig. 4) shows that the highest ESP value on the molecular surface appears near the H element, with a value of 35.98 kcal mol−1, and the strongest positive charge is observed in the region surrounding the H atom. This suggests that the H atoms can easily attract electrophilic reagents for reactions, making these sites prime targets for electrophilic attack. This charge concentration also marks the main reaction center in the molecule, helping explain its chemical behavior. The F atom shows strong negative charge (−25.93 kcal mol−1), making it likely to form bonds with the positively charged H atoms. By mapping the electron distribution of C4F4H4, the location where the reaction occurs can be pinpointed, where the positive H sites grab electrons, while the negative F sites donate them. The charge polarization of a molecule not only explain its reactions, but also provides an important reference for improving reactions and building more effective catalysts. By measuring both the ESP values and surface area, the reactive regions on the molecular surface are further revealed. This provides comprehensive theory for a deeper understanding of the behavior of molecules in supporting chemical reactions.
 |
| | Fig. 4 ESP map and surface area of Prp1 of the C4F4H4 system. | |
3.2 Al/PVDF interfacial reactions at the atomic scale
3.2.1 Reaction mechanism of AlO/HF system.
During the combustion process of Al/PVDF composites, there is a high likelihood of chemical reactions occurring between AlO, a key intermediate product of Al powder combustion, and HF, the primary decomposition product of PVDF. Given this context, a thorough investigation of the interactions between these two compounds is crucial for elucidating the role of PVDF in promoting the combustion process at the microscopic reaction mechanism level. Understanding this process will not only provide insights into the details of the combustion kinetics but also offer theoretical support for optimizing the combustion performance and safety applications of these composites. Fig. 5 illustrates the optimized molecular conformations of Re, IM, TS and Pr in the AlO/HF reaction system, calculated at the B3LYP/6-311+G(3d,2p) level. The reaction system is comprised of one Re configuration, three IM configurations, four TS configurations and one Pr configuration.
 |
| | Fig. 5 Structures of the AlO/HF system calculated at the B3LYP/6-311+G(3d,2p) level, where all bond angles and lengths are given in degrees (°) and angstroms (Å), respectively. | |
Through detailed geometric optimization calculations of the AlO/HF system, the stable initial configuration Re was successfully obtained. Significant interatomic interactions between AlO and HF molecules were observed throughout the reaction system, as demonstrated by the formation of new H–O and Al–F bonds. The creation of these bonds not only signifies the intensification of intermolecular interactions but also precisely reveals the fine-tuning of the interatomic distances, with the H–O and Al–F bond lengths determined to be 0.979 Å and 1.698 Å, respectively. The system shows the complete reaction pathway, ultimately forming the Pr structure. In the initial stage of the reaction, F atoms exhibit strong attraction toward the Al atom, leading to significant atomic rearrangements in the Re structure to form the AlOHF composite. During this process, the H–F bond (0.926 Å) breaks, while an Al–F bond (1.654 Å) and an Al–H bond (1.558 Å) are formed. This pathway overcomes a reaction energy barrier of 16.82 kcal mol−1. The process is referred to as reaction channel (R5). The specific reaction process is as follows:
| | | Re (AlO + HF) → TS1 (AlOH⋯F) → IM1 (AlOHF) | (R5) |
At high temperature, the AlO/HF system continues to react, and the subsequent reaction channels (R6)–(R8) are as follows:
| | | IM3 → TS4 → Pr (AlF + OH) | (R8) |
In channel (R7), the bond angles and bond lengths of the AlHFO complex structure are changed. Given that no bond breaking or reorganization occurs, this pathway overcomes the lowest reaction energy barrier of 3.18 kcal mol−1. It is worth noting that TS4 in the (R8) channel undergoes even more pronounced changes, involving a significant atomic exchange process. This transformation directly results in the formation of an O–H bond (0.979 Å) and an Al–F bond (1.704 Å), overcoming a reaction energy barrier of 70.73 kcal mol−1. Analyzing the reaction process of the entire pathway, the HF molecule successfully removes AlO by breaking the H–F bond and forming a new Al–F bond. This unique Pr configuration not only highlights the complexity and diversity of the intermolecular interactions but also provides new insights into the mechanisms of chemical bond cleavage, reorganization, and atomic rearrangement in the AlO/HF system. Furthermore, this process creates more favorable conditions for subsequent chemical reactions and offers novel approaches and methodologies for the modification and treatment of material surfaces. Table S5 summarizes the rotational constants and vibrational frequencies of the AlO/HF system, while Table S6 presents the Gibbs free energy changes and relative energies of the individual structures within the AlO/HF system, both calculated using the B3LYP/6-311+G(3d,2p) approach. A schematic diagram of the reaction energy level of the AlO/HF system calculated at B3LYP/6-311+G(3d,2p) is shown in Fig. 6. This system shows a simple and complete reaction path, which can be described as follows: Re → TS1 → IM1 → TS2 → IM2 → TS3 → IM3 → TS4 → Pr (Fig. 5).
 |
| | Fig. 6 Potential energy diagram of the AlO/HF system at the B3LYP/6-311+G(3d,2p) level, where the energy is represented in kcal mol−1. | |
3.2.2 Reaction mechanism of Al2O3/HF system.
The optimized molecular geometries of IM, TS and Pr in the Al2O3/HF reaction system, calculated at the B3LYP/6-311+G(3d,2p) level, are presented in Fig. 7. Also, the rotational constants and vibrational frequencies are summarized in Table S7. The system is comprised of six IM structures, seven TS structures and two Pr structures, whose structural features offer critical insights into the Al2O3/HF system interaction mechanism. Analysis of these configurations systematically elucidates the reaction pathways and provides a theoretical foundation for understanding PVDF-enhanced Al combustion. Through comprehensive geometric optimization calculations of the Al2O3/HF structure, multiple reaction pathways were successfully identified.
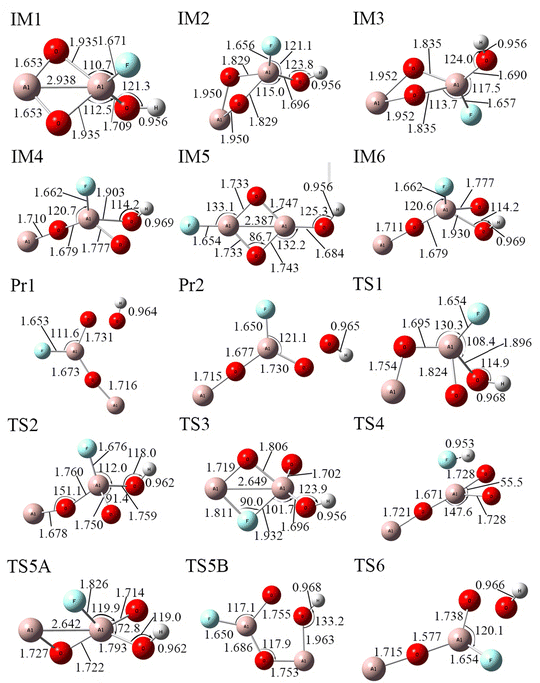 |
| | Fig. 7 Structures of the Al2O3/HF system calculated at the B3LYP/6-311+G(3df,2p) level, where all bond angles and lengths are given in degrees (°) and angstroms (Å), respectively. | |
Notably, the initial configuration of Al2O3 with HF is not observed as the starting point in the simulated reaction process. Instead, all the reaction pathways originate from different Al2O3HF composite states. This phenomenon can be attributed to the inherent instability of the initial structures of Al2O3 and HF alone, as visualized in the TS4 configuration. TS4 represents a transition-state structure, also referred to as an activated complex, which is characterized by its instability.
Fig. 8 presents the potential energy surface diagram for the Al2O3/HF reaction system, revealing four distinct reaction pathways comprising seven elementary reaction channels. In the initial reaction stage, three primary channels ((R9), (R10), and (R11)) dominate the process. These channels all involve conformational transformations of the Al2O3–HF complex, which fundamentally represent energy minimization through molecular rearrangement to form thermodynamically stabilized configurations. The specific reaction channels proceed as follows:
 |
| | Fig. 8 Potential energy diagram of the Al2O3/HF system at the B3LYP/6-311+G(3d,2p) level, where the energy is represented in kcal mol−1. | |
At high temperature, the Al2O3HF composite structure undergoes a complexation reaction. During the reaction, the Al–O bonds (IM1: 1.709 Å, IM2: 1.696 Å and IM3: 1.690 Å) in Al2O3 are cleaved, leading to the formation of Al–F bonds (Pr1: 1.653 Å and Pr2: 1.650 Å) and O–H bonds (Pr1: 0.964 Å and Pr2: 0.965 Å). The overall reaction can be described as the decomposition of Al2O3HF. The reaction channel is as follows:
| | | IM4 → TS4 (Al2O3⋯H⋯F) → IM6 | (R12) |
| | | IM5 → TS5B → Pr1 (Al2O2F⋯OH) | (R14) |
| | | IM6 → TS6 → Pr2 (Al2O2F⋯OH) | (R15) |
The (R13) channel preserves the characteristic conformational transition of the Al2O3–HF complex. In contrast, the (R12) channel demonstrates dissociative behavior, where cleavage of both H–O and Al–F bonds enables the complete detachment of the H and F atoms from the Al2O3 framework, yielding free atomic species. This process exhibits a substantially higher transition state energy, reflecting the significant energy required to overcome strong ligand bonding.
Both the (R14) and (R15) channels induce structural dissociation of the Al2O3–HF complex, yet display markedly different reaction energetics. The (R14) pathway involves Al–O bond rupture, followed by recombination to form AlO2F and OH, overcoming the substantial activation barrier of 120.22 kcal mol−1. Remarkably, the (R15) channel produces identical products through an alternative pathway with a dramatically lower barrier of only 6.57 kcal mol−1. This significant difference suggests that the (R15) channel is energetically more favorable and more likely to occur. The product formation in both the (R14) and (R15) channels verifies the etching effect of HF toward Al2O3 oxides. The exceptionally low activation energy of the (R15) channel provides a molecular-level explanation for the remarkable efficiency of HF in Al surface treatment processes.
The Gibbs free energy change and relative energies of the individual structures in the Al2O3/HF system were calculated at the same energy levels, and the results are detailed in Table S8. A complete reaction path for the Al2O3/HF system thermal decomposition system can be expressed as follows (Fig. 7):
| | | IM1 → TS1 → IM4 → TS4 → IM6 → TS6 → Pr2 | (R16) |
| | | IM2 → TS2 → IM4 → TS4 → IM6 → TS6 → Pr2 | (R17) |
| | | IM3 → TS3 → IM5 → TS5A → IM6 → TS6 → Pr2 | (R18) |
| | | IM3 → TS3 → IM5 → TS5B → Pr1 | (R19) |
Four distinct reaction pathways (R16), (R17), (R18), and (R19) were identified for the decomposition of Al2O3HF, with energy barriers of 114.92, 102.87, 170.29, and 143.97 kcal mol−1, respectively. Based on the energy barriers, it is evident that the reaction is most likely to proceed through pathway (R17) due to its significantly lower energy barrier. The Al2O3 radicals can react with HF, the primary decomposition product of PVDF, thereby facilitating the decomposition of Al2O3.
3.2.3 Reaction kinetics.
The AlO/HF reaction primarily involves two processes, i.e., bipartite complexation and Al–O bond abstraction. The temperature dependence of the effective single-molecule rate constants can be described by a simple chemical activation mechanism, which includes the complexation and decomposition processes associated with AlOHF. The rate constants ki (i = 1, 2, 3, 4) for the different reaction channels are expressed as follows:| | | IM1 → TS2 → IM2 (k2) | (R21) |
| | | IM2 → TS3 → IM3 (k3) | (R22) |
The Al2O3/HF reaction mainly consists of two processes, bipartite complexation and Al–O bond abstraction. The rate constants ki (i = 5, 6, 7, 8, 9, 10, 11) for different channels are expressed as follows:
| | | IM1 → TS1 → IM4 (k5) | (R24) |
| | | IM2 → TS2 → IM4 (k6) | (R25) |
| | | IM3 → TS3 → IM5 (k7) | (R26) |
| | | IM4 → TS4 → IM5 (k8) | (R27) |
| | | IM5 → TS5A → IM6 (k9) | (R28) |
| | | IM5 → TS5B → Pr1 (k10) | (R29) |
| | | IM6 → TS6 → Pr2 (k11) | (R30) |
The single-molecule rate constants of the elementary reactions involved in the AlO/HF and Al2O3/HF reaction systems were calculated using the TST method in the temperature range of 300–3000 K. The detailed parameters of the AlO/HF and Al2O3/HF reaction systems are shown in Tables S9 and S10, respectively. The parameters required to fit the Arrhenius expression, such as Ea and A for each reaction channel, were calculated at the B3LYP/6-311+G(3d,2p) level of theory, as shown in Table S11.
Fig. 9 shows the reaction rate constants calculated for different channels of the AlO/HF (Fig. 9(a)) and Al2O3/HF (Fig. 9(b)) systems at the B3LYP/6-311+G(3d,2p) level. As shown in Fig. 9(a), the reaction rate constants of k1, k2 and k3 show a clear increasing trend with an increase in temperature during the AlO/HF reaction. At T = 300–3000 K, k3 > k2 > k1 > k4, indicating that the AlO/HF reaction is mainly carried out through (R24), and the total reaction rate is dominated by k1, k2, k4. This aligns with the calculated energy barriers. Channel (R24) exhibits the lowest reaction barrier, making its pathway the most thermodynamically favorable, while channel (R25) has the highest barrier due to the energy costs associated with bond cleavage and structural reorganization. The AlO/HF complexation reaction is carried out through channel (R22), which reacts to form AlOHF, and the reaction rate constant is fitted to the Arrhenius expression, as follows: k1 (T) = 2.17 × 1010exp(−13.71 kcal mol−1/RT). The subsequent reaction pathway involves the stepwise thermal decomposition of AlOHF, culminating in the formation of AlF products.
 |
| | Fig. 9 (a) Reaction rate constant diagrams for the AlO/HF system. (b) Reaction rate constant diagrams for the Al2O3/HF system. | |
Based on transition state theory calculations, the fitted Arrhenius expressions for the reaction rate constants involved are: k2 (T) = 1.75 × 1013exp(−17.17 kcal mol−1/RT), k3 (T) = 1.26 × 1013exp(−2.07 kcal mol−1/RT) and k4 (T) = 1.78 × 1014exp(−69.80 kcal mol−1/RT). A notable trend was observed near 3000 K, with the reaction rate k4 exceeding k1, indicating the preferential formation of AlF and OH at elevated temperatures. This suggests that increasing the combustion flame temperature of Al/PVDF systems can effectively promote removal of the Al oxide layer.
The reaction of the Al2O3/HF system proceeds through four pathways, namely reaction path P1 to reaction path P4. The fitted Arrhenius expressions for the total reaction rate constants of these four pathways in the temperature range of 300–3000 K are as follows: k5 (T) = 3.28 × 1012exp(−59.64 kcal mol−1/RT), k6 (T) = 5.42 × 1013exp(−47.55 kcal mol−1/RT), k7 (T) = 3.68 × 1012exp(−23.62 kcal mol−1/RT), k8 (T) = 2.17 × 1013exp(−47.66 kcal mol−1/RT), k9 (T) = 4.53 × 1013exp(−139.19 kcal mol−1/RT), k10 (T) = 1.33 × 1014exp(−119.58 kcal mol−1/RT) and k11 (T) = 1.77 × 1013exp(−6.60 kcal mol−1/RT). The rate constants k5, k8 and k11 represent the complexation reaction kinetics for reaction pathway P1, corresponding to reaction channels (R9), (R12) and (R15), respectively. In pathway P1, the k11 > k8 > k5 pattern proves that (R15) is the main channel, where k8 or k5 controls the overall reaction speed. The reaction rate constants k6, k8 and k11 in pathway P2 (where k6 specifically corresponds to reaction channel (R10)) exhibit distinct temperature-dependent behaviors across the range of 300–3000 K. While k11 demonstrates the most pronounced acceleration with an increase in temperature, the rate constants k6 and k8 maintain remarkably similar trends in their temperature-dependent variations, suggesting parallel reaction mechanisms for these two channels. These kinetic results reveal the consistent dominance of reaction channel (R15) across multiple pathways. In pathway P2, the temperature-dependent behavior of the rate constants (k6, k8 and k11) confirms (R15) as the primary channel. Similarly, in pathway P3, the reaction rates are k7, k9 and k11. These match channels (R11), (R13) and (R15), respectively. In the range of 300–3000 K, the rate follows the order of k11 > k7 > k9. This proves that pathway P3 also mainly uses channel (R15). This consistent preference for (R15) across different pathways suggests its particularly favorable reaction mechanism under these conditions. Pathway P4 has reaction rates k7 and k10. These match channels (R11) and (R14), respectively. The results show k7 > k10. This means that pathway P4 mainly uses channel (R11).
Notably, across the temperature range of 300–3000 K, the comparative analysis of the rate constants for pathways P1, P2, and P3 reveals that k11 exhibits significantly greater growth kinetics than the other rate constants. This demonstrates that all three pathways (P1–P3) predominantly proceed through reaction channel (R15), which corresponds to the OH precipitation step. This critical step initiates the rupture of the Al2O3 oxide layer in the Al2O3/HF system. The kinetic dominance of (R15) further substantiates the superior performance of PVDF in enhancing the combustion characteristics of modified Al powders.
3.2.4 ESP analysis.
A detailed electronic distribution and reactive site study of the product configurations of AlO/HF and Al2O3/HF systems was carried out by ESP analysis. ESP analysis is an important quantum chemical tool to visualize the charge distribution on the surface of a molecule, which in turn reveals the likelihood of the interaction of the molecule with nucleophilic and electrophilic reagents. The ESP surface polarity and the surface area plotted for different ESP ranges are shown in Fig. 10.
 |
| | Fig. 10 (a) ESP map and surface area of Pr in the AlO/HF system. (b) ESP map and surface area of Pr1 in the Al2O3/HF system. (c) ESP map and the surface area of Pr2 in the Al2O3/HF system. | |
In the Pr configuration of the AlO/HF system, the ESP analysis reveals that the highest ESP value on the molecular surface is located near the Al atom, reaching 32.58 kcal mol−1. This result indicates that the lone pair electrons around the Al atom create the strongest positive charge. This high electron density area easily attracts positive ions or molecules for reactions. The large positive charge area near the Al atom makes it highly reactive. Alternatively, the O atom has the strongest negative charge (−33.58 kcal mol−1). This negative area signifies a region of lower electron density, which readily attracts negatively charged ions or molecules. The Al and O atoms in the AlO/HF system show clear opposite behaviors, where Al readily donates electrons, while O strongly attracts them. This natural charge separation directly controls how bonds form and break in the system, explaining its unique reactivity patterns.
The ESP analysis of the final Al2O3/HF structures (Pr1 and Pr2) shows strong positive charges near the Al atoms, measuring 73.58 kcal mol−1 for Pr1 and 74.94 kcal mol−1 for Pr2. These high ESP values regions clearly mark the most reactive sites in both configurations. The extensive surface area of the high ESP regions near the Al atoms further confirms their pivotal role in the reaction. These results indicate that the exceptionally high electron density around the Al atoms makes them prime targets for nucleophilic reagents. The high ESP values of the Al atoms underscore their high reactivity, particularly in interactions with HF, where they facilitate chemical bond formation by providing electrons. The lowest ESP values are observed near the F atoms, measuring −27.33 kcal mol−1 and −28.69 kcal mol−1, respectively. Although the low ESP regions near the F atoms occupy a smaller surface area, their strong nucleophilicity renders them significant in the reaction. As a highly electronegative element, the F atoms exhibit low ESP values, reflecting their strong nucleophilic character. In interactions with Al atoms, the F atoms play a crucial role in promoting the breaking and reorganization of chemical bonds by attracting electrons.
4. Conclusions
In this study, the microscopic mechanism of PVDF pyrolysis and alumina elimination was revealed by theoretical calculations. Calculations using the B3LYP/6-311+G(d,p) method show that the C4F4H4 structure generates HF through a specific pathway, while the AlO/HF and Al2O3/HF reaction paths were computed using the B3LYP/6-311+G(3d,2p) basis set. Combined with the TST theory and ESP analysis, the reaction active sites and kinetic parameters were clarified, providing theoretical support for the application of Al/PVDF composites. Based on these analyses, the following conclusions can be drawn:
(1) In the C4F4H4 molecular system, the migration of the H1 atom is the key reaction step initiating HF formation. Kinetic analysis reveals that HF production predominates in the temperature range of 300–800 K, consistent with experimental observations, confirming this interval as the optimal thermodynamic window for HF generation. PVDF decomposes violently at high temperatures (>800 K), with a significant increase in HF yield. The reaction kinetic analysis showed that the reaction rate increased exponentially at 300–3000 K, which is in accordance with the transition state theory. The ESP analysis confirms that the strong positive charge of H atoms and the strong negative charge of F atoms jointly promote the formation of H–F bonds.
(2) Quantum chemical calculations reveal the reaction mechanisms of the AlO/HF and Al2O3/HF systems. The AlO/HF system undergoes three IM structures and four TS structures, and ultimately generates AlF and OH through H–F bond breaking and Al–F and O–H bond formation, with the (R7) channel dominating and overcoming the lowest reaction energy barrier of 3.18 kcal mol−1. The Al2O3/HF system is more complex, with six IM structures, seven TS structures and two Pr structures in four pathways, generating Al2O2F and OH through Al–O bond breaking and Al–F and O–H bond formation, with the (R15) channel (6.57 kcal mol−1) dominating the pathway. ESP analysis shows that the Al atoms are strongly positively charged and the O or F atoms are negatively charged, driving electrostatic interactions between the atoms.
(3) PVDF significantly improves Al combustion by generating HF, which removes the oxide layer and facilitates Al–F bond formation, with the reaction rate increasing markedly at higher temperatures. These results provide a theoretical basis for optimizing Al/PVDF composites, suggesting that a tailored PVDF coating thickness and reduced Al particle size could enhance the HF release efficiency and Al–F bond formation, thereby accelerating the combustion kinetics. Additionally, pre-treatment of Al surfaces to weaken the oxide layer or doping PVDF with catalytic additives may further improve their reactivity at lower temperatures.
Author contributions
Yajing Xun: writing – original draft, software, data curation, validation, visualization. Lijuan Yan: writing – review & editing, conceptualization, methodology. Yunlan Sun: resources, project administration. Xiaojing Li: validation. Hongjie Feng: validation. Kunru Ma: supervision.
Conflicts of interest
There are no conflicts to declare.
Data availability
Supplementary information is available. The supplementary information file contains all the table data information of the article. See DOI: https://doi.org/10.1039/d5cp02638e.
All data included in this study are available from the corresponding author upon request.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 52376093) and the Science Research Project of the Hebei Education Department (QN2025064).
Notes and references
- Y. C. Chiang, C. C. Shih and S. H. Tung,
et al.
, Polymer, 2018, 155, 146–151 CrossRef CAS.
- Q. Guo, Y. Huang and M. Xu,
et al.
, J. Membr. Sci., 2022, 664, 121115 CrossRef CAS.
- G. Magdy, A. H. Hassanin and I. Kandas,
et al.
, Mater. Chem. Phys., 2024, 325, 129760 CrossRef CAS.
- E. Korczeniewski, P. Bryk and P. Kowalczyk,
et al.
, Chem. Eng. J., 2024, 482, 148777 CrossRef CAS.
-
S. Rahmani, K. Hooshyari, S. Jamalpour, et al., Advanced Fluoropolymer Nanocomposites, Woodhead Publishing, 2023, pp. 213–245 Search PubMed.
- A. Chen, B. Wu and L. Li,
et al.
, Fuel, 2023, 331, 125726 CrossRef CAS.
- S. Ahmad, M. Nawaz and S. Mohammad,
et al.
, Surf. Coat. Technol., 2024, 476, 130172 Search PubMed.
- C. Ma, Y. Li and Y. Gao,
et al.
, Ceram. Int., 2024, 50, 24433–24441 CrossRef CAS.
- D. Wang, M. Zhong and J. Xue,
et al.
, Fuel, 2022, 327, 125199 Search PubMed.
- J. Wang, J. Chen and W. Cao, FirePhysChem, 2024, 4, 216–223 CrossRef.
- A. Koul, A. Ojha and P. Vimal,
et al.
, FirePhysChem, 2024, 4, 131–138 CrossRef.
- Q. Chen, X. Fu and W. Yang,
et al.
, Def. Technol., 2025, 46, 59–70 CrossRef.
- Z. Yang, H. Liu and M. Li,
et al.
, Def. Technol., 2025 DOI:10.1016/j.dt.2025.07.017.
- S. Sukumaran, S. Chatbouri and D. Rouxel,
et al.
, J. Intell. Mater. Syst. Struct., 2021, 32, 746–780 CrossRef CAS.
- G. W. Wang, D. Wang and X. L. Dong,
et al.
, Chem. Eng. J., 2017, 315, 509–515 CrossRef CAS.
- H. Li, J. Zhang and F. Zhao,
et al.
, Fuel, 2025, 390, 134689 CrossRef CAS.
- Y. C. Riwa, A. T. Banyikwa and R. Costa,
et al.
, Resour. Chem. Mater., 2025, 100124 Search PubMed.
- B. Ameduri, Prog. Polym. Sci., 2022, 133, 101591 Search PubMed.
- J. Ji, L. Liang and H. Xu,
et al.
, Combust. Flame, 2022, 238, 111925 Search PubMed.
- Y. Li, J. Li and B. Wang,
et al.
, Surf. Coat. Technol., 2022, 429, 127912 Search PubMed.
- D. W. Kim, K. T. Kim and T. S. Min,
et al.
, Sci. Rep., 2017, 7, 4659 CrossRef PubMed.
- J. B. DeLisio, X. Hu and T. Wu,
et al.
, J. Phys. Chem. B, 2016, 120, 5534–5542 Search PubMed.
- W. Z. Sun, Y. N. Li and W. K. Li,
et al.
, Chin. J. Explos. Propellants, 2021, 44(6), 856–864 Search PubMed.
- Y. Chen, W. Zhang and J. Xu,
et al.
, ACS Appl. Nano Mater., 2024, 7(11), 13347–13357 CrossRef CAS.
- S. Zhang, Y. Sun and J. Chen,
et al.
, Chin. J. Aeronaut., 2023, 36, 334–349 CrossRef.
- R. Padhye, A. J. Aquino and D. Tunega,
et al.
, ACS Appl. Mater. Interfaces, 2017, 9, 24290–24297 CrossRef CAS PubMed.
- M. N. Bello, A. Williams and I. Shancita,
et al.
, J. Mater. Sci., 2020, 55, 14229–14242 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CAS.
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- F. C. Destro, R. Fournet and R. Bounaceur,
et al.
, Combust. Flame, 2024, 270, 113731 Search PubMed.
-
A. Miyoshi, GPOP software, rev. 2022.01.20m1, available from the author Search PubMed.
- R. Klicka and L. Kubácek, Chemom. Intell. Lab. Syst., 1997, 39, 69–75 CAS.
- A. Amr, A. H. Salas and N. Al-Harbi,
et al., Alexandria Eng. J., 2023, 67, 461–472 CrossRef.
- R. Sundberg, Chemom. Intell. Lab. Syst., 1998, 41, 249–252 CrossRef CAS.
- L. Tian and F. Chen, J. Comput. Chem., 2012, 33, 580–592 Search PubMed.
- L. Tian and S. Manzetti, Struct. Chem., 2014, 25, 1521 CrossRef.
- S. Manzetti and L. Tian, J. Phys. Org. Chem., 2013, 26, 473–483 Search PubMed.
- Y. Ji, C. T. Jafvert and N. N. Zyaykina,
et al.
, J. Cleaner Prod., 2022, 367, 133112 CrossRef CAS.
- C. A. T. Soto, J. M. Ramos and A. C. C. Junior,
et al.
, Spectrochim. Acta A, 2013, 114, 475–485 CrossRef PubMed.
- H. Subramanian and M. P. Sibi, Asian J. Org. Chem., 2023, 12(6), 202300175 CrossRef.
- Z. Jin, Q. Jiang and C. Luo,
et al.
, Sustainable Mater. Technol., 2025, 43, e01195 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ac,
Lijuan
Yan
*ac,
Yunlan
Sun
ac,
Lijuan
Yan
*ac,
Yunlan
Sun










