
Therapeutic upregulation of DNA repair pathways: strategies and small molecule activators
Juhyung
Song†
 a,
Cheoljun
Park†
a,
Francis E. B.
Cabanting
b and
Yong Woong
Jun
*a
a,
Cheoljun
Park†
a,
Francis E. B.
Cabanting
b and
Yong Woong
Jun
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, Republic of Korea 43131. E-mail: ywjun@kaist.ac.kr
bDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, Republic of Korea 43131
First published on 11th October 2024
Abstract
DNA repair activity diminishes with age and genetic mutations, leading to a significantly increased risk of cancer and other diseases. Upregulating the DNA repair system has emerged as a potential strategy to mitigate disease susceptibility while minimizing cytotoxic side effects. However, enhancing DNA repair activity presents significant challenges due to the inherent inefficiency in activator screening processes. Additionally, pinpointing a critical target that can effectively upregulate overall repair processes is complicated as the available information is somewhat sporadic. In this review, we discuss potential therapeutic targets for upregulating DNA repair pathways, along with the chemical structures and properties of reported small-molecule activators. We also elaborate on the diverse mechanisms by which these targets modulate repair activity, highlighting the critical need for a comprehensive understanding to guide the development of more effective therapeutic strategies.
Introduction
The integrity of genomic DNA is persistently challenged by endogenous cellular processes, such as oxidative metabolism generating reactive oxygen species (ROS).1 If left unrepaired, accumulated DNA damage can lead to permanent genetic changes, resulting in mutagenesis, carcinogenesis, and neuro-degenerative diseases.2 To counteract these threats, the human body possesses sophisticated DNA repair systems to address varied DNA lesions (Fig. 1). DNA repair systems include base excision repair (BER), which repairs specific damaged bases via base excision by glycosylases; nucleotide excision repair (NER), which repairs bulkier lesions by removing a short single-stranded segment of DNA; homologous recombination (HR), which repairs double-strand breaks (DSBs) using a homologous sister chromatid; and nonhomologous end joining (NHEJ), which repairs DSBs through direct ligation.3,4 | ||
| Fig. 1 Illustration of DNA lesions, their repair pathways, and associated repair proteins. Proteins in blue represent those activatable by small molecules. | ||
Unfortunately, the efficiency and fidelity of DNA repair systems decline with age.5 Additionally, individuals with genetic mutations may be born with inherently attenuated levels of DNA repair activity, which can lead to significantly elevated risks of diseases, including cancer. Among the best characterized examples are individuals carrying BRCA-1 and BRCA-2 mutations, who have a 44–75% chance of developing breast cancer and an 18–40% risk of ovarian cancer over their lifetime.6 These relationships highlight the potential for upregulating DNA repair mechanisms as a strategy to reduce the likelihood of diseases in susceptible individuals.7,8
Despite the pharmaceutical focus on developing small molecules with therapeutic effects, there are currently just over 100 activator candidates in clinical phases II/III and only three FDA-approved activators: arimoclomol (chaperone activator), mitapivat (pyruvate kinase activator), and omaveloxolone (NRF2 activator).9,10 This reflects the challenging nature of identifying effective activators. Unlike active site binders, which can be systematically designed based on the structural information of proteins to bind to active sites and block interactions with substrates, activators must bind to allosteric sites to upregulate catalytic processes. The identification of these sites relies on random compound screening, which is often inefficient. Moreover, the potential efficacy of identified activators remains uncertain.9 While inhibiting a DNA repair enzyme often effectively attenuates the entire DNA repair process, upregulating a single DNA repair enzyme does not necessarily enhance overall repair activity, particularly if the enzymatic activity is not the rate-limiting step. Although current development efforts focus on direct activation of DNA repair enzymes, such as OGG1 and MTH1, the intricate interplay among varied proteins involved in DNA repair processes suggest that enhancing a critical component is pivotal to achieving significant upregulation of the overall repair process.11–14
In this review, we aim to provide a comprehensive overview of potential therapeutic targets for upregulating DNA repair activity. These targets include not only the DNA repair enzymes but also other proteins that enhance DNA repair by activating repair enzymes, increasing their expression levels, altering the local chromatin environment to improve enzyme access, and modulating global cellular conditions to minimize DNA damage. We also present a list of reported activators for these targets, along with their properties and observed efficacy.
8-Oxoguanine DNA glycosylase 1 (OGG1)
One of the most prevalent forms of oxidative DNA damage is 8-oxoguanine (8-oxoG), which results from ROS generated during cellular metabolism.15 Additionally, ROS can generate 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) under reductive conditions.16,17 8-Oxoguanine DNA glycosylase 1 (OGG1), a bifunctional enzyme catalyzing both a glycosylation reaction to remove the oxidized bases and an AP lyase reaction via β-elimination, is responsible for recognizing and excising those damaged guanines from DNA.18,19 As OGG1 performs the first, and also the rate-determining step in the BER pathway of the oxidized bases, it has been suggested that upregulating OGG1 may enhance overall BER activity in cells.9Several small molecules have been identified as OGG1 activators (Fig. 2a). In 2018, Bohr et al. identified six compounds using in vitro fluorescence-based 8-oxoG cleavage assay.11 Under oxidative stress conditions induced by 48-hour incubation with 0.6 mM paraquat (PQ) in the A549 cell line, treatment with 30 μM of compound B, D, and F was found to decrease 8-oxoG levels and prevent nuclear condensation. Among these, compound F emerged as the most effective activator in preventing the accumulation of 8-oxoG and PQ-induced cytotoxicity with an EC50 of 5.0 μM, the concentration at which 50% of maximal activity was observed.12
 | ||
| Fig. 2 Illustration of the DNA repair mechanisms of (a) OGG1, (b) MTH1, and (c) BRCA1, along with the chemical structures of reported activators of each enzyme. | ||
Subsequently, Rumsey et al. extended the in vitro screening process, identifying 13 candidates (EC50 = 0.35–6.4 μM), four of which overlapped with the previous report.12 Furthermore, incubating A549 cells with either compound F (30 μM) or A (50 μM) for 48 hours led to an increase in OGG1 content by 142.3% and 227.4%, respectively, which is significantly higher compared to 88.3% increase in the control group treated with 0.6 mM PQ. Additionally, a substantial increase (10–50%) in DNA ligase III protein levels was also noted upon the treatment with >30 μM of compound A, B, D, or F. In 2022, Helleday et al. revealed the mechanism of compound C using in vitro X-ray crystallography technique, demonstrating that it upregulates OGG1 enzyme activity by interacting with Phe 319 and Gly 42 on OGG1, leading to a ten-fold increase in enzymatic activity through the generation of a novel β,δ-lyase function.13
Coenzyme Q10 (CoQ10) and its reduced form (CoQH2) also have been shown to upregulate OGG1 activity at micromolar concentrations, resulting in over 400% in human fibroblast cells and over 200% in human lymphocyte enhanced OGG1 activity.20,21
Human MutT homolog 1 (MTH1)
While OGG1 excises 8-oxoG from DNA, human MutT homolog 1 (MTH1) cleanses the oxidatively damaged cellular nucleotide pool. This nucleotide surveillance enzyme recognizes 8-oxo-dGTP and hydrolyzes the triphosphate moiety between the α and β phosphates to convert it into 8-oxo-dGMP, preventing its misincorporation into DNA.22 Overexpression of MTH1 in mismatch repair (MMR)-defective cancer cells has been shown to reduce genetic mutations and DNA damage responses, suggesting that MTH1 activators could lower mutagenesis.23,24In 2022, Kool et al. discovered that nilotinib, a tyrosine kinase inhibitor, directly upregulates the activity of MTH1 using a previously developed in vitro MTH1 activity assay based on a chimeric ATP-linked nucleotide (Fig. 2b).14,25 Further structural optimization led to the development of compounds with potent upregulation of MTH1 in vitro and in human cell lines. SU0448, a benzylic morpholine substituent, exhibited up to a 10-fold increase in MTH1 activity at 10 μM in vitro (EC50 = 5.5 μM), while showing moderate toxicity (IC50 = 10–38 μM) and less than 30% inhibition or activation across all tested kinases. The preliminary docking studies in silico suggest that these activators wrap around the substrate pocket, interacting with the triphosphate unit of 8-oxo-dGTP.
Breast cancer type 1 susceptibility protein (BRCA1)
The breast cancer susceptibility gene 1 (BRCA1), a tumor suppressor gene, plays an important role in DSB repair, BER of oxidative DNA damage, and the expression of NER genes (XPC, DDB2, and GADD45).26–28 Mutation in BRCA1 limits these DNA repair processes, significantly increasing the lifetime risk for developing breast cancer and ovarian cancer (they have a 44–75%, and 18–40% chance, respectively).6,29BRCA1-mutated cancers, which often contain “triple-negative” TP53 mutations (lacking estrogen receptor, progesterone receptor, and HER2/NEU oncogene), tend to be resistant to existing chemoprevention agents, resulting in poor outcome of the treatment.7 Thus, upregulating the DNA repair system in the presence of mutated BRCA1 is proposed as a strategy to compensate for the inhibitory effect of mutant BRCA1 on DNA repair activity.In 2014, Ford et al. identified acetohexamide and benserazide from the LOPAC1280 library as DNA repair activators (Fig. 2c).7 These compounds showed a dose-dependent increase in the repair of an oxidatively damaged GFP reporter gene in BRCA1-mutant SUM149PT cells but not in wild-type BRCA1 MCF7 breast cancer cells, implying the restoration of DNA repair activity. Acetohexamide and beserazide increased the repair activity in SUM149PT cells by up to 90% (at 1 μM) and 60% (at 100 μM), respectively. Benserazide delayed the rate of tumor formation and reduced tumor growth in mice in a dose-dependent manner; administration of benserazide (250 mg kg−1) through i.p. injection increased tumor resistance/survival rate in mice by up to 70%.
Curcumin, a natural polyphenol from turmeric, shows notable inhibition on antiproliferative and apoptotic effects, and the formation of ROS. Curcumin has also been shown to attenuate DNA damage by upregulating the expression of BRCA1, BRCA2, and ERCC1.30 Treatment with 5 μM curcumin in bone marrow cells increased the expression of BRCA1 and BRCA2 over 170%, activating subsequent NER and HR processes.
The Sirtuin protein (SIRT)
Sir2 proteins, members of sirtuin (SIRT) family, regulate various physiological responses including cell fate, stress, and aging through the deacetylation of specific substrate proteins.31 Seven sirtuins (SIRT1–7) have been identified in human cells so far, each with distinct roles.32SIRT1, a human homologue of Sir2, is implicated in DNA damage response and repair. Upon DNA damage, it increases cell survival rate through binding to and deacetylating the p53 tumor suppressor, which mediates gene expression of proteins involved in NER (see transcriptional activators section below).33 SIRT1 also enhances DNA repair capacity under DNA damaging conditions by binding to and deacetylating the repair protein Ku70, which is critical for DSB repair through NHEJ pathway.34,35 Overexpression of SIRT1 in a mouse model has been shown to increase the survival rate by approximately 46% and decreased the frequency of fatal thymic lymphoma upon γ-irradiation by around 45% compared to controls.36
SIRT3, which plays a crucial role in regulating mitochondrial function, also upregulates DNA repair capacity through the deacetylation of various proteins. SIRT3 is involved in the deacetylation of OGG1, which stabilizes the enzyme and enhances its ability to excise 8-oxoG lesions.37 Additionally, SIRT3 deacetylates Lys 56 on histone H3 (H3K56), thereby recruiting p53-binding protein 1 (53BP1) to promote NHEJ.38 Furthermore, SIRT3 has been reported to stimulate the DNA damage response, significantly reducing the number of DSB in non-small cell lung cancer (NSCLC) cells.39
Considerable efforts have been made for the development of sirtuin activators, which can be broadly classified into two categories: natural compounds and synthetic sirtuin-activating compounds (STACs). Resveratrol, a well-known polyphenolic compound, acts as an allosteric activator with an EC1.5 value of 46.3 μM in vitro, representing the concentration needed to achieve a 50% increase in activity (Fig. 3). Oleic acid, a natural fatty acid found in various animal and vegetable sources, has been reported to increase the activity of SIRT6 with an EC50 of 90 μM in vitro.40 Resveratrol, a well-known polyphenolic compound, acts as an allosteric activator of SIRT1 with an EC1.5 value of 46.3 μM in vitro. In contrast, synthetic STACs, as developed by Westphal et al. show substantially greater potency with EC1.5 values ranging from 0.16 to 2.9 μM in vitro, underscoring their enhanced effectiveness compared to natural compounds.41
 | ||
| Fig. 3 Illustration of relationship between SIRT proteins and DNA repair activities, along with the chemical structures of SIRT-activators. | ||
AMP-activated protein kinase (AMPK)
AMP-activated protein kinase (AMPK) plays a pivotal role in modulating cellular metabolic pathways and protecting against metabolic stress, such as ROS.42 AMPK activation is intricately linked to DNA repair through BER in several ways. Firstly, AMPK phosphorylates and activates poly (ADP-ribose) polymerase-1 (PARP-1), which facilitates the repair of single strand break (SSB) by recruiting multiple X-ray repair cross-complementing protein 1 (XRCC1) to the site of damage.43 Notably, while PARP-1 inhibition is a well-established therapeutic strategy, its activation using nicotinamide, a coenzyme of PARP-1, enhances DNA repair pathways. This enhancement occurs by opening condensed chromatin structures and forming ADP-ribose polymers, which subsequently recruit DNA repair complexes.44 Additionally, AMPK activation is also associated with the upregulation of OGG1 activity by increasing SIRT3 expression levels in osteoarthritis (OA) chondrocytes.45 Lastly, AMPK phosphorylates p53 at Ser 15, triggering AMPK-dependent cell-cycle arrest and further activation of BER pathway.46,47AMPK activators have been extensively studied as therapeutic agents, and categorized into three groups. The first includes AMP analogs, such as AICAR reported by Beri et al., the first known pharmacological AMPK-activator for acute lymphoblastic leukemia (Fig. 4).48,49 AICAR-mediated AMPK activation inhibits mTORC1, leading to significantly higher OGG1 expression and reduced oxidative DNA damage.
 | ||
| Fig. 4 Illustration of relationship between AMPK and DNA repair activities, along with the chemical structures of AMPK activators. | ||
For instance, treatment with AICAR (2 mM) in the murine proximal tubular epithelial cells under high glucose conditions (25 mM) resulted in a 5-fold increase in OGG1 levels compared to untreated controls. Furthermore, AICAR administration (2 mg kg−1, 5 days a week for 4 weeks) to diabetic mice halved the total amount of 8-oxoG in the kidney.50
The second group consists of compounds that bind directly to the β-subunit of AMPK. Cool et al. identified the first example of this group containing a thienopyridone core, A769662. This compound exhibited significantly higher potency in vitro (EC50 = 0.116 ± 0.025 μM) than the conventional AMPK activator, AMP (EC50 = 6 ± 3 μM).51,52 Furthermore, administration of A769662 activated AMPK, thereby upregulating the level of SIRT3 and OGG1, leading to decreased amount of mitochondrial DNA damage in human knee OA chondrocyte.53
The third group comprises activators that indirectly stimulate AMPK by affecting the AMP/ATP ratio, disturbing respiratory chain reactions, and interfering with carbohydrate uptake or ATP generation.43 These are primarily polyphenols and alkaloids such as resveratrol, metformin, and curcumin. Interestingly, these activators have also been implicated in other DNA repair pathways. For instance, metformin, a first-line oral glucose-lowering agent for type 2 diabetes, increases BER activity via significantly upregulating XRCC1 and p53 levels.47 Resveratrol, a natural phytoestrogen known for modulating the rate of senescence, increases OGG1 activity and expression, thereby reducing DNA damage markers, such as 8-oxoG and γH2AX by approximately 30% in human peritoneal mesothelial cells treated with 0.5 μM.54 Polyphenols from green tea have been shown to expedite the repair of UV-induced DNA damage (pyrimidine dimer) in NER-proficient mice by enhancing NER gene levels. Treatment with green tea polyphenols (0.5% w/v) reduced the number of cyclobutene pyrimidine dimer-positive cells by half compared to control.55
Transcriptional activators (TA)
Transcriptional activators (TAs) are proteins that bind to specific genes, boosting their transcription by facilitating the formation of transcription initiation complexes.56,57 TAs can also augment DNA repair systems by disengaging chromatin and recruiting factors that stimulate DNA repair processes.58 This leads to preferential DNA repair of the transcribed strand of genes and promoter surrounding sequences.59,60Potent TAs, such as Gal4-VP16 and the retinoic acid receptor (RAR), have been found to stimulate NER processes to remove DNA damage in promoter regions in vivo, with an observed 2- to 5-fold increase.58 Interestingly, this DNA repair activation is not directly mediated by the transcription machinery but rather results from removing barriers that restrict the access and recruiting DNA repair factors to the lesion.
TAp63γ binds to a group of p53-target genes, increasing the expression levels of the p53 tumor suppressor proteins which enhance NER of UV-induced DNA damage through strong transcriptional induction of DDB2, XPC, and GADD45 at both mRNA and protein levels.61,62 Treatment with thymidine dinucleotides (pTpT), a known activator of the p53 tumor suppressor protein, in skin fibroblasts exhibited nuclear accumulation of p53, upregulating a set of genes involved in DNA repair (ERCC3 and GADD45) and cell cycle inhibition (SD11), thus enhancing repair of UV-induced DNA damage (Fig. 5a).63 Additionally, pTpT-treatment has been shown to enhance the repair of DNA damage caused by chemical carcinogens such as benzo[a]pyrene, commonly found in food cooked at high temperatures.64,65
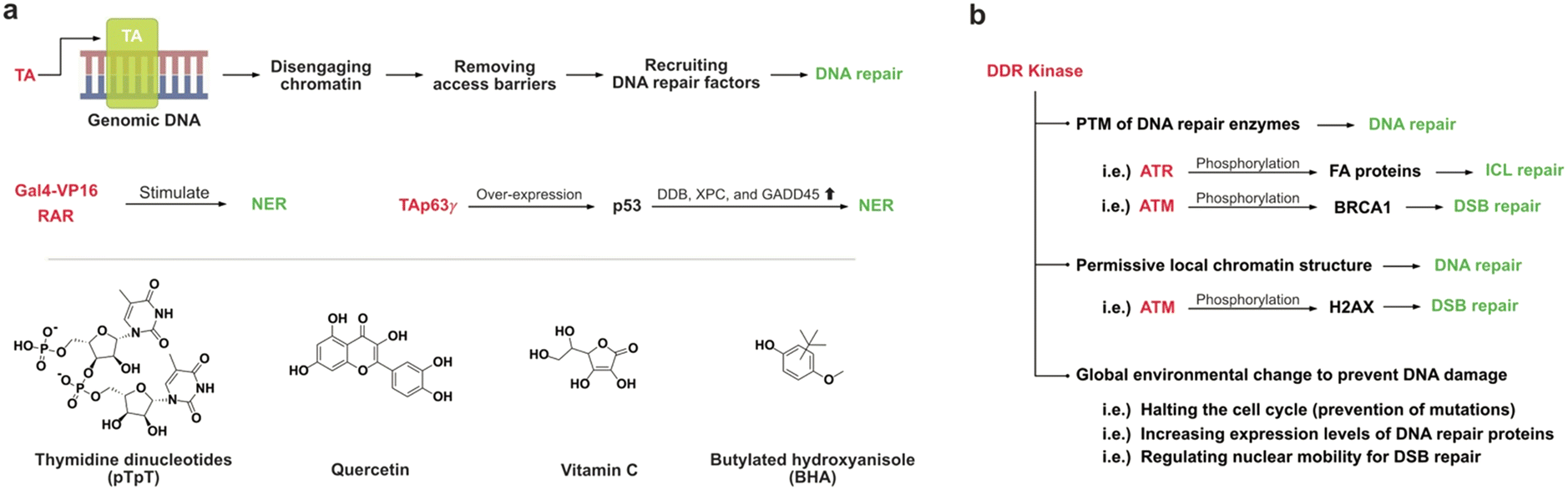 | ||
| Fig. 5 (a) Illustration of mechanisms that transcriptional activators modulate DNA repair activities, along with the chemical structures of their activators. (b) Three different mechanisms that DDR kinases modulate DNA repair activities. | ||
Nuclear factor erythroid 2-related factor 2 (NRF2), critical in the cellular response to oxidative stress, upregulates OGG1 expression by binding to the OGG1 promoter.66 Common antioxidants, such as vitamin C and butylated hydroxyanisole (BHA), interact with the promotor region of NRF2, increasing NRF2 levels that consequently upregulate OGG1 expression levels and decrease the amount of 8-oxoG in MCF-10A cells.67
Administration of quercetin (50 mg kg−1), a widely recognized bioflavonoid found in fruits and vegetables, reversed the initially decreased level of NRF2 in mice treated with the DNA alkylating agent 1,2-dimethylhydrazine (20 mg kg−1). This treatment increased the expression levels of OGG1, apurinic/apyrimidinic endonuclease 1 (APE1), and XRCC1 by up to 2-fold, leading to an approximately 15% AP site reduction.68
DNA damage response (DDR) kinases
DNA damage response (DDR) kinases, including DNA-dependent protein kinases (DNA-PKcs), ataxia telangiectasia-mutated kinase (ATM), and ATM and Rad3-related proteins (ATR), play a crucial role in regulating multiple DNA repair mechanisms at DNA lesions.69 These kinases enhance the efficiency of DNA repair through several different ways (Fig. 5b).First, these kinases phosphorylate DNA repair enzymes, a post-translational modification (PTM) that modulates their repair activities, including the repair of interstrand crosslinks (ICLs) by ATR.70 ATR phosphorylates several Fanconi anemia (FA) proteins, including FANCD2, FANCI, FANCA, and FANCG, which are essential components of the FA complex involved in ICL repair.71,72
Second, DDR kinases initiate complex changes in the local chromatin structure near DNA lesions, creating a permissive environment for DNA repair by recruiting additional DDR factors. For instance, ATM phosphorylates the histone variant H2AX at a conserved serine residue (Ser 139), producing γH2AX at DSBs.73 A complex involving γH2AX recruits additional ATM molecules to the flanking chromatin regions, facilitating the propagation of H2AX phosphorylation and further recruitment of numerous chromatin-modifying, DDR signaling, and DNA repair proteins.69,74
Finally, in addition to activating DNA repair through direct regulation of repair proteins, DDR kinases also act at a global level to alter the overall cellular environment, making it more conducive to repair. It behaves through (i) halting the cell cycle to provide time for DNA damage repair before DNA replication, (ii) increasing the gene expression levels of DNA repair proteins mediated through p53 regulation, and (iii) regulating global nuclear mobility to promote DSB repair processes.69
To our best knowledge, there is a lack of documented data on small molecule activators of DDR kinases. However, several reports describe the upregulation of DNA repair activity through the overexpression of proteins that interact with these kinases. Nijmegen breakage syndrome 1 (NBS1) protein, which forms the MRN complex along with MRE11 and RAD50, is essential for the activation of ATM in response to DSBs.75 The MRN complex facilitates the recruitment of ATM to DSB sites, initiating downstream repair processes. Overexpression of NBS1 in G0 phase U2OS cells exhibited increased chromatin association of HR factors, namely RAD51 and RPA. DNA topoisomerase-binding protein 1 (TopBP1) activates ATR by interacting with ATR-interacting protein (ATRIP) at DSB site to facilitate repair.76 Experiments using Xenopus laevis extracts have shown that the addition of exogenous TopBP1 can induce ATP activation.77
Given the complex network of interactions among DDR components, a thorough understanding and organization of this network are essential for designing and developing small molecule activators of DDR kinases.78
Conclusion
Given that the extension of ‘healthspan’ becomes an increasingly important goal in modern medicine, the decline in DNA repair activity with aging represents a significant obstacle, particularly in cancer prevention. The growing body of evidence supports the significance of upregulating DNA repair system as a promising approach to mitigate the risk of age-associated diseases that result from the accumulation of DNA damage. Despite the notable advances in identifying small molecule activators for DNA repair enzymes, the field is still in its infancy.One of the foremost challenges in this area is the inherent difficulty of identifying small-molecule activators. As discussed, targeting allosteric sites of enzymes precludes the use of structural information for rational drug design, as these sites are often less well-defined. Moreover, the highly optimized nature of protein structures, which has been evolved over millions of years, makes further enhancement via allosteric modulation a daunting task. In this context, the integration of artificial intelligence (AI) into activator discovery holds great promise, offering new avenues for upregulating biological processes.
The difficulty of upregulating overall DNA repair activity through the activation of individual enzymes represents another critical challenge. The intricate interplay between varied DNA repair pathways and their regulatory networks demands a nuanced and systemic approach to drug discovery. This review has highlighted potential therapeutic targets for DNA repair upregulation, along with the chemical structures and efficacy of reported small-molecule activators. These insights should serve as a valuable foundation for future research and development in this area.
While upregulating the DNA repair system is a promising therapeutic strategy, it can paradoxically lead to side effects in certain individuals. For instance, in patients undergoing anticancer therapies like chemotherapy or radiation, which target DNA damage, enhanced DNA repair activity may promote resistance to these treatments. Furthermore, excessive activation of DNA repair processes could provoke genetic instability due to frequent nick generation and may trigger an inflammatory response, resulting in tissue damage.
It is noteworthy that while DNA repair systems prevent genetic mutations by repairing DNA damage before replication, they cannot reverse mutations that have already occurred. Therefore, the elderly and susceptible individuals may require regular administration of DNA repair activators to mitigate mutations from ongoing DNA damage. This necessitates taking into consideration the long-term safety and low toxicity of these activators in drug development. In this regard, some natural supplements highlighted in this review may offer a promising starting point, given their favorable safety profiles and potential efficacy. While the challenges ahead in this field are substantial, advancing beyond these initial steps is crucial to fully harness the potential of DNA repair upregulation, aiming to enhance the quality of life that typically diminishes with age.
Data availability
This manuscript is a review article and does not include any new data set. All data discussed in this review are available from the original sources cited throughout the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) [RS-2024-00346077, RS-2024-00399739].Notes and references
- S. Ragu, N. Droin, G. Matos-Rodrigues, A. Barascu, S. Caillat, G. Zarkovic, C. Siberchicot, E. Dardillac, C. Gelot and J. Guirouilh-Barbat, Cell Death Differ., 2023, 30, 1349–1365 CrossRef.
- R. Huang and P.-K. Zhou, Signal Transduct. Tar., 2021, 6, 254 CrossRef PubMed.
- C. J. Lord and A. Ashworth, Nature, 2012, 481, 287–294 CrossRef.
- Y. W. Jun and E. T. Kool, Acc. Chem. Res., 2022, 55, 3495–3506 CrossRef PubMed.
- D. B. Lombard, K. F. Chua, R. Mostoslavsky, S. Franco, M. Gostissa and F. W. Alt, Cell, 2005, 120, 497–512 CrossRef PubMed.
- S. Chen and G. Parmigiani, J. Clin. Oncol., 2007, 25, 1329–1333 CrossRef PubMed.
- E. Alli, D. Solow-Cordero, S. C. Casey and J. M. Ford, Cancer Res., 2014, 74, 6205–6215 CrossRef CAS PubMed.
- E. Alli and J. M. Ford, Mol. Cell. Oncol., 2015, 2, e979685 CrossRef PubMed.
- L. Dow, A. Case, M. Paustian, B. Pinkerton, P. Simeon and P. C. Trippier, RSC Med. Chem., 2023, 14, 2206–2230 RSC.
- A. Mullard, Nat. Rev. Drug Discovery, 2024, 23, 88–95 CrossRef CAS.
- B. A. Baptiste, S. R. Katchur, E. M. Fivenson, D. L. Croteau, W. L. Rumsey and V. A. Bohr, Free Radical Biol. Med., 2018, 124, 149–162 CrossRef CAS.
- G. Tian, S. R. Katchur, Y. Jiang, J. Briand, M. Schaber, C. Kreatsoulas, B. Schwartz, S. Thrall, A. M. Davis, S. Duvall, B. A. Kaufman and W. L. Rumsey, Sci. Rep., 2022, 12, 14685 CrossRef.
- M. Michel and T. Helleday, et al. , Science, 2022, 376, 1471–1476 CrossRef CAS.
- Y. Lee, Y. Onishi, L. McPherson, A. M. Kietrys, M. Hebenbrock, Y. W. Jun, I. Das, S. Adimoolam, D. Ji, M. G. Mohsen, J. M. Ford and E. T. Kool, ACS Chem. Biol., 2022, 17, 2074–2087 CrossRef CAS PubMed.
- Y.-K. Tahara, A. M. Kietrys, M. Hebenbrock, Y. Lee, D. L. Wilson and E. T. Kool, ACS Chem. Biol., 2019, 14, 2606–2615 CrossRef.
- S. Gao, Y. Tahara, E. T. Kool and M. M. Greenberg, Nucleic Acids Res., 2024, 52, 7437–7446 CrossRef.
- M. A. Lovell and W. R. Markesbery, Nucleic Acids Res., 2007, 35, 7497–7504 CrossRef.
- M. Dizdaroglu, P. Jaruga, M. Birincioglu and H. Rodriguez, Free Radical Biol. Med., 2002, 32, 1102–1115 CrossRef PubMed.
- S. D. Bruner, D. P. Norman and G. L. Verdine, Nature, 2000, 403, 859–866 CrossRef.
- D. Schniertshauer, D. Gebhard, H. van Beek, V. Nöth, J. Schon and J. Bergemann, DNA Repair, 2020, 87, 102784 CrossRef PubMed.
- M. Tomasetti, R. Alleva and A. R. Collins, FASEB J., 2001, 15, 1425–1427 CrossRef PubMed.
- E. Markkanen, DNA Repair, 2017, 59, 82–105 CrossRef.
- P. Rai, J. J. Young, D. G. Burton, M. G. Giribaldi, T. T. Onder and R. A. Weinberg, Oncogene, 2011, 30, 1489–1496 CrossRef PubMed.
- M. T. Russo, M. F. Blasi, F. Chiera, P. Fortini, P. Degan, P. Macpherson, M. Furuichi, Y. Nakabeppu, P. Karran and G. Aquilina, Mol. Cell. Biol., 2004, 24, 465–474 CrossRef.
- D. Ji, A. A. Beharry, J. M. Ford and E. T. Kool, J. Am. Chem. Soc., 2016, 138, 9005–9008 CrossRef.
- T. Saha, J. K. Rih, R. Roy, R. Ballal and E. M. Rosen, J. Biol. Chem., 2010, 285, 19092–19105 CrossRef PubMed.
- A.-R. Hartman and J. M. Ford, Nat. Genet., 2002, 32, 180–184 CrossRef.
- R. Scully, S. Ganesan, K. Vlasakova, J. Chen, M. Socolovsky and D. M. Livingston, Mol. Cell, 1999, 4, 1093–1099 CrossRef PubMed.
- E. Alli, V. B. Sharma, P. Sunderesakumar and J. M. Ford, Cancer Res., 2009, 69, 3589–3596 CrossRef.
- X. Chen, J. Wang, Z. Fu, B. Zhu, J. Wang, S. Guan and Z. Hua, Sci. Rep., 2017, 7, 17724 CrossRef PubMed.
- S.-I. Imai, C. M. Armstrong, M. Kaeberlein and L. Guarente, Nature, 2000, 403, 795–800 CrossRef PubMed.
- R. A. Frye, Biochem. Biophys. Res. Commun., 2000, 273, 793–798 CrossRef PubMed.
- W. Y. Chen, D. H. Wang, R. C. Yen, J. Luo, W. Gu and S. B. Baylin, Cell, 2005, 123, 437–448 CrossRef.
- J. Jeong, K. Juhn, H. Lee, S.-H. Kim, B.-H. Min, K.-M. Lee, M.-H. Cho, G.-H. Park and K.-H. Lee, Exp. Mol. Med., 2007, 39, 8–13 CrossRef.
- P. Pace, G. Mosedale, M. R. Hodskinson, I. V. Rosado, M. Sivasubramaniam and K. J. Patel, Science, 2010, 329, 219–223 CrossRef.
- P. Oberdoerffer, S. Michan, M. McVay, R. Mostoslavsky, J. Vann, S.-K. Park, A. Hartlerode, J. Stegmuller, A. Hafner and P. Loerch, Cell, 2008, 135, 907–918 CrossRef PubMed.
- Y. Cheng, X. Ren, A. S. Gowda, Y. Shan, L. Zhang, Y. Yuan, R. Patel, H. Wu, K. Huber-Keener and J. Yang, Cell Death Dis., 2013, 4, e731–e731 CrossRef.
- A. Sengupta and D. Haldar, DNA Repair, 2018, 61, 1–16 CrossRef.
- K. Cao, Y. Chen, S. Zhao, Y. Huang, T. Liu, H. Liu, B. Li, J. Cui, J. Cai and C. Bai, J. Cancer, 2021, 12, 5464–5472 CrossRef.
- J. L. Feldman, J. Baeza and J. M. Denu, J. Biol. Chem., 2013, 288, 31350–31356 CrossRef PubMed.
- J. C. Milne and C. H. Westphal, et al. , Nature, 2007, 450, 712–716 CrossRef.
- D. Garcia and R. J. Shaw, Mol. Cell, 2017, 66, 789–800 CrossRef PubMed.
- M. Szewczuk, K. Boguszewska, J. Kaźmierczak-Barańska and B. T. Karwowski, Mol. Biol. Rep., 2020, 47, 9075–9086 CrossRef.
- D. Surjana, G. M. Halliday and D. L. Damian, J. Nucleic Acids, 2010, 2010, 157591 CrossRef.
- L.-Y. Chen, Y. Wang, R. Terkeltaub and R. Liu-Bryan, Osteoarthr. Cartil., 2018, 26, 1539–1550 CrossRef.
- R. G. Jones, D. R. Plas, S. Kubek, M. Buzzai, J. Mu, Y. Xu, M. J. Birnbaum and C. B. Thompson, Mol. Cell, 2005, 18, 283–293 CrossRef.
- I. D. Turacli, T. Candar, E. B. Yuksel, S. Kalay, A. K. Oguz and S. Demirtas, Biochimie, 2018, 154, 62–68 CrossRef.
- G. M. Leclerc, G. J. Leclerc, G. Fu and J. C. Barredo, J. Mol. Signaling, 2010, 5, 1–13 CrossRef PubMed.
- J. E. Sullivan, K. J. Brocklehurst, A. E. Marley, F. Carey, D. Carling and R. K. Beri, FEBS Lett., 1994, 353, 33–36 CrossRef.
- S. L. Habib, A. Yadav, D. Kidane, R. H. Weiss and S. Liang, Cell Cycle, 2016, 15, 3048–3059 CrossRef.
- O. Göransson, A. McBride, S. A. Hawley, F. A. Ross, N. Shpiro, M. Foretz, B. Viollet, D. G. Hardie and K. Sakamoto, J. Biol. Chem., 2007, 282, 32549–32560 CrossRef.
- B. Cool, B. Zinker, W. Chiou, L. Kifle, N. Cao, M. Perham, R. Dickinson, A. Adler, G. Gagne and R. Iyengar, Cell Metab., 2006, 3, 403–416 CrossRef.
- R. Liu-Bryan, Y. Wang and R. Terkeltaub, Osteoarthr. Cartil., 2015, 23, A157–A158 CrossRef.
- J. Mikuła-Pietrasik, A. Kuczmarska, B. Rubiś, V. Filas, M. Murias, P. Zieliński, K. Piwocka and K. Książek, Free Radical Biol. Med., 2012, 52, 2234–2245 CrossRef.
- S. K. Katiyar, M. Vaid, H. van Steeg and S. M. Meeran, Cancer Prev. Res., 2010, 3, 179–189 CrossRef.
- M. Ptashne, Nature, 1988, 335, 683–689 CrossRef PubMed.
- S. Björklund, G. Almouzni, I. Davidson, K. P. Nightingale and K. Weiss, Cell, 1999, 96, 759–767 CrossRef.
- P. Frit, K. Kwon, F. Coin, J. Auriol, S. Dubaele, B. Salles and J.-M. Egly, Mol. Cell, 2002, 10, 1391–1401 CrossRef PubMed.
- I. Mellon, G. Spivak and P. C. Hanawalt, Cell, 1987, 51, 241–249 CrossRef CAS PubMed.
- Y. Tu, S. Tornaletti and G. Pfeifer, EMBO J., 1996, 15, 675–683 CrossRef PubMed.
- J. Liu, M. Lin, C. Zhang, D. Wang, Z. Feng and W. Hu, DNA Repair, 2012, 11, 167–176 CrossRef.
- S. Adimoolam and J. M. Ford, DNA Repair, 2003, 2, 947–954 CrossRef.
- M. S. Eller, T. Maeda, C. Magnoni, D. Atwal and B. A. Gilchrest, Proc. Natl. Acad. Sci., 1997, 94, 12627–12632 CrossRef.
- T. Maeda, M. S. Eller, M. Hedayati, L. Grossman and B. A. Gilchrest, DNA Repair, 1999, 433, 137–145 CrossRef.
- Y. W. Jun, M. Kant, E. Coskun, T. A. Kato, P. Jaruga, E. Palafox, M. Dizdaroglu and E. T. Kool, ACS Cent. Sci., 2023, 9, 1170–1179 CrossRef PubMed.
- Q. Shang, C. Pan, X. Zhang, T. Yang, T. Hu, L. Zheng, S. Cao, C. Feng, X. Hu and X. Chai, J. Biol. Chem., 2023, 299, 102798 CrossRef.
- B. Singh, A. Chatterjee, A. M. Ronghe, N. K. Bhat and H. K. Bhat, BMC Cancer, 2013, 13, 1–9 CrossRef.
- S. G. Darband, S. Sadighparvar, B. Yousefi, M. Kaviani, F. Ghaderi-Pakdel, A. Mihanfar, Y. Rahimi, K. Mobaraki and M. Majidinia, Life Sci., 2020, 253, 117584 CrossRef CAS PubMed.
- B. M. Sirbu and D. Cortez, Cold Spring Harbor Perspect. Biol., 2013, 5, a012724 Search PubMed.
- P. R. Andreassen, A. D. D'Andrea and T. Taniguchi, Genes Dev., 2004, 18, 1958–1963 CrossRef CAS.
- M. Ishiai, H. Kitao, A. Smogorzewska, J. Tomida, A. Kinomura, E. Uchida, A. Saberi, E. Kinoshita, E. Kinoshita-Kikuta and T. Koike, Nat. Struct. Mol. Biol., 2008, 15, 1138–1146 CrossRef.
- J. Wilson, K. Yamamoto, A. Marriott, S. Hussain, P. Sung, M. Hoatlin, C. Mathew, M. Takata, L. Thompson and G. Kupfer, Oncogene, 2008, 27, 3641–3652 CrossRef PubMed.
- O. Fernandez-Capetillo, A. Lee, M. Nussenzweig and A. Nussenzweig, DNA Repair, 2004, 3, 959–967 CrossRef PubMed.
- A. Al-Hakim, C. Escribano-Diaz, M.-C. Landry, L. O'Donnell, S. Panier, R. K. Szilard and D. Durocher, DNA Repair, 2010, 9, 1229–1240 CrossRef.
- H. Zhou, K. Kawamura, H. Yanagihara, J. Kobayashi and Q.-M. Zhang-Akiyama, J. Radiat. Res., 2017, 58, 487–494 CrossRef.
- D. A. Mordes and D. Cortez, Cell Cycle, 2008, 7, 2809–2812 CrossRef PubMed.
- A. E. Burrows and S. J. Elledge, Genes Dev., 2008, 22, 1416–1421 CrossRef.
- A. Bensimon, R. Aebersold and Y. Shiloh, FEBS Lett., 2011, 585, 1625–1639 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |