
Novel sulfonamides unveiled as potent anti-lung cancer agents via tumor pyruvate kinase M2 activation†
Rudradip
Das
 ,
Deep Rohan
Chatterjee
,
Saumya
Kapoor
,
Het
Vyas
and
Amit
Shard
*
,
Deep Rohan
Chatterjee
,
Saumya
Kapoor
,
Het
Vyas
and
Amit
Shard
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research-Ahmedabad (NIPER-A), Opposite Airforce station, Palaj, Gandhinagar, Gujarat – 382355, India. E-mail: amit@niperahm.res.in
First published on 11th July 2024
Abstract
This rational pursuit led to the identification of a novel sulfonamide derivative as a potent anti-lung cancer (LC) compound. Considering these results, we synthesized 38 novel sulfonamide derivatives with diverse skeletal structures. In vitro cytotoxicity assays revealed a potent and selective antiproliferative effect against A549 cells after evaluating a panel of cancer cell lines. Compound 9b has emerged as a potent activator of tumor pyruvate kinase M2 (PKM2), a protein known to play a critical role in LC. Apoptosis assays and cell cycle analysis demonstrated early apoptosis and G2 phase arrest. In silico studies demonstrated interactions between compound 9b and the activator binding site of PKM2. Surface plasmon resonance (SPR) experiments strongly indicated that 9b has a high affinity (Kd of 1.378 nM) for PKM2. Furthermore, the increase in reactive oxygen species and decrease in lactate concentration suggested that compound 9b has significant anticancer effects. Notably, the increase in particle size following treatment with 9b suggested the tetramerization of PKM2. This work provides insights that might advance efforts to develop effective non-platinum anticancer agents.
Introduction
Lung cancer comprises a diverse array of molecularly and histologically heterogeneous subtypes. Lung cancer can be divided into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). The incidence of SCLC is decreasing, but that of NSCLC is continually on the rise, and the most frequent histological type among NSCLCs is adenocarcinoma.1–3 According to data furnished in 2020 by the World Health Organization (WHO), lung cancer is the second most common cancer.4 It is a leading cause of death, accounting for 18.4% of all cancer deaths worldwide.5 For NSCLC, the 5-year relative survival rate is only 28%.6 Due to late-stage diagnosis and a dearth of treatment options, lung cancer has long been regarded as a difficult clinical problem.7 In patients with advanced lung adenocarcinoma, tyrosine kinase inhibitors (TKIs) of the epithelial growth factor receptor (EGFR), such as gefitinib and erlotinib, are commonly used. However, clinical studies have shown that only 10% of patients respond to gefitinib or erlotinib.8 Combination chemotherapy slightly improves survival but increases toxicity.9–11Metabolic disruption, especially in glucose metabolism, is a distinguishing feature of NSCLC. Cancer cells often redirect a significant portion of glucose metabolism reactions toward glycolysis, even in oxygen-rich environments. Given the heterogeneity of NSCLC, there is a pressing need for the development of novel molecules targeting additional validated targets against this disease.12
Pyruvate kinase M2 (PKM2), a rate-limiting enzyme of the glycolytic pathway, is directly related to the onset and progression of different diseases13a–d including lung. Mutated PKM2, which is prevalently dimeric, can aid in the treatment of lung cancers. A study by Heiden et al. confirmed this observation.13b Additionally, overcoming acquired drug resistance could be achieved by suppressing the cellular energy supply through targeted intervention in glycolysis.14 The choice of PKM2 modulation over other kinases to mitigate cancer can be justified. In 2018, Liu et al. reported that PKM2 is highly upregulated in lung cancer. This study was performed in adenocarcinomic human alveolar basal epithelial cells (A549).15 The positive correlation between secreted PKM2 and tumor malignancy has been investigated and established.16
Thus, PKM2 is one of the reasons behind lung cancer invasion, progression, and migration.16 Lung cancer cells, like those of other cancers, selectively express tumor PKM2. This enzyme helps direct glucose metabolites into biosynthetic pathways that promote cell growth. By influencing the balance between oxidative phosphorylation and glycolysis, PKM2 activity provides cancer cells with a metabolic advantage under various conditions.
Studies on PKM2 in lung cancer have revealed a relationship between tumor aggressiveness and prognosis for patients. Patients with lung cancer who exhibit high levels of PKM2 expression typically have a worse prognosis and are more resistant to treatment. Considering the importance of the target, there has been a growing interest among drug discovery scientists in the last decade, as evidenced by the increasing number of research reports.17a–c Specifically, PKM2 activators such as TP-1454 have been tested in clinical trials for cancer treatment [NCT04328740].18
In addition, mitapivat, a PKR activator, has been approved by the US FDA for the treatment of hemolytic anemia.19 Small molecules are widely used as probes in biology and drugs in medicine.20 Among small molecules in medicine, sulfonamides21 are the basis of many drugs collectively known as sulfa drugs.22
The global sulfonamide market size was US$ 107.34 Mn in 2022, and this value is expected to increase by 4.5% by 2032.23a–c These sulfonamides are medicinally significant scaffolds present in antimicrobial agents such as sulfamethazine and sulfamethizole and in anticonvulsants such as sultiame. Thiazide diuretics such as hydrochlorothiazide, metolazone, and indapamide contain this moiety. Sulfonamide-based derivatives have been explored for a while now to alleviate different types of cancers. Some USFDA-approved drugs include belinostat for peripheral T-cell lymphoma, pazopanib for malignant neoplasms of the kidney and soft tissue sarcoma, dabrafenib for unresectable and malignant melanoma, amsacrine for acute adult leukemia, pictilisib for solid cancers and non-Hodgkin's lymphoma, and ABT-199 for acute myeloid leukemia and chronic lymphocytic leukemia.24 Reports surfacing recently have indicated that sulfonamides have the potential to be effective against lung cancers.25 These compounds act as tubulin inhibitors and microtubule disruptors to inhibit lung cancer.26–31 Benzodioxine-based sulfonamides reported by Zhang et al. in 2017 were able to act as PKM2 activators.32 Recently, in 2021, dithiocarbamate-based sulfonamides were furnished by Yin et al.33 Structurally, well-known PKM2 activators such as DASA-58 (3-(4-(2,3-dihydrobenzo[b]dioxin-6-ylsulfonyl)-1,4-diazepan-1-ylsulfonyl)aniline) have sulfonamides.34 Thus, sulfonamides have been shown to activate PKM2. Generating diverse skeletal scaffolds efficiently accelerates the exploration of new territories in chemical space. To successfully synthesize compound libraries with potential applications in oncology, chemical features commonly found in natural products are recommended. This involves incorporating sp3 and sp2-hybridized basic nitrogen atoms, along with the integration of innovative skeletal structures. Here, imidazopyri(mi)dines were the heterocyclics covalently stitched to sulfonamides. Imidazopyri(mi)dine derivatives are attractive scaffolds for drug discovery because these compounds possess a variety of pharmacological properties, such as gamma-aminobutyric acid (GABA) A antagonistic35 and fms-like tyrosine kinase 3 (FLT3) inhibitory activities.36,37 Zolpidem38 and alpidem39 are some of the approved drugs with imidazopyridines in their structures and are widely used as sedatives and hypnotics. In addition, imidazopyri(mi)dine-containing drugs, including P38α kinase inhibitors,40 are utilized to treat rheumatoid arthritis40 and Crohn's disease.41 Imidazopyridines are reported to act as phosphoinositide 3-kinase (PI3K) inhibitors.42 These scaffolds are purine analogs that can also be utilized to treat cancer (Fig. 1).43 Both imidazopyrimidines and imidazopyridines have been developed as anticancer agents.44 Recently, one report claimed that compounds having these scaffolds are effective in breast cancer and act as PKM2 activators according to in vitro results.43 Notably, these compounds also possess a piperazine linker between them.45–47 This moiety provides a larger surface area and structural rigidity, thereby resulting in hydrogen bond donor and acceptor properties that may increase water solubility, oral bioavailability, and improve ADMET properties.48 Moreover, piperazine-containing compounds are known to be approved for an array of drugs ranging from ranolazine,49 which is a sodium blocker used to treat chronic angina, to cyclizine (antihistamine),50 amoxapine (prescribed to alleviate depression),51 and imatinib (used to treat chronic myeloid leukemia).52
 | ||
| Fig. 1 Chemical structures of USFDA-approved drugs containing sulfonamides (I–VII), USFDA-approved drugs containing sulfonamides for cancer treatment (VIII–XIII), imidazopyrimidine-based (XIV–XVI), and piperazine-based drugs (XVII–XIX) for the treatment of various diseases. | ||
Thus, designing a hybrid molecule with sulfonamides and imidazopyri(mi)dines with a piperazine bridge will synergistically promote the anticancer effects of these two significant scaffolds with piperazine, improving attributes such as cell permeability,53 water solubility, and oral bioavailability.48 In an attempt to improve the pharmacological properties of high-affinity PKM2 inhibitors,54a we replaced the thiazole moiety with sulfonamides. As we report here, during these studies, against A549 cells, these compounds showed exceptionally high anti-LC activity.
The synthesis of 38 novel sulfonamide derivatives based on imidazopyrimidine and imidazopyridine was swiftly followed by spectroscopic characterization using 1H, 13C, and 19F NMR and HRMS. Single-crystal X-ray diffraction (SC X-ray diffraction) and energy-dispersive X-ray spectroscopy (EDS) were employed for further structural confirmation and elemental analysis. Compound purity was evaluated using HPLC. Field emission scanning electron microscopy (FESEM) provided valuable insights into the surface morphology. In docking studies with the activator binding site of PKM2 (PDB id 3GR4), the compounds showed promising scores compared to other binding sites of PKM2. Lactate dehydrogenase-coupled enzyme assays indicated that the majority of the series were PKM2 activators. Surface plasmon resonance (SPR) confirmed the affinity of compound 9b for PKM2. An in vitro cytotoxicity study was performed in a panel of cancer cell lines. The whole series was evaluated against the human oral squamous cell carcinoma cell line CAL-27, the human breast cancer cell line MCF-7, the human colorectal cancer cell line COLO-205 and adenocarcinomic human alveolar basal epithelial cells (A549). The compounds were not sufficiently active in other cell lines (detailed in the ESI†) but were significantly potent against A549 cells. The test revealed compound 9b to be the most potent and nontoxic to the normal lung epithelial cell line BEAS-2B, demonstrating minimal cytotoxicity and reinforcing its cancer specificity (Table 1). These traits have been observed in previous reports stating that sulfonamides are particularly effective in lung cancers.54b–d Apoptosis assay and cell cycle analysis conducted with compound 9b supported this hypothesis. The increase in reactive oxygen species and decrease in lactate concentration indicated that 9b had significant anticancer effects. The increased particle size after treatment with 9b suggested that PKM2 tetramerization and elevated pyruvate concentration were common traits observed with PKM2 activators. Density functional theory (DFT) studies revealed that molecule 9b possessed a very low HOMO–LUMO gap, indicative of its high reactivity, a characteristic associated with anticancer agents, as reported by Manicum et al.55 Molecular dynamics (MD) simulation studies established that molecule 9b stabilized the activator binding site of PKM2. Molecular electrostatic potential (MESP) studies demonstrated that molecule 9b was predominantly electrophilic and capable of binding with nucleophilic sites in proteins. Taken together, these findings underscore the significant potential of these novel compounds for development into potent oncotherapeutic agents for NSCLC treatment.
| Compound code | Z | Y | Isolated yield in % | IC50 (μM) – A549 | IC50 (μM) – BEAS-2B | AC50 (PKM2) in μM | IC50 (PKM2) in μM | Docking score-activator binding site (kCal mol−1) |
|---|---|---|---|---|---|---|---|---|
| a This combined data set was analyzed for compound selection from the entire series, in case of further studies. The results of the LDH-coupled enzyme assay and the cytotoxicity assay are expressed as the mean ± SEM of triplicate measurements. IC50 is a quantitative measure that indicates how much of a particular inhibitory substance (e.g. drug) is needed to inhibit, in vitro, a given biological process or biological component by 50%. AC50 = concentration at which 50% of maximum activity is observed. | ||||||||
| 9a | N | Ph-Me | 63 | 1.00 ± 0.365 | 39.55 ± 5.35 | 0.1049 ± 0.058 | −8.59 | |
| 9b | N | Ph | 68 | 0.91 ± 0.314 | 59.46 ± 4.53 | 0.03007 ± 0.083 | −8.09 | |
| 9c | N | Thiophene | 72 | 1.47 ± 0.141 | 44.99 ± 6.76 | 0.0935 ± 0.060 | −7.80 | |
| 9d | N | 4-CF3-Ph | 78 | 1.23 ± 1.217 | 15.87 ± 3.77 | 0.2109 ± 0.071 | −8.16 | |
| 9e | N | 4-Br-Ph | 71 | 1.33 ± 0.069 | 20.81 ± 4.57 | 0.5305 ± 0.019 | −8.24 | |
| 9f | N | 4-F-Ph | 80 | 1.48 ± 0.140 | 19.97 ± 5.42 | 0.7034 ± 0.047 | −8.83 | |
| 9g | N | 4-OMe-Ph | 66 | 1.48 ± 0.054 | 28.43 ± 5.86 | 0.5354 ± 0.0081 | −8.48 | |
| 9h | N | 2,5-Dichloro-Ph | 74 | 1.26 ± 0.16 | 26.46 ± 8.21 | 0.6478 ± 0.049 | −8.20 | |
| 9i | N | 2,4,6-Trimethyl-Ph | 81 | 1.06 ± 0.267 | 51.35 ± 8.55 | 0.7253 ± 0.021 | −9.33 | |
| 9j | N | 5-Br,2-Ome-Ph | 69 | 1.54 ± 0.111 | 39.07 ± 8.62 | 0.6428 ± 0.041 | −7.18 | |
| 9k | N | 3-Br-Ph | 62 | 1.14 ± 0.498 | 38.98 ± 8.17 | 0.6340 ± 0.076 | −8.75 | |
| 9l | N | Cyclohexyl | 63 | 1.54 ± 0.087 | 39.04 ± 8.73 | 0.7829 ± 0.031 | −7.54 | |
| 9m | N | 5-Br-Thiophene | 60 | 1.60 ± 0.048 | 36.47 ± 7.00 | 0.6806 ± 0.030 | −7.87 | |
| 9n | N | Cyclopropyl | 70 | 1.76 ± 0.069 | 53.45 ± 6.84 | 0.0923 ± 0.059 | −7.07 | |
| 9o | N | 2-Cyano-Ph | 61 | 1.72 ± 0.059 | 46.32 ± 5.04 | 0.5153 ± 0.002 | −8.53 | |
| 9p | N | 2-Cl,4-F-Ph | 75 | 1.72 ± 0.070 | 16.78 ± 5.3 | 0.6121 ± 0.415 | −8.87 | |
| 9q | N | 5-Cl-Thiophene | 77 | 1.75 ± 0.043 | 9.29 ± 3.76 | 0.4372 ± 0.089 | −7.81 | |
| 9r | N | 4′-OMe-Biphenyl | 79 | 1.60 ± 0.061 | 9.13 ± 4.81 | 0.4347 ± 0.566 | −8.26 | |
| 9s | N | 4-NO2-Ph | 66 | 1.66 ± 0.048 | 66.92 ± 6.01 | 0.0388 ± 0.089 | NA | |
| 10a | CH | Ph-Me | 75 | 1.67 ± 0.113 | 51.15 ± 5.82 | 0.7955 ± 0.055 | −7.24 | |
| 10b | CH | Ph | 74 | 1.69 ± 0.074 | 44.68 ± 4.49 | 0.1410 ± 0.630 | −7.18 | |
| 10c | CH | Thiophene | 69 | 1.14 ± 0.277 | 30.47 ± 8.41 | 0.1340 ± 0.079 | −8.75 | |
| 10d | CH | 4-CF3-Ph | 69 | 1.30 ± 0.145 | 61.36 ± 9.97 | 0.550 ± 0.044 | −7.54 | |
| 10e | CH | 4-Br-Ph | 64 | 1.74 ± 0.074 | 64.24 ± 5.33 | 0.1462 ± 0.060 | −7.87 | |
| 10f | CH | 4-F-Ph | 69 | 1.68 ± 0.065 | 52.22 ± 8.50 | 0.382 ± 0.056 | −7.07 | |
| 10g | CH | 4-OMe-Ph | 79 | 1.83 ± 0.053 | 46.05 ± 5.58 | 0.0416 ± 0.048 | −8.53 | |
| 10h | CH | 2,5-Dichloro-Ph | 72 | 1.50 ± 0.084 | 45.95 ± 9.06 | 0.6158 ± 0.065 | −7.81 | |
| 10i | CH | 2,4,6-Trimethyl-Ph | 80 | 1.68 ± 0.039 | 11.47 ± 3.59 | 0.1806 ± 0.021 | −9.10 | |
| 10j | CH | 5-Br,2-OMe-Ph | 70 | 1.44 ± 0.084 | 30.92 ± 8.10 | 0.3285 ± 0.035 | −6.50 | |
| 10k | CH | 3-Br-Ph | 77 | 1.67 ± 0.120 | 39.90 ± 5.16 | 0.6060 ± 0.064 | −8.73 | |
| 10l | CH | Cyclohexyl | 78 | 1.65 ± 0.068 | 60.57 ± 7.60 | 0.889 ± 0.049 | −5.14 | |
| 10m | CH | 5-Br-Thiophene | 62 | 1.67 ± 0.049 | 45.69 ± 10.74 | 0.1382 ± 0.063 | −8.17 | |
| 10n | CH | Cyclopropyl | 78 | 1.24 ± 0.171 | 50.54 ± 7.04 | 0.432 ± 0.017 | −7.40 | |
| 10o | CH | 2-Cyano-Ph | 66 | 1.51 ± 0.105 | 55.67 ± 2.88 | 0.1465 ± 0.071 | −8.64 | |
| 10p | CH | 2-Cl,4-F-Ph | 62 | 0.95 ± 0.366 | 37.80 ± 4.73 | 0.2046 ± 0.071 | −9.35 | |
| 10q | CH | 5-Cl-Thiophene | 77 | 1.32 ± 0.170 | 48.90 ± 7.90 | 0.5517 ± 0.017 | −6.12 | |
| 10r | CH | 4′-OMe-Biphenyl | 73 | 1.23 ± 0.216 | 56.52 ± 5.19 | 0.341 ± 0.036 | −8.13 | |
| 10s | CH | 4-NO2-Ph | 65 | 1.46 ± 0.118 | 20.09 ± 6.58 | 0.6492 ± 0.089 | NA | |
| Doxorubicin | 1.64 ± 0.101 | 24.67 ± 7.36 | ||||||
| DASA-58 (Std. PKM2 activator) | 1.46 ± 0.27 | 35.23 ± 3.36 | 0.020 ± 0.010 | −8.279 | ||||
| L-Phenylalanine (Std. PKM2 inhibitor) | 0.055 ± 0.042 | |||||||
Results and discussion
In silico docking studies
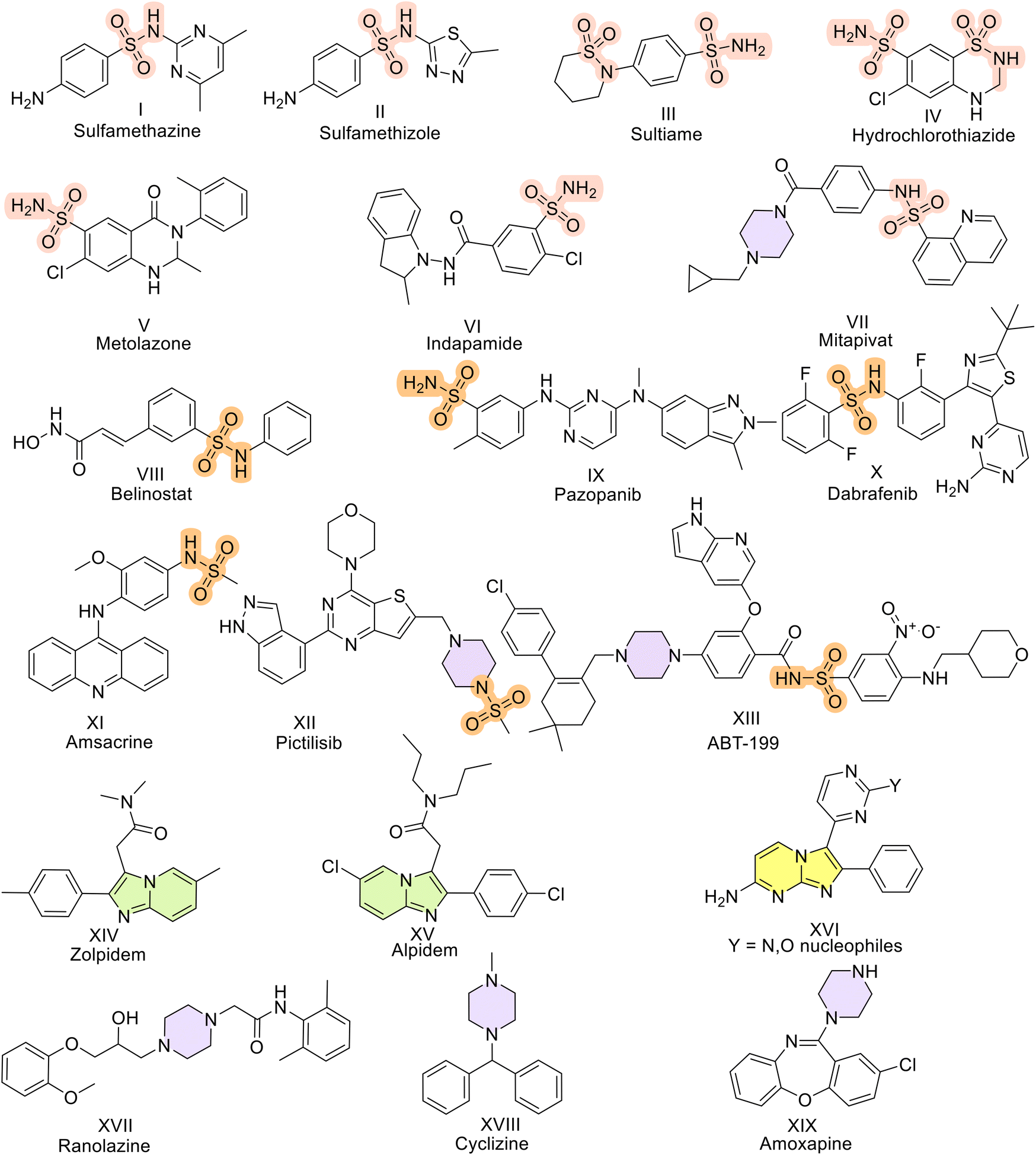
By conducting docking studies at different sites of PKM2 (PDB id – 3GR4) to obtain insights into the binding affinities of the compounds, we observed that the molecules docked into the activator binding site with binding affinities as promising as −9.35 kCal mol−1. The majority of the series displayed binding affinities above −7 kCal mol−1. Here, DASA-58 was used as the standard since it has been established as a small molecule activator of pyruvate kinase M2 (PKM2). It activates PKM2, which increases pyruvate kinase activity and glycolysis. If we consider molecule 9b (binding affinity of −8.09 kCal mol−1), the reason behind this high affinity can be explained. Compounds 9b and DASA-58 bind to the same binding pocket of PKM2. Residues such as Leu 353, Asp 354, Gln 393, Leu 394, Glu 397, Met 30, and Leu 27 are present in the vicinity of both 9b and DASA-58. Further analysis revealed that 9b and DASA-58 have similar interactions owing to their structural similarity. 9b and DASA-58 have common H-bonding interactions with Tyr 390 of the C chain and Leu 353 of the D chain of PKM2. The oxygen of the carbonyl and sulfonyl functionalities of 9b and the sulfonyl group of DASA-58 are responsible for such interactions. This intermolecular H-bonding is known to stabilize ligands in the pocket of the protein (in this case, the activator binding site). Phe 26 of the C chain, which has a pi–pi stacking interaction with the phenyl group, is common to 9b and DASA-58. This noncovalent interaction between the phenyl rings containing pi-orbitals contributes significantly to the high binding affinity of 9b for the activator binding pocket (Fig. 2). The presence of a sulfonamide group in 9b and DASA-58 and the presence of piperazine, a structurally similar scaffold to the 1,4-diazepine ring present in DASA-58, result in high binding affinity. The binding affinity studies subsequently corroborated the results of other in vitro studies.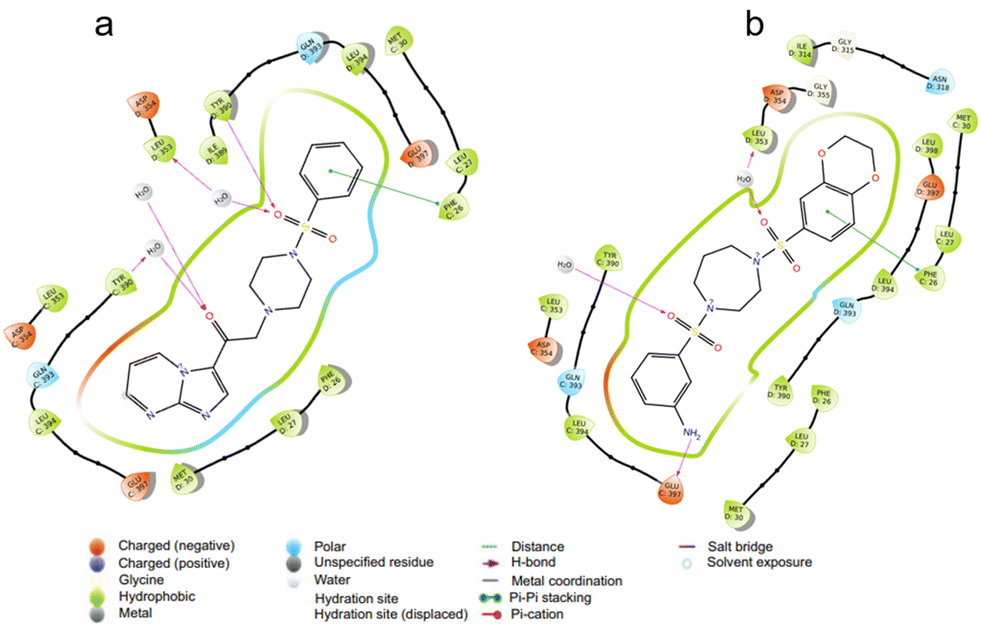 | ||
| Fig. 2 The interactions of ligands a) 9b and b) DASA-58 when docked at the activator binding site (DASA-58 binding site) of the PKM2 protein (PDB id 3GR4) are illustrated, highlighting favorable interactions represented by various colors. | ||
Synthesis of imidazopyri(mi)dine-based sulfonamides
Previously, our group reported imidazopyri(mi)dine-based piperazine derivatives that functioned as PKM2 modulators. Based on that foundation and positive results from in silico docking studies, we synthesized imidazopyri(mi)dine-based sulfonamide derivatives with a piperazine linker, hypothesizing that these shall be PKM2 modulators. There are two parts to the synthesis. The first section involves the preparation of 2-chloro-1-(imidazo[1,2-a]pyri(mi)din-3-yl)ethan-1-one, which is then used in the second section to synthesize the final compounds. Condensation of commercially available 2-aminopyri(mi)dines 1a–b with N,N-dimethylformamide dimethyl acetal (DMF-DMA) and subsequent cyclization of the resulting N,N-dimethyl-N′-(pyri(mi)din-2-yl)formimidamide 2a–b with 1,3-dichloroacetone 3 yielded the scaffold 2-chloro-1-(imidazo[1,2-a]pyri(mi)din-3-yl)ethan-1-one 4a–b. All the compounds were purified by column chromatography.Next, in a separate reaction vessel, commercially available substituted sulfonyl chloride 6 was reacted with Boc-protected piperazine 5. The free nitrogen from the piperazine formed a covalent bond with the sulfur of the substituted sulfonyl chloride, resulting in the dissipation of HCl and the formation of 7. The lone pair of electrons on the secondary amine of piperazine attacks sulfonyl chloride and forces the chlorine to leave. This adduct 7 was then deprotected by TFA, leaving the protected piperazine nitrogen free. This nitrogen from the deprotected species 8 formed an adduct with the scaffold 2-chloro-1-(imidazo[1,2-a]pyri(mi)din-3-yl)ethan-1-one 4a–b, with the dissipation of HCl. These novel imidazopyri(mi)dine-based sulfonamides (9a–s and 10a–s) (Scheme 1) were used for further evaluation. FE-SEM and EDS analyses were also performed (data detailed in the Fig. S46 of the ESI†).
 | ||
| Scheme 1 Synthetic procedure for compounds 9a–9s and 10a–10s. a) Generation of the intermediate 2-chloro-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one 4a–b from 2-aminopyrimidine 2a–b through Schiff base formation. b) Elaboration of imidazopyrimidine-flanked sulfonamide derivatives employing 2-chloro-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one 4a–b as pivotal intermediates. | ||
Single-crystal X-ray diffraction analysis confirmed the chemical structure
To achieve complete and unambiguous structural resolution at the crystallographic level, 10q crystals (although we tried to crystallize different compounds, 10q could be successfully crystallized) were obtained for X-ray diffraction analysis. Fig. 3 shows the elucidated structure of compound 10q, and Table S1† summarizes the related crystal data, data collection, and structure refinement details. The analysis revealed that the compound crystallizes in a triclinic format with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), having an asymmetric unit containing one molecule with analogous geometry and bonding parameters. Tables S4 and S5 (ESI†) summarize the selected bond lengths and angles for the compound.
(no. 2), having an asymmetric unit containing one molecule with analogous geometry and bonding parameters. Tables S4 and S5 (ESI†) summarize the selected bond lengths and angles for the compound.
 | ||
| Fig. 3 Single crystal X-ray crystallography of compound 10q displayed at the 50% probability level, visualized using ORTEP3 software. The crystallographic data has been deposited with the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2331542. | ||
Lactate dehydrogenase-coupled enzyme assays reveal PKM2 modulation
This cell-free assay determines the percentage relative activity of pyruvate kinase M2 (PKM2) by assessing the extent of the conversion of NADH to NAD+ through differences in absorbance over a stipulated period. This assay (Fig. 4) aids in determining whether the compounds synthesized are PKM2 activators or inhibitors. DASA-58 is a small molecule activator of pyruvate kinase M2 (PKM2). It activates PKM2, which increases pyruvate kinase activity and glycolysis and is thus kept as a standard PKM2 activator in this assay. Similarly, L-phenylalanine is a PKM2 inhibitor, as has been previously reported for the inhibition of PKM2.56,57 A PKM2 activator can convert PKM2 dimers (responsible for malignancy) into tetramers (maintaining homeostasis) and mitigate overexpressed PKM2 dimers, thereby decreasing cancer incidence. PKM2 inhibitors block the formation of PKM2 dimers, altogether curbing cancer. This is also the preliminary step in determining the structure–activity relationship of the series furnished. The difference in the position and nature of the functional groups is inherently responsible for the difference in the extent and type of PKM2 modulation. Analysis of the results from this assay revealed that the majority of the compounds were PKM2 activators, and only a few emerged as PKM2 inhibitors (9l, 9m, 9s, 10h, 10m, 10q, and 10r). From the results, it is evident that in the case of imidazopyrimidine-based sulfonamides, if the sulfonyl group is attached to an unsubstituted phenyl ring (9b), the AC50 is the lowest. It increases when the phenyl ring is substituted or replaced by other systems, such as thiophene, cyclohexane, or cyclopropyl. Imidazopyridine-based sulfonamides do not follow this trend at all. Here, the compound with the lowest AC50 is the one where the sulfonyl group is attached to the phenyl group with a methoxy group (an electron-donating group) at the para position. Thus, the effect of electron-donating groups is visible in imidazopyridine-based derivatives. The compounds in which electron-withdrawing groups (such as cyano or trifluoromethyl groups) are attached displayed high AC50 values. This preliminary evaluation of PKM2 modulation can be used to establish a foundation for further in vitro studies.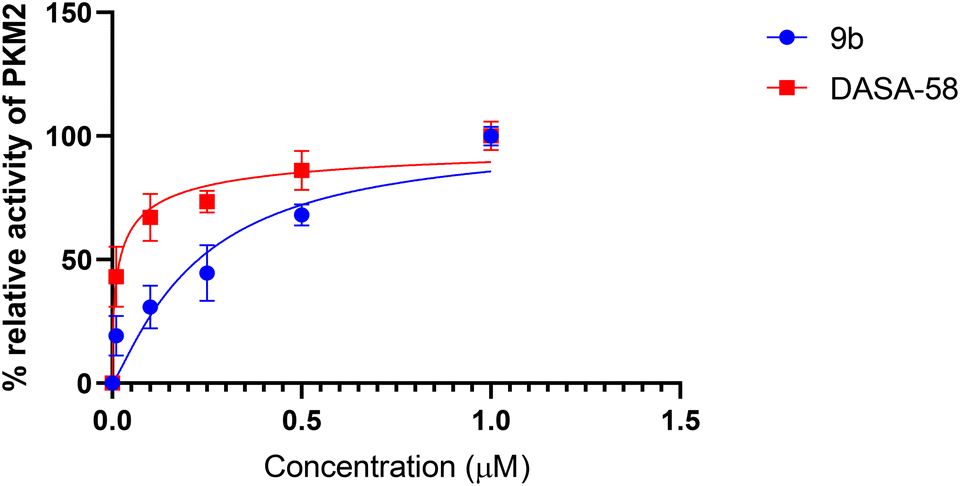 | ||
| Fig. 4 Lactate dehydrogenase-coupled enzyme assay comparing the activity of compound 9b with the standard PKM2 activator DASA-58, illustrating concentration-dependent activation presented as % relative activity of PKM2. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Surface plasmon resonance demonstrates the high affinity of 9b for PKM2
When an incoming photon of light strikes a metal surface, surface plasmon resonance happens (usually a gold surface). Part of the light energy links via the metal coating at a specific angle of incidence with the electrons in the metal surface layer, which are subsequently excited and move. The electron motions, which are now known as plasmons, travel parallel to the metal surface. An electric field with a range of about 300 nm from the metal surface to sample solution border is produced by the plasmon oscillation in turn. Utilizing a high-refractive index glass prism in the Kretschmann geometry of the attenuated total reflection technique, incident light is used in a commercial SPR biosensor setup. The refractive index of the substance in close proximity to the metal surface determines the predetermined SPR angle at which resonance occurs under the conditions of a constant light source wavelength. Plasmon cannot thus occur when there is a little change in the refractive index of the sensing medium (for example, due to biomolecule attachment). Thus, variations in the refracted light collected on a detector are measured to achieve detection. Furthermore, the surface concentration may be measured by recording resonance angle variations or keeping an eye on the intensity of refracted light.54e Here, the interaction between the immobilized PKM2 and the synthesized compound 9b was determined by SPR at an immobilized PKM2 concentration of 50 μG mL−1. Compound 9b has a tremendous affinity for PKM2 and exhibited a Kd value of 1.378 nM (Fig. 5a). This low Kd value is an indication of the affinity of 9b for immobilized PKM2. Thus, 9b was confirmed to interact with PKM2 through in silico docking studies and SPR analysis. In addition, it was observed that compound 9b had a concentration-dependent response at 90 seconds (Fig. 5b). | ||
| Fig. 5 a) Graphical representation showing the interaction between compound 9b and PKM2, with a dissociation constant (Kd) of 1.378 nM. b) A sensogram was generated for the interaction between compound 9b and PKM2, demonstrating a concentration-dependent response at 90 seconds. | ||
This denoted the completion of the cycle at 90 seconds from one baseline to the next. The insights approximately 9b inspired us to proceed with this compound for further biological studies.
Antiproliferative activity
The antiproliferative activity of the sulfonamides was evaluated using a cytotoxicity assay with Alamar blue dye on the A549 cell line. The whole series of compounds showed promising results, with IC50 values ranging from 0.91 μM to 1.83 μM, in a concentration-dependent manner. Compound 9b was the most potent among the compounds in the series, with an IC50 as low as 0.91 μM (Fig. 6). Doxorubicin, a well-known anticancer drug, as well as DASA-58, a standard PKM2 activator was used as the standards. Doxorubicin had an IC50 value of 1.64 μM, and DASA-58's IC50 value was 1.46 μM. Thus, a study performed in triplicate indicated that the compounds exhibited good cytotoxicity against lung cancer cells. These results prompted us to evaluate other attributes, such as apoptotic ability, as well as their ability to arrest the cell cycle. The series was also evaluated against a panel of other cancer cell lines such as COLO-205, MCF-7, and CAL-27 along with A549. The results indicated potency selectively against the A549 cell line (data is detailed in Fig. S41–S45 of the ESI†). | ||
| Fig. 6 The cytotoxicity assay results conducted with the human adenocarcinoma cell line A549 revealed compound 9b to exhibit the highest potency. Doxorubicin and DASA-58 were utilized as the positive standards. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Cancer specificity of potent compounds
Potent compounds were evaluated against the human normal lung epithelial cell line BEAS-2B to establish their cancer specificity (Fig. 7 and S42 of the ESI†). The percentage of cell viability of the compounds against this cell line indicated that these compounds were nontoxic to the normal lung epithelial cell line BEAS-2B at concentrations identical to the IC50 values of the compounds against the human lung epithelial adenocarcinoma cell line A549. This observation confirmed their cancer specificity and motivated us to evaluate these molecules further. | ||
| Fig. 7 The cytotoxicity assay results conducted with the human normal bronchial epithelial cell line BEAS-2B revealed compound 9b to exhibit cancer specificity. Doxorubicin and DASA-58 were utilized as the positive standards. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Apoptosis assay
The most potent compound, 9b, as evaluated by a cytotoxicity assay performed against the A549 cell line, was subjected to an apoptosis assay. The same cell line was inoculated with compound 9b (at the same concentration as the IC50 value) to assess the apoptotic ability of the compound. The control group and the treated group were compared through flow cytometry. The control group had 91.83% live cells and 3.46% dead cells, whereas in the 9b-treated group, the percentage of live cells decreased to 56.43%, and the percentage of early apoptotic cells increased to 30.91%. This finding clearly indicates that the compound is effective against lung cancer. To confirm our findings, we proceeded with cell cycle analysis (Fig. 8). | ||
| Fig. 8 The apoptosis assay conducted on A549 cells treated with compound 9b revealed a decrease in the percentage of live cells from 91.83% in the control to 56.43% upon treatment with 9b, accompanied by a percentage of early apoptotic cells at 30.91%. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Cell cycle analysis
This study was designed to investigate whether compound 9b can arrest the cell cycle. Additionally, insights about the phase arrested by the compound was computed. The control group and treated group were analyzed by flow cytometry. The results revealed a difference between the groups. In the control group, 68.48% of the cells were in the G1 phase of the cell cycle, which was reduced to 4.00% after treatment with 9b, and the percentage of cells in the S and G2 phases increased concomitantly. This decrease in the G1 phase and increase in the G2 and S phases indicated the ability of 9b to arrest the cancer cell cycle predominantly in the G2 phase and in the S phase (Fig. 9). | ||
| Fig. 9 The results of cell cycle analysis comparing compound 9b-treated A549 cell line with untreated cells revealed a significant arrest in the G2 phase. In untreated cells, the distribution was 68.48% in the G1 phase, 14.88% in the S phase, and 15.89% in the G2 phase, while 9b-treated cells exhibited 4.001% in G1 phase, 21.96% in the S phase, and 73.36% in G2 phase. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
ROS detection assay
ROS induction is a hallmark of the successful development of anticancer compounds. Fluorometric evaluation of the ROS generated after treatment with 9b (0.91 μM) in comparison to the untreated group revealed a time-dependent increase in ROS. This steady increase in ROS with time (Fig. 10) in turn results in early apoptosis and ultimately cancer cell death. ROS-induced apoptosis may be the mechanism by which 9b affects A549 cells in vitro. ROS enhancement may also activate glycolysis as reported in studies earlier through PKM2 activation.54f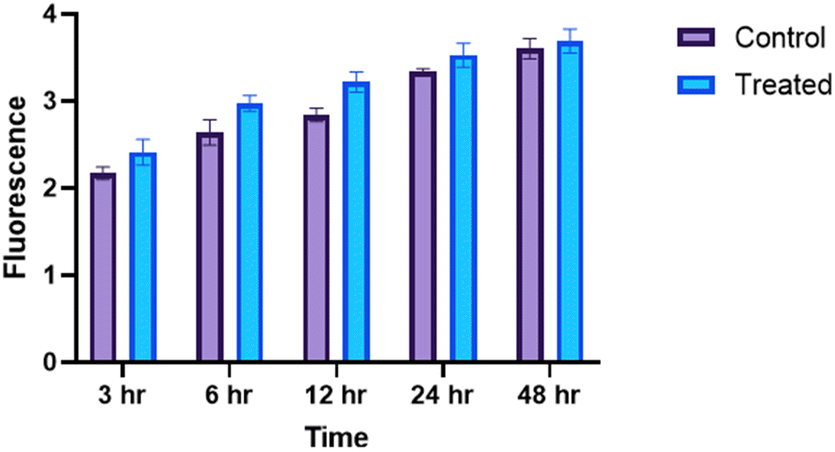 | ||
| Fig. 10 The ROS assay conducted on A549 cells treated with compound 9b revealed a time-dependent increase in ROS concentration compared to the untreated cells. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Lactate release assay
Colorimetry was used to assess lactate release from A549 cells treated with 9b. Compared to that in the untreated group, the quantity of lactate produced by the cells in the 9b-treated group was significantly lower. Because high cellular lactate levels promote improper cell signaling during carcinogenesis, an undesired positive feedback loop is initiated, increasing glucose uptake, glycolysis, lactate generation, and release and decreasing mitochondrial activity and clearance. Thus, the decrease in lactate content by 9b (Fig. 11) is an intriguing discovery that may point to this compound's anticancer ability. | ||
| Fig. 11 The lactate release assay conducted on A549 cells treated with compound 9b revealed a significant decrease in lactate levels compared to the untreated cells. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
Pyruvate assay
Compound 9b was able to increase ROS and decrease lactate, which makes it a viable candidate for further anticancer evaluation. Previously, docking studies showed the promising binding affinity of 9b at the activator binding site of PKM2. In addition, an LDH-coupled enzyme assay indicated that 9b is a PKM2 activator. To further investigate the mechanism by which 9b operates, we performed an in vitro pyruvate assay. We observed a significant increase in the amount of pyruvate in the treated group in contrast to that in the control group (Fig. 12). This increase aligns with previous studies conducted by other groups, where PKM2 activators increased the pyruvate concentration13,58 and decreased the lactate concentration. Thus, 9b is in unison with the proven notion and exhibits attributes in agreement with a PKM2 activator. | ||
| Fig. 12 The pyruvate assay conducted on A549 cells treated with compound 9b revealed a significant increase in pyruvate levels compared to the untreated cells. Experimental data are presented as mean ± standard deviation, performed in triplicate. Statistical significance was assessed using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, where p < 0.05 was considered statistically significant. The significance levels are indicated in the figures as *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. | ||
PKM2 tetramerization with 9b
It is known that if a compound activates PKM2, its administration should result in the tetramerization of PKM2, thereby increasing the particle size of PKM2. This phenomenon can be tested. We evaluated the tetramerization potential of 9b by determining the size distributions of the particles using a particle size analyzer. According to these findings, 9b increased particle size in a manner similar to that of the well-known PKM2 activator DASA-58. Notably, DASA-58 was less effective than 9b (Fig. 13). | ||
| Fig. 13 a) A simplified operational flow diagram illustrating the process of dynamic light scattering. b) The particle size of PKM2 was increased by treatment with 9b and DASA-58 compared to the control. Upon comparison between 9b and DASA-58, it was noted that 9b induced a greater increase in the particle size of PKM2 compared to DASA-58. Experimental data are presented as mean ± standard deviation, performed in triplicate. | ||
Density functional theory studies and molecular electrostatic potential calculations
The data gathered thus far indicated that 9b can have a profound cytotoxic effect on A549 cells. The structural attributes that could have been behind this observation need to be explored. Thus, DFT, MESP, and MD studies were performed to obtain a profound picture of the structural properties responsible and the effect that 9b can have on PKM2 over a stipulated time.The gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) was measured in eV. This difference may correspond to the bioactivity of the molecule. We calculated the HOMO–LUMO gap of compound 9b, i.e., ΔEHOMO–LUMO, and found it to be 0.142 eV. The HOMO of compound 9b was found to be focused on the piperazine ring. The LUMO of compound 9b was focused exclusively on the imidazopyrimidine ring. The small gap indicates that 9b is more reactive. Other reactivity parameters were calculated, including the global electrophilicity index (ω), chemical hardness (η), and electronic chemical potential (μ). 9b had a global electrophilicity index of 0.600 eV, a chemical hardness of 0.142 eV, and an electronic chemical potential of 0.413 eV. The global electrophilicity index is a measure of a molecule's ability to accept electrons, and 9b has a promising value of 0.600 eV. The low value of ΔEHOMO–LUMO indicated the high reactivity of the molecule (Fig. 14a and b). This is also an indication that 9b might bind favorably with PKM2 and cause significant alterations in the normal metabolic functions of the cancer cells, which might induce death or prevent further development.
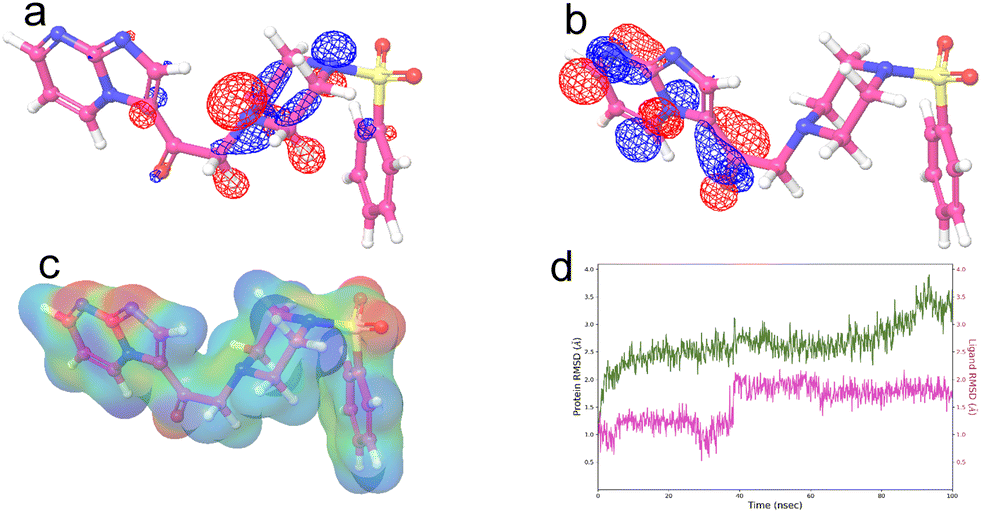 | ||
| Fig. 14 a) DFT studies used to compute the HOMO and b) LUMO of compound 9b. The HOMO of compound 9b was observed to be focused on the piperazine ring. The LUMO of the compound was concentrated on the imidazopyrimidine ring. Other reactivity parameters, such as the global electrophilicity index (ω), chemical hardness (η), and electronic chemical potential (μ), were calculated. For 9b, the global electrophilicity index (ω) was 0.600 eV, the chemical hardness (η) was 0.142 eV, and the electronic chemical potential (μ) was 0.413 eV. c) MESP was performed to determine the electrophilic and nucleophilic sites present within a molecule. The red color denotes the electrophilic region, and the blue color denotes the nucleophilic region, while the other colors, such as green, yellow, and orange, denote the intermediate range between blue and red. It was observed that the electrophilic sites were distributed around the boundary of the molecule. The nucleophilic sites were only present around the nitrogens of the imidazopyrimidine moiety and the sulfonyl group between the piperazine ring and the phenyl ring. d) The RMSD graph of 9b at the activator binding site of the PKM2 protein (PDB id 3GR4) after a molecular dynamics simulation (holo) of 100 ns, which reveals the efficient stabilization of ligand 9b bound at the activator binding site after a 60 ns timeframe. | ||
With this observation from DFT studies, we turned our attention to computing the MESP for 9b. The MESP is known to correlate with a compound's pharmacological activity. This experiment was carried out to determine the electrophilic and nucleophilic sites that exist within a molecule. Red represents the nucleophilic region, blue represents the electrophilic region, and other colors, such as green, yellow, and orange, represent the intermediate range between the two. Nucleophilic sites were found around the nitrogen of the imidazopyrimidine moiety and the sulfonyl group. The electrophilic sites, on the other hand, were distributed around the molecular boundaries.
This observation is a significant addition to what we have witnessed thus far. The abundance of electrophilic sites consolidates the possibility of biological nucleophiles on macromolecules interacting with xenobiotic electrophiles to covalently bind with them (Fig. 14c). Specifically, a reaction with proteins can have a definite cytotoxic effect.
Molecular dynamic simulation studies
To investigate the molecular mechanisms involved in 9b binding to the activator binding pocket of human pyruvate kinase M2, a 100 ns molecular dynamic simulation was performed (PDB ID 3GR4). The RMSD of the holo system (9b–PKM2 complex) was between 1.5 and 3 Å. The apo system (only PKM2 without 9b) had an RMSD between 1.5 and 3.5 Å (Fig. 14d). The lower RMSD value is indicative of better stabilization of PKM2 when 9b is present as a ligand in comparison to the system where 9b is absent. The stabilization effect is visible from the 20 ns timeframe.Experimental section
Chemical synthesis
Initially, 3.0 equivalents of DMF-DMA were applied to 2-aminopyri(mi)dine 1a–b in the presence of methanol at 80 °C for 4 hours. The consumption of all of the 2-aminopyri(mi)dine 1a–b was used to determine when the reaction had finished by thin layer chromatography (TLC). Without additional purification, the reaction mixture was concentrated under vacuum pressure and used to furnish the Schiff bases 2a–b. The finished product was exposed to 1,3-dichloroacetone 3 for four hours at 70 °C in the presence of the aprotic solvent acetonitrile.59 The imidazopyri(mi)dine-substituted chloroacetophenone adducts 4a–b were purified by column chromatography and used as a starting material for further reactions after the reaction was finished. Next, three steps were taken to complete the synthesis. A round-bottomed flask was first filled with a mixture of substituted sulfonyl chlorides 6 (1 equiv.) and Boc-protected piperazine 5 (1.05 equiv.) in 4 mL of dichloromethane. Next, triethylamine (1.2 equiv.) was added, and the mixture was continuously stirred at room temperature for two hours. TLC was used to monitor the reaction's development (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate at a ratio of 70:30). The reaction mixture was first extracted with ethyl acetate, after which it was washed with water, dried over anhydrous sodium sulfate, and filtered, and the solvent was then evaporated using a rotary evaporator. Crude product 7 then underwent a deprotection process. Trifluoroacetic acid (TFA) and DCM (3:7) were used to dissolve the dry intermediate, which was then agitated at room temperature for two hours. DCM and TFA were evaporated on a rotary evaporator after deprotection was confirmed by TLC (hexane:ethyl acetate at a ratio of 50:50). DMSO was subsequently added to the deprotected intermediate 8, which was then dissolved. Then, the previously synthesized imidazopyri(mi)dine-substituted chloroacetophenone adducts 4a–b (1 equiv.) and activated K2CO3 (1.2 equiv.) were added, and the mixture was agitated at 80 °C for two hours.43 The reaction mixture was poured over crushed ice after it had finished, as determined by TLC (in 100% ethyl acetate), and then extracted using a DCM–water solvent system. To obtain the product as a solid, the extracted organic layer was passed over anhydrous sodium sulfate and purified by column chromatography (with a hexane:ethyl acetate mobile phase at a ratio of 30:70). The 1H, 13C, and 19F NMR spectra and HRMS data were used to comprehensively characterize products 9a–s and 10a–s.
:ethyl acetate at a ratio of 70:30). The reaction mixture was first extracted with ethyl acetate, after which it was washed with water, dried over anhydrous sodium sulfate, and filtered, and the solvent was then evaporated using a rotary evaporator. Crude product 7 then underwent a deprotection process. Trifluoroacetic acid (TFA) and DCM (3:7) were used to dissolve the dry intermediate, which was then agitated at room temperature for two hours. DCM and TFA were evaporated on a rotary evaporator after deprotection was confirmed by TLC (hexane:ethyl acetate at a ratio of 50:50). DMSO was subsequently added to the deprotected intermediate 8, which was then dissolved. Then, the previously synthesized imidazopyri(mi)dine-substituted chloroacetophenone adducts 4a–b (1 equiv.) and activated K2CO3 (1.2 equiv.) were added, and the mixture was agitated at 80 °C for two hours.43 The reaction mixture was poured over crushed ice after it had finished, as determined by TLC (in 100% ethyl acetate), and then extracted using a DCM–water solvent system. To obtain the product as a solid, the extracted organic layer was passed over anhydrous sodium sulfate and purified by column chromatography (with a hexane:ethyl acetate mobile phase at a ratio of 30:70). The 1H, 13C, and 19F NMR spectra and HRMS data were used to comprehensively characterize products 9a–s and 10a–s.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-tosylpiperazin-1-yl)ethan-1-one (9a). Appearance: pale orange solid; yield: (63%); 1H NMR (500 MHz, CDCl3): δ 9.85 (dd, J = 6.9, 2.1 Hz, 1H), 8.79 (dd, J = 4.2, 2.1 Hz, 1H), 8.73 (s, 1H), 7.66–7.60 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.18 (dd, J = 6.9, 4.2 Hz, 1H), 3.72 (s, 2H), 3.08 (s, 4H), 2.71 (t, J = 5.0 Hz, 4H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 187.2, 153.6, 151.0, 144.7, 143.9, 136.47, 132.0, 129.8, 127.8, 121.0, 111.4, 52.6, 45.9, 40.9, 21.5; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H22N5O3S+: 400.1438, found: 400.1439. Chromatographic purity: 99.81%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-(phenylsulfonyl)piperazin-1-yl)ethan-1-one (9b). Appearance: yellow solid; yield: (68%); 1H NMR (500 MHz, CDCl3): δ 9.84 (dd, J = 6.9, 2.1 Hz, 1H), 8.79 (dd, J = 4.2, 2.1 Hz, 1H), 8.70 (s, 1H), 7.76 (m, J = 2.0 Hz, 1H), 7.74 (q, J = 2.0 Hz, 1H), 7.66–7.61 (m, 1H), 7.58–7.54 (m, 2H), 7.19 (dd, J = 6.8, 4.2 Hz, 1H), 3.74 (s, 2H), 3.15–3.08 (m, 4H), 2.72 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.1, 153.6, 151.0, 144.5, 136.4, 135.1, 133.0, 129.3, 129.1, 127.7, 121.0, 111.5, 64.5, 52.5, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H20N5O3S+: 386.1281, found: 386.1282. Chromatographic purity: 100%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)ethan-1-one (9c). Appearance: buff solid; yield: (72%); 1H NMR (500 MHz, CDCl3): δ 9.86 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.74 (s, 1H), 7.65 (dd, J = 5.0, 1.3 Hz, 1H), 7.54 (dd, J = 3.7, 1.3 Hz, 1H), 7.21–7.16 (m, 2H), 3.75 (s, 2H), 3.17 (s, 4H), 2.78–2.73 (m, 4H); 13C NMR (125 MHz, CDCl3): δ 187.1, 153.7, 151.1, 144.7, 136.4, 135.4, 132.6, 132.4, 127.8, 121.0, 111.4, 64.7, 52.5, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C16H18N5O3S2+: 392.0846, found: 392.0857. Chromatographic purity: 99.57%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-((4-(trifluoromethyl)phenyl)sulfonyl)piperazin-1-yl)ethan-1-one (9d). Appearance: pale yellowish solid; yield: (78%); 1H NMR (500 MHz, CDCl3): δ 9.84 (d, J = 8.9 Hz, 1H), 8.82–8.77 (m, 1H), 8.70 (s, 1H), 7.89 (d, J = 8.3 Hz, 2H), 7.83 (d, J = 8.3 Hz, 2H), 7.18 (dd, J = 6.9, 4.2 Hz, 1H), 3.75 (s, 2H), 3.14 (s, 4H), 2.74 (t, J = 4.8 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.9, 153.7, 151.1, 144.6, 139.0, 136.4, 128.2, 128.2, 126.4, 126.3, 126.3, 126.3, 121.0, 111.5, 64.5, 52.5, 45.9; 19F NMR (471 MHz, CDCl3): δ −63.11; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H19F3N5O3S+: 454.1155, found: 454.1156. Chromatographic purity: 99.81%.
2-(4-((4-Bromophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9e). Appearance: dark yellow solid; yield: (71%); 1H NMR (500 MHz, CDCl3): δ 9.85 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.72 (s, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 3.74 (s, 2H), 3.15–3.07 (m, 4H), 2.73 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.7, 136.4, 134.2, 132.5, 129.2, 128.2, 121.0, 111.4, 64.6, 52.5, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H19BrN5O3S+: 464.0386, found: 464.0401. Chromatographic purity: 99.80%.
2-(4-((4-Fluorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9f). Appearance: yellow buff solid; yield: (80%); 1H NMR (500 MHz, CDCl3): δ 9.85 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.72 (s, 1H), 7.78 (dd, J = 8.9, 5.2 Hz, 2H), 7.26–7.21 (m, 2H), 7.18 (dd, J = 6.9, 4.2 Hz, 1H), 3.74 (s, 2H), 3.11 (s, 4H), 2.73 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.7, 136.4, 131.3, 130.5, 130.4, 116.6, 116.4, 111.5, 64.7, 52.5, 45.9; 19F NMR (471 MHz, CDCl3): δ −104.54; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H19FN5O3S+: 404.1187, found: 404.1180. Chromatographic purity: 99.74%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-((4-methoxyphenyl)sulfonyl)piperazin-1-yl)ethan-1-one (9g). Appearance: pale orange solid; yield: (66%); 1H NMR (500 MHz, CDCl3): δ 9.85 (dd, J = 6.9, 2.1 Hz, 1H), 8.79 (dd, J = 4.2, 2.1 Hz, 1H), 8.74 (s, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 7.01 (d, J = 8.9 Hz, 2H), 3.90 (s, 3H), 3.72 (s, 2H), 3.08 (s, 4H), 2.71 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.2, 163.2, 153.6, 151.0, 144.7, 136.4, 129.9, 126.5, 121.0, 114.3, 111.4, 64.9, 55.6, 52.6, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H22N5O4S+: 416.1387, found: 416.1386. Chromatographic purity: 95.17%.
2-(4-((2,5-Dichlorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9h). Appearance: pale yellow solid; yield: (74%); 1H NMR (500 MHz, CDCl3): δ 9.88 (dd, J = 6.9, 2.1 Hz, 1H), 8.81 (dd, J = 4.2, 2.1 Hz, 1H), 8.78 (s, 1H), 8.03 (dt, J = 5.1, 1.5 Hz, 1H), 7.48 (d, J = 1.4 Hz, 1H), 7.47 (d, J = 1.4 Hz, 1H), 7.20 (dd, J = 6.9, 4.2 Hz, 1H), 3.76 (s, 2H), 3.41 (dd, J = 6.5, 3.6 Hz, 4H), 2.71 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.2, 153.7, 151.0, 144.6, 137.2, 136.5, 133.7, 133.4, 133.2, 131.7, 131.7, 130.5, 111.5, 64.8, 53.0, 45.7; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H18Cl2N5O3S+: 454.0502, found: 454.0506. Chromatographic purity: 99.34%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-(mesitylsulfonyl)piperazin-1-yl)ethan-1-one (9i). Appearance: white solid; yield: (81%); 1H NMR (500 MHz, CDCl3): δ 9.88 (dd, J = 6.9, 2.1 Hz, 2H), 8.80 (s, 1H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 7.01–6.94 (m, 2H), 3.73 (s, 2H), 3.36–3.21 (m, 4H), 2.67 (d, J = 5.0 Hz, 4H), 2.31 (s, 6H), 1.58 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 187.4, 160.4, 153.6, 144.8, 142.6, 140.4, 136.4, 132.0, 130.8, 120.4, 111.4, 65.6, 60.4, 53.8, 52.7, 44.2, 30.9, 29.7, 23.8, 21.8, 14.2; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C21H26N5O3S+: 428.1751, found: 428.1761. Chromatographic purity: 99.51%.
2-(4-((5-Bromo-2-methoxyphenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9j). Appearance: off white solid; yield: (69%); 1H NMR (500 MHz, CDCl3): δ 9.88 (dd, J = 6.9, 2.1 Hz, 1H), 8.83–8.79 (m, 2H), 8.00 (d, J = 2.5 Hz, 1H), 7.63 (dd, J = 8.8, 2.6 Hz, 1H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 3.92 (s, 3H), 3.75 (s, 2H), 3.34 (t, J = 4.9 Hz, 4H), 2.70 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.3, 156.0, 153.6, 151.1, 144.8, 137.2, 136.5, 134.0, 127.7, 121.1, 114.1, 112.4, 111.4, 65.0, 56.3, 53.2, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H21BrN5O4S+: 494.0492, found: 494.0479. Chromatographic purity: 97.09%.
2-(4-((3-Bromophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9k). Appearance: pale orange solid; yield: (62%); 1H NMR (500 MHz, CDCl3): δ 9.85 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.71 (s, 1H), 7.90 (t, J = 1.8 Hz, 1H), 7.76 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.69 (ddd, J = 7.9, 1.7, 1.0 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 3.75 (s, 2H), 3.13 (s, 4H), 2.74 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.6, 138.0–135.6 (m), 130.6 (d, J = 17.4 Hz), 126.3, 123.3, 121.0, 111.5, 64.5, 52.5, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H19BrN5O3S+: 464.0386, found: 464.0370. Chromatographic purity: 96.12%.
2-(4-(Cyclohexylsulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9l). Appearance: pale orange solid; yield: (63%); 1H NMR (500 MHz, CDCl3): δ 9.91 (s, 1H), 8.85 (s, 1H), 8.81 (dd, J = 4.2, 2.1 Hz, 1H), 7.20 (dd, J = 6.9, 4.2 Hz, 1H), 3.76 (s, 2H), 3.54–3.41 (m, 4H), 2.92 (ddd, J = 12.2, 8.6, 3.4 Hz, 1H), 2.69 (t, J = 4.8 Hz, 4H), 2.19–2.08 (m, 2H), 1.89 (dt, J = 13.2, 3.2 Hz, 2H), 1.80–1.66 (m, 2H), 1.51 (qd, J = 12.5, 3.5 Hz, 2H), 1.42–1.10 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 187.4, 153.6, 151.1, 144.8, 136.5, 121.1, 111.4, 65.2, 61.4, 54.0, 46.1, 26.0, 25.2, 25.1; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H26N5O3S+: 392.1751, found: 392.1729. Chromatographic purity: 95.51%.
2-(4-((5-Bromothiophen-2-yl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9m). Appearance: pale yellow solid; yield: (60%); 1H NMR (500 MHz, CDCl3): δ 9.86 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.3, 2.1 Hz, 1H), 8.74 (s, 1H), 7.29 (d, J = 3.9 Hz, 1H), 7.19 (dd, J = 6.8, 4.2 Hz, 1H), 7.14 (d, J = 3.9 Hz, 1H), 3.76 (s, 2H), 3.17 (t, J = 4.9 Hz, 4H), 2.76 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.7, 136.4, 136.3, 132.7, 130.8, 121.0, 120.4, 111.5, 64.6, 52.4, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C16H17BrN5O3S2+: 469.9951, found: 469.9972. Chromatographic purity: 97.27%.
2-(4-(Cyclopropylsulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9n). Appearance: pale orange solid; yield: (70%); 1H NMR (500 MHz, CDCl3): δ 9.90 (dd, J = 6.8, 2.1 Hz, 1H), 8.85 (s, 1H), 8.82 (dd, J = 4.2, 2.1 Hz, 1H), 7.21 (dd, J = 6.9, 4.2 Hz, 1H), 3.78 (s, 2H), 3.47–3.39 (m, 4H), 2.74 (t, J = 4.9 Hz, 4H), 1.18 (dt, J = 4.8, 1.5 Hz, 2H), 1.05–0.99 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 187.3, 153.6, 151.1, 144.8, 136.5, 121.1, 111.4, 65.0, 52.9, 46.0, 25.2, 4.3; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C15H20N5O3S+: 350.1281, found: 350.1281. Chromatographic purity: 97.98%.
2-((4-(2-(Imidazo[1,2-a]pyrimidin-3-yl)-2-oxoethyl)piperazin-1-yl)sulfonyl)benzonitrile (9o). Appearance: buff yellow solid; yield: (61%); 1H NMR (500 MHz, CDCl3): δ 9.86 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.74 (s, 1H), 8.02 (dd, J = 7.9, 1.3 Hz, 1H), 7.92 (dd, J = 7.4, 1.5 Hz, 1H), 7.79 (td, J = 7.7, 1.5 Hz, 1H), 7.74 (td, J = 7.6, 1.4 Hz, 1H), 7.20 (dd, J = 6.9, 4.2 Hz, 1H), 3.76 (s, 2H), 3.33 (t, J = 5.0 Hz, 4H), 2.74 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.6, 139.2, 136.5, 135.7, 133.0 (d, J = 8.8 Hz), 130.4, 121.0, 116.3, 111.5, 111.1, 64.7, 52.6, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H19N6O3S+: 411.1234, found: 411.1235. Chromatographic purity: 98.91%.
2-(4-((2-Chloro-4-fluorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9p). Appearance: pale yellow solid; yield: (75%); 1H NMR (500 MHz, CDCl3): δ 9.87 (ddd, J = 6.9, 2.2, 0.7 Hz, 1H), 8.80 (dd, J = 4.2, 2.1 Hz, 1H), 8.78 (s, 1H), 8.06 (dd, J = 8.9, 5.9 Hz, 1H), 7.29 (dd, J = 8.2, 2.5 Hz, 1H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 7.15–7.09 (m, 1H), 3.75 (s, 2H), 3.38 (t, J = 4.9 Hz, 4H), 2.71 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.8, 154.6, 151.1, 144.8, 136.4, 134.2, 132.0, 121.0, 119.9, 119.7, 114.4, 114.3, 111.4, 64.9, 54.0, 46.0; 19F NMR (471 MHz, CDCl3) δ 103.52; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H18ClFN5O3S+: 438.0797, found: 438.0806. Chromatographic purity: 95.98%.
2-(4-((5-Chlorothiophen-2-yl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (9q). Appearance: off white solid; yield: (77%); 1H NMR (500 MHz, CDCl3): δ 9.87 (dd, J = 6.9, 2.1 Hz, 1H), 8.81 (dd, J = 4.2, 2.1 Hz, 1H), 8.75 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.20 (dd, J = 6.9, 4.2 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 3.77 (s, 2H), 3.17 (d, J = 5.2 Hz, 4H), 2.76 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.0, 153.7, 151.1, 144.7, 137.8, 136.4, 133.3, 132.0, 127.2, 121.0, 111.5, 64.6, 52.4, 46.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C16H17ClN5O3S2+: 426.0456, found: 426.0464. Chromatographic purity: 99.64%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-((4′-methoxy-[1,1′-biphenyl]-4-yl)sulfonyl)piperazin-1-yl)ethan-1-one (9r). Appearance: white solid; yield: (79%); 1H NMR (500 MHz, CDCl3): δ 9.84 (dd, J = 6.9, 2.1 Hz, 1H), 8.79 (dd, J = 4.2, 2.1 Hz, 1H), 8.72 (s, 1H), 7.78 (d, J = 8.5 Hz, 2H), 7.70 (d, J = 8.5 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.17 (dd, J = 6.9, 4.2 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 3.88 (s, 3H), 3.73 (s, 2H), 3.15 (s, 4H), 2.74 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 187.1, 160.1, 153.6, 145.7, 144.6, 136.4, 132.8, 131.6, 128.5, 128.3, 127.2, 114.5, 111.4, 64.7, 55.4, 52.6, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C25H26N5O4S+: 492.1700, found: 492.1677. Chromatographic purity: 100%.
1-(Imidazo[1,2-a]pyrimidin-3-yl)-2-(4-((4-nitrophenyl)sulfonyl)piperazin-1-yl)ethan-1-one (9s). Appearance: yellow solid; yield: (66%); 1H NMR (500 MHz, CDCl3): δ 9.84 (dd, J = 6.9, 2.1 Hz, 1H), 8.80 (dd, J = 4.3, 2.1 Hz, 1H), 8.71 (s, 1H), 8.44–8.40 (m, 2H), 7.98–7.93 (m, 2H), 7.19 (dd, J = 6.9, 4.2 Hz, 1H), 3.75 (s, 2H), 3.18 (d, J = 5.1 Hz, 4H), 2.75 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.9, 153.7, 150.7, 144.7, 141.4, 136.4, 128.9, 124.4, 120.9, 111.5, 64.6, 52.4, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C18H19N6O5S+: 431.1132, found: 431.1148. Chromatographic purity: 99.85%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-tosylpiperazin-1-yl)ethan-1-one (10a). Appearance: dark yellow solid; yield: (75%); 1H NMR (500 MHz, CDCl3): δ 9.61 (dt, J = 6.9, 1.2 Hz, 1H), 8.52 (s, 1H), 7.76 (dt, J = 8.9, 1.2 Hz, 1H), 7.64 (d, J = 1.9 Hz, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.54–7.49 (m, 1H), 7.34 (s, 1H), 7.33 (d, J = 1.9 Hz, 1H), 7.10 (td, J = 7.0, 1.3 Hz, 1H), 3.71 (s, 2H), 3.09 (s, 4H), 2.71 (t, J = 4.9 Hz, 4H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 186.2, 148.7, 143.8, 143.5, 132.0, 129.7, 129.4, 128.7, 127.9, 122.7, 117.7, 115.3, 64.7, 52.6, 45.9, 21.5; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C20H23N4O3S+: 399.1485, found: 399.1478. Chromatographic purity: 97.59%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-(phenylsulfonyl)piperazin-1-yl)ethan-1-one (10b). Appearance: yellow solid; yield: (74%); 1H NMR (500 MHz, CDCl3): δ 9.68–9.58 (m, 1H), 8.52 (s, 1H), 7.78 (dd, J = 8.2, 1.5 Hz, 3H), 7.66–7.62 (m, 1H), 7.59–7.52 (m, 3H), 7.12 (td, J = 6.9, 1.3 Hz, 1H), 3.75 (s, 2H), 3.14 (s, 4H), 2.74 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 148.8, 143.4, 139.1, 135.7, 133.0, 132.9, 130.8, 130.4, 129.5, 128.7, 117.8, 115.4, 111.2, 64.5, 52.6, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H21N4O3S+: 385.1329, found: 385.1323. Chromatographic purity: 97.26%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-(thiophen-2-ylsulfonyl)piperazin-1-yl)ethan-1-one (10c). Appearance: white solid; yield: (69%); 1H NMR (500 MHz, CDCl3): δ 9.62 (d, J = 7.0 Hz, 1H), 8.53 (s, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.64 (dd, J = 5.1, 1.3 Hz, 1H), 7.55–7.53 (m, 1H), 7.52 (dd, J = 7.2, 1.6 Hz, 1H), 7.16 (dd, J = 5.0, 3.7 Hz, 1H), 7.11 (td, J = 6.9, 1.3 Hz, 1H), 3.76 (s, 2H), 3.18 (s, 4H), 2.76 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.0, 148.8, 143.4, 135.4, 132.6, 132.4, 129.5, 128.7, 127.8, 122.7, 117.8, 115.4, 64.5, 52.8, 46.7; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C17H19N4O3S2+: 391.0893, found: 391.0885. Chromatographic purity: 100%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-((4-(trifluoromethyl)phenyl)sulfonyl)piperazin-1-yl)ethan-1-one (10d). Appearance: pale yellowish solid; yield: (69%); 1H NMR (500 MHz, CDCl3): δ 9.61 (d, J = 6.9 Hz, 1H), 8.50 (s, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.82 (d, J = 8.2 Hz, 2H), 7.77 (dt, J = 9.0, 1.1 Hz, 1H), 7.53 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.11 (td, J = 6.9, 1.3 Hz, 1H), 3.74 (s, 2H), 3.14 (s, 4H), 2.74 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 148.8, 143.9, 139.0, 134.8, 134.6, 129.5, 128.7, 128.3, 126.3, 122.7, 117.8, 115.4, 64.4, 52.5, 45.9; 19F NMR (471 MHz, CDCl3): δ −63.12; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C20H20F3N4O3S+: 453.1203, found: 453.1199. Chromatographic purity: 99.75%.
2-(4-((4-Bromophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10e). Appearance: dark yellow solid; yield: (64%); 1H NMR (500 MHz, CDCl3): δ 9.63 (dt, J = 6.9, 1.2 Hz, 1H), 8.53 (s, 1H), 7.79 (dt, J = 9.0, 1.2 Hz, 1H), 7.75–7.68 (m, 2H), 7.66–7.61 (m, 2H), 7.54 (ddd, J = 9.0, 6.9, 1.4 Hz, 1H), 7.13 (td, J = 6.9, 1.3 Hz, 1H), 3.75 (s, 2H), 3.13 (s, 4H), 2.75 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.0, 148.7, 143.4, 134.2, 132.4, 129.4, 129.2, 128.7, 128.1, 122.7, 117.7, 115.3, 64.5, 52.5, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H20BrN4O3S+: 463.0434, found: 463.0422. Chromatographic purity: 98.04%.
2-(4-((4-Fluorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10f). Appearance: yellow buff solid; yield: (69%); 1H NMR (500 MHz, CDCl3): δ 9.61 (dt, J = 6.9, 1.3 Hz, 1H), 8.51 (s, 1H), 7.82–7.75 (m, 3H), 7.52 (ddd, J = 8.6, 6.9, 1.3 Hz, 1H), 7.23 (t, J = 8.5 Hz, 2H), 7.11 (td, J = 6.9, 1.3 Hz, 1H), 3.74 (s, 2H), 3.14–3.09 (m, 4H), 2.73 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.0, 166.3, 164.3, 148.7, 143.4, 131.4, 130.5, 130.4, 129.4, 128.7, 117.7, 116.5, 116.3, 115.3, 64.5, 52.5, 45.9; 19F NMR (471 MHz, CDCl3): δ −104.7; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H20FN4O3S+: 403.1235, found: 403.1228. Chromatographic purity: 95.48%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-((4-methoxyphenyl)sulfonyl)piperazin-1-yl)ethan-1-one (10g). Appearance: pale orange solid; yield: (79%); 1H NMR (500 MHz, CDCl3): δ 9.70–9.56 (m, 1H), 8.55 (s, 1H), 7.84–7.75 (m, 1H), 7.71 (d, J = 8.9 Hz, 2H), 7.54 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.12 (td, J = 6.9, 1.2 Hz, 1H), 7.02 (d, J = 8.9 Hz, 2H), 3.91 (s, 3H), 3.74 (s, 2H), 3.11 (s, 4H), 2.74 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.1, 163.2, 143.4, 129.9, 129.4, 128.7, 126.6, 117.7, 115.3, 114.3, 64.7, 55.6, 52.6, 46.0; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C20H23N4O4S+: 415.1435, found: 415.1427. Chromatographic purity: 99.32%.
2-(4-((2,5-Dichlorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10h). Appearance: white solid; yield: (72%); 1H NMR (500 MHz, CDCl3): δ 9.64 (dt, J = 6.9, 1.2 Hz, 1H), 8.57 (s, 1H), 8.02 (t, J = 1.4 Hz, 1H), 7.81–7.76 (m, 1H), 7.53 (ddd, J = 8.9, 6.9, 1.3 Hz, 1H), 7.47 (d, J = 1.3 Hz, 2H), 7.12 (td, J = 6.9, 1.3 Hz, 1H), 3.75 (s, 2H), 3.45–3.37 (m, 4H), 2.71 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.1, 148.7, 143.5, 137.2, 133.6, 133.3, 133.2, 131.7, 130.9, 130.5, 130.0, 129.4, 128.7, 122.7, 117.8, 115.3, 64.7, 52.9, 45.8; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H19Cl2N4O3S+: 453.0549, found: 454.0506. Chromatographic purity: 99.71%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-(mesitylsulfonyl)piperazin-1-yl)ethan-1-one (10i). Appearance: white solid; yield: (80%); 1H NMR (500 MHz, CDCl3): δ 9.64 (dd, J = 20.1, 6.8 Hz, 1H), 8.61 (s, 1H), 7.81 (dd, J = 16.4, 9.1 Hz, 1H), 7.56 (dt, J = 15.9, 8.0 Hz, 1H), 7.15 (dt, J = 27.0, 6.9 Hz, 1H), 6.97 (s, 2H), 3.74 (s, 2H), 3.28 (t, J = 5.0 Hz, 4H), 2.69 (t, J = 4.9 Hz, 4H), 2.64 (s, 6H), 2.32 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 186.4, 148.7, 143.6, 142.6, 140.4, 132.8, 131.2, 129.4, 128.7, 122.8, 117.3, 115.3, 65.0, 52.3, 44.2, 23.0, 20.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C22H27N4O3S+: 427.1798, found: 427.1794. Chromatographic purity: 98.31%.
2-(4-((5-Bromo-2-methoxyphenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10j). Appearance: off white solid; yield: (70%); 1H NMR (500 MHz, CDCl3): δ 9.64 (dt, J = 6.9, 1.2 Hz, 1H), 8.57 (s, 1H), 8.00 (d, J = 2.5 Hz, 1H), 7.78 (dt, J = 8.9, 1.2 Hz, 1H), 7.62 (dd, J = 8.8, 2.6 Hz, 1H), 7.53 (ddd, J = 8.8, 6.9, 1.3 Hz, 1H), 7.12 (td, J = 6.9, 1.3 Hz, 1H), 6.90 (dd, J = 8.8, 5.0 Hz, 1H), 3.91 (s, 3H), 3.75 (s, 2H), 3.34 (t, J = 5.0 Hz, 4H), 2.70 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.2, 156.0, 148.7, 143.5, 137.1, 134.1, 129.4, 128.7, 127.8, 122.8, 117.8, 115.3, 114.0, 112.4, 64.8, 56.3, 53.2, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C20H22BrN4O4S+: 493.0540, found: 493.0531. Chromatographic purity: 99.18%.
2-(4-((3-Bromophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10k). Appearance: dark yellow solid; yield: (77%); 1H NMR (500 MHz, CDCl3): δ 9.64 (dt, J = 7.0, 1.2 Hz, 1H), 8.52 (s, 1H), 7.92 (t, J = 1.8 Hz, 1H), 7.85–7.75 (m, 2H), 7.75–7.69 (m, 1H), 7.55 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.13 (td, J = 6.9, 1.3 Hz, 1H), 3.77 (s, 2H), 3.16 (d, J = 6.0 Hz, 4H), 2.76 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 148.8, 143.3, 137.2, 136.0, 130.6, 130.6, 129.5, 128.7, 126.3, 123.3, 122.7, 117.8, 115.4, 64.4, 52.5, 46.0; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H20BrN4O3S+: 463.0434, found: 463.0423. Chromatographic purity: 99.09%.
2-(4-(Cyclohexylsulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10l). Appearance: pale orange solid; yield: (78%); 1H NMR (500 MHz, CDCl3): δ 9.66 (d, J = 6.9 Hz, 1H), 8.62 (s, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.54 (ddd, J = 8.8, 7.0, 1.4 Hz, 1H), 7.13 (td, J = 6.9, 1.3 Hz, 1H), 3.76 (s, 2H), 3.48–3.45 (m, 4H), 2.92 (tt, J = 12.1, 3.5 Hz, 1H), 2.69 (t, J = 4.8 Hz, 4H), 2.13 (d, J = 12.7 Hz, 2H), 1.89 (d, J = 13.1 Hz, 2H), 1.71 (d, J = 11.9 Hz, 1H), 1.51 (dd, J = 12.5, 3.5 Hz, 2H), 1.32–1.14 (m, 3H); 13C NMR (125 MHz, CDCl3): δ 185.7, 143.5, 129.4, 128.8, 122.8, 117.8, 115.3, 65.0, 61.4, 53.6, 46.1, 29.6, 26.6, 25.2, 25.1; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H27N4O3S+: 391.1798, found: 391.1792. Chromatographic purity: 99.68%.
2-(4-((5-Bromothiophen-2-yl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10m). Appearance: pale yellow solid; yield: (62%); 1H NMR (500 MHz, CDCl3): δ 9.63 (dt, J = 6.9, 1.2 Hz, 1H), 8.53 (s, 1H), 7.78 (dt, J = 9.0, 1.2 Hz, 1H), 7.53 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.28 (d, J = 3.9 Hz, 1H), 7.23–7.08 (m, 2H), 3.76 (s, 2H), 3.18 (t, J = 5.0 Hz, 4H), 2.76 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 148.8, 143.4, 136.3, 132.7, 130.8, 129.5, 128.7, 122.7, 120.3, 117.8, 112.2, 64.4, 52.4, 46.0; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C17H18BrN4O3S2+: 468.9998, found: 468.9987. Chromatographic purity: 95.82%.
2-(4-(Cyclopropylsulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10n). Appearance: pale orange solid; yield: (78%); 1H NMR (500 MHz, CDCl3): δ 9.67 (dt, J = 6.9, 1.2 Hz, 1H), 8.63 (s, 1H), 7.81 (dt, J = 8.9, 1.2 Hz, 1H), 7.58–7.54 (m, 1H), 7.15 (td, J = 6.9, 1.3 Hz, 1H), 3.80 (s, 2H), 3.43 (t, J = 4.9 Hz, 4H), 2.76 (t, J = 4.9 Hz, 4H), 2.29 (tt, J = 8.0, 4.8 Hz, 1H), 1.22–1.16 (m, 2H), 1.05–0.99 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 186.2, 148.7, 143.5, 129.8, 128.7, 123.4, 117.8, 115.4, 64.8, 53.3, 46.0, 25.5, 4.3; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C16H21N4O3S+: 349.1329, found: 349.1328. Chromatographic purity: 95.43%.
2-((4-(2-(Imidazo[1,2-a]pyridin-3-yl)-2-oxoethyl)piperazin-1-yl)sulfonyl)benzonitrile (10o). Appearance: white solid; yield: (66%); 1H NMR (500 MHz, CDCl3): δ 9.64 (dt, J = 6.9, 1.2 Hz, 1H), 8.55 (s, 1H), 8.07–8.01 (m, 1H), 7.96–7.91 (m, 1H), 7.75 (td, J = 7.6, 1.4 Hz, 3H), 7.55 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.13 (td, J = 6.9, 1.3 Hz, 1H), 3.77 (s, 2H), 3.36 (t, J = 4.9 Hz, 4H), 2.77 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 148.8, 143.4, 139.9, 135.7, 133.0, 132.9, 130.8, 129.5, 128.7, 124.5, 117.8, 116.1, 115.4, 111.2, 64.5, 52.1, 45.9; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C20H20N5O3S+: 410.1281, found: 410.1277. Chromatographic purity: 98.95%.
2-(4-((2-Chloro-4-fluorophenyl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10p). Appearance: pale yellowish solid; yield: (62%); 1H NMR (500 MHz, CDCl3): δ 9.64 (d, J = 6.9 Hz, 1H), 8.57 (s, 1H), 8.05 (dd, J = 8.9, 5.8 Hz, 1H), 7.78 (d, J = 9.0 Hz, 1H), 7.53 (ddd, J = 8.8, 6.9, 1.3 Hz, 1H), 7.28 (s, 1H), 7.14–7.09 (m, 2H), 3.75 (s, 2H), 3.44–3.26 (m, 4H), 2.84–2.55 (m, 4H); 13C NMR (125 MHz, CDCl3): δ 186.6, 165.1, 162.8, 148.7, 143.5, 135.0, 132.0, 129.4, 128.7, 122.7, 119.8, 117.7, 116.3, 114.8, 64.4, 52.9, 45.7; 19F NMR (471 MHz, CDCl3): δ −103.60; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H19ClFN4O3S+: 437.0845, found: 437.0837. Chromatographic purity: 100%.
2-(4-((5-Chlorothiophen-2-yl)sulfonyl)piperazin-1-yl)-1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one (10q). Appearance: off white solid; yield: (77%); 1H NMR (500 MHz, CDCl3): δ 9.67–9.64 (m, 1H), 8.56 (s, 1H), 7.80 (dt, J = 8.9, 1.2 Hz, 1H), 7.55 (ddd, J = 8.8, 6.9, 1.4 Hz, 1H), 7.34 (d, J = 4.0 Hz, 1H), 7.14 (td, J = 6.9, 1.3 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 3.78 (s, 2H), 3.20 (t, J = 5.0 Hz, 4H), 2.78 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.0, 148.8, 143.4, 137.7, 133.5, 132.0, 129.5, 128.7, 127.2, 122.7, 117.8, 115.4, 64.5, 52.4, 46.0; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C17H18ClN4O3S2+: 425.0503, found: 425.0498. Chromatographic purity: 95.82%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-((4′-methoxy-[1,1′-biphenyl]-4-yl)sulfonyl)piperazin-1-yl)ethan-1-one (10r). Appearance: white solid; yield: (73%); 1H NMR (500 MHz, CDCl3): δ 9.63 (dt, J = 6.9, 1.1 Hz, 1H), 8.53 (s, 1H), 7.82–7.79 (m, 2H), 7.78 (dt, J = 9.0, 1.2 Hz, 1H), 7.72–7.70 (m, 2H), 7.60–7.56 (m, 2H), 7.53 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.12 (td, J = 6.9, 1.2 Hz, 1H), 7.06–7.03 (m, 2H), 3.90 (s, 3H), 3.76 (s, 2H), 3.17 (s, 4H), 2.76 (t, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 186.0, 160.1, 148.7, 145.6, 143.3, 133.3, 131.6, 129.4, 128.7, 128.5, 128.3, 127.2, 122.7, 117.7, 115.3, 114.5, 64.5, 55.4, 52.6, 46.0; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C26H27N4O4S+: 491.1748, found: 491.1736. Chromatographic purity: 100%.
1-(Imidazo[1,2-a]pyridin-3-yl)-2-(4-((4-nitrophenyl)sulfonyl)piperazin-1-yl)ethan-1-one (10s). Appearance: dark yellow solid; yield: (65%); 1H NMR (500 MHz, CDCl3): δ 9.60 (dt, J = 6.9, 1.2 Hz, 1H), 8.50 (s, 1H), 8.40 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.8 Hz, 2H), 7.79–7.75 (m, 1H), 7.53 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 7.12 (dd, J = 6.9, 1.3 Hz, 1H), 3.74 (s, 2H), 3.17 (t, J = 4.9 Hz, 4H), 2.75 (t, J = 4.9 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 185.9, 150.3, 148.7, 143.4, 141.5, 129.5, 128.9, 128.7, 124.3, 122.6, 117.7, 115.4, 64.4, 53.1, 46.4; HRMS (ESI-Q-TOF) m/z: [M + H]+ calculated for C19H20N5O5S+: 430.1180, found: 430.1185. Chromatographic purity: 97.99%.
FE-SEM and EDS
The overall surface morphological characteristics and size of the synthesized molecules were examined using FE-SEM or field emission scanning electron microscopy (Carl Zeiss, Germany) at an accelerating voltage of 3–5 kV. Energy dispersive X-ray spectroscopy (EDS) was used to perform elemental analysis.The samples were first spread on top of the carbon tape, and after being sputter-coated with gold metal, electron conduction between the sample and the stub surface charge accumulation was successfully eliminated. At various magnifications, 20 μm, 10 μm, and 5 μm micrographs were taken. Following this, EDS analysis was also carried out.
Single-crystal X-ray diffraction
The crystals were prepared by dissolving the powdered compound 10q in a solvent system made of CHCl3:CH3OH at a ratio of 1:1 (1 ml each) in a glass vial (5 ml), left undisturbed for 3 days and allowed to slowly evaporate at room temperature using the slow evaporation method in a dust-free environment. Single crystals of C17H17ClN4O3S2, 10q were collected. A suitable crystal was selected and mounted on a Bruker APEX-II CCD diffractometer equipped with a PHOTON II detector using graphite monochromated MoKα radiation (λ = 0.461 mm−1). The crystal was kept at 299.0 K during data collection. Crystalline data for 10q (M = 424.91 g mol−1): triclinic, space group P (no. 2), a = 6.6727(9) Å, b = 10.1859(14) Å, c = 14.367(2) Å, α = 85.903(5)°, β = 80.316(5)°, γ = 73.428(5)°, V = 922.3(2) Å3, Z = 2, T = 299.0 K, μ(MoKα) = 0.461 mm−1, Dcalc = 1.530 g cm−3, 25692 reflections measured (2.876° ≤ 2Θ ≤ 58.58°), 4319 unique (Rint = 0.0615, Rsigma = 0.0349) were used in all calculations. The final R1 was 0.0600 (I > 2σ(I)), and wR2 was 0.1524 (all data). Using Olex2, the structure was solved with the olex2.solve structure solution program using charge slipping and refined with the ShelXL refinement package using least squares minimization. The final data were visualized with ORTEP-3 software. The crystal and refinement data for the compounds are shown in Table S1.† The full details are reported in the ESI.† The structure of compound 10q was deposited under CCDC deposition number 2331542.
Cell line
The A549 cell line, a lung cancer cell line, COLO-205, a human colorectal cancer cell line, MCF-7, a human breast cancer cell line, and CAL-27, a human oral squamous cell carcinoma cell line was obtained from the American Type Culture Collection (ATCC). The BEAS-2B cell line was a generous gift from Dr. Srivatsava Naidu, Assistant Professor, Centre for Biomedical Engineering, Indian Institute of Technology Ropar, Punjab, India. DMEM, DMEM/F12 (Dulbecco's modified essential medium and Ham's F-12 medium), fetal bovine serum (FBS), penicillin, and streptomycin were obtained from Thermo Fisher Scientific (Gibco) India. Human pyruvate kinase M2 (PKM2, SAE0021) was purchased from Sigma Aldrich (India). Alamar blue dye was purchased from HiMedia Laboratories (India). F-12K medium was used to cultivate the A549 cells. All media contained 100 IU mL−1 penicillin, 100 IU mL−1 streptomycin, and 10% fetal bovine serum (FBS). The cells were cultured in 95% air and 5% CO2 in a CO2 incubator at 37 °C.Antiproliferative activity
The ability of the synthetic chemicals to inhibit A549 cell proliferation was tested. In a 96-well plate, 1 × 104 cells per well were plated and allowed to attach for 24 hours. A459 cells were exposed to various concentrations of 9b (1 μM, 1.5 μM, 2 μM, 2.5 μM, and 3 μM) for 48 hours. A Varioskan LUX multimode microplate reader (Thermo Fisher Scientific) was used to record the fluorescence at 570 nm and emission at 590 nm after an additional 4 hours of incubation with 10 μL (5 mg mL−1) of Alamar blue solution. The percentage of viable cells was estimated using the following equation: the formula for percentage cell viability is (At/Ac) × 100, where At is the absorbance of the test compound and Ac is the control. Media absorbance was measured for cell viability using a Varioskan LUX multimode microplate reader. Cell viability studies were performed following the same protocol with the human normal lung epithelial cell line BEAS-2B to evaluate the cancer specificity of the synthesized series. The protocol followed was the same as that of the studies performed on A549 cells with different sample concentrations (20 μM, 40 μM, 60 μM, 80 μM, and 100 μM). Doxorubicin (D1515-10MG, Sigma Aldrich) and DASA-58 (SML2853-5MG, Sigma Aldrich) were used as standard compounds.60 Similar protocol was followed for evaluating the compounds against COLO-205, MCF-7 and CAL-27 cell line.Pyruvate assay
This assay was designed to evaluate the effect of 9b (0.91 μM) on A549 cells in terms of changes in the amount of pyruvate in the 9b-treated group compared with the untreated group. The assay was performed with a Pyruvate Assay kit (MAK071) procured from Sigma Aldrich according to the manufacturer's instructions. The absorbance measurements were taken using a Varioskan LUX microplate reader at a wavelength of 570 nm (Thermo Fisher Scientific). The experiment was carried out in triplicate.58Lactate release assay
Supernatants from 9b (0.91 μM) treated A549 cell culture (1 × 104 cells per well) were used to assess lactate release. The lactate concentration in the supernatant was determined using a Lactate Assay kit (MAK064) procured from Sigma Aldrich, as directed by the manufacturer. The raw data were normalized to the cell density acquired using the trypan blue procedure. The absorbance measurements were taken using a Varioskan LUX microplate reader at a wavelength of 570 nm (Thermo Fisher Scientific). The experiment was carried out three times.61,62Cell cycle analysis
A549 (1 × 105) cells were seeded in each well and treated with 9b at a concentration of 0.91 μM before being incubated for 24 hours at 37 °C with 95% air and 5% CO2. After incubation, the cells were rinsed with 1× phosphate-buffered saline (PBS) three times, and a pellet was generated. The cells were fixed in 300 μL of ice-cold 70% ethanol and left for 30 minutes. Following incubation, the cells were treated with 250 μL of RNaseA (100 μG mL−1) on ice for 20 minutes after being washed with 1× PBS. The cells were then rinsed with 1× PBS and given 250 μL of 50 μG mL−1 propidium iodide (Thermo Fisher Scientific (Gibco) India) before being left alone for 20 minutes. Using the S3e Cell Sorter device, cell cycle analysis and an apoptosis experiment were carried out (Bio-Rad).63Apoptosis assay
A549 (1 × 105) cells were cultured in each well of a 6-well plate containing F-12K medium. After 24 hours, the cells were treated with 9b at a concentration of 0.91 μM and then incubated for 24 hours at 37 °C with 5% CO2. After 24 hours, the cells were washed with 1× PBS and resuspended for 20 minutes in 100 μL of 1× annexin binding buffer on ice protected from any light source. Next, the cells were stained for 15 minutes at room temperature with 5 μL each of propidium iodide (PI) and an amount of Annexin-V (Thermo Fisher Scientific (Gibco) India) (5 μL of 100 μG mL−1). After the incubation period, 400 μL of 1× annexin binding buffer was added, and then a flow cytometer (Bio-Rad) was used to quantify the fluorescence.64Reactive oxygen species (ROS) detection assay
A549 cells were cultured overnight (1 × 104 cells per well) to detect the extent of ROS in the cell cultures. Cultures treated with 9b (0.91 μM) were compared with the control group using the Fluorometric Intracellular ROS kit purchased from Sigma Aldrich (MAK145). The assay was performed according to the manufacturer's protocol. The fluorescence measurements were taken using a Varioskan LUX microplate reader at an excitation wavelength of 520 nm and an emission wavelength of 605 nm (Thermo Fisher Scientific). The experiment was carried out three times. The assay was planned for 24 hours, with readings measured at 3 h, 6 h, 12 h, 24 h and 48 h.65,66Lactate dehydrogenase (LDH)-coupled enzyme assay
The reported methodology was followed to evaluate the activation and inhibition of pyruvate kinase (PK) using a lactate dehydrogenase-coupled assay. After being dissolved in DMSO, all of the synthesized substances were created at final concentrations of 0.01, 0.1, 0.25, 0.5, and 1 μM. The final concentrations in the reaction mixture were 50 mM Tris-HCl (pH 7.5), 100 mM KCl, 10 mM MgCl2, 0.6 mM PEP, 0.9 mM ADP, 0.12 mM β-NADH, and 8 U mL−1 LDH. All these chemicals were purchased from Sigma Aldrich, India. 9 μL of recombinant human PKM2 (Sigma Aldrich, India) solution (50 pg mL−1) was combined with 1 μL of the desired analyte and maintained at 25 °C for 30 minutes. Then, the pyruvate concentration was assessed after 10 μL of this solution was added to 125 μL of the reaction mixture. By quantifying the decrease in β-NADH (Alfa Aesar) absorbance at 340 nm over 20 minutes, the substances were assessed using a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific).67Surface plasmon resonance (SPR)
The specific SPR angle at which resonance occurs under a constant light source wavelength and a thin metal surface is determined by the refractive index of the material near the metal surface. There are five parts of a sensogram generated from SPR. These are dissociation, equilibrium, association, regeneration, and baseline.68 Here, the PKM2 protein was immobilized on a 50 μg mL−1 carboxymethylated sensor chip. The buffer used was 1.05× PBS-P+. The stock solutions were prepared in an assay running buffer supplemented with DMSO. Once ligand 9b was immobilized on the sensor surface, any remaining binding sites were blocked, and the interaction analysis sensorgram was generated and analyzed. The system used was a Cytiva Biacore™ T200. All the experiments were performed according to the manufacturer's protocol for small- to medium-sized compound libraries (few samples).Determination of particle size by zetasizer
In that order, recombinant PKM2 (250 μg mL−1) and the compound (9b) solution (10 μM) were each filtered through 20 nm (Whatman, Anotop 10) and 0.45 μm hydrophilic PTFE (Millipore) filters. The particle size of the recombinant PKM2 filtrate was measured using a dynamic laser light scattering (DLS) analyzer after exposure to 9b for 30 minutes at room temperature (Zetasizer Nano ZS, Malvern Panalytical, United Kingdom).43Molecular docking studies
The Protein Data Bank (PDB) was used to obtain the crystal structure of the protein human pyruvate kinase M2 (PDB ID: 3GR4), which was subsequently used to mimic the protein structure in this work. The protein structures are refined in terms of bond order, formal charges, missing hydrogen atoms, topologies, and incomplete and terminal amide groups. Beyond the 5 Å range of the heteroatom, water molecules were removed. All possible ionization states for each heteroatom in the protein structure were generated, and the most stable state was selected. The orientations of the retained water molecules changed, and the hydrogen bonds were distributed. Finally, a restricted reduction of the protein structure was carried out using the OPLS2005 force field to realign side-chain hydroxyl groups and lessen potential steric conflicts. The reduction is limited to the provided protein coordinates by a previous root mean square deviation (RMSD) tolerance of 0.3. ChemDraw Ultra version 20.0 was used to construct the ligand structures in CDX format. The LigPrep module of Maestro 12.7.161 was then used to execute these ligands in the Schrödinger suite 2021 after being converted to the mol2 format. Using stereochemical, ionization, and tautomeric variations together with energy minimization, they were converted from 2D to 3D structures, optimized for geometry, desalted, and corrected for missing hydrogen atoms. These ligands' charged groups were neutralized, and their bond ordering was filled. Ionization and tautomerization states were produced between pH 6.8 and 7.2 using the Epik module. In the final step of LigPrep, compounds were decreased using the Schrödinger Impact Program's Optimized Potentials for Liquid Simulations-2005 (OPLS-2005) force field until a root mean square deviation of 1.8 was achieved. The steepest descent method was used for minimization, followed by the conjugate gradient method. After only one low-energy ring conformation per ligand was produced, the optimized ligands were used for docking investigations.69 The generated protein that was used to create the receptor grid had the ligands preserved in its crystal structure. The protein's binding box was modified to have dimensions of 14 × 14 × 14, which restricts the centroid of the docked posture.69 The accuracy of the docking process was evaluated by contrasting an experimental binding mode discovered by X-ray crystallography with the lowest energy pose (binding conformation) of the cocrystallized ligand predicted by the object scoring function Glide score (G score). The higher precision Glide docking method was confirmed by withdrawing the cocrystallized ligand from the protein's binding site and redocking it with its binding site.69Hydrogen bonding interactions and the root mean square deviation (RMSD) between the predicted and actual X-ray crystallographic conformations were used to analyze the data. Using the receptor grid and already-made ligand molecules, the chosen compounds were glide-docked. The favorable interactions between ligand molecules and the receptor were evaluated using Glide ligand docking software (Schrödinger Release 2020-3: Maestro, Schrödinger, LLC, New York, NY, 2023). All docking calculations used the OPLS-2005 force field in extra precision (XP) mode. The docking technique described above was executed in flexible docking mode, which automatically generates conformations for each input ligand. To evaluate the interaction of the ligand with the receptor, a series of hierarchical filters were used to fit the created ligand poses. The initial filter examines the spatial fit of the ligand to the protein active site and assesses the complementarity of ligand–receptor interactions using a grid-based method modeled after the empirical ChemScore function. This approach recognizes beneficial hydrophobic, hydrogen-bonding, and metal–ligation interactions while penalizing steric conflicts.69 Poses that pass these first inspections move on to the method's next stage, which involves evaluating and minimizing an approximation of the nonbonded ligand–receptor interaction energy grid. Finally, the reduced postures were rescored using the Glide score scoring algorithm. The fitness ratings for each ligand in PKM2 were compared together with a summary of the XP-Glide scores of the active compounds. To determine the relative PKM2 modulation activity, the glide scores of the examined compounds were compared to those of the reference substance DASA-58 (a known PKM2 activator).69
Density functional theory studies and molecular electrostatic potential calculations
Using the licensed Schrödinger software program's Jaguar module, a DFT analysis of molecule 9b was conducted. With the Becke 3-Lee–Yang–Parr (B3LYP) technique, which uses the 6-31+G(d) basis set on the atoms, the geometries were optimized using hybrid functionals. The optimized geometries underwent FMO analysis and MESP. FMO analysis provides insight into the mechanism of action of this molecule. The high activity of the molecule is correlated with the tiny gap between the HOMO and LUMO.70Molecular dynamic simulation studies
To evaluate the stability of the docked 9b/PKM2 (PDB ID – 3GR4) complex, a 100 ns MD simulation analysis was conducted. In an explicit solvent system with an OPLS3 force field, the complex was examined using Schrödinger 2023's Desmond module. The molecular system was solvated utilizing crystallographic water (TIP3P) molecules under orthorhombic periodic boundary conditions. By injecting Na+ as a counter ion, the system was neutralized, and the overlapping water molecules were eliminated. An ensemble (NPT) of Nose–Hoover thermostats and a barostat were employed to maintain consistent temperatures (300 K) and pressures (1 bar) of the systems. A hybrid energy minimization method that used conjugate gradient techniques after 1000 steps of steepest descent was applied. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) approach with restricted memory and a convergence threshold gradient of 1 kCal mol−1 was applied to reduce energy. The smooth particle mesh Ewald method was used for long-range electrostatic interactions, with a cutoff radius of 9 Å for Van der Waals and Coulomb interactions. For bonded, near-bonded, and far-bonded interactions with 2, 2, and 6 femtoseconds (fs), respectively, multiple time step reference system propagator algorithm (RESPA) integrations were used in the dynamics analysis. The data were collected every 100 picoseconds (ps), and the resulting trajectory was assessed using the Maestro 12.7.161 graphical user interface.70Crystallographic data
CCDC contains the supplementary crystallographic data for this paper. The CCDC deposition number is 2165838. The crystallographic data are from Amit Shard CCDC 2331542: Experimental Crystal Structure Determination, 2021, DOI: https://doi.org/10.5517/ccdc.csd.cc2j8511.Statistical analysis
GraphPad Prism software, version 8.0 (GraphPad Prism Software, Inc., La Jolla, CA) was used for all the statistical analyses in this work. The experimental data are shown as the mean ± standard deviation, n = 3. The level of significance was tested using Student's t-test or two-way analysis of variance with Tukey's multiple comparisons test, and p < 0.05 was considered to indicate statistical significance. Tested significance is displayed in the figures as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0005.Conclusion
Imbibing inspiration from the previous literature and synthesizing molecules that provide the advantage of tunability, allowing for a precise design tailored to physiological conditions, is a rational drug discovery approach. By incorporating specific chemical attributes into their structure, these molecules enhance their interaction with targeted biological molecules. The modulation of mutated oncogenic proteins, including tumor PKM2, through rationally developed molecules represents an underexplored frontier in research. The original aim of this study was to investigate whether substituting thiazole with sulfonamide could maintain its affinity for PKM2 while improving the physicochemical properties of the compound. Although we observed that sulfonamides indeed strongly activated PKM2, to our delight, this effect was accompanied by a potent anti-LC effect.To design the ligands, drawing upon prior knowledge and experience in designing modulators against PKM2, we initiated a hybridization approach. This hybridization was inspired by the reputable roles of sulfonamides and imidazopyri(mi)dines in drug discovery. Imidazopyri(mi)dines were hybridized with sulfonamides, resulting in novel sulfonamide-based imidazopyri(mi)dines through established and reliable reactions, enhancing skeletal diversity. Using in silico tools, we assessed whether the synthesized molecules effectively modulated PKM2 at the desired site and screened for promiscuous PAINS. This computational approach was followed for the synthesis of 38 derivatives. This was followed by full spectroscopic characterization and purity analysis for biological investigation. The chemical structure was fully elucidated by single-crystal X-ray diffraction (XRD). Furthermore, thorough wet laboratory experiments were performed to determine the efficacy, selectivity, and mode of action of these compounds. Our novel imidazopyri(mi)dine-based sulfonamides exhibited success in alleviating human adenocarcinoma in vitro. The LDH-coupled enzyme assay established the majority of the series as PKM2 activators with AC50 values comparable to those of DASA-58. Notably, compound 9b was the most potent, inducing early apoptosis and G2 phase arrest in the A549 cell cycle. Further investigations revealed significant anticancer effects of 9b, including increased reactive oxygen species and decreased lactate concentrations. Particle size enhancement post-treatment indicated tetramerization of PKM2, aligning with common traits of PKM2 activators. Structural features were explored through DFT, MESP, and MD studies, revealing a low HOMO–LUMO gap and an abundance of electrophilic sites in 9b—attributes indicative of promising anticancer agents with high reactivity. MD studies demonstrated that 9b stabilizes PKM2 after binding to the activator binding site. SPR analysis confirmed the strong affinity of 9b for PKM2, establishing it as a potential lead compound with promising drug-binding kinetics. By enhancing its potential established by an exploratory SAR, especially in lung cancer mitigation using biophysical techniques, we propose furthering 9b for preclinical studies. The molecules can be furthered for in vivo studies in lung cancer models to evaluate the effectivity in animal system. Also, in-depth molecular pathway can be furnished to elucidate the mechanism of action of the compound 9b in lung cancer.
Abbreviations
| ACN | Acetonitrile |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| ATCC | American Type Culture Collection |
| ALK | Anaplastic lymphoma kinase |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
| B3LYP | Becke 3-Lee–Yang–Parr |
| CAGR | Compound annual growth rate |
| CCDC | Cambridge Crystallographic Data Centre |
| CHCl3 | Chloroform |
| DASA-58 | 3-[[4-(2,3-Dihydro-1,4-benzodioxin-6-ylsulfonyl)-1,4-diazepanyl]sulfonyl]aniline |
| DCM | Dichloromethane |
| DMEM/F12 | Dulbecco's modified essential medium and Ham's F-12 medium |
| DFT | Density function theory |
| DMF | Dimethylformamide |
| DMF-DMA | N,N-Dimethylformamide dimethyl acetal |
| DMSO | Dimethyl sulfoxide |
| EDAX | Energy-dispersive X-ray analysis |
| EDS | Energy dispersive X-ray spectroscopy |
| EGFR | Epithelial growth factor receptor |
| FBS | Fetal bovine serum |
| FDA | Food and Drug Administration |
| FESEM | Field emission scanning electron microscopy |
| FLT3 | fms-like tyrosine kinase 3 |
| FMO | Frontier molecular orbital high-performance liquid chromatography |
| HRMS | High-resolution mass spectrometry |
| LC | Lung cancer |
| LUMO | Lowest unoccupied molecular orbital |
| MD | Molecular dynamics |
| MESP | Molecular electrostatic potential |
| MMP | Matrix metalloproteinase |
| NMR | Nuclear magnetic resonance |
| NPT | Nose–Hoover thermostats |
| NSCLC | Non-small cell lung cancer |
| PBS | Phosphate-buffered saline |
| PI | Propidium iodide |
| PI3K | Phosphoinositide 3-kinase inhibitors |
| PK | Pyruvate kinase |
| PKR | Pyruvate kinase R |
| PKM2 | Pyruvate kinase M2 |
| RESPA | Reference system propagator algorithm |
| RMSD | Root mean square deviation |
| RBM39 | RNA-binding motif protein 39 |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| SCLC | Small cell lung cancer |
| SC X-RD | Single crystal X-ray diffraction |
| SEM | Scanning electron microscopy |
| SPR | Surface plasmon resonance |
| TCPS | Tissue culture polystyrene |
| TKIs | Tyrosine kinase inhibitors |
| TLC | Thin layer chromatography |
| USFDA | United States Food and Drug Administration |
| WHO | World Health Organization |
| XP | Extra precision |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Amit Shard: conceptualization, supervision and funding acquisition. Rudradip Das: investigation, methodology, writing – original draft, validation. Deep Rohan Chatterjee: investigation. Saumya Kapoor: investigation. Het Vyas: investigation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the director, NIPER-ahmedabad, for support and encouragement. The authors also acknowledge support from the Dept. of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India for this research. The authors are also grateful to Dr. Yogendra S. Padwad, Principal Scientist, Dept. of Pharmacology and Toxicology, CSIR-IHBT, Palampur, Himachal Pradesh, India, for his support and SPR studies. The authors heartfully thank Dr. Srivatsava Naidu, Assistant Professor, Centre for Biomedical Engineering, Indian Institute of Technology Ropar, Punjab, India, for providing the human normal lung epithelial cell line BEAS-2B as a gift. Amit Shard acknowledges the funding received from the Indian Council of Medical Research (ICMR), Govt. of India (File no: 5/4/8-29/CD/AS/2022-NCD-II). The authors would like to acknowledge the Central Instrumentations Lab, University of Hyderabad for the Single Crystal X-ray Diffraction Facility. Generative AI and AI-assisted technologies were only used in the writing process to improve the readability and language of the manuscript. The communication number for publication is NIPER-a/02/2024/1015.References
- E. K. Chung, S. H. Yong, E. H. Lee, E. Y. Kim, Y. S. Chang and S. H. Lee, Tuberc. Respir. Dis., 2023, 86, 1–13 CrossRef PubMed.
- W. D. Travis, E. Brambilla, M. Noguchi, A. G. Nicholson, K. R. Geisinger, Y. Yatabe, D. G. Beer, C. A. Powell, G. J. Riely and P. E. Van Schil, J. Thorac. Oncol., 2011, 6, 244–285 CrossRef PubMed.
- J. A. Barta, C. A. Powell and J. P. Wisnivesky, Ann. Glob. Health, 2019, 85, 1–16 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Ca-Cancer J. Clin., 2023, 73, 17–48 CrossRef PubMed.
- K. C. Thandra, A. Barsouk, K. Saginala, J. S. Aluru and A. Barsouk, Contemp. Oncol., 2021, 25, 45–52 CAS.
- American Society of Clinical Oncology, Lung Cancer – Non-Small Cell: Statistics, https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics#:~:text=ForNSCLC%2Cthe5-year,thesubtypeoflungcancer.
- F. R. Hirsch, G. V. Scagliotti, J. L. Mulshine, R. Kwon, W. J. Curran, Y.-L. Wu and L. Paz-ares, Lancet, 2017, 389, 299–311 CrossRef CAS PubMed.
- B. E. Johnson and P. A. Jänne, Cancer Res., 2005, 65, 7525–7529 CrossRef CAS PubMed.
- B. T. Hennessy, E. O. Hanrahan and O. S. Breathnach, Onco Targets Ther, 2003, 8, 270–277 CAS.
- M. Gogishvili, T. Melkadze, T. Makharadze, D. Giorgadze, M. Dvorkin, K. Penkov, K. Laktionov, G. Nemsadze, M. Nechaeva, I. Rozhkova, E. Kalinka, C. Gessner, B. Moreno-Jaime, R. Passalacqua, S. Li, K. McGuire, M. Kaul, A. Paccaly, R. G. W. Quek, B. Gao, F. Seebach, D. M. Weinreich, G. D. Yancopoulos, I. Lowy, G. Gullo and P. Rietschel, Nat. Med., 2022, 28, 2374–2380 CrossRef CAS PubMed.
- C. K. Lee, L. Davies, Y. L. Wu, T. Mitsudomi, A. Inoue, R. Rosell, C. Zhou, K. Nakagawa, S. Thongprasert, M. Fukuoka and S. Lord, J. Natl. Cancer Inst., 2017, 109, 279 Search PubMed.
- J.-Q. Xu, Y.-L. Fu, J. Zhang, K.-Y. Zhang, J. Ma, J.-Y. Tang, Z.-W. Zhang and Z.-Y. Zhou, Front. Pharmacol., 2022, 13, 1037341 CrossRef CAS PubMed.
- (a) M. Rihan and S. Sharma, Toxicol. Appl. Pharmacol., 2024, 485, 116905 CrossRef CAS PubMed; (b) D. Anastasiou, Y. Yu, W. J. Israelsen, J. K. Jiang, M. B. Boxer, B. S. Hong, W. Tempel, S. Dimov, M. Shen, A. Jha, H. Yang, K. R. Mattaini, C. M. Metallo, B. P. Fiske, K. D. Courtney, S. Malstrom, T. M. Khan, C. Kung, A. P. Skoumbourdis, H. Veith, N. Southall, M. J. Walsh, K. R. Brimacombe, W. Leister, S. Y. Lunt, Z. R. Johnson, K. E. Yen, K. Kunii, S. M. Davidson, H. R. Christofk, C. P. Austin, J. Inglese, M. H. Harris, J. M. Asara, G. Stephanopoulos, F. G. Salituro, S. Jin, L. Dang, D. S. Auld, H. W. Park, L. C. Cantley, C. J. Thomas and M. G. Van der Heiden, Nat. Chem. Biol., 2012, 8, 839–847 CrossRef CAS PubMed; (c) M. Kusuma, S. Arora, S. Kalra, A. Chaturvedi, M. Heuser and R. Kumar, Curr. Top. Med. Chem., 2021, 21, 2258–2271 CrossRef CAS PubMed; (d) S. Arora, G. Joshi, A. Chaturvedi, M. Heuser, S. Patil and R. Kumar, J. Med. Chem., 2021, 65, 1171–1205 CrossRef PubMed.
- M. B. D. Aldonza, J.-Y. Hong and S. K. Lee, Exp. Mol. Med., 2017, 49, e286 CrossRef CAS PubMed.
- S. An, L. Huang, P. Miao, L. Shi, M. Shen, X. Zhao, J. Liu and G. Huang, Onco Targets Ther, 2018, 11, 2097–2109 CrossRef PubMed.
- C. Wang, S. Zhang, J. Liu, Y. Tian, B. Ma, S. Xu, Y. Fu and Y. Luo, Cell Rep., 2020, 30, 1780–1797 CrossRef CAS PubMed.
- (a) J. Zhang, C. Zhang, X. Liu, N. Sun, C. Zhang, R. Li and Z. Zhang, Transl. Cancer Res., 2020, 9, 3293 CrossRef CAS PubMed; (b) S. Zhu, Y. Guo, X. Zhang, H. Liu, M. Yin, X. Chen and C. Peng, Cancer Lett., 2021, 503, 240–248 CrossRef CAS PubMed; (c) B. Rathod, S. Chak, S. Patel and A. Shard, RSC Med. Chem., 2021, 12, 1121–1141 RSC.
- S. Sommakia, Y. Matsumura, C. Allred, S. Pathi, E. Tyagi, J. Foulks, A. Siddiqui and S. Warner, Eur. J. Cancer, 2022, 174, S47 CrossRef.
- H. Al-Samkari and E. J. van Beers, Ther. Adv. Hematol., 2021, 12, 1–11 Search PubMed.
- S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64–66 CrossRef CAS PubMed.
- R. M. I. Elsamra, M. S. Masoud and A. M. Ramadan, Sci. Rep., 2022, 12, 20192 CrossRef CAS PubMed.
- R. Das, G. Tambe and A. Shard, Results Chem., 2023, 100950 CrossRef CAS.
- (a) S. Ghosh, Sulphonamides Market Outlook 2022–2032, https://www.futuremarketinsights.com/reports/sulphonamides-market; (b) İ. Gulçin and P. Taslimi, Expert Opin. Ther. Pat., 2018, 28, 541–549 CrossRef PubMed; (c) Y. Wan, G. Fang, H. Chen, X. Deng and Z. Tang, Eur. J. Med. Chem., 2021, 226, 113837 CrossRef CAS PubMed.
- G. H. Liu, T. Chen, X. Zhang, X. L. Ma and H. S. Shi, MedComm, 2022, 3, 181 CrossRef PubMed.
- Y. Wan, G. Fang, H. Chen, X. Deng and Z. Tang, Eur. J. Med. Chem., 2021, 226, 113837 CrossRef CAS PubMed.
- L. A. Jaragh-alhadad, M. S. Ali, M. S. Moustafa, G. I. Harisa, F. K. Alanazi and S. Karnik, J. Mol. Struct., 2022, 1268, 133699 CrossRef CAS.
- E. Sisco and K. L. Barnes, ACS Med. Chem. Lett., 2021, 12, 1030–1037 CrossRef CAS PubMed.
- A. Vicente-Blázquez, M. González, R. Álvarez, S. Del Mazo, M. Medarde and R. Peláez, Med. Res. Rev., 2019, 39, 775–830 CrossRef PubMed.
- L. A. Jaragh-alhadad, M. S. Ali, M. S. Moustafa, G. I. Harisa, F. K. Alanazi and S. Karnik, J. Mol. Struct., 2022, 1268, 133699 CrossRef CAS.
- R. J. Man, D. J. Tang, X. Y. Lu, Y. T. Duan, X. X. Tao, M. R. Yang, L. L. Wang, B. Z. Wang, C. Xu and H. L. Zhu, MedChemComm, 2016, 7, 1759–1767 RSC.
- M. González, M. Ovejero-Sánchez, A. Vicente-Blázquez, R. Álvarez, A. B. Herrero, M. Medarde, R. González-Sarmiento and R. Peláez, Int. J. Mol. Sci., 2021, 22, 1907 CrossRef PubMed.
- Q.-Z. Zhang, D. Zhang, F. Huang, C.-Y. Ke, Q. Pan and X.-L. Zhang, Curr. Org. Synth., 2017, 14, 578–581 CrossRef CAS.
- R. Li, X. Ning, J. He, Z. Lin, Y. Su, R. Li and Y. Yin, Bioorg. Chem., 2021, 108, 104653 CrossRef CAS PubMed.
- S. L. Warner, K. J. Carpenter and D. J. Bearss, Future Med. Chem., 2014, 6, 1167–1178 CrossRef CAS PubMed.
- K. Yakoub, S. Jung, C. Sattler, H. Damerow, J. Weber, A. Kretzschmann, A. S. Cankaya, M. Piel, F. Rösch and A. S. Haugaard, J. Med. Chem., 2018, 61, 1951–1968 CrossRef CAS PubMed.
- B. Frett, N. McConnell, C. C. Smith, Y. Wang, N. P. Shah and H. Li, Eur. J. Med. Chem., 2015, 94, 123–131 CrossRef CAS PubMed.
- V. Bavetsias, S. Crumpler, C. Sun, S. Avery, B. Atrash, A. Faisal, A. S. Moore, M. Kosmopoulou, N. Brown and P. W. Sheldrake, J. Med. Chem., 2012, 55, 8721–8734 CrossRef CAS PubMed.
- K. G. Lloyd and B. Zivkovic, Pharmacol., Biochem. Behav., 1988, 29, 781–783 CrossRef CAS PubMed.
- A. Durand, J. P. Thénot, G. Bianchetti and P. L. Morselli, Drug Metab. Rev., 1992, 24, 239–266 CrossRef CAS PubMed.
- C. Peifer, G. Wagner and S. Laufer, Curr. Top. Med. Chem., 2006, 6, 113–149 CrossRef CAS PubMed.
- S. L. Colletti, J. L. Frie, E. C. Dixon, S. B. Singh, B. K. Choi, G. Scapin, C. E. Fitzgerald, S. Kumar, E. A. Nichols and S. J. O'Keefe, J. Med. Chem., 2003, 46, 349–352 CrossRef CAS PubMed.
- O. Kim, Y. Jeong, H. Lee, S.-S. Hong and S. Hong, J. Med. Chem., 2011, 54, 2455–2466 CrossRef CAS PubMed.
- S. Patel, C. Globisch, P. Pulugu, P. Kumar, A. Jain and A. Shard, Eur. J. Pharm. Sci., 2022, 170, 106112 CrossRef CAS PubMed.
- A. Kamal, G. B. Kumar, V. L. Nayak, V. S. Reddy, A. B. Shaik, R. Rajender and M. Kashi Reddy, MedChemComm, 2015, 6, 606–612 RSC.
- M. Al-Ghorbani, M. A. Gouda, M. Baashen, O. Alharbi, F. A. Almalki and L. V. Ranganatha, Pharm. Chem. J., 2022, 56, 29–37 CrossRef CAS.
- R.-H. Zhang, H.-Y. Guo, H. Deng, J. Li and Z.-S. Quan, J. Enzyme Inhib. Med. Chem., 2021, 36, 1165–1197 CrossRef CAS PubMed.
- Z. Mao, X. Zheng, Y. Qi, M. Zhang, Y. Huang, C. Wan and G. Rao, RSC Adv., 2016, 6, 7723–7727 RSC.
- X. Peng, Q. Chen, B. Han, H. Zhang, J. Li and Q. Zhang, in Privileged Scaffolds in Drug Discovery, ed. B. Yu, N. Li and C. Fu, Elsevier, 2023, pp. 273–299 Search PubMed.
- D. T. Nash and S. D. Nash, Lancet, 2008, 372, 1335–1341 CrossRef CAS PubMed.
- C. L. Faingold, in Histamine II and Anti-Histaminics: Chemistry, Metabolism and Physiological and Pharmacological Actions, Springer, Berlin, 1978, pp. 561–573 Search PubMed.
- S. G. Jue, G. W. Dawson and R. N. Brogden, Drugs, 1982, 24, 1–23 CrossRef CAS PubMed.
- M. Deininger, E. Buchdunger and B. J. Druker, Blood, 2005, 105, 2640–2653 CrossRef CAS PubMed.
- S. Zheng, K. Lavrenyuk, N. G. Lamson, K. C. Fein, K. A. Whitehead and K. N. Dahl, ACS Biomater. Sci. Eng., 2019, 6, 367–374 CrossRef PubMed.
- (a) R. Das, P. Pulugu, A. A. Singh, D. R. Chatterjee, S. Baviskar, H. Vyas, S. K. Behera, A. Srivastava, H. Kumar and A. Shard, J. Med. Chem., 2024, 1–20 CAS; (b) K. Fukuoka, J. Usuda, Y. Iwamoto, H. Fukumoto, T. Nakamura, T. Yoneda, N. Narita, N. Saijo and K. Nishio, Invest. New Drugs, 2001, 19, 219–227 CrossRef CAS PubMed; (c) M. Schröder, M. Petrova, Z. Vlahova, G. M. Dobrikov, I. Slavchev, E. Pasheva and I. Ugrinova, Biomedicines, 2022, 10, 1353 CrossRef PubMed; (d) D. T. Kahraman, A. Karaküçük-İyidoğan, Y. Saygideger, E. E. Oruç-Emre, T. Taskin-Tok, E. Başaran, S. Ilhan, B. S. Demir, A. Üren and H. Bayram, New J. Chem., 2022, 46, 2777–2791 RSC; (e) J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef CAS; (f) Z. G. Movahed, M. Rastegari-Pouyani, M. Hossein Mohammadi and K. Mansouri, Biomed. Pharmacother., 2019, 112, 108690 CrossRef PubMed.
- A.-L. E. Manicum, H. Louis, G. E. Mathias, E. C. Agwamba, F. P. Malan, T. O. Unimuke, W. J. Nzondomyo, S. A. Sithole, S. Biswas and S. Prince, Inorg. Chim. Acta, 2023, 546, 121335 CrossRef CAS.
- F. Almouhanna, B. Blagojevic, S. Can, A. Ghanem and S. Wölfl, Cancer Metab., 2021, 9, 1–10 CrossRef PubMed.
- H. P. Morgan, F. J. O'Reilly, M. A. Wear, J. R. O'Neill, L. A. Fothergill-Gilmore, T. Hupp and M. D. Walkinshaw, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5881–5886 CrossRef CAS PubMed.
- B. Kim, C. Jang, H. Dharaneeswaran, J. Li, M. Bhide, S. Yang, K. Li and Z. Arany, J. Clin. Invest., 2018, 128, 4543–4556 CrossRef PubMed.
- K. K. Vasu, C. S. Digwal, A. N. Pandya, D. H. Pandya, J. A. Sharma, S. Patel and M. Agarwal, Bioorg. Med. Chem. Lett., 2017, 27, 5463–5466 CrossRef CAS PubMed.
- N. Sharma, G. Arya, R. M. Kumari, N. Gupta and S. Nimesh, Bio Protoc., 2019, 9, e3131 Search PubMed.
- M. Xu, S. Chen, W. Yang, X. Cheng, Y. Ye, J. Mao, X. Wu, L. Huang and J. Ji, Cell. Physiol. Biochem., 2018, 47, 151–160 CrossRef CAS PubMed.
- M. Cai, J. Wan, K. Cai, H. Song, Y. Wang, W. Sun and J. Hu, Cancers, 2022, 15, 87 CrossRef PubMed.
- H. J. Yun, S. Jin, J. Park, E.-W. Lee, H.-T. Lee, Y. H. Choi, B. W. Kim and H. J. Kwon, J. Microbiol. Biotechnol., 2022, 32, 918 CrossRef CAS PubMed.
- T. Rajavel, P. Packiyaraj, V. Suryanarayanan, S. K. Singh, K. Ruckmani and K. P. Devi, Sci. Rep., 2018, 8, 2071 CrossRef PubMed.
- J. Yu, Y. Yang, S. Li and P. Meng, Exp. Ther. Med., 2021, 22, 1–9 Search PubMed.
- X. Wu, X. Li, Z. Li, Y. Yu, Q. You and X. Zhang, J. Med. Chem., 2018, 61, 11280–11297 CrossRef CAS PubMed.
- J. Chen, J. Xie, Z. Jiang, B. Wang, Y. Wang and X. Hu, Oncogene, 2011, 30, 4297–4306 CrossRef CAS PubMed.
- H. H. Nguyen, J. Park, S. Kang and M. Kim, Sensors, 2015, 15, 10481–10510 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- H. Wang, X. Fan, P. P. Xie, S. Yang, P. Pigeon, Y. Xiong, S. Gai, X. Qi, J. Wang, Q. Zhang and W. Li, J. Med. Chem., 2023, 67(2), 1209–1224 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular docking studies of synthesized compounds, copies of NMR and HRMS spectra, and supplementary data. CCDC 2165838. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4md00367e |
| This journal is © The Royal Society of Chemistry 2024 |