
Photon and phonon powered photothermal catalysis
Chang
Xu†
a,
Qijun
Tang†
 ab,
Wenguang
Tu
*a and
Lu
Wang
*a
ab,
Wenguang
Tu
*a and
Lu
Wang
*a
aSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, 518172, Guangdong, P. R. China. E-mail: tuwenguang@cuhk.edu.cn; lwang@cuhk.edu.cn
bSchool of Chemistry and Materials Science, University of Science and Technology of China, 230026, Hefei, Anhui, P. R. China
First published on 8th May 2024
Abstract
Photo-thermal catalysis, leveraging light as an energy source, has emerged as a groundbreaking approach in driving chemical reactions. This method uniquely combines photonic and phononic elements of solar energy, offering enhanced reaction rates and altered selectivity under moderate conditions. This work delves into the core mechanisms of photo-thermal catalysis, focusing on the conversion processes and synergistic effect of photons and phonons, including the key advancements in applications such as catalytic CO2 and CH4 conversion, NH3 synthesis, and plastic upcycling. A guideline for future investigations into mechanisms and application development of photothermal catalysis could be paved.
Broader contextBy integrating traditional photocatalysis and thermocatalysis, photothermal catalysis has attracted significant attention from academia and industry. In a typical photothermal catalytic process, the photon represents the light, and the phonon represents the heat. Thus, the so-called photothermal catalysis can be driven either by phonons or photons and phonons. Unfortunately, the roles of photons and phonons in the photothermal catalytic process remain unclear due to the limited understanding of photothermal catalysis. In this perspective, we divide the recent advancement of photothermal catalysis into two distinct kinds, namely photon to phonon and photon and phonon, respectively, and provide an overview of the research from the viewpoint of photons and phonons. The reviewed work covers a wide range of reactions, including CO2 hydrogenation, CH4 conversion, NH3 synthesis, and plastic upcycling. Such understanding could significantly contribute to unraveling the intricate mechanisms of photothermal catalysis from a fundamental physical chemistry perspective, thereby inspiring innovative design strategies for efficient photothermal catalysts in the future. |
1. Introduction
Over the past few decades, a marked escalation in global energy demand has been observed, especially for the transportation and industrial sectors, in which the consumption of fossil fuels is dominated.1 This reliance on fossil fuels has precipitated substantial pollution challenges, notably through the emission of greenhouse gases such as carbon dioxide (CO2) and methane (CH4).2 Thus, a global strategic plan is to use renewable energy to replace traditional fossil fuels and eliminate the carbon footprint.3 A diverse array of renewable energy options, including wind energy,4 geothermal energy,5 wave energy,6 and solar energy,7 have emerged and are increasingly gaining traction.Solar energy, in particular, is considered a clean, plentiful, and perpetual energy resource. Utilization of solar energy has been extensively explored in areas such as photocatalysis,8 photovoltaics,9 and solar water heating.10 Photocatalysis, a process to convert solar energy into chemical energy, has attracted considerable attention due to its mild reaction conditions and it is environmentally benign.11 However, the efficiency of photocatalysis still falls short of practical application benchmarks. For instance, most photocatalysts are limited to absorbing ultraviolet light, which constitutes roughly 5% of the solar spectrum.12 Additionally, the activation energy in photocatalysis is often insufficient to initiate reactions, especially when compared to traditional thermocatalysis, which typically operates under more extreme conditions (e.g., high temperatures and pressures).
Addressing this challenge, photothermal catalysis has emerged as a novel paradigm. Photothermal catalysis involves the mutual conversion between photons and phonons, both of which play several roles during the reaction. The photons have to be absorbed by the catalyst, and then (1) excite the charge carriers to participate in the reaction or tune the acidity and basicity of the active sites, (2) excite the charge carriers to recombine and convert into phonons via thermalization, and (3) generate phonons via localized surface plasmon resonance (LSPR).13–15 The thermal energy of photothermal catalysis is an embodiment of phonons, which could be provided externally or converted from photons and decrease the reaction barrier by elevating the temperature.16 Due to these advantages, photothermal catalysis is receiving increasing attention, as evidenced by a growing body of related research (refer to Fig. 1). Although some reviews have introduced photothermal catalysis via the simple classification of photothermal catalysis into photo-assisted thermocatalysis, thermal-assisted photocatalysis, and photothermal-synergistic catalysis,14,17–20 it is necessary to provide one review to deeply understand the mechanisms of photothermal catalysis and highlight the recent advancements of photothermal catalysis.
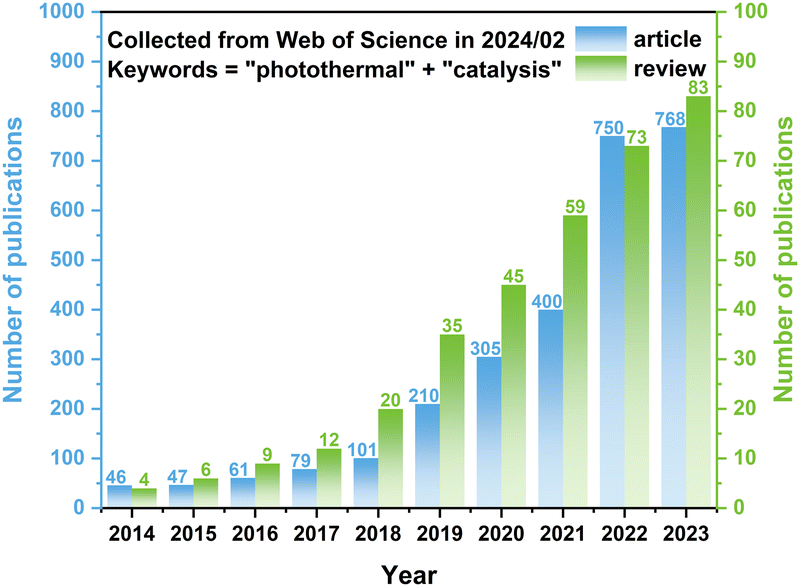 | ||
| Fig. 1 Number of publications (articles and reviews) related to “photothermal” and “catalysis”. The data were collected from the Web of Science in February 2024. | ||
This review will explore the mechanisms underlying photothermal catalysis, focusing on the conversion of photons to phonons and the synergistic interplay between them. It will then categorize and summarize applications of photothermal catalysis, including CO2 and CH4 conversion, NH3 synthesis, and plastic upcycling. The review concludes with insights and future perspectives on photothermal catalysis, aiming to deepen understanding and foster further advancements in this field.
2. Mechanism of photothermal catalysis
Photothermal catalysts have garnered widespread application across a variety of catalytic domains, notably in CO2 hydrogenation,21 dry reforming of methane,22 and NH3 synthesis,23 among others. The cornerstone of their broad utilization lies in the maximal energy conversion efficiency inherent in the photothermal process. This process adeptly converts electromagnetic energy into either heat or charge carriers with notable efficiency, both of which significantly influence catalytic activity. The operational mechanisms of photothermal catalysis can be broadly categorized into two distinct types, as illustrated in Fig. 2: photon → phonon and photon + phonon. It is critical to acknowledge that these two mechanisms often operate concurrently, albeit to varying extents, within a given photothermal catalytic process.24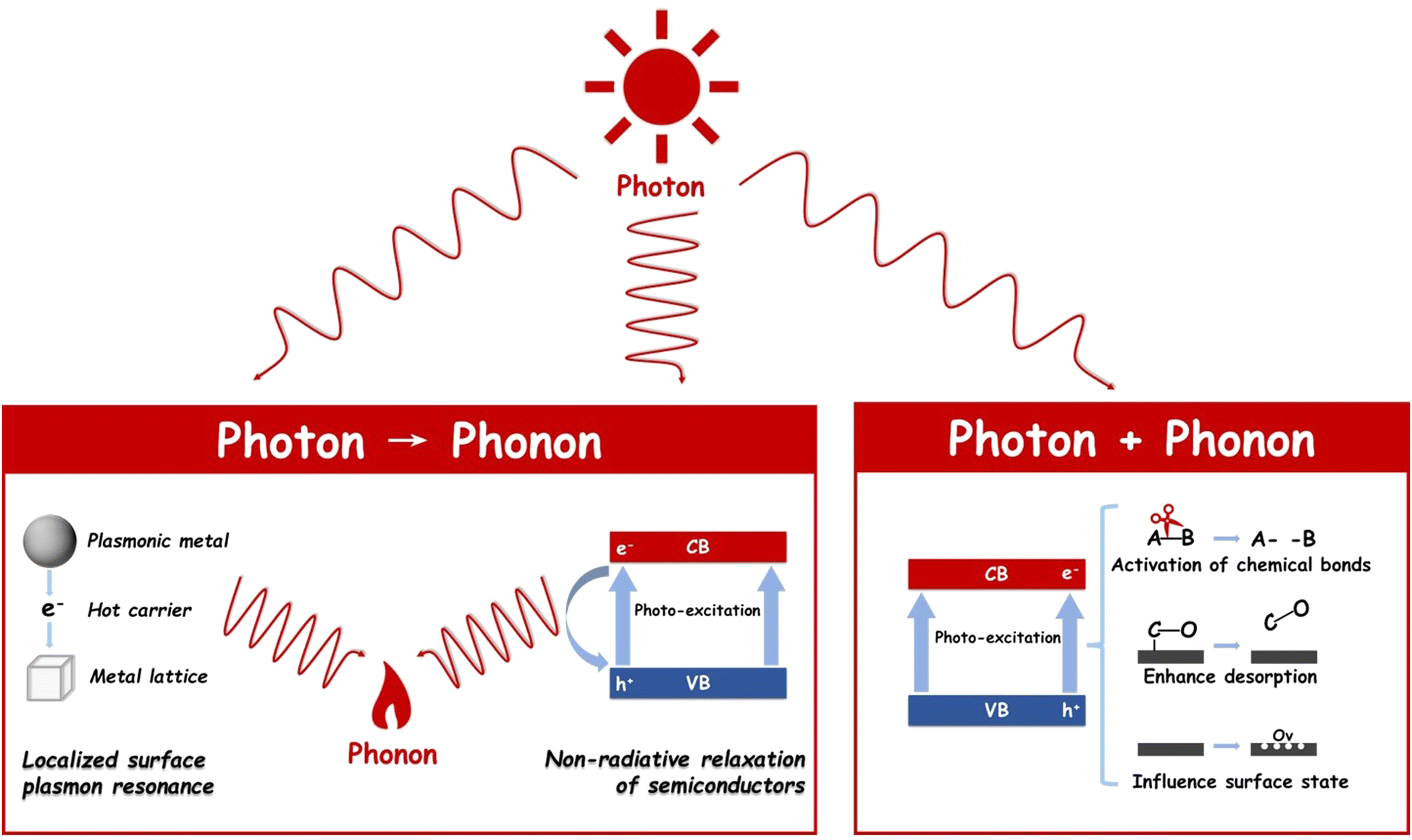 | ||
| Fig. 2 Mechanism of photothermal catalysis including photons → phonons, and photons + phonons. | ||
2.1 Photon → Phonon
The term ‘photon→ phonon’ encapsulates the process whereby electromagnetic energy is transmuted into thermal energy. This conversion can be efficiently facilitated through mechanisms such as the localized surface plasmon resonance (LSPR) effect or non-radiative relaxation in semiconductors. These processes underscore the interaction between light and matter in the realm of photothermal catalysis, offering an in-depth understanding of energy transformation at the nanoscale.Upon excitation, excited electrons undergo decay through either a radiative pathway, manifesting as re-emitted photons, or a non-radiative pathway, which involves electron–electron collisions and electron–hole pair recombination.29 Notably, when plasmon energy is non-radiatively dissipated within the metallic nanoparticles—a process also referred to as ‘Landau damping’30—energetic ‘hot’ charge carriers are generated within the plasmonic structure.28 These carriers, if sufficiently energized, can escape the plasmonic nanoparticles, injecting into adjacent surface adsorbates or semiconductors, thereby triggering surface chemical reactions at a femtosecond (fs) scale.31
Alternatively, ‘hot’ electrons that do not contribute to surface reaction charge migration are transformed into metal lattice phonons via electron-lattice collisions, leading to a rise in metal lattice temperature at the picosecond (ps) level.32 Subsequently, this thermal energy diffuses into the surrounding environment at the nanosecond (ns) level through phonon–phonon scattering.33 This heat transfer, stemming from the photon-to-phonon conversion, not only facilitates mass transfer but also enhances reaction rates from a thermodynamic perspective.
In summary, LSPR encompasses three concurrent processes: the enhancement of the local electric field, the generation of energetic hot charge carriers, and a localized heating effect. Collectively, these mechanisms significantly boost the reaction rate.34,35
2.2 Photon + Phonon
As previously discussed, phonon (or heat) can come from the LSPR effect, non-radiative relaxation of semiconductors, and external heating. All sources of phonon (or heat) will synergistically contribute to the thermal catalytic process including providing the driving force for the reaction, lowering the activation energy, facilitating the breaking of chemical bonds, and enhancing the adsorption and desorption of reactants and intermediates.37–39In photothermal catalysis, photons play pivotal roles not only in transforming themselves into phonons but also in influencing and altering the thermal catalysis process by modulating the surface properties of the catalyst or influencing the reaction pathway. For example, Ouyang's group used 2D black In2O3−x nanosheets for photothermal CO2 reduction. The photo-induced oxygen vacancies not only enhanced light harvesting but also improved the chemical adsorption of CO2 molecules. Therefore, both high activity (103.21 mmol gcat−1 h−1) and CO selectivity (nearly 100%) were achieved on In2O3−x nanosheets. A similar phenomenon was also discovered by Li's group that under light illumination, oxygen vacancies in CeO2 are created by photo-induced electrons during dry reforming of methane (DRM), thereby replenishing the consumed oxygen and maintaining stable CO2 thermo-activation.40 Moreover, electron–hole pairs can induce alterations in the catalyst's surface state through the introduction of charged species (electrons and holes), redox reactions, or the creation of defects, indirectly impacting catalytic performance.41 Time-dependent density functional theory (TDDFT) calculations and experimental characterization by Singh and colleagues demonstrated that the surface frustrated Lewis pairs (FLPs) on In2O3−x(OH)y, constituted of a Lewis acid (coordinately unsaturated proximal In) and a Lewis base (InOH surface), were enhanced under light illumination.13 This enhancement is corroborated by experimental findings showing a reduction of 21 kJ mol−1 in the activation energy of the Reverse Water Gas Shift Reaction (RWGS) in the presence of light.42 Besides, in our recent research,43 it was uncovered by experimental results and DFT calculations that the introduction of light would enable a new pathway of the RWGS reaction in a tandem ethane dehydrogenation and CO2 hydrogenation system. As a result, the ethane dehydrogenation reaction was shifted with the rate of ethylene 11.5 mmol g−1 h−1 at 650 °C and the CO production rate increased to 5.5 mmol g−1 h−1 at 650 °C.
In a word, the combination of phonon and photon will provide a stronger driving force for the reaction, facilitate the breaking of chemical bonds, enhance the adsorption and desorption of reactants and intermediates as well as change the surface properties of catalysts, achieving synergistic enhancement of catalytic reactions through photo and thermal cooperation.
2.3 Other parameters
Due to the optimal exploitation of the photo-thermal effect in catalytic processes, the intense light absorption, efficient charge carrier generation, and high capacity for thermal energy production or transformation of photothermal catalysts should be considered as well.24 Thus, the effect of the physicochemical properties of the materials and the corresponding temperature measurement methods will be introduced.In theory, the quantity of hot carriers is positively correlated with the size of plasmonic nanoparticles (NPs).46 However, since the dissipation of most energy by scattering or heating, the energy of generated hot carriers is always close to the Fermi level state. In contrast, smaller NPs could generate fewer electron–hole pairs, with higher energy states and notably shorter lifetimes (on the order of femtoseconds).47 The elevated energy of hot electrons increases the probability of populating acceptor states or overcoming Schottky barriers, which is paramount in photothermal catalysis. Nevertheless, a smaller NP size does not necessarily enhance overall photothermal performance. This diminution may result in inefficient charge carrier separation, attributed to spatial confinement.48,49 On the other hand, increasing NP size could trigger a red shift in the resonant frequency of plasmonic NPs, facilitating a more efficient absorption of visible and IR regions.49,50 In addition, the size could also influence the heating capability of plasmonic structures. The plasmon-heating effect in metallic nanospheres scales quadratically with the NP radius. However, larger plasmonic NPs tend to undergo radiative decay more readily, diminishing localized heat dissipation.51 Consequently, achieving an optimal balance between these size-dependent effects is crucial in the design of plasmonic NP-based catalysts.
Morphology is another key parameter determining the optical properties of nanostructures. Bar shapes, bipyramids, or branched structures exhibiting a red shift in their primary localized LSPR absorption peaks are thus better suited for harnessing the low-energy regions of the solar spectrum.52–54 In addition, the efficiency in hot carrier generation is also intimately related to the morphology. Geometries that foster strong, uneven electric fields and pronounced confinement effects can produce high-energy hot electrons and holes. Other geometries, such as thin films or nanosphere structures, are the opposite.55,56 Moreover, the morphology also plays a pivotal role in determining heat production within plasmonic nanostructures.57 Specifically, sharp, flat, or elongated geometries have been shown to facilitate more intense heat generation compared to spherical counterparts.58 This underscores the importance of carefully considering nanostructure morphological aspects in designing and optimizing plasmonic materials for specific applications.
Among them, metal/semiconductor hybrid materials have emerged as a particularly extensively researched system. An ideal semiconductor support for photothermal catalysts is characterized by high surface area and robust broadband optical absorption, which could facilitate the generation of charge carriers or elevate local temperatures. Typically, these structures incorporate narrow bandgap metal oxide semiconductors, which are rich in defects and possess mid-bandgap states, significantly enhancing light absorption in the low-energy regions.24,34,59,60 For instance, the subsurface oxygen defects could electronically interact with active sites by embedding metallic In into In2O3 to enhance photothermal catalytic CO2 reduction.61 Under the effect of electron-delocalization of O–In–(O)Vo–In–In structural units at the interface, the electrons in the subsurface oxygen defects are extracted and accumulate at the active sites of the surface. This phenomenon enhances the electronic coupling with CO2 and stabilizes the intermediate, increasing the turnover frequency of CO2 reduction to 7615 h−1.
In addition, materials with high porosity and high surface area (MOFs and zeolites) are a relatively underexplored avenue in photothermal catalysis, but are emerging as a promising field of research.17,62,63 A pioneering study by Jiang's group introduced Pt/PCN-224(M) composites, which integrated Pt nanocrystals and porphyrinic MOFs, marking the first report of the photothermal effect in MOFs.64 The transfer of hot electrons from Pt to PCN-224(M) leads to a decrease in electron density on the Pt surface. This dynamic can be finely adjusted to optimize catalytic performance by carefully balancing the influences of the Schottky junction and plasmonic effects through modulation of light intensity. This synergistic mechanism improves the overall photocatalytic performance, surpassing the individual contributions from each component.
Core–shell structured hybrid materials capitalize on their bifunctional nature to offer tunable optical, electronic, and thermal properties, presenting a compelling approach for the development of materials with enhanced photothermal performance.64 A notable example is the work of Kumar's group, which developed Pt@TiO2–AuNPs catalysts.65 Au plasmonic nanoparticles (NPs) have been enveloped in a TiO2 coating with Pt NPs to form a core–shell structure. Compared to bare Au NPs, the inclusion of a thin TiO2 layer leads to an increased quantum yield, attributable to an expansion in surface area, a red-shift and broadening of the LSPR peak, and an augmented light absorption capability. Furthermore, the strategic placement of Pt NPs plays a crucial role in capturing hot electrons from the CB of TiO2, effectively restraining electron–hole recombination and thereby enhancing the overall reaction rate.
2.4 Temperature measurement method
Accurate temperature measurement of photothermal catalysis is crucial for distinguishing between thermal and non-thermal contributions to the overall process. Here, some common temperature measurement methods have been overviewed.(i) Directly measuring the temperature of bulk catalysts using a thermocouple in real-time.
(ii) IR thermography grounded in the Planck blackbody emission principle, could estimate the local temperature by the emitted energy of a body.17
(iii) Tip-enhanced Raman spectroscopy (TERS) enables mapping local temperatures at the nanometer scale by calculating the ratio of anti-Stokes and Stokes Raman signal intensities.66,67 Ozin's group has probed the precision local temperature of Pd@Nb2O5 by exploring Stokes and anti-Stokes Raman bands, as high as 470 °C.68
(iv) Arrhenius plots are a fundamental tool for assessing the impact of temperature on chemical reaction rates, which exists as a crucial parameter, activation energy (Ea). Utilizing this principle, our group investigated the mechanistic pathways driving the photothermal catalysis reaction.34 Comparing activation barriers under both light and dark conditions at consistent temperatures, it is possible to determine the impact of photon-induced charge carriers on the reaction rates. In the case of the RWGS reaction, the Arrhenius plot revealed two nearly parallel lines for dark and light. The similar activation energies imply the same reaction mechanism, indicating that solar energy was mainly converted into thermal energy (Photon→ Phonon) to enhance the catalytic performance.
(v) Based on the equilibrium constants to calculate the local temperature of catalysts. Due to the localized nature of heat generation, a significant temperature gradient exists between the catalyst and its surroundings. He's group calculated the local temperature of Ni catalysts based on the equilibrium constants and constituent content of different product gases when photothermal CO2 hydrogenation reactions reached the equilibrium state.69
(vi) The measured temperature may require infrared calibration. Take the core–shell structured Ni@p-SiO2 catalyst of He's group as an example.69 Compare the FTIR spectrum of the SiO2 shell and theoretical spectra of the blackbody radiation, revealing that the SiO2 shell could absorb the infrared light radiated from the Ni (the heat source). However, the calculated contribution of thermal radiation to the total heat dissipation of Ni NPs over this reaction could reach up to 43%, indicating that the measured local temperature should be increased by 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K.
K.
3. Photothermal catalysis for reaction systems
In conventional photocatalytic processes, the infrared region, which constitutes approximately 43% of solar radiation's total energy, is often overlooked due to it having insufficient energy to facilitate electronic transitions in most semiconductors. However, there are significant photothermal conversion effects in some suitable narrow-band semiconductor materials or some specific metals. In the context of metallic substances, this conversion is attributed to the relaxation of intense electric fields generated by local surface plasmon resonance under illumination. This phenomenon not only induces localized elevated temperatures but also generates high-energy ‘hot electrons’. Conversely, in semiconductor materials, analogous conversion processes are driven by either Auger recombination or Shockley–Reed–Hall mechanisms, wherein surplus energy is transformed into thermal energy via phonon vibrations.24With the further exploration and advancement of photothermal effects, photothermal catalysis as an emerging catalytic paradigm has been proposed, promising enhanced utilization of renewable solar energy. This innovative approach involves the integration of photocatalysis and thermocatalysis, harnessing both photo- and thermo-effects in a unified catalytic system. This integration facilitates the conversion of solar energy into thermal and chemical forms.70 To date, photothermal catalysis has been applied in various reactions with different internal mechanisms (“photon → phonon” and “photon + phonon”), as shown in Tables 1 and 2.
| Catalysts | Catalytic performance | Reaction conditions | Ref. |
|---|---|---|---|
| Cu2ZnAl0.5Ce5Zr0.5Ox | CO: 248.5mmol g−1 h−1 |
Flow reactor, CO2/H2 ratio = 1:1, (459 °C) 40 mL min−1, simulate solar light source, ∼2 suns |
88 |
| HzIn2O3−x(OH)y | CO: 433.68 μmol g−1 h−1 | Flow reactor, CO2/H2 ratio = 1:1, (300 °C) 4 mL min−1, 300 W Xe lamp, ∼6 suns |
34 |
| CH3OH: 23.03μmol g−1h− |
|||
| 0.35Ru@Ni2V2O7 | CO2 conversion: 93.5%, | Batch reactor, pressure: 1 bar, (350 °C), 0.5 h CO2/H2 ratio = 1:4, 300 W Xe lamp, 2.0 W cm−2 |
79 |
| CH4: 114.9 mmol g−1 h−1 | |||
| Ni@p-SiO2-30 | CO2 conversion: 54%, | Batch reactor, pressure: 1 bar, (579 °C), 1 h CO2/H2 ratio = 1:1, 300 W Xe lamp, 2.8 W cm−2 |
69 |
| CO selectivity: 83%, | |||
| CH4: 20.6 mol g−1 h−1 | |||
| CoFe-650 alloy/Al2O3 | CO2 conversion: 78.6%, | Batch reactor, pressure: 1.8 bar, CO2/H2 ratio = 1:4, 300 W Xe lamp, 5.2 W cm−2 (320 °C), 2 h |
91 |
| CO selectivity: 4.97%, | |||
| CH4 selectivity: 59.77%, | |||
| C2+ selectivity: 35.26% | |||
| hm-Ni/Al2O3 | CO: 9614.3 mmolgNi−1 min−1 | Flow reactor, 89.5 mL min−1, CH4/CO2/N2 ratio = 3:3:4, 500 W Xe lamp, 345.6 kW cm−2, 706 °C |
107 |
| H2: 8573.0 mmol gNi−1 min−1 | |||
| Rh/LaNiO3 | CO: 527.6 mmol h−1 gRh−1 | Flow reactor, 50 mL min−1, CH4/CO2/Ar ratio = 1:1:3, 300 W Xe lamp, 3.5 W cm−2, 440 °C |
108 |
| H2: 452.3 mmol h−1 gRh−1 | |||
| Rh/SrTiO3 | CO: 4.5 μmolmin−1 |
Flow reactor, 10 mL min−1, CH4/CO2/Ar ratio = 1:1:98, 50W Hg–Xe lamp |
109 |
| H2: 4.4 μmol min−1 | |||
| Cu-CNN/Pd-BDCNN | CO: 852.6 μmol g−1 h−1 | Flow reactor, 10 mL min−1, CH4/CO2/Ar ratio = 1:1:8, 300 W Xe lamp, 300 mW cm−2 |
110 |
| H2: 844.1 μmol g−1 h−1 | |||
| AuRu0.31 | NH3: 101.4 μmol g−1 h−1 | N2 pressure: 2 bar, water, 300 W Xe lamp, 400 mW cm−2 | 118 |
| Au@Ui0-66 powder | NH3: 10.81 mmol g−1 h−1 | N2, water (K2SO4), 300 W Xe lamp, 400 mW cm−2 | 119 |
| Cu96Fe4 | NH3: 1342 μmol g−1 h−1 | N2, water, 300 W Xe lamp, 250 mW cm−2 | 120 |
| MoO3−x | AQE: 1.24% (808 nm) | N2, water, 300 W Xe lamp, | 121 |
| SACE: 0.057% | |||
| AgNP-Co | Δ stress at break: 2.5 Mpa | LDPE, blue light (270 mW cm−2), 55 °C, 120 h | 129 |
| AgNPs | Δ stress at break: 16 Mpa | PECA, 300 W Xe lamp, 600 mWcm−2 (445nm), 140 °C, 1 h |
130 |
| CNT-PDA | PET conversion: 100% | PET, Uv light 300 W, 600 mW cm−2, 180 °C, 2 h, mPET: 5 g, | 131 |
| BHET yield: 30.69% | |||
| Co SSCs | PET conversion: 100% | PET, simulated sunlight, 0.74 W cm−2, 180 °C, 3 h, mPET: 0.5 g | 132 |
| BHET yield: 82.6% |
| Catalysts | Catalytic performance | Reaction conditions | Ref. |
|---|---|---|---|
| 10Cu5Ga/CeO2 | CO: 111.2 mmol g−1 h−1 | Flow reactor, CO2/H2 ratio = 1:1, 20 mL min−1, 300 W Xe lamp, 1.95 W cm−2, 320 °C |
63 |
| ZnFe2O4 | CH3OH: 1757 μmol g−1 h−1 | Flow reactor, 6.5 bar, CO2/H2 ratio = 1:3, 8 mL min−1, with external heat, LED white light,1.83 W cm−2, 254 °C |
76 |
| Ni–Ru/HZSM-5 | CH4: 6.76 mmol g−1 h−1 | Flow reactor, CO2/H2 ratio = 1:4, 5 mL min−1, with external heat, 130 W Xe lamp, 3.5 W cm−2, 300 °C |
62 |
| CoFe2O4 | CO2 conversion: 12.9% | Flow reactor, CO2/H2 ratio = 1:4, 2.5 mL min−1, with external heat, 300 W Xe lamp, 2 W cm−2, 300 °C |
92 |
| C2+: 1.1 mmolg−1h−1 |
|||
| C2+ selectivity: 29.8% | |||
| Ni3Fe1 | CO: 0.63 mol g−1 h−1 | Flow reactor, 1.8 bar, 75 mL min−1, CH4/CO2/Ar ratio = 1:1:2, 500 W Xe lamp, 3.62 W cm−2, 350 °C |
111 |
| H2: 0.33 mol g−1 h−1 | |||
| Ni–CeO2–CePO4 | CO: 4 mmol g−1 h−1 | Flow reactor, 10 mL min−1, CH4/CO2/Ar ratio = 1:1:2, four Prizmatix LEDs, (UV, blue, green, red and a beam combiner) 48.1 kW cm−2, 350 °C |
112 |
| H2: 4 mmol g−1 h−1 | |||
| Pt/TiO2 | CO: 1499.2 mmol g−1 h−1 | Flow reactor, 20 mL min−1, CH4/CO2/Ar ratio = 48:48:4, with external heat, 300 W Xe lamp, 5 W cm−2, 700 °C |
113 |
| H2: 1107.1 mmol g−1 h−1 | |||
| Ni/Ga2O3 | CO: ∼200 μmol g−1 h−1 | Flow reactor, 10 mL min−1, CH4/CO2/Ar ratio = 1:1:4, with external heat, 300 W Xe lamp, 2.7 W cm−2, 391 °C |
114 |
| H2: ∼188 μmol g−1 h−1 | |||
| Ni/TiO2 | NH3: 55.7 μg g−1 h−1 | Flow reactor, N2/H2 = 1:3, 30 mL min−1, with external heat, 300 W Xe lamp, 400 °C |
120 |
| Ru–Cs/MgO | NH3: 4464 μmol g−1 h−1 | Flow reactor, N2/H2 = 1:3, 75 mL min−1, with external heat, blue LED, 4.7 W cm−2, 333 °C |
121 |
| Cs10Ru2@ST | NH3: 3580 μmol g−1 h−1 | Flow reactor, N2/H2 = 1:3, 40 mL min−1, with external heat, 300 W Xe lamp, 1 sun, 350 °C |
122 |
| Ru/C | NH3: 1750 μmol g−1 h−1 | Flow reactor, N2/H2 = 1:3, 40 mL min−1, with external heat, 300 W Xe lamp, 5.0 W cm−2, 350 °C |
123 |
| g-C3N4 | PS conversion: 100%, benzaldehyde selectivity: 51%, acetophenone selectivity: 31%, benzoic acid selectivity: 18% | PS, flow reactor, WHSV = 0.9h−1, 30 mL acetonitrile, 10bar O2, 300 W Xe lamp, 120 °C, 8 h, mPS: 500 mg |
134 |
| DEG-ligated TiO2 | PET conversion: 100% | PET, batch reactor, 30 mL CH3CN, 10bar O2, simulated sunlight, 0.65 W cm−2, 190 °C, 0.5 h, mPET: 500 mg |
135 |
| BHET yield: 85% | |||
| Ni–Ti–Al | Hydrogen production: 34 mol kg−1 | LDPE, batch reactor, inert atmosphere N2 simulated sunlight, 500 °C | 136 |
| Jet fuel selectivity: 80% | |||
| Ru/TiO2 | LDPE conversion: 100%, | LDPE, batch reactor, N2/H2 = 3:7, 30 bar Xe lamp, 3.00 W cm−2, 220 °C, 3 h, mLDPE: 80 mg |
137 |
| C5–C21 selectivity: 86% |
3.1 CO2 hydrogenation
The escalating concentration of atmospheric CO2 (surpassing 420 ppm) represents a critical challenge for human sustainability.71 To overcome this issue, substantial research efforts have been devoted to converting CO2 into fuel via solar-related catalytic technologies. Among these, photothermal catalysis shows considerable potential in facilitating CO2 conversion under relatively mild conditions.Since the seminal report in 2014,72,73 photothermal hydrogenation of CO2 has emerged as a vibrant and rapidly growing field of research. This approach has demonstrated its versatility across a spectrum of applications, including but not limited to the reverse water gas shift (RWGS) reaction,59,74,75 methanol synthesis,34,76–78 the Sabatier reaction,69,79–81 and the production of C2+ compounds.82–84 Each of these applications showcases the adaptability of photothermal technology and underscores its significance in addressing the global CO2 challenge.
mmol g−1 h−1. This advanced material design approach markedly enhanced both catalytic stability and activity, promising to revolutionize the field of photothermal catalysis and pave the way for its industrial application.
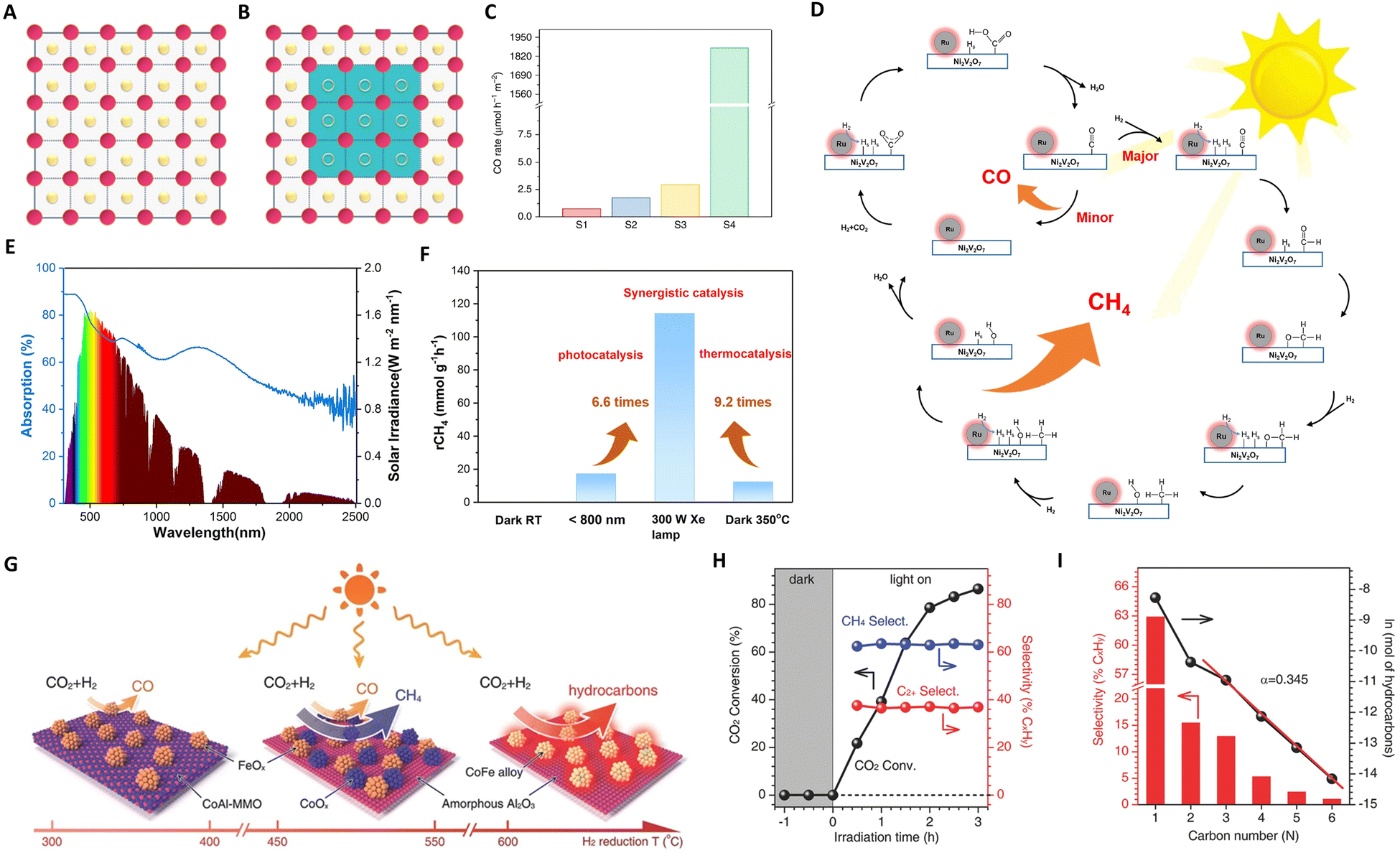 | ||
| Fig. 3 (A) and (B) Graphical representation of the original and treated In2O3, wherein the blue region, pink dots, yellow dots, and yellow circles represent the amorphous phase, In atoms, O atoms, and [O], respectively. The scale bars are the same for all images, and green squares indicate the formation of an amorphous phase. (C) Photocatalytic evaluation of black indium oxide. These figures have been reproduced from ref. 59 with permission from Springer Nature, copyright 2020. (D) The proposed catalytic reaction mechanism for the Ru@NVO catalyst. (E) UV-vis-IR absorption spectrum of the NVO film. (F) Average CH4 production rates over Ru@NVO at room temperature under the different conditions. These figures have been reproduced from ref. 79 with permission from Elsevier, copyright 2021. (G) Illustration of the different CoFe-x catalysts formed by hydrogen reduction of a CoFeAl-LDH nanosheet precursor at different temperatures. (H) The time course of CO2 conversion and product selectivity for CO2 hydrogenation over CoFe-650 under UV-vis irradiation. (I) The hydrocarbon product distribution obtained over CoFe-650 under UV-vis irradiation for 2 h. These figures have been reproduced from ref. 91 with permission from John Wiley & Sons, copyright 2017. | ||
Wang's group recently made a significant breakthrough by developing a new black indium oxide tandem catalyst for photothermal CO2 conversion to methanol.34 This novel catalyst, benefiting from surface site engineering, exhibits enhanced absorbance and concentration of surface [O], thereby overcoming the thermodynamic limitations typically associated with conventional methanol synthesis. This catalyst operates on a tandem reaction scheme, wherein CO, a by-product from the Reverse Water Gas Shift (RWGS) reaction, is utilized as an in situ feedstock for methanol formation. This tandem process transforms the traditionally competing RWGS and methanol synthesis into a cohesive reaction pathway within a flow reactor system. Under sunlight illumination, methanol selectivity of up to 33.24% and 49.23% at low (50%) and high (75%) hydrogen concentrations, respectively, was achieved. This development signals a promising advance towards the realization of a solar refinery for sustainable methanol production.
In the context of photothermal CO2 methanation, ruthenium (Ru) catalysts are widely acknowledged for their efficacy, as evidenced by recent studies.89 However, the scarcity and high cost of Ru have spurred the search for alternative catalysts that either minimize or eliminate the use of noble metals.90 In this vein, Zou's group developed a Ru@Ni2V2O7 catalyst, demonstrating remarkable methanation efficiency.79 This catalyst operates via a synergistic mechanism between Ru and Ni2V2O7, as shown in Fig. 3(D) and (E). Here, Ru clusters act as ‘nano-heaters,’ enhancing local thermal conditions (photon → phonon), which facilitate H2 activation and H2O desorption. The hydrogen atoms generated interact with CO2 on the oxygen-vacancy-rich surface of Ni2V2O7, leading to efficient CO2 methanation. The Ru@Ni2V2O7 catalyst achieves a CO2 conversion efficiency of 3.5%, approaching the thermodynamic equilibrium limit for thermo-catalytic CO2 methanation. Furthermore, drawing inspiration from the greenhouse effect, He and Ozin reported a nickel nanocrystal encapsulated in nano-porous silica (Ni@p-SiO2).69 This configuration enables heating of the nickel core through non-radiative relaxation of photogenerated electrons. The insulation and infrared shielding of the silica sheath yield a supra-photothermal effect, safeguarding the Ni core against sintering and coking. This design has led to industrially relevant rates of conversion, selectivity, and durability.
In 2018, Zhang's group introduced an innovative approach by employing CoFe alloy catalysts for photothermal CO2 hydrogenation to hydrocarbons.91 These catalysts, comprising CoFe alloys supported on alumina derived from layered double hydroxide nanosheets, exhibited high efficiency in the process. A critical aspect of their methodology involved modulating the reduction temperature between 300 °C and 700 °C, as shown in Fig. 3(G). This modification in reduction temperature allowed precise control over the CoFe-x surface chemistry, shifting product selectivity from CO to CH4, and ultimately to C2+ hydrocarbons. Notably, the CoFe-650 catalyst, characterized by discrete alloy nanoparticles, demonstrated significant C–C coupling selectivity in CO2 hydrogenation. As depicted in Fig. 3(H) and (I), under Xe lamp irradiation, the C2+ selectivity reached an impressive 35%, with an overall conversion rate of 78.6%. This represents a pioneering achievement in the field, marking the first report of such a high yield in C2+ hydrocarbon synthesis via photothermal CO2 hydrogenation.
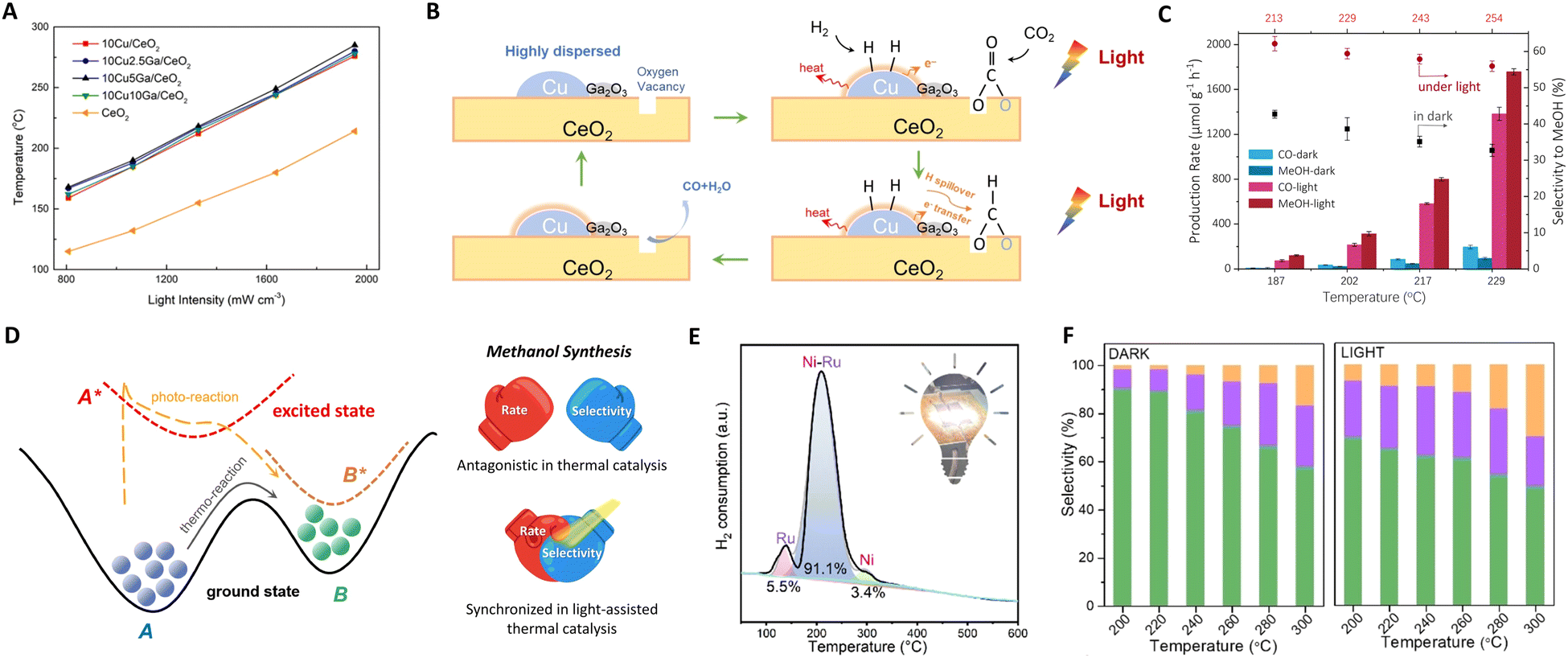 | ||
| Fig. 4 (A) Solar-driven CO production over CuGaCe catalysts under different light intensities. (B) Proposed reaction mechanism for photothermal RWGS over CuGa/CeO2. These figures have been reproduced from ref. 63 with permission from Elsevier, copyright 2021. (C) Catalytic performance on ZnFe2O4. (D) Sketch of a thermodynamic and photodynamic chemical reaction pathway. These figures have been reproduced from ref. 76 with permission from Elsevier, copyright 2023. (E) The deconvolution results of the H2-TPR profile of the fresh Ni–Ru/HZSM-5. This figure has been reproduced from ref. 62 with permission from American Chemical Society, copyright 2023. (F) The product gas selectivity of CoFe2O4 under light and dark conditions. This figure has been reproduced from ref. 92 with permission from John Wiley & Sons, copyright 2023. | ||
Additionally, Ozin's group explored zinc iron oxide spinel as a model photothermal catalyst, proposing an alternative strategy for methanol synthesis.76 Though introducing photon energy could shift the reaction pathway to a new photo-thermodynamic excited equilibrium state (photon + phonon), breaking the limitations of traditional thermodynamic equilibrium. Surface oxygen vacancies of ZnFe2O4 are crucial for trapping and concentrating photoinduced charge carriers as well as for CO2 capture and activation at the same sites. This is because the reaction process primarily yields CO and methanol, both deriving from a common intermediate: bidentate carbonate anchored at oxygen vacancy sites. Thus, ZnFe2O4 could facilitate the transformation of feed gas through excited state intermediates, circumventing thermodynamic equilibrium constraints (photon effect) to improve methanol selectivity and yield, as shown in Fig. 4(C) and (D). By fine-tuning photon energy, approximately 80% of the photo-induced charge carriers participate in the chemical reaction before recombination, achieving a 17.5 times increase in methanol production rate to 1757.1 μmol g−1 h−1.
In a recent study, Ozin et al. introduced a composite material comprising bimetallic nickel–ruthenium nanoparticles supported on protonated zeolite (Ni–Ru/HZSM-5).62 The black Ni nanoparticles generate local heating effects, endowing Ni–Ru/HZSM-5 with exceptional light-harvesting capabilities. The bimetallic Ni–Ru nanoparticles, with a photothermal Ni core and Ru corona, provide the necessary thermal energy to enhance synergistic photothermal and hydrogen atom transfer effects, as illustrated in Fig. 4(E). The efficiency of this photothermal process was demonstrated in the CO2 methanation reaction, comparing CH4 formation rates under identical temperatures with and without light illumination. Under purely thermal conditions, the CH4 formation rate was 1.88 mmol g−1 h−1. However, under a 24 h light illumination reaction, this rate increased to 6.76 mmol g−1 h−1, while maintaining a CH4 selectivity of 89.4%. These results offer critical insights into the impact of photothermal heating and enable a high selectivity associative CO2 methanation reaction pathway.
Furthering their contributions, Ozin's group developed a CoFe–CoFe2O4 alloy spinel nanocomposite for CO2 hydrogenation under atmospheric pressure, as shown in Fig. 4(F).92 In this system, the CoFe2O4 spinel is instrumental in facilitating the RWGS reaction, converting feed gas into CO, which then serves as a synthon for Fischer–Tropsch Synthesis (FTS) over the CoFe alloy. This results in a high C2–C4 rate and selectivity of 29.8%. Additionally, the concept of charge asymmetric active sites in catalysts has been explored for CO2 reduction to C2+ products.93,94 These asymmetric active-metal sites generate diverse charge distributions on adjacent adsorbed C1 intermediates, reducing electrostatic repulsion and promoting C–C coupling reactions.95,96 It is posited that the asymmetry of these catalytic active sites, along with key intermediates, not only catalyzes the C–C coupling reaction but also inhibits the hydrogenation of C1 intermediates into C1 products, through a synergistic mechanism at adjacent sites.97
3.2 Dry reforming of methane
The dry reforming of methane (DRM, eqn (1)) reaction represents a sustainable approach for mitigating CO2 emissions, thereby addressing concurrent climate and energy crises.98 This process transforms greenhouse gases (CO2 and CH4) into syngas (CO and H2), which can subsequently be utilized in the Fischer–Tropsch process for high-value hydrocarbon production.99,100 However, traditional DRM reactions necessitate the cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2 (805 kJ mol−1) and the C–H bond in CH4 (434 kJ mol−1), requiring thermal energy inputs and thus leading to substantial energy consumption.101–103
O bond in CO2 (805 kJ mol−1) and the C–H bond in CH4 (434 kJ mol−1), requiring thermal energy inputs and thus leading to substantial energy consumption.101–103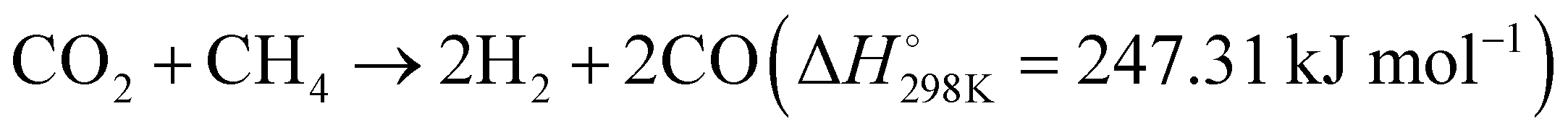 | (1) |
In response to these challenges, photothermal catalysis has been increasingly applied in this domain, offering a cleaner and milder alternative to conventional high-temperature processes.104–106 The integration of photothermal catalysis in DRM reactions presents a significant advancement for converting CO2 and CH4 into valuable chemical feedstocks. This approach enhances the overall energy efficiency of the process and contributes to the reduction of greenhouse gas emissions, aligning with global environmental and energy sustainability goals.
 | ||
| Fig. 5 (A) and (B) Production rates and apparent activation energy of H2 and CO under irradiation or heating over Rh/LaNiO3. These figures have been reproduced from ref. 108 with permission from John Wiley & Sons, copyright 2023. (C) Tentative mechanistic aspects of the photocatalytic DRM by Rh/STO. (D) The temperature dependence of DRM activity under dark and light irradiation conditions. These figures have been reproduced from ref. 109 with permission from Springer Nature, copyright 2020. | ||
Additionally, Miyauchi et al. reported a Rh/SrTiO3 catalyst, which effectively promotes methane reforming under UV light irradiation at low temperatures.109 The introduction of rhodium ions into the SrTiO3 crystal forms impurity levels above the valence band, enhancing visible light absorbability. This catalyst efficiently utilizes photon energy for methane conversion with a high quantum efficiency of 5.9%. Under UV light, the excited electrons in the conduction band of STO are transferred to rhodium particles, acting as an electron acceptor in the DRM reaction and achieving efficient charge separation, as illustrated in Fig. 5(C) and (D). Electrons in Rh reduce CO2 to CO, while photogenerated holes in the valence band of SrTiO3 migrate to the interface of rhodium nanoparticles, reacting with CH4 and O2− to produce H2. This innovative principle has potential for application in a variety of uphill reactions, where photon energy can be harnessed to overcome the limitations imposed by traditional thermodynamics and high thermal energy requirements. This approach enables the extraction of valuable products from diverse carbon resources, effectively pushing the boundaries of chemical conversion processes.
Moreover, the dual-reaction-sites catalyst Cu-CNN/Pd-BDCNN has been reported by Yin's group.110 The strategic incorporation of copper and palladium in this catalyst serves a dual purpose: Cu and Pd act as electron and hole acceptors, respectively, while concurrently enhancing light absorption and charge separation within the catalyst. This innovative design significantly facilitates the reaction process. Remarkably, this catalyst has demonstrated a high production rate of approximately 800 μmol g−1 h−1 under light irradiation, without the need for external heating. This result underscores the efficacy and potential of dual-reaction-site strategies in the realm of photothermal catalysis, suggesting a promising avenue for future developments in this field.
 | ||
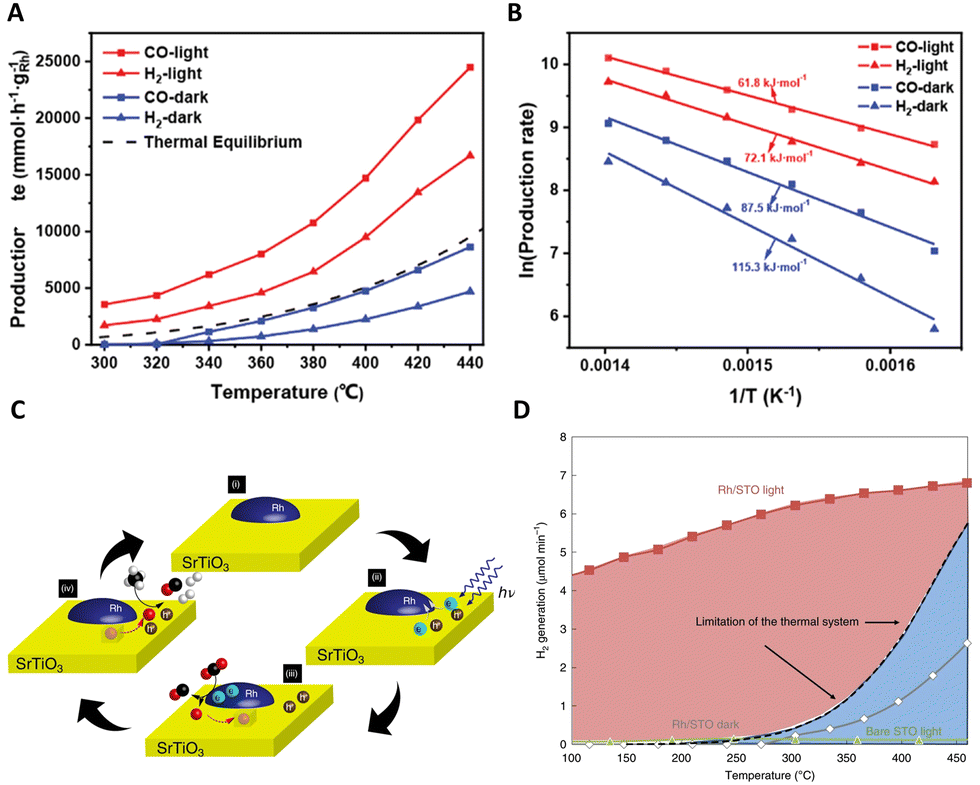
| Fig. 6 (A) In situ DRIFT spectra for photo-driven DRM reaction on Ni3Fe1. (B) Schematic illustration for the photo-driven DRM reaction over the Ni3Fe1 nanoalloys. These figures have been reproduced from ref. 111 with permission from John Wiley & Sons, copyright 2022. (C) Wavelength dependence of the observed photothermal catalytic activity. This figure has been reproduced from ref. 112 with permission from Springer Nature, copyright 2023. (D) The schematic diagram of external thermal promotion effect on the behaviors of hot carriers. This figure has been reproduced from ref. 113 with permission from Elsevier, copyright 2022. | ||
In an exemplary study, Ozin's group has significantly enhanced the photothermal dry reforming performance of Ni–CeO2 by selectively phosphating the CeO2 nanorod support surface.112 This modification led to a remarkable increase in the H2:CO ratio of the produced gas from 0.44 to 0.95 under 48.1 kW m−2 white light irradiance at 350 °C, maintaining high stability over 50 h. A notable aspect of this research is the exploration of the disparity in catalytic efficiency between thermal and photo-assisted conditions. This exploration was conducted through the application of various light wavelengths, including ultraviolet (UV), blue, red, and green, as shown in Fig. 6(C). It was observed that blue light facilitated the most significant temperature increase, suggesting a predominantly photothermal effect. In contrast, UV light elicited a response more characteristic of photochemical processes, likely attributable to the absorption properties of the CeO2–CePO4 support. The effects under green and red light were linked to the excitation of nickel plasmon resonance. Further insights were provided by DRIFTS analysis, which confirmed an enhanced conversion of CO2 under illumination. This was evidenced by a more pronounced CO2 stretching peak at 2354 cm−1. This study not only underscores the intricacy inherent in photothermal catalysis but also illuminates the vast potential of light manipulation in optimizing catalytic processes.
In their innovative research, Sun et al. delved into the dynamics of energetic hot carriers within photothermal catalytic systems, particularly focusing on the influence of external thermal effects.113 Their study unveils a dual behavior of these carriers, which could be promoted at low to medium temperatures but experience suppression at higher temperatures due to external heating. This phenomenon is crucial as the production of thermal energy not only boosts the generation of energetic hot carriers but also reduces the redox potentials of reactants, thereby facilitating their involvement in reactions geared towards specific products, as depicted in Fig. 6(D). A critical finding of this work is the observation that at high reaction temperatures, which exceed the threshold of conventional thermal catalysis, active sites on the metal surface become predominantly occupied by thermal catalysis. This occupancy limits the availability of these sites for hot carriers, thereby reducing the effectiveness of photo-mediated catalysis. In contrast, at lower and medium temperatures where the active sites are not fully saturated, there is a noticeable improvement in the utilization of both photo-induced energetic hot carriers and thermal energy. This synergistic interaction between solar and thermal energies is most effective at medium temperatures. This aspect of the study illuminates the intricate interplay within photothermal catalysis, providing valuable insights into the efficient and practical use of solar energy in industrial applications.
Complementing these findings, Zhou's group conducted a study exploring the interplay between two primary competing reactions in the photothermal dry reforming of methane over Ni/Ga2O3.114 By manipulating light irradiation, they observed a reversal in the direction of electron transfer from Ga2O3 to Ni, resulting in the formation of Ni0 sites. These sites are instrumental in generating a substantial quantity of hot electrons through the electronic inter-band transition of Ni, thereby significantly enhancing both the formation and desorption of H2. As a result of this process, the H2/CO ratio in the production gas markedly increased from 0.55 to nearly 1. This discovery provides profound insights into the role of light irradiation in photothermal catalytic reactions and paves the way for optimizing the proportion of H2 in C–H activation and hydrogen production processes.
3.3 NH3 synthesis
The Haber–Bosch process, one of the seminal discoveries of the 20th century,115 revolutionized the production of ammonia (NH3). This innovation laid the foundation for the mass production of NH3, which has been used to manufacture fertilizers, explosives, and other nitrogenous chemicals.116 The core methodology of NH3 synthesis has remained unchanged for more than a century. Approximately 95% of global NH3 production still relies on the traditional Haber–Bosch method.117 However, the process is inherently constrained by thermodynamic limitations. To achieve viable industrial performance, the reactors must operate under high temperatures (400 °C to 500 °C) and pressures (100 bar to 200 bar),17 posing significant energy and environmental challenges. In sharp contrast, photocatalytic NH3 production offers an alternative route with less energy input and ambient pressure. Thus, photothermal catalysis emerges as a promising solution, particularly in enhancing the efficiency and selectivity of NH3 production, signifying a potential paradigm shift in this critical chemical process.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage. A notable advancement in this field was made by Xiong and colleagues, who developed a novel AuRu core-antenna nanostructure. This innovation has been shown to catalyze the dissociation of N2 into NH3 effectively, achieving a notable production rate of 101.4 μmol g−1 h−1 using the LSPR effect in pure water under mild conditions.118 The design integrates a gold (Au) core to harvest light across a broad spectrum, with ruthenium (Ru) antennae serving as active sites for N2 fixation. As illustrated in Fig. 7(A)–(C), the N2 fixation process exhibits a near-linear dependence on light intensity, underscoring the role of plasmonic hot electrons in driving the reduction of N2 to ammonia. This correlation is further supported by the full extinction spectra range of the AuRu nanostructures, indicating a high utilization efficiency of incident light. Specifically, the apparent quantum efficiencies (AQEs) at 350 nm and 550 nm were determined to be 0.21% and 0.17%, respectively, signifying the pivotal role of photon energy in the N2 fixation process facilitated by the plasmonic AuRux catalysts. Expanding on this concept, Wang's group explored the use of metal–organic framework (MOF) membranes as nanoreactors to disperse and confine gold nanoparticles (AuNPs), achieving direct plasmonic photocatalytic nitrogen fixation under ambient conditions.119 The AuNPs generate hot electrons under visible irradiation, which are then directly injected into surface-adsorbed N2 molecules. These molecules are activated by a strong, localized surface plasmon resonance field, which facilitates electron transfer, energy transfer, and localized-electric-field polarization effects. This results in a higher apparent quantum efficiency and lower apparent activation energy under stronger irradiation.
N bond cleavage. A notable advancement in this field was made by Xiong and colleagues, who developed a novel AuRu core-antenna nanostructure. This innovation has been shown to catalyze the dissociation of N2 into NH3 effectively, achieving a notable production rate of 101.4 μmol g−1 h−1 using the LSPR effect in pure water under mild conditions.118 The design integrates a gold (Au) core to harvest light across a broad spectrum, with ruthenium (Ru) antennae serving as active sites for N2 fixation. As illustrated in Fig. 7(A)–(C), the N2 fixation process exhibits a near-linear dependence on light intensity, underscoring the role of plasmonic hot electrons in driving the reduction of N2 to ammonia. This correlation is further supported by the full extinction spectra range of the AuRu nanostructures, indicating a high utilization efficiency of incident light. Specifically, the apparent quantum efficiencies (AQEs) at 350 nm and 550 nm were determined to be 0.21% and 0.17%, respectively, signifying the pivotal role of photon energy in the N2 fixation process facilitated by the plasmonic AuRux catalysts. Expanding on this concept, Wang's group explored the use of metal–organic framework (MOF) membranes as nanoreactors to disperse and confine gold nanoparticles (AuNPs), achieving direct plasmonic photocatalytic nitrogen fixation under ambient conditions.119 The AuNPs generate hot electrons under visible irradiation, which are then directly injected into surface-adsorbed N2 molecules. These molecules are activated by a strong, localized surface plasmon resonance field, which facilitates electron transfer, energy transfer, and localized-electric-field polarization effects. This results in a higher apparent quantum efficiency and lower apparent activation energy under stronger irradiation.
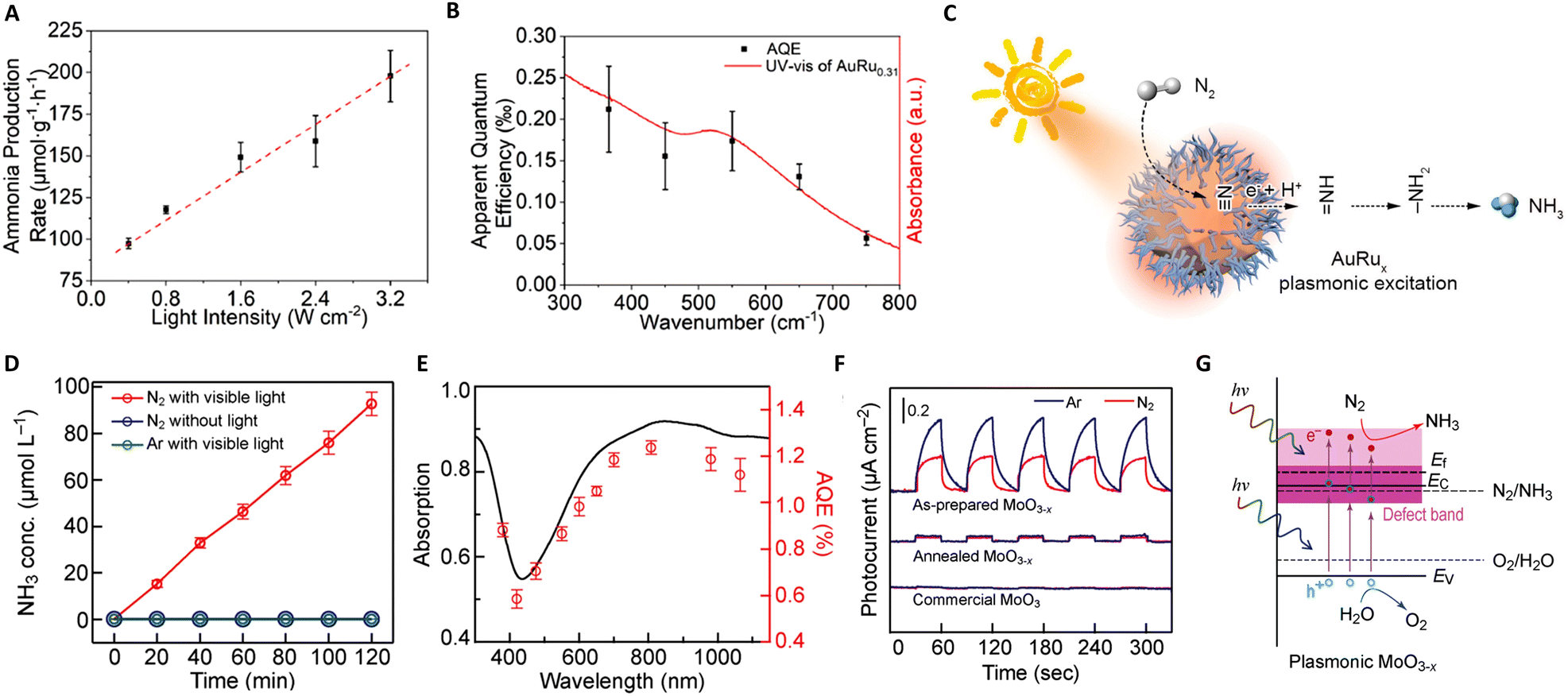 | ||
| Fig. 7 (A) and (B) Photocatalytic ammonia production rates and calculated AQEs by AuRu0.31. (C) The schematic diagram for the photo driven NH3 synthesis over the AuRu0.31 nanostructure. These figures have been reproduced from ref. 118 with permission from American Chemical Society, copyright 2019. (D) Photothermal catalytic NH3 production of MoO3−x under different conditions. (E) Action spectrum of the MoO3−x spheres for N2 photo-fixation. (F) Photocurrent responses of serial MoO3−x spheres under an Ar and N2 atmosphere, respectively. (G) Schematics illustrating the band structures of the plasmonic MoO3−x photocatalyst. These figures have been reproduced from ref. 121 with permission from John Wiley & Sons, copyright 2021. | ||
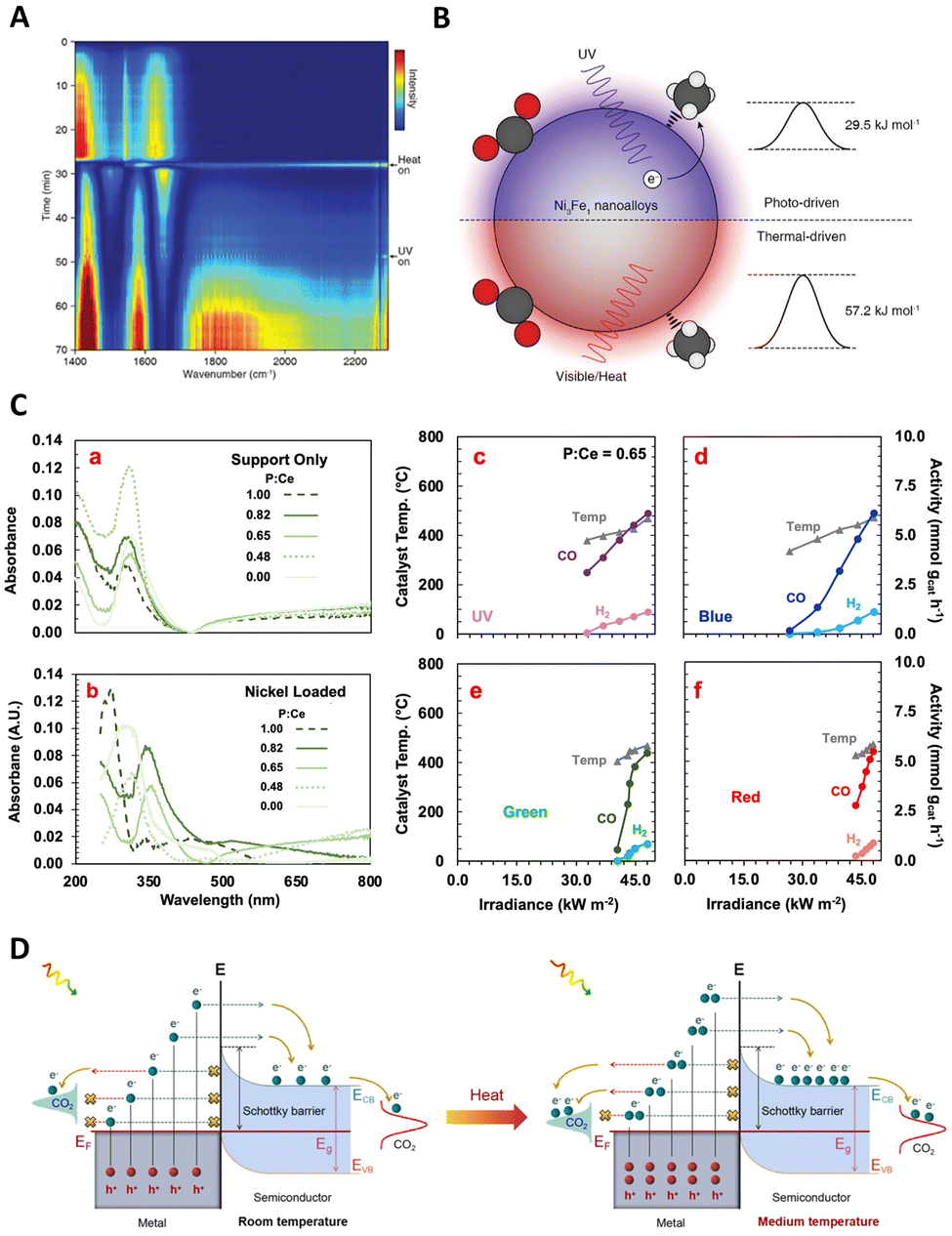
Furthermore, the utilization of hybrid materials in this domain has also been investigated. For instance, Wang and Zhang et al. developed a low-cost, plasmon-assisted Cu-based porous catalyst, Cu96Fe4, which achieved an NH3 production rate of 342 μmol gcat−1 h−1 without the need for sacrificial agents.120 The Cu framework generates hot electrons through surface plasmon resonance, while the Fe atoms on the surface act as active sites for the efficient adsorption and activation of N2. The efficient polarization of N2 over the Fe atoms significantly decreases the reaction barrier, contributing directly to the N2 fixation process. Nevertheless, the efficiency of these systems is often limited by the inability of hot electrons to cross the Schottky barrier, leading to their recombination with holes within the metal nanocrystal. Addressing this challenge, Wang's group introduced a Schottky-barrier-free plasmonic semiconductor, MoO3−x, which demonstrates efficient N2 fixation.121 N2 molecules are chemisorbed and activated at the oxygen vacancy (OV) sites in MoO3−x. The abundance of electrons induced by the OVs in the conduction band creates a strong plasmon resonance on the surface of MoO3−x, visible in Fig. 7(D)–(G). This unique semiconductor permits the free transport of plasmonic hot charge carriers and minimizes electron–hole recombination through defect states introduced by the OVs. As a result, this system exhibits a superior N2 fixation ability, an apparent quantum efficiency of 1.24%, and a notable solar-to-ammonia conversion efficiency of 0.057%. This breakthrough in Schottky-barrier-free construction paves a new path for the rational design of efficient photo-thermal catalysts.
Furthering this approach, Liu et al. reported a Ru-based catalyst (Ru–Cs/MgO), demonstrating efficient ammonia production with high reaction rates and yields.123 As depicted in Fig. 8(A) and (C), the creation and control of thermal gradients via photothermal heating of the catalyst surfaces play a crucial role in this system. The non-isothermal gradient between the catalyst surface and its bottom promotes higher temperatures at the surface, facilitating N2 scission, while cooler temperatures at the bottom preserve NH3 yield. A negative gradient aligns thermophoretic forces with the reactant flow, effectively removing NH3 from the hottest region and preventing its decomposition. Conversely, a positive gradient under the same absolute value yields lower reaction rates and product yields. These findings highlight the potential of light-induced thermal gradients as thermodynamic pumps, enhancing the catalytic activity in photo-thermal systems. Additionally, García and colleagues developed Cs-promoted ruthenium nanoparticles supported on strontium titanate (CsyRux@ST) for photothermal catalysis of NH3 synthesis.124 Operating under 350 °C and ambient pressure, this system achieves an impressive NH3 production rate, surpassing dark conditions by 68%. This enhanced performance is attributed to the combined effects of photo-induced hot carrier and local thermal mechanisms at the irradiated Ru nanoparticles, facilitating N2 activation and hydrogenation.
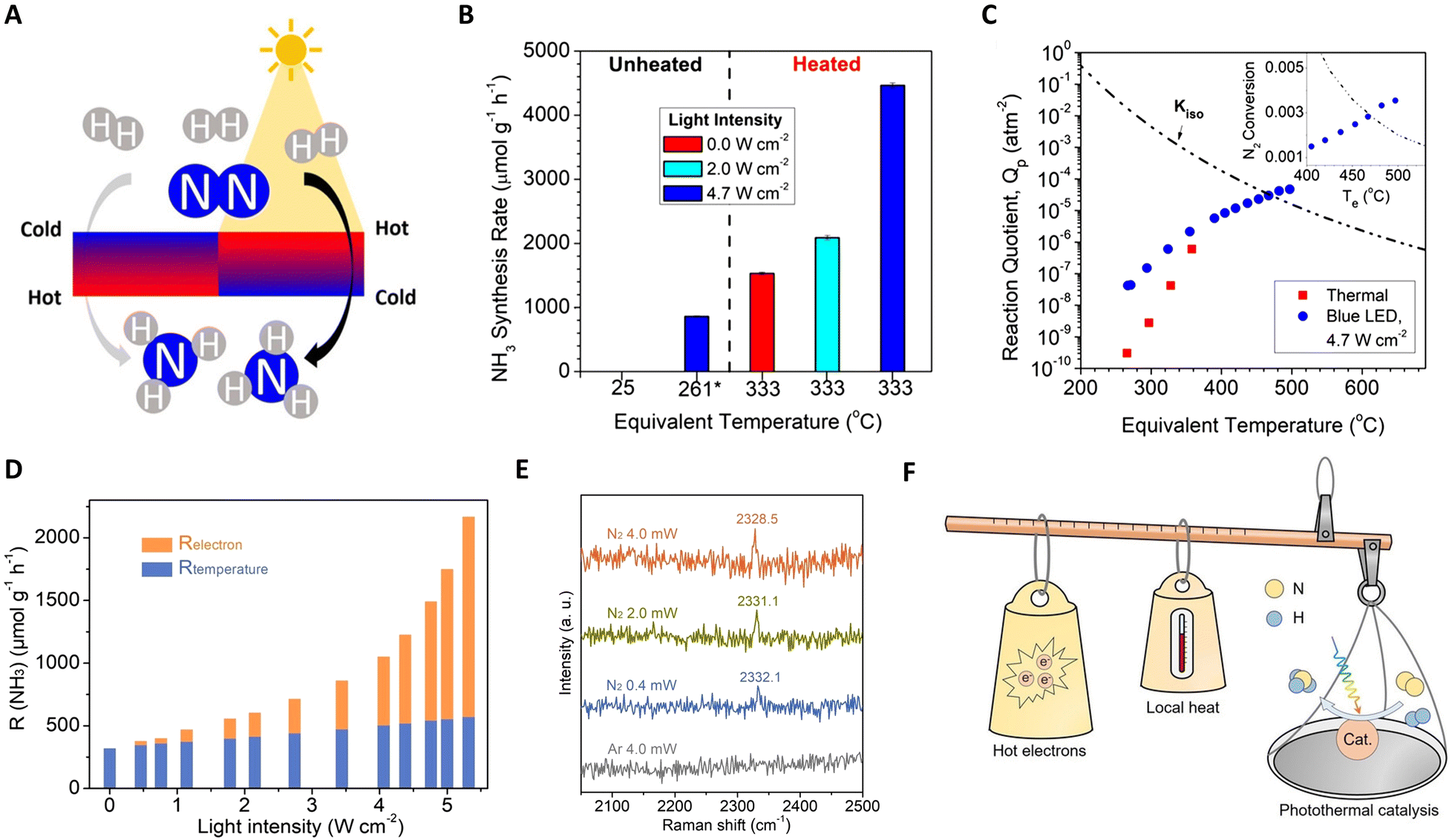 | ||
| Fig. 8 (A) A schematic diagram for photo-driven NH3 synthesis over Ru–Cs/MgO. (B) NH3 synthesis rates under dark and additionally illuminated conditions. (C) Dependence of NH3 synthesis and thermal gradients on illumination. These figures have been reproduced from ref. 123 with permission from American Chemical Society, copyright 2019. (D) Photothermal catalytic ammonia production rate contribution from photo-induced hot electrons (Relectron) and actual reaction temperature (Rtemperature) as a function of full light intensity at Tapparent = 350°C. (E) In situ Raman spectra over the Ru/C catalyst. (F) A schematic diagram for the photo-driven NH3 synthesis over Ru–Cs/MgO. These figures have been reproduced from ref. 125 with permission from John Wiley & Sons, copyright 2023. | ||
While photothermal catalysis is a promising green technology, differentiating the impacts of hot electrons and local heating remains a challenge. Zhang and collaborators tackled this issue in ammonia synthesis using a Ru-loaded carbon catalyst as a model.125 They quantified the contributions of photo-induced hot electrons and local heating effects separately using Le Chatelier's principle (non-thermal contribution (Relectron) = total photo-thermal rate (RPTC) − pure thermal effects (Rtemperature)), as shown in Fig. 8(D)–(F). Intriguingly, with increasing light intensity, Relectron transitions from a linear to a super-linear relationship, confirming non-thermal effects in the system. Under specific conditions (5.0 W cm−2 light intensity and 350 °C), hot-electron contributions accounted for 73.6% of the total NH3 production rate. Complementary analyses, including DRIFTS, in situ Raman, and kinetic experiments, further revealed that light facilitates NH3 synthesis at lower temperatures by injecting photo-induced hot-electrons into the unoccupied anti-bonding orbitals of adsorbed N2, aiding in NN bond cleavage.
3.4 Plastic upcycling
The escalating global crisis of plastic waste accumulation, posing substantial economic and health threats,126 has prompted a keen interest in the catalytic upcycling of plastics.127 Photothermal catalysis, an emergent technology, harnesses electromagnetic and thermal energies.71,128 This method stands out as a sustainable alternative to energy-intensive thermocatalysis by utilizing renewable solar energy. Consequently, photothermal catalysis achieves superior catalytic performance under mild conditions, enhancing its industrial applicability. Moreover, the confluence of light and heat within this technique could potentially alter reaction pathways, steering the transformation of waste plastics into higher-value chemicals.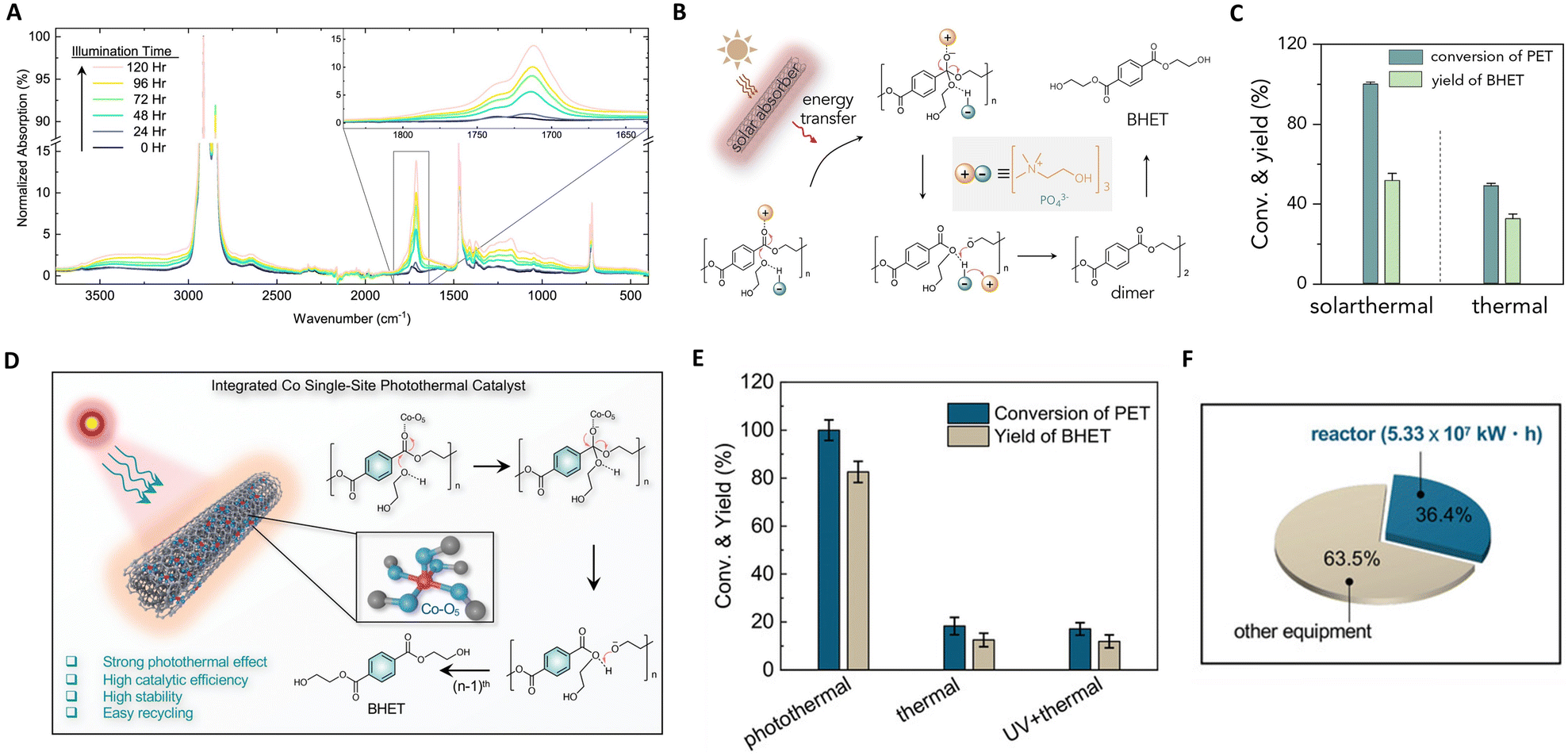 | ||
| Fig. 9 (A) Infrared spectra of PE-AgNP-Co as a function of illumination time. This figure has been reproduced from ref. 131 with permission from IOP Publishing, copyright 2019. (B) The proposed mechanism of the photothermal upcycling of PET. (C) The proposed mechanism of the photothermal upcycling of PET. These figures have been reproduced from ref. 133 with permission from Elsevier, copyright 2022. (D) Integrated functionalities of Co SSCs and the photothermal catalytic mechanism of PET glycolysis over Co SSCs. (E) Conversions and yields of upcycling of PET over Co SSCs under different reaction conditions. (F) Energy consumption evaluation of Co SSCs. These figures have been reproduced from ref. 134 with permission from John Wiley & Sons, copyright 2022. | ||
Recent focus has shifted towards photothermal upcycling of plastics, treating waste as a source of recoverable chemicals rather than material for CO2 mineralization. Chen et al. developed a multi-walled carbon nanotube modified by polydopamine (CNT-PDA) for PET depolymerization.133 Under solar irradiation, CNT-PDA serves as a light absorber, converting solar energy into thermal energy to drive PET depolymerization, as depicted in Fig. 9(B) and (C). The absorbers first efficiently convert sunlight to thermal energy, enabling PET depolymerization into BHET via a nucleophilic addition–elimination mechanism in the presence of an organic catalyst (cholinium phosphate). Solar thermal catalysis exhibited thrice the recycling efficiency of thermal catalysis due to localized heating effects. In the same vein, they introduced cobalt single-site catalysts (Co SSCs) on CNT-PDA for photothermally catalyzing polyester upcycling.134 The Co-O5 single-site coordinates with polyester carbonyl groups, enhancing nucleophilic addition–elimination processes, as shown in Fig. 9(D) and (E). The space-time yield of Co SSCs was significantly higher than general catalysts, and PET conversion and bis(2-hydroxyethyl) terephthalate yield in photothermal catalysis surpassed those in thermal catalysis (5.4 and 6.6 times). Technical economic analysis revealed that recycling 105 tons of waste PET through photothermal catalysis consumes 146.4 GW h of electrical energy and reduces CO2 emissions by 7.44 × 104 tons. Thus, an efficient photothermal catalytic system for plastic recycling has immense potential for waste plastic valorization.
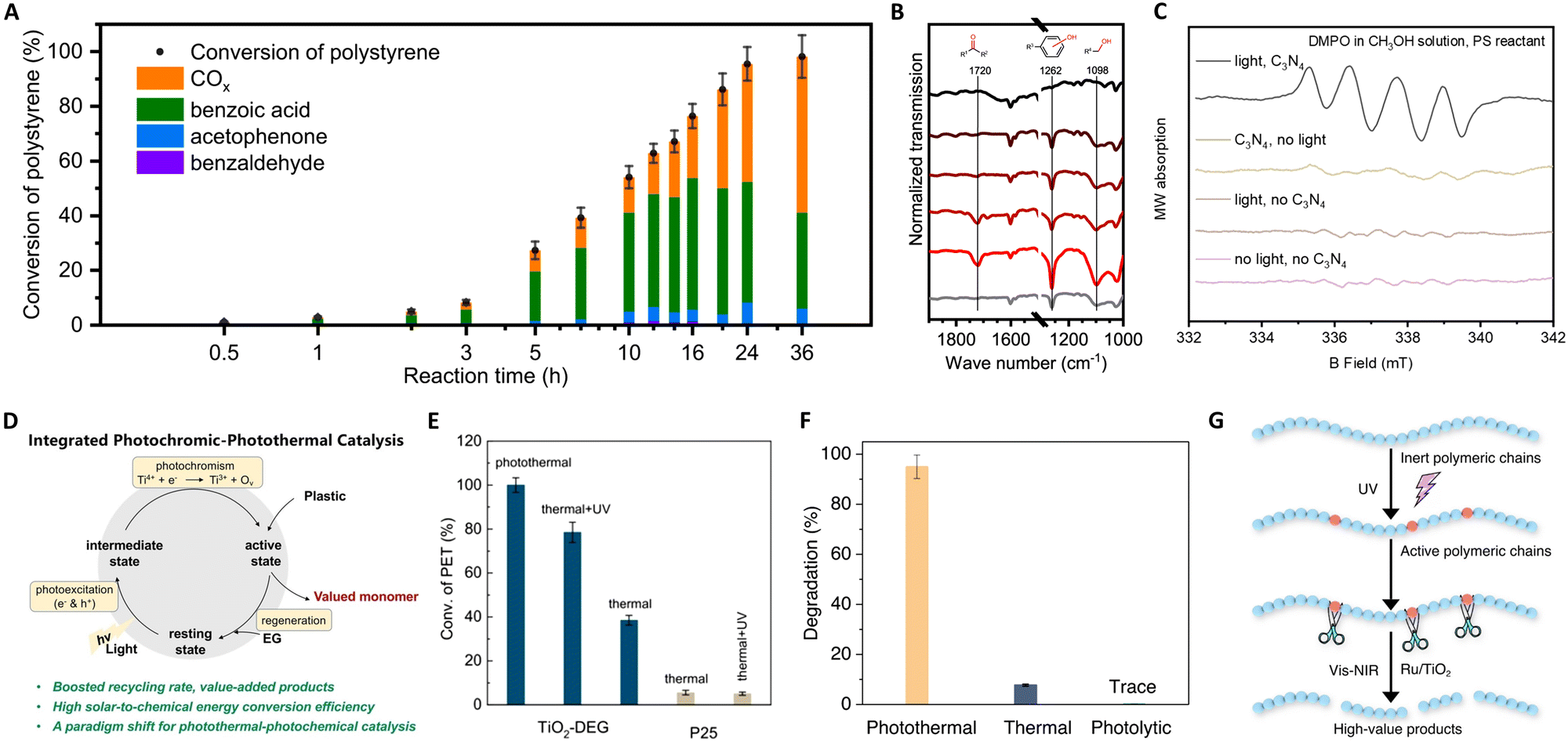 | ||
| Fig. 10 (A) Time-evolution of different products for polystyrene photothermal catalytic reaction at 150 °C. (B) The infrared (IR) transmission spectra of the polystyrene reactant and the recovered polystyrene after reactions with light irradiation for 0 h, 0.5 h, 2 h, 5 h, and 10 h and without light irradiation for 10 h. (C) EPR spectra under different illumination and catalyst conditions. These figures have been reproduced from ref. 136 with permission from Springer Nature, copyright 2022. (D) Schematic illustration of the proposed photothermal reaction mechanism. (E) Catalytic performance evaluation of TiO2-DEG via PET glycolysis. These figures have been reproduced from ref. 137 with permission from John Wiley & Sons, copyright 2023. (F) Degradation percentages of LDPE under photothermal (300 °C), thermal (300 °C), and photolysis under ambient temperature conditions. (G) Schematic illustration of the proposed photothermal reaction mechanism. These figures have been reproduced from ref. 139 with permission from Springer Nature, copyright 2023. | ||
Furthermore, Zeng's group pioneered the use of concentrated solar energy for photothermal catalytic upcycling of polyolefin waste, devising a photothermal catalytic pyrolysis apparatus capable of reaching 500 °C.138 Their results with a Ni–Ti–Al catalyst demonstrated significant hydrogen production (34 mol kg−1) and jet fuel selectivity (80%), attributed to the interplay between photogenerated electrons in plasmonic metal Ni and the carrier transfer capability of non-plasmonic semiconductor TiO2. This method offers an eco-friendly approach to the high-value recycling of waste plastics into hydrogen and jet fuel. In a notable advancement, Zhang's group reported on the photothermal pyrolysis of polyolefin plastics (LDPE, HDPE, UHMWPE, PP, and commercial LDPE bags) using a Ru/TiO2 catalyst.139 This method significantly outperforms traditional thermal catalysis, exhibiting a tenfold increase in efficiency under photothermal conditions compared to dark conditions. The key to this process is the use of UV light to activate the polymeric chains, which are then efficiently decomposed from polyolefin plastics into lower molecular-weight molecules by Ru nanoparticles, as shown in Fig. 10(F) and (G). Additionally, the application of Vis and NIR light irradiation induces localized heating, melting the polymers for optimal catalyst contact, and facilitating C–C bond scission on Ru sites. Remarkably, this approach enables the complete conversion of waste polyolefins into valuable liquid fuels, mainly gasoline (86%) and diesel-range hydrocarbons (C5–C21), within a short 3-hour timeframe. Crucially, the system demonstrates high efficiency under concentrated sunlight, representing a significant stride towards sustainable, solar-driven plastic waste recycling.
4. Conclusions and perspective
Photothermal catalysis, a process energized by both photon and phonon interactions, has recently emerged as a cutting-edge strategy for achieving sustainable and clean energy and chemical production. In this innovative approach, thermal energy, conveyed through phonons, can be supplied externally or generated from photons, effectively reducing the reaction barrier by increasing temperature. This conversion of electromagnetic to thermal energy is efficiently facilitated via mechanisms such as localized surface plasmon resonance (LSPR) or non-radiative relaxation in semiconductors, distinguishing it from conventional photocatalysis and thermocatalysis. The former often encounters limitations in photon utilization efficiency, particularly within the visible light spectrum, whereas the latter demands severe reaction conditions.However, photothermal catalysis harnesses the synergistic effects of photons and phonons to markedly improve catalytic reactions. Utilizing renewable solar irradiation, this method enables the transformation or coupling of photons with phonons, showcasing several advantages: (i) milder reaction conditions compared to thermocatalysis, (ii) enhanced reaction rates and selectivity, and (iii) increased stability and minimized sintering of the active phase. These collective benefits highlight the significant potential of photothermal catalysis in producing solar fuels.
However, some existing problems and challenges should also be attended to and solved. (i) In-depth understanding of the mechanism of photothermal catalysis is key to controlling the reaction and selectivity of the desired products. However, the difficulty in the detection of some transitional species and a significant lack of understanding of the nature of reaction intermediates are the barriers to the next step. (ii) Economic photothermal catalysts with a strong ability to absorb lower-energy regions in the electromagnetic spectrum are highly desired. In general, most photocatalyst materials show light absorption around 300–500 nm, with weak visible light absorption above 600 nm. Unfortunately, the energy of solar light is mainly in the latter region. Improving the utilization of a wider solar spectrum is worth consideration. Due to the aim of pilot plants and industrialization, the cost and mild reaction conditions of catalysts are also critical. (iii) The xenon lamps’ light intensity is around 20–50 times higher than that of natural light. Direct utilization of solar energy is challenging, highlighting the need for designing ideal reactors that are capable of harnessing solar energy effectively.
This review delves into the mechanisms and applications of photothermal catalysis, examining both photon and phonon contributions. This analysis aims to not only clarify the field but also to inspire future research. (i) For the mechanism research, integrating advanced in situ characterization techniques and computational calculations promises to enrich the understanding and enable direct observations from both photon and phonon perspectives. For instance, femtosecond infrared laser pulses were used to observe the reaction dynamics of photothermal catalysis via the desorption and oxidation of CO on Ru(0001), thus revealing the slower CO desorption coupling to phonons (∼20 ps) and the faster oxidation of CO driven by the hot electrons (∼3 ps).140
(ii) In the realm of catalyst design, tuning the surface of catalysts to black has been identified as a promising strategy for directly converting UV-VIS-IR light into thermal energy.34,59 Concurrently, the expanding comprehension of photochemical and thermal interactions within nanostructured catalysts will advocate for the continuous exploration and development of materials characterized by broadband absorption and high responsiveness. Moreover, high temperatures can potentially skew the equilibrium of the reaction system in gas-phase heterogeneous exothermic reactions.24 Therefore, the strategic implementation of thermal management within catalyst systems emerges as a critical factor in maintaining the delicate equilibrium necessary for optimal catalytic function. For instance, the advent of 3D-printed metallic and ceramic structures, serving as conduits for fluid, signifies a breakthrough in enhancing thermal energy and mass transfer within catalytic flow systems.141 This inspiring idea of heat management for the fabrication of photothermal catalysts may offer a novel paradigm in catalyst design.
(iii) Continuous flow operation is better suited for large-scale photothermal processes. Elongated and reflective parabolic surfaces were used to focus light onto a transparent tube through which the reactant flows. Nevertheless, the inherent variability of solar illumination due to natural day/night cycles and changes in solar angle presents significant challenges for the industrial application of photothermal catalysis. Hence, the development of reactors for capturing, focusing, and storing solar energy is of paramount importance. For example, IMDEA Energy in Spain realized the solar tower fuel plant,142 which seamlessly integrates a solar tower concentrating facility, a solar reactor, and a gas-to-liquid unit. Within this framework, the 50-kW solar reactor demonstrated its capability by performing 62 consecutive redox cycles over 9 days, culminating in the production of approximately 5191 L of syngas. Furthermore, the thermal radiation of high-temperature catalytic reactions seems to be beneficial in improving the overall catalytic performance. Our research group recently proposed thermal radiative catalysis that utilizes high temperature and its corresponding thermal radiation to facilitate ethane dehydrogenation reaction, whereas the catalytic performance will not be influenced by external light sources.143 These notable accomplishments underscore the vast potential of electromagnetic energy in powering large-scale chemical manufacturing processes and serve as a beacon for future research directions.
Photothermal catalysis represents a broad definition of reactions where both thermal energy and electromagnetic energy (photons and phonons) are involved, necessitating an unambiguous classification. Such comprehensive insights into the interplay between photons and phonons are expected to lead to novel catalytic paradigms, inspire innovative catalyst preparation methods, and design targeted reactors. These kinds of high energy efficiency and precise process control approaches are poised to improve the overall catalytic performance further. Ultimately, we foresee that photothermal systems will achieve record performances, potentially opening a new pathway for sustainable chemical and fuel production.
Author contributions
Chang Xu: conceptualization, validation, formal analysis, investigation, data curation, writing – original draft, and writing – review and editing. Qijun Tang: conceptualization, validation, formal analysis, investigation, data curation, writing – original draft, and writing – review and editing. Wenguang Tu*: writing – original draft, writing – review and editing, resources, funding acquisition, supervision, and project administration. Lu Wang*: writing – original draft, writing – review and editing, resources, funding acquisition, supervision, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors appreciate the support from the National Key R&D Program of China (grants no. 2021YFF0502000), the National Natural Science Foundation of China (grants no. 52102311 and 52202306), Shenzhen Natural Science Foundation 2022, Provincial Talent Plan (grants no. 2023TB0012 and 2023TB0006), the University Development Fund (grants no. UDF01001721), the Program from Guangdong Introducing Innovative and Entrepreneurial Teams (2019ZT08L101 and RCTDPT-2020-001), the Shenzhen Key Laboratory of Eco-materials and Renewable Energy (ZDSYS20200922160400001), and the Shenzhen Natural Science Foundation (GXWD20201231105722002-20200824163747001). Dedicated to Professor Geoffrey A. Ozin's 80th birthday.Notes and references
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- S. Paraschiv and L. S. Paraschiv, Energy Rep., 2020, 6, 237–242 CrossRef.
- K. K. Jaiswal, C. R. Chowdhury, D. Yadav, R. Verma, S. Dutta, K. S. Jaiswal, B. Sangmesh and K. S. K. Karuppasamy, Energy Nexus, 2022, 7, 100118 CrossRef CAS.
- M. S. Nazir, A. J. Mahdi, M. Bilal, H. M. Sohail, N. Ali and H. M. N. Iqbal, Sci. Total Environ., 2019, 683, 436–444 CrossRef CAS.
- J. W. Lund and A. N. Toth, Geothermics, 2021, 90, 101915 CrossRef.
- A. F. d O. Falcão, Renewable Sustainable Energy Rev., 2010, 14, 899–918 CrossRef.
- N. Kannan and D. Vakeesan, Renewable Sustainable Energy Rev., 2016, 62, 1092–1105 CrossRef.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- O. A. Al-Shahri, F. B. Ismail, M. A. Hannan, M. S. H. Lipu, A. Q. Al-Shetwi, R. A. Begum, N. F. O. Al-Muhsen and E. Soujeri, J. Cleaner Prod., 2021, 284, 125465 CrossRef.
- S. Faisal Ahmed, M. Khalid, M. Vaka, R. Walvekar, A. Numan, A. Khaliq Rasheed and N. Mujawar Mubarak, Therm. Sci. Eng. Prog., 2021, 25, 100981 CrossRef.
- C. Xu, P. Ravi Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868–3902 RSC.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206–1214 CrossRef CAS PubMed.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem. Catal., 2022, 2, 52–83 CrossRef CAS.
- P. C. Angelomé, H. Heidari Mezerji, B. Goris, I. Pastoriza-Santos, J. Pérez-Juste, S. Bals and L. M. Liz-Marzán, Chem. Mater., 2012, 24, 1393–1399 CrossRef.
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304452 CrossRef CAS PubMed.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- S. Fang and Y. H. Hu, Chem. Soc. Rev., 2022, 51, 3609–3647 RSC.
- S. Du, X. Bian, Y. Zhao, R. Shi and T. Zhang, Chem. Res. Chin. Univ., 2022, 38, 723–734 CrossRef CAS.
- J. Zhang, H. Chen, X. Duan, H. Sun and S. Wang, Mater. Today, 2023, 68, 234–253 CrossRef CAS.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- D. Takami, J. Tsubakimoto, W. Sarwana, A. Yamamoto and H. Yoshida, ACS Appl. Energy Mater., 2023, 6, 7627–7635 CrossRef CAS.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen, J. Li, S. Liu, J. Zhao and L. Zhang, Chem, 2019, 5, 2702–2717 CAS.
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC.
- A. Bansal, J. S. Sekhon and S. S. Verma, Plasmonics, 2014, 9, 143–150 CrossRef CAS.
- M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2010, 132, 9383–9391 CrossRef CAS PubMed.
- N. Jiang, X. Zhuo and J. Wang, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS PubMed.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- X. Li, D. Xiao and Z. Zhang, New J. Phys., 2013, 15, 023011 CrossRef CAS.
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Chem. Sci., 2021, 12, 5701–5719 RSC.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS.
- Z. Zhang, C. Mao, D. M. Meira, P. N. Duchesne, A. A. Tountas, Z. Li, C. Qiu, S. Tang, R. Song, X. Ding, J. Sun, J. Yu, J. Y. Howe, W. Tu, L. Wang and G. A. Ozin, Nat. Commun., 2022, 13, 1512 CrossRef CAS PubMed.
- A. Gellé, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 120, 986–1041 CrossRef PubMed.
- X. Yang and D. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- V. Jain, R. K. Kashyap and P. P. Pillai, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- J. Zhou, H. Liu and H. Wang, Chin. Chem. Lett., 2023, 34, 107420 CrossRef CAS.
- F. Pan, X. Xiang, Z. Du, E. Sarnello, T. Li and Y. Li, Appl. Catal., B, 2020, 260, 118189 CrossRef CAS.
- B. Xie, D. Hu, P. Kumar, V. V. Ordomsky, A. Y. Khodakov and R. Amal, Joule, 2024, 8, 312–333 CrossRef CAS.
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Phys. Chem. Chem. Phys., 2015, 17, 14623–14635 RSC.
- Z. Zhang, H. Tian, J. Sun, D. M. Meira, M. Zhang, X. Ding, D. Ji, C. Qiu, Z. Lu, L. Sun, Y. Zhang, W. Tu, Y. Zhou, X. Yang, J. Howe, L. Wang, S.-Y. Tong and Z. Zou, Chem. Catal., 2023, 3, 100644 CrossRef CAS.
- J. J. Mock, M. Barbic, D. R. Smith, D. A. Schultz and S. Schultz, J. Chem. Phys., 2002, 116, 6755–6759 CrossRef CAS.
- J. Zhang, H. Liu, Z. Wang and N. Ming, Adv. Funct. Mater., 2007, 17, 3295–3303 CrossRef CAS.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- S. Dal Forno, L. Ranno and J. Lischner, J. Phys. Chem. C, 2018, 122, 8517–8527 CrossRef CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- Q. Zhang, W. Li, C. Moran, J. Zeng, J. Chen, L.-P. Wen and Y. Xia, J. Am. Chem. Soc., 2010, 132, 11372–11378 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- B. J. Wiley, Y. Chen, J. M. McLellan, Y. Xiong, Z.-Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS PubMed.
- B. J. Wiley, Y. Xiong, Z.-Y. Li, Y. Yin and Y. Xia, Nano Lett., 2006, 6, 765–768 CrossRef CAS PubMed.
- M. J. Mulvihill, X. Y. Ling, J. Henzie and P. Yang, J. Am. Chem. Soc., 2010, 132, 268–274 CrossRef CAS PubMed.
- H. Zhang and A. O. Govorov, J. Phys. Chem. C, 2014, 118, 7606–7614 CrossRef CAS.
- L. V. Besteiro and A. O. Govorov, J. Phys. Chem. C, 2016, 120, 19329–19339 CrossRef CAS.
- G. Baffou, R. Quidant and F. J. García de Abajo, ACS Nano, 2010, 4, 709–716 CrossRef CAS PubMed.
- G. Baffou, R. Quidant and C. Girard, Appl. Phys. Lett., 2009, 94, 153109 CrossRef.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- K. Manthiram and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 3995–3998 CrossRef CAS PubMed.
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef PubMed.
- X. Yan, M. Cao, S. Li, P. N. Duchesne, W. Sun, C. Mao, R. Song, Z. Lu, X. Chen, W. Qian, R. Li, L. Wang and G. A. Ozin, J. Am. Chem. Soc., 2023, 145, 27358–27366 CrossRef CAS PubMed.
- B. Deng, H. Song, K. Peng, Q. Li and J. Ye, Appl. Catal., B, 2021, 298, 120519 CrossRef CAS.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- D. Kumar, C. H. Park and C. S. Kim, ACS Sustainable Chem. Eng., 2018, 6, 8604–8614 CrossRef CAS.
- M. V. Balois, N. Hayazawa, F. C. Catalan, S. Kawata, T.-A. Yano and T. Hayashi, Anal. Bioanal. Chem., 2015, 407, 8205–8213 CrossRef CAS PubMed.
- W. Sun, G. Zhong, C. Kubel, A. A. Jelle, C. Qian, L. Wang, M. Ebrahimi, L. M. Reyes, A. S. Helmy and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 6329–6334 CrossRef CAS PubMed.
- J. Jia, H. Wang, Z. Lu, P. G. O’Brien, M. Ghoussoub, P. Duchesne, Z. Zheng, P. Li, Q. Qiao, L. Wang, A. Gu, A. A. Jelle, Y. Dong, Q. Wang, K. K. Ghuman, T. Wood, C. Qian, Y. Shao, C. Qiu, M. Ye, Y. Zhu, Z. H. Lu, P. Zhang, A. S. Helmy, C. V. Singh, N. P. Kherani, D. D. Perovic and G. A. Ozin, Adv. Sci., 2017, 4, 1700252 CrossRef PubMed.
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng, A.-B. Xu, S. Tang, A. Tavasoli, M. Peng, W. Liu, A. S. Helmy, L. He, G. A. Ozin and X. Zhang, Nat. Energy, 2021, 6, 807–814 CrossRef CAS.
- S. Li, P. Miao, Y. Zhang, J. Wu, B. Zhang, Y. Du, X. Han, J. Sun and P. Xu, Adv. Mater., 2021, 33, e2000086 CrossRef PubMed.
- J. Sun, W. Sun, L. Wang and G. A. Ozin, Acc. Mater. Res., 2022, 3, 1260–1271 CrossRef CAS.
- L. B. Hoch, T. E. Wood, P. G. O’Brien, K. Liao, L. M. Reyes, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400013 CrossRef PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. Garcia, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, e1903915 CrossRef PubMed.
- Y. F. Xu, P. N. Duchesne, L. Wang, A. Tavasoli, F. M. Ali, M. Xia, J. F. Liao, D. B. Kuang and G. A. Ozin, Nat. Commun., 2020, 11, 5149 CrossRef CAS PubMed.
- Y.-F. Xu, A. A. Tountas, R. Song, J. Ye, J.-H. Wei, S. Ji, L. Zhao, W. Zhou, J.-H. Chen, G. Zhao, X. Yao, M. M. Sain, D.-B. Kuang and G. A. Ozin, Joule, 2023, 7, 738–752 CrossRef CAS.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- X. Yu, X. Ding, Y. Yao, W. Gao, C. Wang, C. Wu, C. Wu, B. Wang, L. Wang and Z. Zou, Adv. Mater., 2024, e2312942, DOI:10.1002/adma.202312942.
- Y. Chen, Y. Zhang, G. Fan, L. Song, G. Jia, H. Huang, S. Ouyang, J. Ye, Z. Li and Z. Zou, Joule, 2021, 5, 3235–3251 CrossRef CAS.
- H. Jiang, L. Wang, H. Kaneko, R. Gu, G. Su, L. Li, J. Zhang, H. Song, F. Zhu, A. Yamaguchi, J. Xu, F. Liu, M. Miyauchi, W. Ding and M. Zhong, Nat. Catal., 2023, 6, 519–530 CrossRef CAS.
- Z. Lu, Y. Xu, Z. Zhang, J. Sun, X. Ding, W. Sun, W. Tu, Y. Zhou, Y. Yao, G. A. Ozin, L. Wang and Z. Zou, J. Am. Chem. Soc., 2023, 145, 26052–26060 CrossRef CAS PubMed.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- S. Ning, H. Ou, Y. Li, C. Lv, S. Wang, D. Wang and J. Ye, Angew. Chem., Int. Ed., 2023, 62, e202302253 CrossRef CAS PubMed.
- O. Martin, A. J. Martin, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferre and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- T. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. Huang, F. M. Ali, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521 CrossRef PubMed.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- Y. Li, X. Bai, D. Yuan, C. Yu, X. San, Y. Guo, L. Zhang and J. Ye, Nat. Commun., 2023, 14, 3171 CrossRef CAS PubMed.
- J. Ren, S. Ouyang, H. Xu, X. Meng, T. Wang, D. Wang and J. Ye, Adv. Energy Mater., 2017, 7, 1601657 CrossRef.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- R. Song, Z. Li, J. Guo, P. N. Duchesne, C. Qiu, C. Mao, J. Jia, S. Tang, Y. F. Xu, W. Zhou, L. Wang, W. Sun, X. Yan, L. Guo, D. Jing and G. A. Ozin, Angew. Chem., Int. Ed., 2023, 62, e202304470 CrossRef CAS PubMed.
- J. Zhu, W. Shao, X. Li, X. Jiao, J. Zhu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2021, 143, 18233–18241 CrossRef CAS PubMed.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- W. H. Li, J. Yang and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213318 CrossRef CAS PubMed.
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS PubMed.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- L. Mascaretti, A. Schirato, T. Montini, A. Alabastri, A. Naldoni and P. Fornasiero, Joule, 2022, 6, 1727–1732 CrossRef.
- K. Yang, Z. Gu, Y. Long, S. Lin, C. Lu, X. Zhu, H. Wang and K. Li, Green Energy Environ., 2021, 6, 678–692 CrossRef CAS.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS PubMed.
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2015, 6, 494–497 CrossRef.
- T. Xie, Z.-Y. Zhang, H.-Y. Zheng, K.-D. Xu, Z. Hu and Y. Lei, Chem. Eng. J., 2022, 429, 132507 CrossRef CAS.
- Z. Zhu, W. Guo, Y. Zhang, C. Pan, J. Xu, Y. Zhu and Y. Lou, Carbon Energy, 2021, 3, 519–540 CrossRef CAS.
- Q. Li, H. Wang, M. Zhang, G. Li, J. Chen and H. Jia, Angew. Chem., Int. Ed., 2023, 62, e202300129 CrossRef CAS PubMed.
- Q. Hu, Y. Li, J. Wu, Y. Hu, H. Cao and Y. Yang, Adv. Energy Mater., 2023, 13, 2300071 CrossRef CAS.
- Y. Yao, B. Li, X. Gao, Y. Yang, J. Yu, J. Lei, Q. Li, X. Meng, L. Chen and D. Xu, Adv. Mater., 2023, 35, e2303654 CrossRef PubMed.
- S. Shoji, X. Peng, A. Yamaguchi, R. Watanabe, C. Fukuhara, Y. Cho, T. Yamamoto, S. Matsumura, M.-W. Yu, S. Ishii, T. Fujita, H. Abe and M. Miyauchi, Nat. Catal., 2020, 3, 148–153 CrossRef CAS.
- W. Zhou, B. H. Wang, L. Tang, L. Chen, J. K. Guo, J. B. Pan, B. Lei, B. Hu, Z. J. Bai, M. Tulu, Z. X. Li, X. Wang, C. T. Au and S. F. Yin, Adv. Funct. Mater., 2023, 33, 2214068 CrossRef CAS.
- J. Zhao, X. Guo, R. Shi, G. I. N. Waterhouse, X. Zhang, Q. Dai and T. Zhang, Adv. Funct. Mater., 2022, 32, 2204056 CrossRef CAS.
- A. Tavasoli, A. Gouda, T. Zahringer, Y. F. Li, H. Quaid, C. J. Viasus Perez, R. Song, M. Sain and G. Ozin, Nat. Commun., 2023, 14, 1435 CrossRef CAS PubMed.
- J. Zhang, Y. Li, J. Sun, H. Chen, Y. Zhu, X. Zhao, L.-C. Zhang, S. Wang, H. Zhang, X. Duan, L. Shi, S. Zhang, P. Zhang, G. Shao, M. Wu, S. Wang and H. Sun, Appl. Catal., B, 2022, 309, 121263 CrossRef CAS.
- Z. Rao, Y. Cao, Z. Huang, Z. Yin, W. Wan, M. Ma, Y. Wu, J. Wang, G. Yang, Y. Cui, Z. Gong and Y. Zhou, ACS Catal., 2021, 11, 4730–4738 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- D. Mateo, A. Sousa, M. Zakharzhevskii and J. Gascon, Green Chem., 2024, 26, 1041–1061 RSC.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- L. W. Chen, Y. C. Hao, Y. Guo, Q. Zhang, J. Li, W. Y. Gao, L. Ren, X. Su, L. Hu, N. Zhang, S. Li, X. Feng, L. Gu, Y. W. Zhang, A. X. Yin and B. Wang, J. Am. Chem. Soc., 2021, 143, 5727–5736 CrossRef CAS PubMed.
- T. Hou, L. Chen, Y. Xin, W. Zhu, C. Zhang, W. Zhang, S. Liang and L. Wang, ACS Energy Lett., 2020, 5, 2444–2451 CrossRef CAS.
- H. Bai, S. H. Lam, J. Yang, X. Cheng, S. Li, R. Jiang, L. Shao and J. Wang, Adv. Mater., 2022, 34, e2104226 CrossRef PubMed.
- S. Wang, W. Yu, S. Xu, K. Han and F. Wang, ACS Sustainable Chem. Eng., 2021, 10, 115–123 CrossRef.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706–1711 CrossRef CAS PubMed.
- Y. Peng, J. Albero, A. Franconetti, P. Concepcion and H. Garcia, ACS Catal., 2022, 12, 4938–4946 CrossRef CAS PubMed.
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L. Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304452 CrossRef CAS PubMed.
- S. Lee, Y. R. Lee, S. J. Kim, J.-S. Lee and K. Min, Chem. Eng. J., 2023, 454, 140470 CrossRef CAS.
- C. Xing, H. Cai, D. Kang and W. Sun, Adv. Energy Sustainability Res., 2023, 4, 2300015 CrossRef CAS.
- Y. Li, Q. Guan, G. Huang, D. Yuan, F. Xie, K. Li, Z. Zhang, X. San and J. Ye, Adv. Energy Mater., 2022, 12, 2202459 CrossRef CAS.
- H. Ge, Y. Kuwahara, K. Kusu, Z. Bian and H. Yamashita, Appl. Catal., B, 2022, 317, 121734 CrossRef CAS.
- Y. Wang, A. Zavabeti, F. Haque, B. Y. Zhang, Q. Yao, L. Chen, D. Chen, Y. Hu, N. Pillai, Y. Liu, K. A. Messalea, C. Yang, B. Jia, D. M. Cahill, Y. Li, C. F. McConville, J. Z. Ou, L. Kong, X. Wen and W. Yang, Mater. Today, 2022, 55, 21–28 CrossRef CAS.
- G. Firestone, H. Huang, J. R. Bochinski and L. I. Clarke, Nanotechnology, 2019, 30, 475706 CrossRef CAS PubMed.
- H. Huang, G. Firestone, D. Fontecha, R. E. Gorga, J. R. Bochinski and L. I. Clarke, Nanoscale, 2020, 12, 904–923 RSC.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305–1317 CrossRef CAS.
- Y. Liu, X. Wang, Q. Li, T. Yan, X. Lou, C. Zhang, M. Cao, L. Zhang, T. K. Sham, Q. Zhang, L. He and J. Chen, Adv. Funct. Mater., 2022, 33, 2210283 CrossRef.
- Y. Liu, C. Li, H. Yu and J. Guo, Chem, 2022, 8, 2586–2588 CAS.
- R. Cao, M. Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- Y. Liu, C. Zhang, J. Feng, X. Wang, Z. Ding, L. He, Q. Zhang, J. Chen and Y. Yin, Angew. Chem., Int. Ed., 2023, 62, e202308930 CrossRef CAS PubMed.
- H. Luo, D. Yao, K. Zeng, J. Li, S. Yan, D. Zhong, J. Hu, H. Yang and H. Chen, Fuel Process. Technol., 2022, 230, 107205 CrossRef CAS.
- Y. Miao, Y. Zhao, G. I. N. Waterhouse, R. Shi, L. Z. Wu and T. Zhang, Nat. Commun., 2023, 14, 4242 CrossRef CAS PubMed.
- M. Bonn, S. Funk, Ch Hess, N. D. Denzler and C. Stampfl, Science, 1999, 285, 1042–1045 CrossRef CAS PubMed.
- E. Hansjosten, A. Wenka, A. Hensel, W. Benzinger, M. Klumpp and R. Dittmeyer, Chem. Eng. Process., 2018, 130, 119–126 CrossRef CAS.
- S. Zoller, E. Koepf, D. Nizamian, M. Stephan, A. Patane, P. Haueter, M. Romero, J. Gonzalez-Aguilar, D. Lieftink, E. de Wit, S. Brendelberger, A. Sizmann and A. Steinfeld, Joule, 2022, 6, 1606–1616 CrossRef CAS PubMed.
- X. Ding, Z. Zhang, J. Sun, J. Y. Y. Loh, D. Ji, J. Lu, C. Liu, L. Zhao, W. Liu, J. Zhao, S. Tang, M. Safari, H. Cai, W. Tu, N. P. Kherani, Z. Hu, G. A. Ozin, Z. Zou and L. Wang, Joule, 2023, 7, 2318–2334 CrossRef CAS.
Footnote |
| † C. X., and Q. T. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |