
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoton-upconverters for blue organic light-emitting diodes: a low-cost, sky-blue example†
Le
Yang
 *ac,
Xian Wei
Chua
ab,
Zhihong
Yang
a,
Xiangpeng
Ding
a,
Yong
Yu
a,
Ady
Suwardi
a,
Meng
Zhao
a,
Karen Lin
Ke
a,
Bruno
Ehrler
d and
Dawei
Di
e
*ac,
Xian Wei
Chua
ab,
Zhihong
Yang
a,
Xiangpeng
Ding
a,
Yong
Yu
a,
Ady
Suwardi
a,
Meng
Zhao
a,
Karen Lin
Ke
a,
Bruno
Ehrler
d and
Dawei
Di
e
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Singapore 138634, Singapore. E-mail: yang_le@imre.a-star.edu.sg
bCavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB30HE, UK
cDepartment of Materials Science and Engineering, National University of Singapore, Singapore 117575, Singapore
dCenter for Nanophotonics, AMOLF, Science Park 104, 1098 XG, Amsterdam, The Netherlands
eState Key Laboratory of Modern Optical Instrumentation, College of Optical Science and Engineering, International Research Center for Advanced Photonics, Zhejiang University, Hangzhou, 310027, China
First published on 24th January 2022
Abstract
In the research ecosystem's quest towards having deployable organic light-emitting diodes with higher-energy emission (e.g., blue light), we advocate focusing on fluorescent emitters, due to their relative stability and colour purity, and developing design strategies to significantly improve their efficiencies. We propose that all triplet–triplet annihilation upconversion (TTA-UC) emitters would make good candidates for triplet fusion-enhanced OLEDs (“FuLEDs”), due to the energetically uphill nature of the photophysical process, and their common requirements. We demonstrate this with the low-cost sky-blue 1,3-diphenylisobenzofuran (DPBF). Having satisfied the criteria for TTA-UC, we show DPBF as a photon upconverter (Ith 92 mW cm−2), and henceforth demonstrate it as a bright emitter for FuLEDs. Notably, the devices achieved 6.5% external quantum efficiency (above the ∼5% threshold without triplet contribution), and triplet-exciton-fusion-generated fluorescence contributes up to 44% of the electroluminescence, as shown by transient measurements. Here, triplet fusion translates to a quantum yield (ΦTTA-UC) of 19%, at an electrical excitation of ∼0.01 mW cm−2. The enhancement is meaningful for commercial blue OLED displays. We also found DPBF to have decent hole mobilities of ∼0.08 cm2 V−1 s−1. This additional finding can lead to DPBF being used in other capacities in various printable electronics.
Organic light-emitting diodes (OLEDs) occupy an increasing market share in our daily appliances for display, particularly owing to their flexibility, compact/ultrathin form factor and better colour quality. The research ecosystem recognises the remaining gap in producing competent blue OLED pixels in full-colour organic display technology,1 due to blue being on the higher-energy end of the visible spectrum. Since the 1st generation fluorescent OLEDs which typically have low efficiencies,2,3 2nd (phosphorescent) and 3rd (thermally activated delayed fluorescence (TADF)) generation organic emitters have become promising candidates;4–6 blue OLEDs made from corresponding phosphorescent or TADF emitters have also produced increasingly impressive devices.7–10 We advocate here a critical tri-factor of competent red-green-blue (RGB) displays, that an OLED (of any colour) must satisfy three intrinsic criteria: (1) performance or efficiency; (2) stability; and (3) colour purity and gamut. The challenge with blue OLED pixels is the difficulty to tick all three boxes in one single system.
Most 2nd and 3rd generation blue OLEDs have achieved very high device efficiencies, way exceeding their 1st generation fluorescent counterparts which have a quantum efficiency bottleneck imposed by electrically excited excitons. In typical fluorescent OLEDs,2,3 the spontaneous formation of spin-dependent excitons upon charge injection usually follows a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 emissive singlet to dark triplet excitons, leading to an intrinsic internal quantum efficiency (IQE) threshold of only 25%. Phosphorescent and TADF OLEDs subsequently broke this bottleneck, by utilising all dark triplets as emissive states, in principle leading to 100% IQE.4,5 However, 2nd and 3rd generation blue OLEDs, having overcome factor (1), often suffer from less satisfactory factors (2) and (3) – their shelf-life may be shorter than fluorescent emitters, and it is difficult to achieve as deep a blue and as narrow an emission spectrum as many fluorescent emitters.
:3 emissive singlet to dark triplet excitons, leading to an intrinsic internal quantum efficiency (IQE) threshold of only 25%. Phosphorescent and TADF OLEDs subsequently broke this bottleneck, by utilising all dark triplets as emissive states, in principle leading to 100% IQE.4,5 However, 2nd and 3rd generation blue OLEDs, having overcome factor (1), often suffer from less satisfactory factors (2) and (3) – their shelf-life may be shorter than fluorescent emitters, and it is difficult to achieve as deep a blue and as narrow an emission spectrum as many fluorescent emitters.
We advocate developing strategies with fluorescent OLEDs, where the emission site originates from fluorescent molecules, focusing on overcoming their efficiency bottlenecks. Sensitisers and dual-dopants have been excellent strategies, lifting fluorescent device efficiencies significantly, meanwhile preserving the colour features of the fluorescent emitter and ensuring better device stability by having exciton formation and emission occurring on different dopants in the system.11–15 They all involve an energetically downhill energy transfer process, i.e., the injected energy and exciton formed on the sensitizer (phosphorescent, TADF or spin-state interconversion material) must be even higher than the emitted (output) blue light from the accepter/emitter.
Here, we promote an alternative single-dopant strategy, using triplet–triplet annihilation (TTA), or triplet fusion, as the favourable energetically uphill pathway towards building better blue fluorescent OLEDs. It facilitates the formation of higher-energy output in devices. A simple selection step, under optical excitation (before making an electronic device), can help to determine if an emitter is suitable. From our experience with 9,10-diphenylanthracene (DPA), a common blue TTA-UC emitter16–18 used in “FuLEDs” (triplet fusion enhanced OLEDs), we seek to establish that any blue TTA-UC emitter would make a suitable candidate for a blue FuLED as they share similar selection criteria.
In TTA upconversion (TTA-UC), two low-energy photons are converted into a higher-energy photon, in a system comprising a triplet sensitizer and a triplet acceptor/emitter.19 Here, the triplet acceptor/emitter should meet the energetic requirement of E(S1) ≤ or ≈ 2E(T1), among other criteria such as a high fluorescence quantum yield, a short singlet lifetime, and a long triplet lifetime. This two-for-one TTA-UC for light emission can also be translated into triplet fusion in conventional fluorescent OLEDs to enhance efficiencies (Fig. 1).20–24 Triplet excitons that form on the emitter upon charge injection are energetically suitable to undergo triplet fusion and generate emissive singlets. Hence, in an ideal scenario where all triplet–triplet encounters lead to emissive singlets, the theoretical IQE ceiling for a FuLED is calculated to be 62.5%.24 This also predicts that the proportion of electroluminescence (EL) from the triplet fusion pathway can be up to 60%, decaying via a slow, delayed channel. In contrast, the fraction of EL from direct singlet recombination occurs promptly on a timescale of nanoseconds. Note that EQE and IQE are related by an outcoupling factor ηoutcoupling, which depends on the dipole alignment in the organic layers and the geometry of the device, and on the refractive indices of the organic materials. Assuming a planar smooth device with isotropic emission, the ηoutcoupling is typically ∼0.2 on glass substrates with one reflective electrode.25,26 As such, we would expect the 25% fluorescent IQE limit to correspond to ∼5% EQE, and the 62.5% FuLED IQE threshold, to a ∼12.5% EQE ceiling. Although triplet fusion enhancement may seem less efficient than dual-dopant/sensitizer strategies for (blue) fluorescent OLEDs, it is still an important mechanism for blue pixels in practical display technologies because: (a) it requires only 1 dopant for ease of fabrication and optimisation; and (b) it involves an energetically uphill photophysical process, encouraging formation of higher-energy (blue) emission at a low operating voltage.
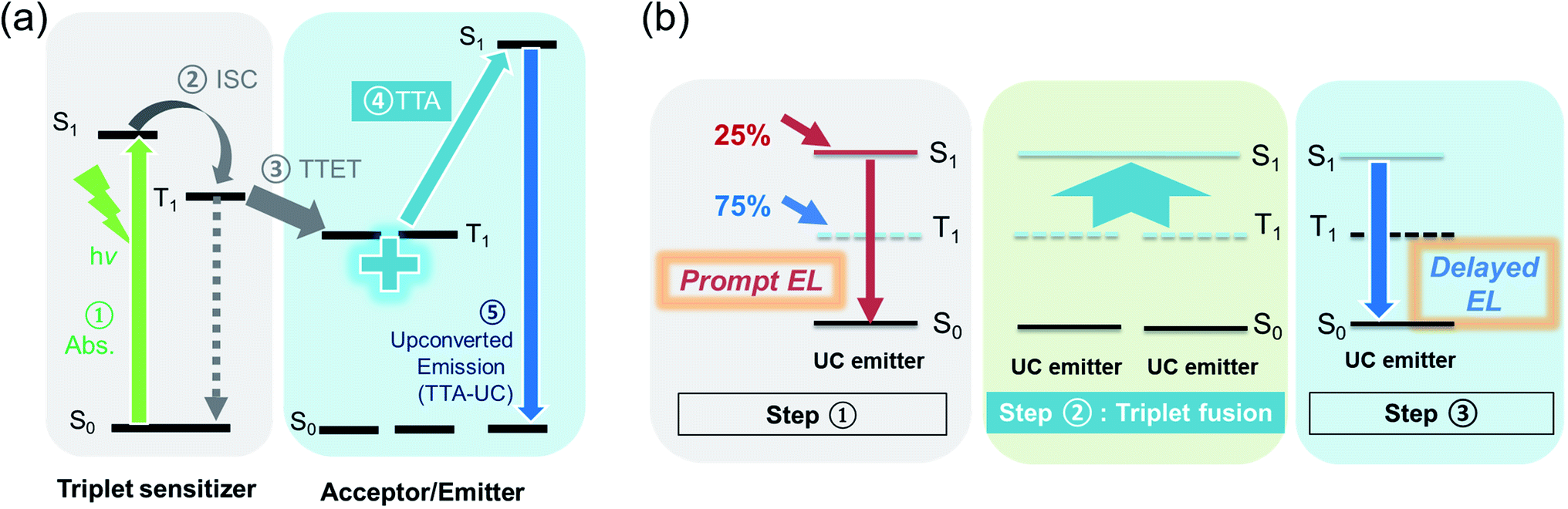 | ||
| Fig. 1 Analogous photophysical processes for TTA emission in an optically and electrically excited system. (a) Simplified photophysical processes leading to TTA in a photon upconversion system, involving a triplet sensitizer and a triplet acceptor. (b) Simplified photophysical processes for triplet fusion in a triplet-fusion enhanced OLED (FuLED). Step 1 consists of spontaneous exciton formation when charges are injected into the device. It follows a spin statistical ratio of 25% (emissive) singlets to 75% (dark) triplets. Prompt emission occurs when the singlets immediately recombine radiatively. Step 2 illustrates TTA or triplet fusion, where two triplets encounter and (ideally) form one singlet. Step 3 shows the fusion-generated singlet emission via a slower, delayed channel (“delayed EL”). | ||
1,3-Diphenylisobenzofuran (DPBF, Fig. 2a inset) is a commercially available, low-cost organic molecule, typically used as a synthetic precursor in Diels–Alder reactions,27–29 and as a sensor for singlet oxygen due to its specific reactivity.27,30,31 The discovery and use of DPBF for its singlet fission properties have been championed by J. Michl et al.,32,33 where an excited singlet exciton splits into two triplet excitons (each with around half of the initial singlet energy). Michl and coworkers demonstrated that singlet fission in DPBF is highly efficient at low temperatures and in certain polymorphs, controlled by the method of film deposition.34,35 Energetically, singlet fission requires a large singlet–triplet separation in the organic molecule, where ideally E(S1) ≥ or ≈ 2E(T1). In DPBF, according to Michl et al.,32–37E(S1) ∼ 2.9 eV and E(T1) ∼ 1.4–1.5 eV. Such energetics of DPBF are also in principle suitable for TTA-UC. However, to date, only Cao et al. has incorporated DPBF experimentally in a hetero TTA-UC system with DPA.38
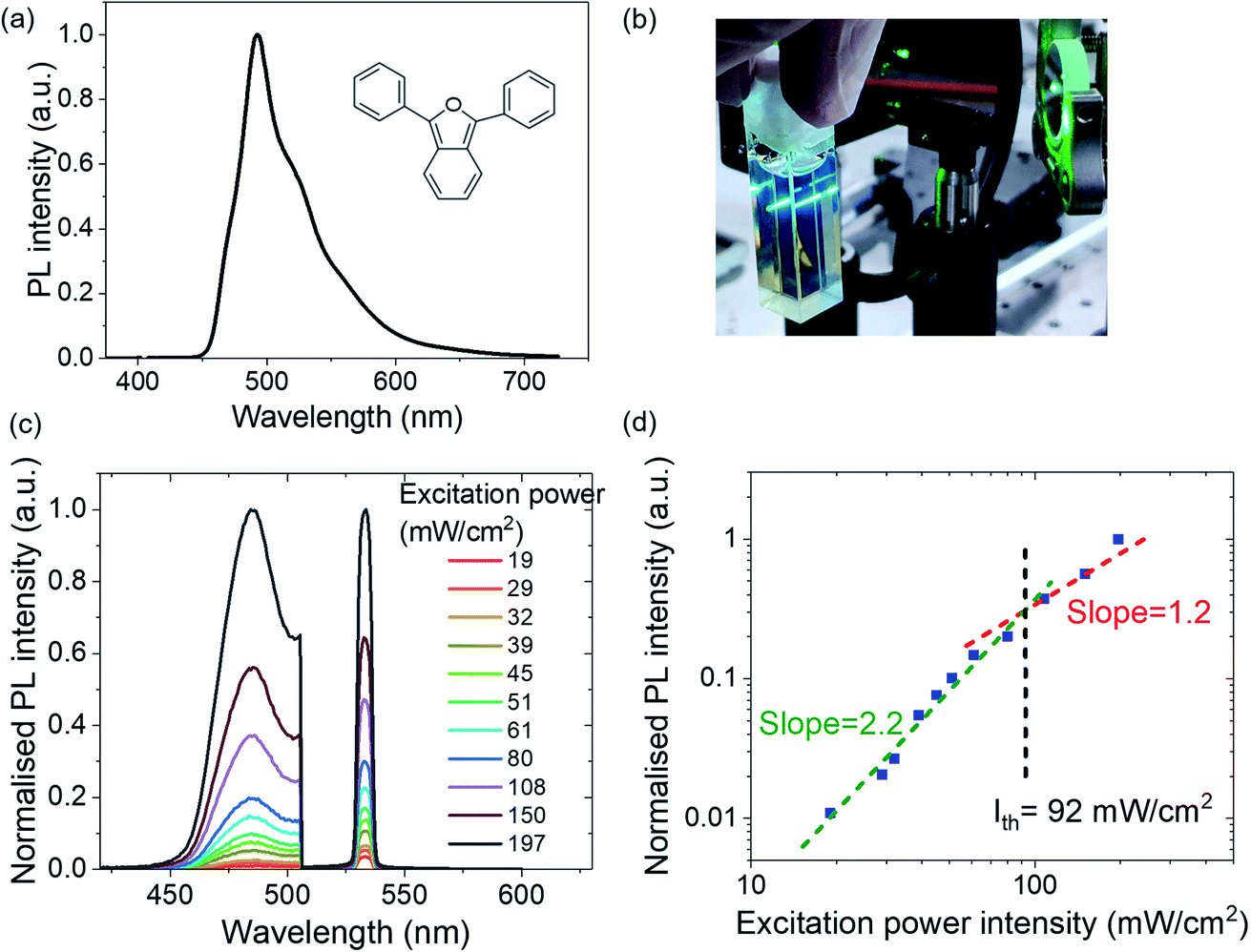 | ||
| Fig. 2 DPBF TTA upconversion behaviour. (a) PL spectrum of DPBF in a blend film of PVK:DPBF under a 407 nm laser. Inset: molecular structure of DPBF. (b) Photograph of DPBF undergoing upconverted emission (via TTA) under CW 532 nm laser excitation. Sky-blue emission is visibly observed. (c) DPBF PL intensity under various excitation laser (CW 532 nm) power densities. Note that the laser peak and DPBF PL peaks are normalised independently, with each peak's highest measurement being standardised at 1.0, to provide a relative comparison within each emission peak. (d) DPBF PL intensity as a function of excitation laser power intensity, to observe the typical TTA-UC dual-regime behaviour. | ||
DPBF is not widely reported as an organic semiconductor in optoelectronic devices. In this paper, we show that verifying DPBF as a TTA-UC material can lead to it being selected as a triplet fusion-enhanced emitter in a blue OLED, meanwhile observing that it aids hole-transport in optoelectronic devices. First, we note that DPBF is highly luminescent when doped in a polymer film, peaking at ∼485 nm (Fig. 2a). Its singlet decay is captured in time-correlated single photon counting (TCSPC) trace in Fig. S1, ESI,† showing a short singlet lifetime of a few nanoseconds.
We then note green-to-blue photon upconversion in a solution of platinum octaethylporphyrin (PtOEP) and DPBF (or PtOEP:DPBF for short; Fig. 2b). Here,conventional phosphorescent PtOEP acts as a triplet sensitizer, absorbing strongly in the green region and has a strong heavy-atom effect for singlet-triplet spin-mixing. DPBF acts as the triplet acceptor or emitter (Fig. 1a), allowing the lower-lying triplet energy to be harvested by TTA-UC as higher-lying emissive singlets relax radiatively to produce sky-blue light. To further confirm the TTA behaviour, we observe the anticipated two-regime relationship between upconverted emission intensity and excitation laser power for typical TTA-UC behaviour (Fig. 2c and d).17,39 Using a continuous wave laser of 532 nm, we clearly observe the upconverted signal at ∼485 nm. In Fig. 2d, the intensity of the upconverted PL (blue squares) shows a clear transition of the slope from quadratic (slope = 2.2) to linear (slope = 1.2). At lower excitation densities, DPBF molecules are in weak annihilation, and the triplet decay is primarily through quasi-first order kinetics and the upconverted PL has a quadratic intensity dependence on the excitation power (green dashed line). Here the upconversion has not reached its maximum efficiency. At higher optical excitation densities, triplet–triplet energy transfer (TTET) and bimolecular TTA would dominate, leading to a linear intensity-dependence of the emission (red dashed line). In this regime, the UC should be tapering towards a maximum. We estimate the power intensity required to enter this regime, Ith, to be ∼92 mW cm−2 from the deflection point of the two regimes. This is lower than, though in the same order of magnitude as, Cao's 115 mW cm−2.38 To provide a benchmark against well-known TTA-UC systems, such as with DPA, see the ESI† Section 2 (Fig. S2), for a relative comparison between DPBF and DPA as upconverters. But even without analysing or verifying the TTA-UC behaviour, generating blue light from a green laser source, Fig. 2b, can qualitatively show that the emitter is a potential blue triplet fusion OLED candidate – by making a simple solution of PtOEP and DPBF, directing a green laser on it and visually observing blue emission.
With that, we then use DPBF as a bright sky-blue emitter in OLEDs, resulting in >6% EQE at about 100 cd m−2. Considering DPBF's suitable exciton energies for triplet fusion, and referring to previous studies on triplet fusion-enhanced OLEDs by our team and by others,20,24,38 we study the effect of triplet fusion in DPBF OLEDs (or FuLEDs24) (Fig. 1b). The FuLEDs are fabricated via a multilayer solution-processable route, where all four organic layers are spin-coated from solutions, with a structure of ITO/PEDOT:PSS/poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(4,4′-(N-(4-sec-butylphenyl)diphenylamine)] (TFB)/poly(9-vinylcarbazole) (PVK):DPBF/bathophenanthroline (Bphen)/LiF/Al. We present here the first demonstration of a DPBF-based OLED, with its electroluminescence (EL) spectrum and device performance shown in Fig. 3a–c. With ∼6.5% EQE consistently across a wide current range, i.e., above the ∼5% EQE threshold expected of simple fluorescent OLEDs, it is the first indication of triplet utilisation. Moreover, DPBF satisfies the criteria to be a FuLED emitter:24 (1) high PL quantum yield (>0.7 in polymer:DPBF films); (2) short singlet lifetime (2–6 ns); (3) relatively long triplet lifetime (hundreds of ns to μs); and (4) 2E(T1) ≥ or ≈ E(S1).
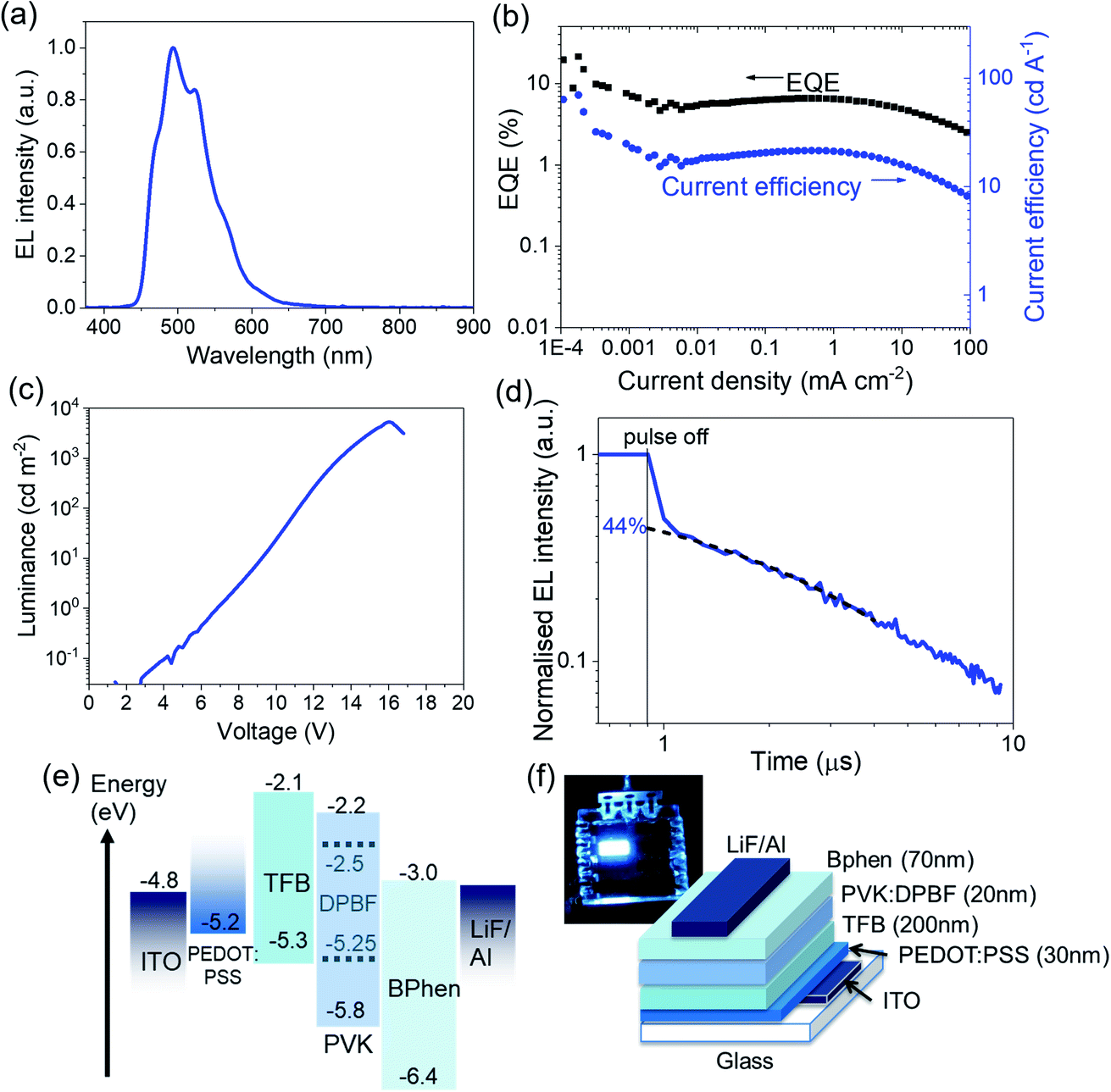 | ||
| Fig. 3 Triplet fusion in DPBF FuLEDs. (a) EL spectrum of a DPBF FuLED. (See Section 6, ESI† on suggested explanations in the differences between the EL and PL spectra for DPBF) (b) EQE and current efficiency vs. current density curves for a DPBF FuLED. (c) Luminance against voltage characterisation curve for a DPBF FuLED. (d) Transient EL of a DPBF FuLED held at 1 mA cm−2. (e) The HOMO–LUMO energy alignment of materials in the device stack. (f) Illustration of the device stack and a photograph of a lit-up DPBF FuLED. | ||
We note that the highest EQE occurs at a low current density, and we speculate that the efficiency roll-off at higher current densities is mainly due to charge imbalance and triplet-charge annihilation from the unoptimised device architecture. This is in good agreement with prior studies.23,24 To confirm the effect of triplet fusion in the FuLED, we next look at transient EL measurement in Fig. 3d. The decay profile shows an initial prompt fluorescence decay due to direct singlet recombination, and a delayed component due to the bimolecular triplet fusion process, characteristic of triplet fusion enhancement in OLEDs.21,23,24,40 The delayed EL component follows a bimolecular decay, another trademark of TTA or triplet fusion, and can be fitted by using eqn (1).24
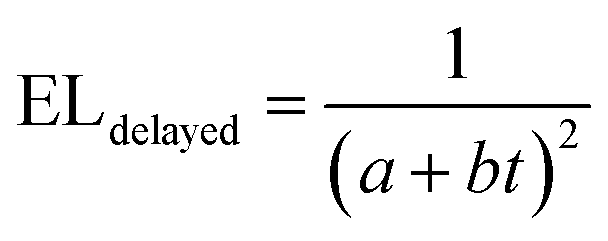 | (1) |
Following the first method to quantify the TTA-UC quantum efficiency in FuLEDs,24 we performed similar calculations. We summarise the calculation method in the ESI,† Section 5. The intrinsic TTA-UC efficiency ηTTA-UC (twice the number of singlets generated by triplet fusion/number of triplets entering the system) is estimated to be 44.8%; and the quantum yield ΦTTA-UC (the number of TTA-generated photons emitted per triplet exciton entering the system, i.e., maximum 50%) is 19%, at an excitation power intensity of ∼0.01 mW cm−2. These values are comparable to those of the deep blue DPA FuLED, and are higher than the efficiency reported for red singlet fission TIPS-pentacene FuLEDs (Table 1).24 It is interesting to note that in the optically excited solution-phase hetero multi-acceptor TTA-UC system, with dual emitters DPBF and DPA, the ΦTTA-UC achieved was comparable at 16%, but at a much higher excitation power intensity of 115 mW cm−2.38 We speculate that processes typically associated with triplet sensitizer molecules (e.g., intersystem crossing, triplet–triplet energy transfer) could be the bottleneck for TTA-UC systems to reach higher efficiencies.
| Emitter in FuLED/TTA-UC | η TTA-UC (%) | Φ TTA-UC (%) | Excitation power intensity (mW cm−2) | |
|---|---|---|---|---|
| DPBF (sky-blue) device | 44.8 | 19.0 | 0.01 (electrically); 92 (optically, sensitized by PtOEP) | This work |
| DPA (deep-blue) device | 49.0 | 19.6 | 0.001 (electrically) | Ref. 24 |
| TIPS-pentacene (red, known singlet fission material) device | 35.1 | 6.8 | 0.001 (electrically) | Ref. 24 |
| DPBF/DPA optical TTA-upconverter system, in solution | N.A. | 16.0 | 115 (optically, sensitized by PtOEP) | Ref. 38 |
Interestingly, we find that DPBF possesses high vertical hole mobility from single-carrier (hole-only) devices. The current–voltage (J–V) curves are analysed with a basic space charge limited current (SCLC) model.41 Most SCLC hole mobility studies of organic semiconductor thin films typically fall in the range of 10−4 cm2 V−1 s−1 or lower, and values in the range of 10−2 cm2 V−1 s−1 are among the best.42 The hole mobilities obtained for non-annealed, amorphous DPBF are considerably satisfactory and reproducible at ∼0.08 cm2 V−1 s−1 (Fig. S3 and Section 3, ESI†). This helps to facilitate hole transport across the device stack; it might explain why the thick hole-blocking layer in our device design is necessary, and even at 70–80 nm the BPhen may still be insufficient for the DPBF device architecture. Meanwhile, this revelation may prompt DPBF to be used as a hole-transport layer on its own in optoelectronic devices.
In conclusion, we have demonstrated the first DPBF-based FuLEDs, prompted by DPBF's well-separated singlet-triplet energies originally studied for singlet fission. Its prominent behaviour as a TTA-UC emitter in solution with PtOEP as a triplet sensitizer serves as a check for suitability as a FuLED (triplet-fusion enhanced OLED) emitter. We find that, using DPBF as an example, blue TTA-UC emitters are suitable blue FuLED emitters as they share similar selection criteria: S1 and T1 energetics, a high blue fluorescence quantum yield, a short singlet lifetime and a relatively long triplet lifetime. A quick check using a PtOEP:DPBF solution and a green excitation laser, producing DPBF's sky blue emission, can serve as an easy step to verify the emitter's suitability in FuLEDs. This step can be applied when working with potential emitters in the future. DPBF as a FuLED candidate has produced efficient sky-blue devices. Transient-EL measurement identified the role of triplets, contributing to up to 44% of the total EL. The intrinsic TTA-UC efficiency (ηTTA-UC) and the quantum yield (ΦTTA-UC) are estimated to be 44.8% and 19% respectively, at a very low excitation power intensity of ∼0.01 mW cm−2. Such enhancements are meaningful towards blue OLED displays as they are still based on fluorescent emitters whose efficiencies can be further improved. Decent hole mobility in the vertical direction (∼0.08 cm2 V−1 s−1) is observed, prompting potential use for charge transport in other devices. Being inexpensive and easily soluble in most common solvents, and if device stability can be ensured with decent encapsulation, DPBF makes a great candidate for applications in solution-processable optoelectronic devices and printable electronics.
Conflicts of interest
There are no conflicts to declare.Author contributions
L. Y. initiated the project, fabricated, and characterised organic light-emitting diodes. L. Y. with D. D. conducted transient EL measurements. L. Y., X. W. C., X. D., K. L. K. and Z. Y. planned and conducted TTA-UC and fusion-related experiments and characterisation. Y. Y., A. S. and M. Z. contributed towards TTA-UC related experiments and discussions. D. D. and B. E. were engaged in in-depth discussion relating to DPBF and photophysics, and provided many useful ideas and suggestions. L. Y. prepared the initial manuscript; all authors contributed to the manuscript.Acknowledgements
L. Y., X. W. C., Z. Y., Y. Y., A. S., and M. Z. thank the Agency of Science, Technology and Research (A*STAR), Singapore, for sponsoring, including the SERC Central Research Fund, and AME Programmatic CPI A18A1b0045, and scholarships. L. Y. also extends appreciation towards the University of Cambridge, especially to Prof Richard Friend for insightful discussions. D. D. acknowledges the National Natural Science Foundation of China (NSFC) (61975180), Zhejiang University Education Foundation Global Partnership Fund, and Cambridge University Department of Physics. B.E. acknowledges the Netherlands Organisation for Scientific Research (NWO). The authors also thank Y. Y. Guo, C. H. Tian, and T. C. Sum for their discussion in the revision stage of the manuscript.References
- A. Monkman, ACS Appl. Mater. Interfaces, 2021 DOI:10.1021/acsami.1c09189
.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS
- J. Huh, M. J. Sung, S. Kwon, Y. Kim and J. Kim, Adv. Funct. Mater., 2021, 31, 2100967 CrossRef CAS
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208–215 CrossRef CAS
- H. Lim, H. J. Cheon, S.-J. Woo, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Adv. Mater., 2020, 32, 2004083 CrossRef CAS PubMed
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed
- L. Yang, V. Kim, Y. Lian, B. Zhao and D. Di, Joule, 2019, 3, 2381–2389 CrossRef CAS
- T. Ye, S. Shao, J. Chen, Z. Chen, L. Wang and D. Ma, J. Appl. Phys., 2010, 107, 054515 CrossRef
- S. Nam, J. W. Kim, H. J. Bae, Y. M. Maruyama, D. Jeong, J. Kim, J. S. Kim, W. Son, H. Jeong, J. Lee, S. Ihn and H. Choi, Adv. Sci., 2021, 8, 2100586 CrossRef CAS PubMed
- J.-H. Kim, F. Deng, F. N. Castellano and J. Kim, Chem. Mater., 2012, 24, 2250–2252 CrossRef CAS
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322 RSC
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS
- D. Y. Kondakov, J. Appl. Phys., 2007, 102, 114504–163102 CrossRef
- D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar and J. P. Spindler, J. Appl. Phys., 2009, 106, 124510 CrossRef
- D. Y. Kondakov, Philos. Trans. R. Soc., A, 2015, 373, 20140321 CrossRef PubMed
- B. H. Wallikewitz, D. Kabra, S. Gélinas and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 22–25 CrossRef
- D. Di, L. Yang, J. M. Richter, L. Meraldi, R. M. Altamimi, A. Y. Alyamani, D. Credgington, K. P. Musselman, J. L. MacManus-Driscoll and R. H. Friend, Adv. Mater., 2017, 29, 1605987 CrossRef PubMed
- N. C. Greenham, R. H. Friend and D. D. C. Bradley, Adv. Mater., 1994, 6, 491–494 CrossRef CAS
- J.-S. Kim, P. K. H. Ho, N. C. Greenham and R. H. Friend, J. Appl. Phys., 2000, 88, 1073 CrossRef CAS
-
P. C. Kierkus, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2001 Search PubMed
- R. Rodrigo, Tetrahedron, 1988, 44, 2093–2135 CrossRef CAS
- D. Tobia and B. Rickborn, J. Org. Chem., 1987, 52, 2611–2615 CrossRef CAS
- P. Carloni, E. Damiani, L. Greci, P. Stipa, F. Tanfani, E. Tartaglini and M. Wozniak, Res. Chem. Intermed., 1993, 19, 395–405 CrossRef CAS
- R. H. Young, K. Wehrly and R. L. Martin, J. Am. Chem. Soc., 1971, 93, 5774–5779 CrossRef CAS
-
J. Michl, A. J. Nozik, X. Chen, J. C. Johnson, G. Rana, A. Akdag and A. F. Schwerin, in Proc. of SPIE, ed. Z. H. Kafafi and P. A. Lane, 2007, vol. 6656, p. 66560E Search PubMed
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed
- J. C. Johnson, A. J. Nozik and J. Michl, J. Am. Chem. Soc., 2010, 132, 16302–16303 CrossRef CAS PubMed
- J. L. Ryerson, J. N. Schrauben, A. J. Ferguson, S. C. Sahoo, P. Naumov, Z. Havlas, J. Michl, A. J. Nozik and J. C. Johnson, J. Phys. Chem. C, 2014, 118, 12121–12132 CrossRef CAS
- A. F. Schwerin, J. C. Johnson, M. B. Smith, P. Sreearunothai, D. Popović, J. Cerný, Z. Havlas, I. Paci, A. Akdag, M. K. MacLeod, X. Chen, D. E. David, M. A. Ratner, J. R. Miller, A. J. Nozik and J. Michl, J. Phys. Chem. A, 2010, 114, 1457–1473 CrossRef CAS PubMed
- J. N. Schrauben, J. L. Ryerson, J. Michl and J. C. Johnson, J. Am. Chem. Soc., 2014, 136, 7363–7373 CrossRef CAS PubMed
- X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334–2338 CrossRef CAS
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC
- C. J. Chiang, A. Kimyonok, M. K. Etherington, G. C. Griffiths, V. Jankus, F. Turksoy and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 739–746 CrossRef CAS
-
M. A. Lampert and P. Mark, Current injection in solids, Academic Press, 1970 Search PubMed
- Y. Li, R. G. Clevenger, L. Jin, K. V. Kilway and Z. Peng, J. Mater. Chem. C, 2014, 2, 7180 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00803j |
| This journal is © The Royal Society of Chemistry 2022 |