
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Annulative π-extension of BODIPYs made easy via gold(I)-catalyzed cycloisomerization†
Jorge
Labella
a,
Gonzalo
Durán-Sampedro
 a,
M. Victoria
Martínez-Díaz
*ab and
Tomás
Torres
*abc
a,
M. Victoria
Martínez-Díaz
*ab and
Tomás
Torres
*abc
aDepartamento de Química Orgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: tomas.torres@uam.es; victoria.martinez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
cIMDEA-Nanociencia, Campus de Cantoblanco, 28049 Madrid, Spain
First published on 12th June 2020
Abstract
Here we report gold(I)-catalyzed cycloisomerization as a new powerful synthetic tool for the preparation of π-extended BODIPY derivatives. The catalytic system PPhF3AuCl/AgSbF6 enables the synthesis of [b]-[2,1]naphtho-fused-BODIPYs (2a–2c) under mild conditions, in excellent yields and short reaction times. The reaction is totally regioselective to the 6-endo-dig product and for the α-position of the BODIPY, which is both the kinetically and thermodynamically favored pathway, as supported by the free energy profile calculated by means of Density Functional Theory (DFT). Moreover, this methodology also allows the synthesis of two new families of [b]-aryl-fused-BODIPYs, namely, [3,4]phenanthro- (2e and 2f) and [1,2]naphtho-fused (2g) BODIPYs. Their molecular and electronic structures were established by NMR and UV-vis spectroscopies as well as single-crystal X-ray diffraction analysis. As can be noted from the X-ray structures, 2a, 2e and 2g present interesting structural differences at both the molecular and packing level. Interestingly, despite being isomers, the UV/vis spectra of 2a and 2g revealed significant differences in their electronic structures. The origin of this finding was studied by Time-Dependent DFT calculations. Calculated DFT Nuclear Independent Chemical Shift (NICS(0)) values also supported the different electronic structures of 2a and 2g.
Introduction
Gold(I)-catalyzed activation of alkynes has emerged over the last decade as a powerful tool for the construction of molecular complexity from readily accessible starting materials.1 Due to relativistic effects, gold(I) exhibits a high affinity for the alkyne π-bonds and forms η2-alkyne complexes, which are susceptible to react with a variety of heteroatom- or carbon-based nucleophiles under mild reaction conditions.2 More recently, this kind of catalysis has gained increased attention in the field of molecular materials, allowing for the efficient synthesis of novel highly conjugated systems, such as enantiopure helicenes,3 polycyclic aromatic hydrocarbons (PAHs)4 or other large heteroaromatic derivatives,5 with absorption bands shifted to red.Porphyrinoids are well-known chromophores that are nowadays widely used in a plethora of applied fields.6 Boron-dipyrromethenes (BODIPYs) and their derivatives, which are related to porphyrinoids and present unique redox and photophysical properties (e.g., excellent photostability, intense absorption profiles, high quantum yields, and small Stokes shifts), hold a privileged position.7 These properties have skillfully been used in a wide range of applications, such as photovoltaic cells,8 photodynamic therapy (PDT),9 OLEDs10 or probes for bioimaging,11 just to name a few. In this context, absorption and emission in the Near-Infrared Region (NIR) is highly desirable. Consequently, much effort has been devoted to developing synthetic methodologies to extend the π-system of BODIPYs. Among them, the fusion at the [a]- or [b]-bond (Scheme 1a) with aromatic rings such as benzene, naphthalene or phenanthrene is particularly promising.12 Despite the ring fusion at the [b]-position results in a higher bathochromic shift and more stability,13 the [b]-aryl-fused-BODIPYs have so far been less explored compared to the [a]-ones due to their more challenging synthesis. However, over the last few years, the post-functionalization of the BODIPY core by annulation has gained increased attention as a new efficient synthetic strategy (Scheme 1b). In this regard, several chemical transformations such as oxidative aromatic coupling,14 one-pot Suzuki–Miyaura–Knoevenagel reaction15 or direct C–H palladium catalyzed annulation,16 have been recently reported to prepare [b]-phenanthro and [b]-naphtho-fused-BODIPYs. However, these methodologies, although elegant, are quite limited in terms of scope, yield, group tolerance and availability of the starting material. Therefore, the efficient and flexible synthesis of [b]-aryl-fused-BODIPYs remains a synthetic challenge.
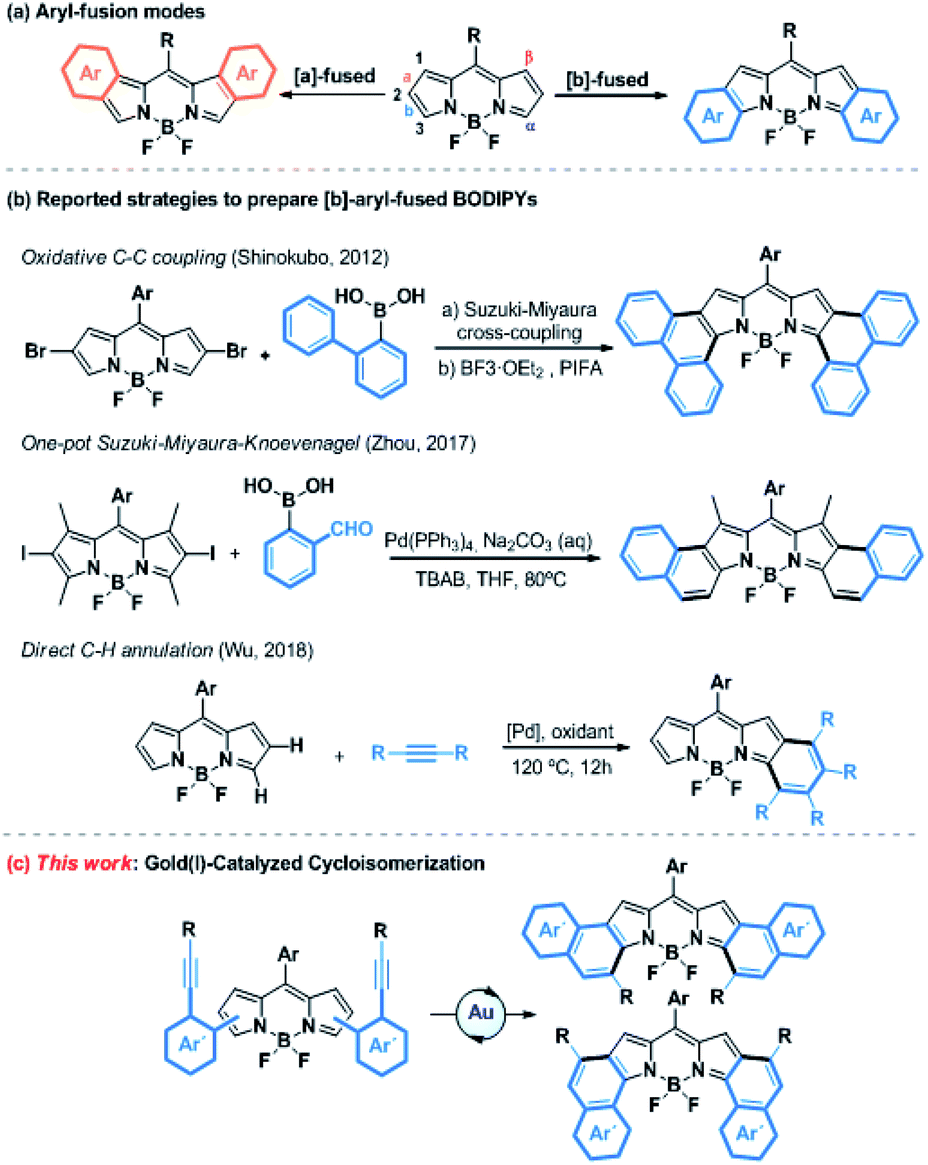 | ||
| Scheme 1 (a) Chemical structures of [a]- and [b]-aryl-fused BODIPYs. (b) Reported post-functionalization reactions of the BODIPY core to prepare [b]-aryl-fused BODIPYs. (c) Strategy described in this work. | ||
Considering the aforementioned features of gold(I)-activation of alkynes, we envisaged the synthesis of [b]-aryl-fused-BODIPYs using such catalysis as a key step. To do that, the well-known metal-catalyzed 6-endo-dig cyclization of ortho-alkynyl-biaryl compounds was selected as the model reaction (Scheme 1c).17 Besides the preparation of [b]-aryl-fused-BODIPYs, this study would provide a new tool for BODIPY functionalization, where the pyrrole rings act as a nucleophile toward gold-activated alkynes, giving rise to further investigations of other gold-catalyzed reactions.
Herein, we report the efficient, versatile and mild synthesis of [b]-naphtho and [b]-phenanthro-fused-BODIPYs using gold(I) catalyzed cyclization as a key step. This method, where several factors such as catalytic conditions and scope have been studied, showed excellent regioselectivity for the 6-endo-dig cyclization product and for the α-position of the BODIPY, whose origin has been investigated by Density Functional Theory (DFT) calculations. Moreover, two new families of BODIPYs, [3,4]-phenanthro- and [1,2]-naphtho-fused BODIPYs, have been synthesized using this methodology and characterized by nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry, and UV-vis spectroscopy as well as single crystal X-ray diffraction analysis. Finally, the structural and electronic differences between these [b]-aryl-fused BODIPYs are discussed, considering the experimental results and supported by DFT and time-dependent (TD)-DFT calculations.
Results and discussion
Optimization and scope of gold-catalyzed cycloisomerization
The 2,6-bis(2-(propynyl)phenyl)-BODIPY 1a, which was prepared from the 2,6-dibromo derivative by Suzuki cross-coupling (see the ESI†), was chosen as the model substrate to evaluate the gold(I)-catalyzed cycloisomerization for forming the [b]-naphtho-fused BODIPY 2a (Table 1). This study was initiated by testing different transition metals, such as In(III), Pt(II) and Au(III), that have been previously used in this kind of cycloisomerization (Entries 1–3), however, the reaction led to a complex mixture of colored products. Therefore a milder catalyst, such as the well-known PPh3AuCl A was used. A initially failed in showing catalytic activity at reflux in toluene or DCE. Gratifyingly, the in situ formation of a cationic gold(I)-complex using 10% of AgSbF6 as a chloride scavenger dramatically increased the activity of A. In fact, under these conditions, after 25 min at 25 °C, 2a was obtained as a major product in 85% yield (Entry 6). Interestingly, when the perfluorinated catalyst B was employed instead of A, the reaction was quantitatively completed after 15 min (Entry 7). This result can be easily explained considering previous studies by Alcarazo et al. on the influence of ligand π-acidity in the metal-catalyzed cycloisomerization of ortho-alkynylated biaryls, where they revealed that electron-poor ancillary ligands substantially enhance the π-acidity of metals and hence their ability to activate alkynes toward nucleophilic attack, which is the rate-determining step.18 Thus, the commercially available PPhF3AuCl catalyst B was selected for further optimization. Toluene as solvent led to longer reaction times and lower yield, probably due to the poorer stabilization of the charged intermediates formed during the reaction (Entry 8).17 Next, the effect of the counterion was also investigated. Chloride scavengers AgOTf (Entry 9) and AgSbF6 gave similar results in terms of yield, albeit the reaction time was slightly shorter in the latter case, as expected given the lower basic character of SbF6− anions.19 Lowering the catalyst loading to 5 mol% (Entry 10) resulted in incomplete conversion of 1a. As expected, the control experiment (Entry 11) confirms that the cationic gold(I) complex is the catalytically active system.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [M] (10%) | AgX (10%) | Solvent | T (°C) | t (min) | Yield (%) |
| a Yields were determined after purification using a chromatography column. b Complex mixture of colored products. c 5% of catalyst loading. Ar = mesityl-. | ||||||
| 1 | PtCl2 | — | DCE | rt → 85 | 480 | —b |
| 2 | InCl3 | — | DCE | rt → 85 | 1440 | —b |
| 3 | AuCl3 | — | DCE | rt | 1440 | —b |
| 4 | PPh3AuCl (A) | — | DCE | rt → 85 | 1440 | 0 |
| 5 | PPh3AuCl (A) | — | Toluene | rt → 115 | 1440 | 0 |
| 6 | PPh3AuCl (A) | AgSbF6 | DCE | rt | 25 | 85 |
| 7 | PPh F 3 AuCl (B) | AgSbF 6 | DCE | rt | 15 | >95 |
| 8 | PPhF3AuCl (B) | AgSbF6 | Toluene | rt | 480 | 52 |
| 9 | PPhF3AuCl (B) | AgOTf | DCE | rt | 25 | 92 |
| 10 | PPhF3AuCl (B)c | AgSbF6c | DCE | rt | 1440 | 74 |
| 11 | — | AgSbF6 | DCE | rt | 1440 | 0 |
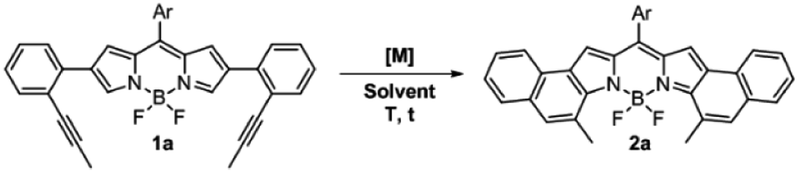
As can be seen from the structure of 2a, the methyl groups at the central benzene ring (Scheme 2, blue benzene) are close and point to the BF2 moiety, so the cycloisomerization could be hindered with bulkier substituents. Moreover, taking the previous work on [b]-naphtho-fused-BODIPYs into account,15 it was expected that the central benzene ring might have a relevant impact on the electronic properties owing to its involvement in both the HOMO and the LUMO of the molecule, unlike the outer benzene ring (Scheme 2, red benzene) which is only involved in the HOMO. For these reasons, we considered it relevant to evaluate the scope of the cycloisomerization with different substituents on the alkyne (Scheme 2).
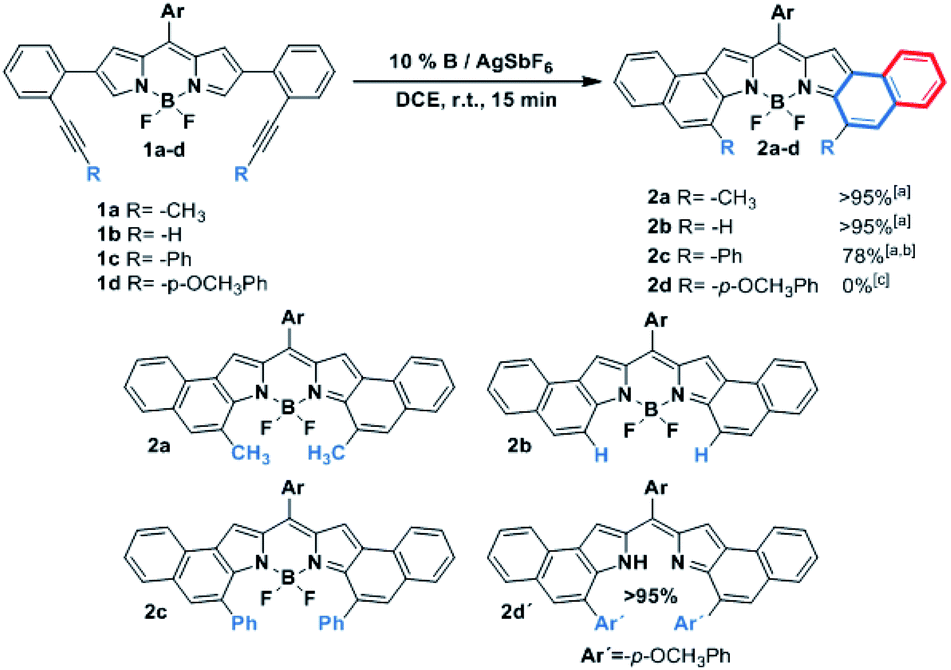 | ||
| Scheme 2 Scope of the cycloisomerization of 1 to form 2 with different substituted alkynes. [a] Yields were determined after purification using a chromatography column. [b] 20% PPh3FAuCl/AgSbF6, 40 °C, DCE and 1 h. [c] not detected, and 2d′ was quantitatively obtained. | ||
Under the optimized conditions, cycloisomerization was first proved with –H as the substituent. As the model reaction, the cyclization product 2b was quantitatively isolated. Subsequently, the reaction with a bulkier group such as phenyl was carried out. Remarkably, in this case 20% of catalyst loading, 1 h and 40 °C were needed for the total disappearance of the starting material, affording 2c in 78% yield. This finding points out to a notable steric hindrance during the cyclization. In the interesting case of para-methoxyphenyl as the substituent, the expected BODIPY 2d was not detected. Instead, the free dipyrromethene 2d′ was the isolated single product. Such results can be rationalized by analysing the X-ray structure of 2d′ (see the ESI†). The steric hindrance between the methoxy moieties results in a slight deviation in the planarity of the dipyrromethene core that enables deborylation (see Fig. S6.1†).
It is important to highlight the pronounced regioselectivity of the reaction. First, only the 6-endo-dig cyclization is detected, over the also conceivable 5-exo-dig mode. These results are in total agreement with other reported cationic gold(I)-catalyzed cycloisomerization reactions,3,5 where total 6-endo-dig selectivity is also observed. In addition, the reaction is completely regioselective to the α-position of the BODIPY over the β-one.
DFT calculations of the reaction mechanism: α-regioselectivity
In order to shed light on the origin of this remarkable regioselectivity, the reaction pathway of the cycloisomerization at both the α- and β-positions was quantitatively explored by means of DFT calculations (see the ESI† for computational details). Fig. 1 depicts the calculated free energy profile of such pathways for the selected model BODIPY (i.e., 2-(2-(propynyl)phenyl)-BODIPY). Furthermore, in order to evaluate the effect of the meso substituent, the free energy profile was computed with both H- and Mes- at the meso position (left and right side of Fig. 1, respectively).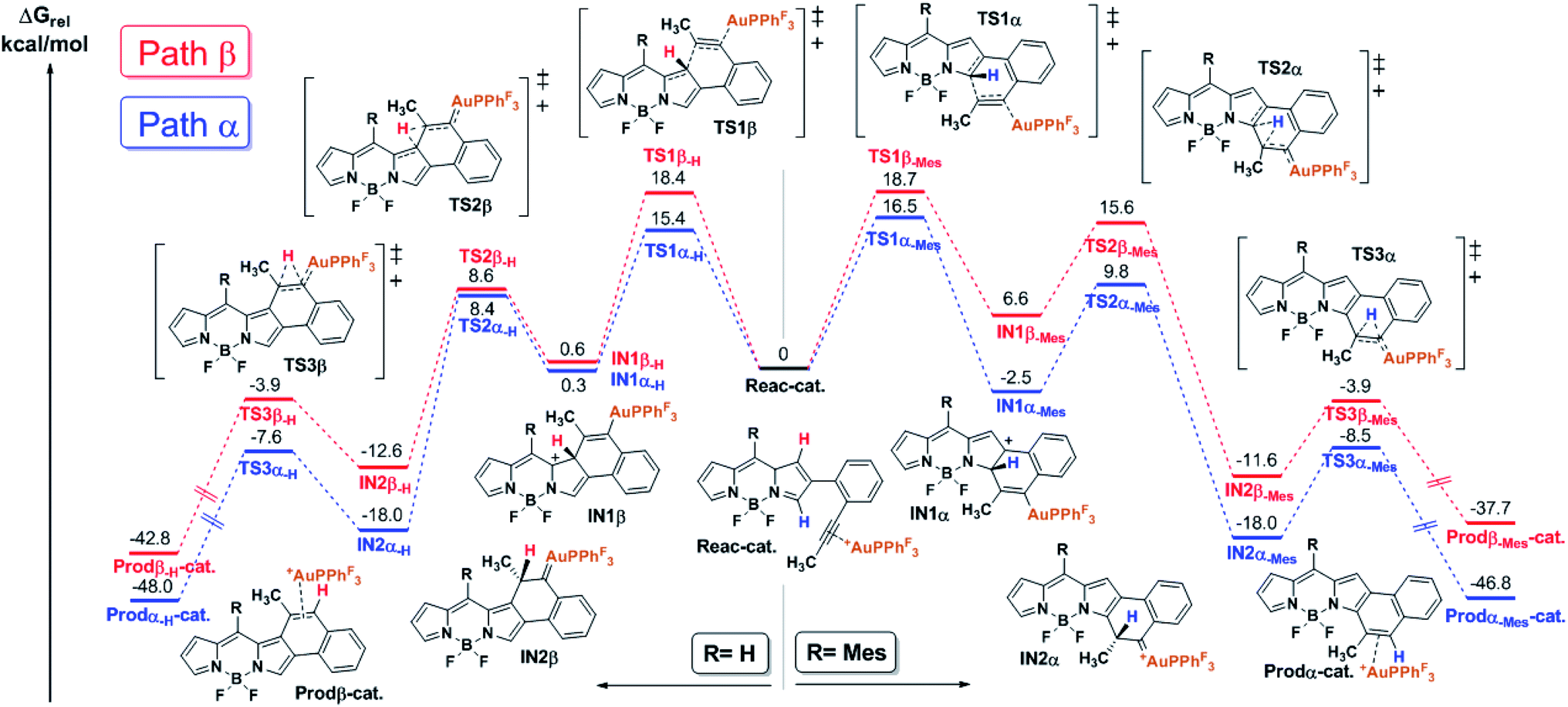 | ||
| Fig. 1 Free energy profiles (kcal mol−1) for the cycloisomerization of 2-(2-(propynyl)phenyl)-BODIPY (model substrate for this study). The red and blue lines represent the calculated profiles for the β- and α-paths, respectively. Such calculated pathways were elucidated with H- (left side) and Mes- (right side) as meso substituents. | ||
The first step of the reaction involves the intramolecular attack of the pyrrole ring by the activated alkyne to form the intermediates IN1α-H/Mes and IN1β-H/Mes through the transition states TS1α-H/Mes and TS1β-H/Mes, respectively. Importantly, TS1α-Mes lies 2.2 kcal mol−1 lower than TS1β-Mes and TS1α-H lies 3.0 kcal mol−1 lower than TS1β-H, suggesting that, regardless of the meso substituent, the nucleophilic attack at the α-position is more favorable. Considering that such an attack is governed by orbital interactions (soft acid and base),2 this result can be rationalized by inspection of the HOMO of the complex Reac-cat (Fig. S6.5†) where, although the α-carbon contributes considerably, the β-carbon is barely involved. Our calculations indicate that this step exhibits the highest energy barrier for both paths and therefore it is expected to have a strong influence on the kinetics of the whole process. Interestingly, if IN1α-Mes is 9.1 kcal mol−1 more stable than IN1β-Mes then the α-attack is exergonic (−2.5 kcal mol−1) while the β-attack is endergonic (6.6 kcal mol−1). However, in the case of IN1α/β-H, both intermediates have similar energy and both processes are slightly endergonic. The next step along the reaction coordinates involves a 1,2-H shift through TS2α/β-H/Mes that does not depend much on the path considered and leads to intermediates IN2α/β-H and IN2α/β-Mes which are respectively 20–26 kcal mol−1 and 15–18 kcal mol−1 more stable than their precursors, and the energies of the following transition states TS3α/β-H/Mes are much lower than those of TS2α/β-H/Mes. Thus, the formation of Au-carbenes IN2α/β-H/Mes is likely to be irreversible. Finally, a second very exergonic 1,2-H shift that requires moderate activation (barriers of 8–11 kcal mol−1) and is not affected by the path or substituent considered, leads to naphtho-fused-BODIPY-Au π-complexes Prodα/β-H/Mes-cat, which after dissociation regenerate the catalyst and deliver the products.
With these result in hand, it can be concluded that, due to the electronic differences between the α- and β-carbons, the nucleophilic attack (rate-determining step) is kinetically favored in the case of the α-cyclization and does not arise from steric interactions with the meso substituent. In the case of Mes-at the meso position, the β-attack is thermodynamically unfavored and such an equilibrium should be shifted to the starting materials which, in turn, evolve into the thermodynamically favored IN1α-Mes. In summary, the α-fused BODIPY is both the kinetic and thermodynamic product, resulting in a theoretical regioselectivity which is in good agreement with the experimentally observed selectivity.
Synthesis of [3,4]phenanthro- and [1,2]naphtho-fused BODIPYs
In order to assess the versatility of our methodology, the synthesis of other fused-BODIPY families was targeted. Firstly, we expanded further the conjugation of 2a by attaching another benzene ring at the [h]-bond of the naphthalene fragment. To this end, the precursor 1e was assembled similarly by Suzuki cross-coupling (see ESI†) and then, was subjected to cycloisomerization under the previously optimized conditions (Scheme 3a). The [3,4]phenanthro-fused BODIPY 2e was isolated also in a quantitative yield, being the first example reported of this highly conjugated family. Moreover, 2e comprises the highest number of consecutively fused benzene rings found in a BODIPY derivative. Interestingly, despite the higher steric hindrance expected during the cyclization compared with 2a, the activity of our catalytic system did not decrease. Additionally, the mono-fused counterpart of 2e and 2f was successfully prepared from 1f following a similar synthetic route (Scheme 3a). | ||
| Scheme 3 Synthesis of (a) [3,4]phenanthro-fused-BODIPYs 2e and 2f, and (b) [1,2]naphtho-fused-BODIPY 2g. [a] Yields were determined after purification using a chromatography column. | ||
Next, we envisioned that the gold(I)-catalyzed cyclization of 1g would afford the [1,2]naphtho-fused-BODIPY 2g (Scheme 3b), which is the isomer of 2a. Surprisingly, this family of BODIPY has remained unreported to date. Initial efforts by the Burgess group yielded only the mono-[1,2]naphtho-fused BODIPY after oxidation of the hydrogenated counterpart with DDQ.20 Semiempirical calculations suggested that the second oxidation is hindered by steric interactions. More recently, a mono-[1,2]naphtho-fused BODIPY was serendipitously reported by Wu et al. in an attempt to prepare [2,1]naphtho BODIPYs via the Scholl reaction.21 Again, the [1,2]-fusion at both pyrrole rings was not achieved. The precursor 1g was prepared also by Suzuki cross-coupling (see the ESI†) using the reported 3,5-dichloro-BODIPY as the starting material. Finally, the cycloisomerization of 1g was carried out leading to 2g in a quantitative yield. As expected, the reaction time is shorter than that in the case of 2a (5 min vs. 15 min). This result can be rationalized considering that the 2,6-carbons are better nucleophiles than 3,5-carbons, which usually behave as electrophiles.7 It should be noted that the synthesis of three different [b]-aryl-fused BODIPYs was readily achieved by just changing the position or the design of the boronic acid.
Characterization and theoretical studies of [1,2], and [2,1]naphtho- and [3,4]phenanthro-fused BODIPYs
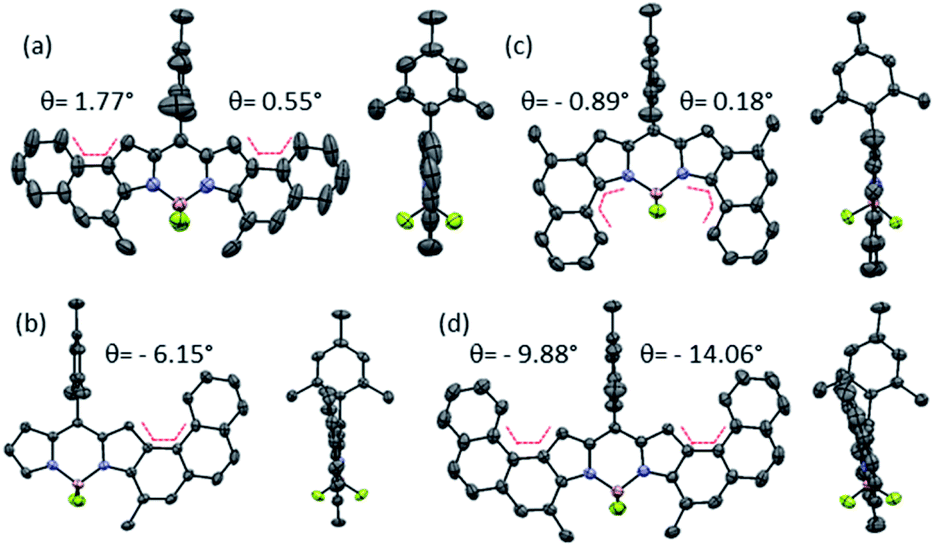 | ||
| Fig. 2 Front (right) and side (left) view X-ray structures of (a) 2a, (b) 2f, (c) 2g, and (d) the most tensioned unit of 2e in the unit cell. Atom's colour code: carbon in grey, nitrogen in blue, iodine in violet, boron in pink, fluorine in green and the angle θ in red. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). 2b and 2c display similar absorption spectra also reaching the maximum at 650 nm. Interestingly, 2e exhibits a Q band bathochromically shifted only by 10 nm relative to that of 2a. From these results, it can be concluded that the absorption maxima of these derivatives are not significantly affected by substitution of the central or outer benzene rings. It is worth noting that the absorption profile of 2g is substantially different from that of its isomer 2a. In this case, the absorption spectrum presents three intense bands at 683, 632 and 548 nm which do not show a clear different trend as a function of the solvent polarity. Moreover, it should be noted that the molar absorption coefficient of these bands (50000, 32000 and 25000 M−1 cm−1, respectively) decreases drastically compared to 2a, which may be attributed to the decreased transition dipole moment, which in the case of BODIPYs, is polarized along the longer molecular axis.24 Considering the emission properties, the quantum yields of these systems fall below 0.1 as a result of the [b]-ring fusion.15
000 M−1 cm−1). 2b and 2c display similar absorption spectra also reaching the maximum at 650 nm. Interestingly, 2e exhibits a Q band bathochromically shifted only by 10 nm relative to that of 2a. From these results, it can be concluded that the absorption maxima of these derivatives are not significantly affected by substitution of the central or outer benzene rings. It is worth noting that the absorption profile of 2g is substantially different from that of its isomer 2a. In this case, the absorption spectrum presents three intense bands at 683, 632 and 548 nm which do not show a clear different trend as a function of the solvent polarity. Moreover, it should be noted that the molar absorption coefficient of these bands (50000, 32000 and 25000 M−1 cm−1, respectively) decreases drastically compared to 2a, which may be attributed to the decreased transition dipole moment, which in the case of BODIPYs, is polarized along the longer molecular axis.24 Considering the emission properties, the quantum yields of these systems fall below 0.1 as a result of the [b]-ring fusion.15
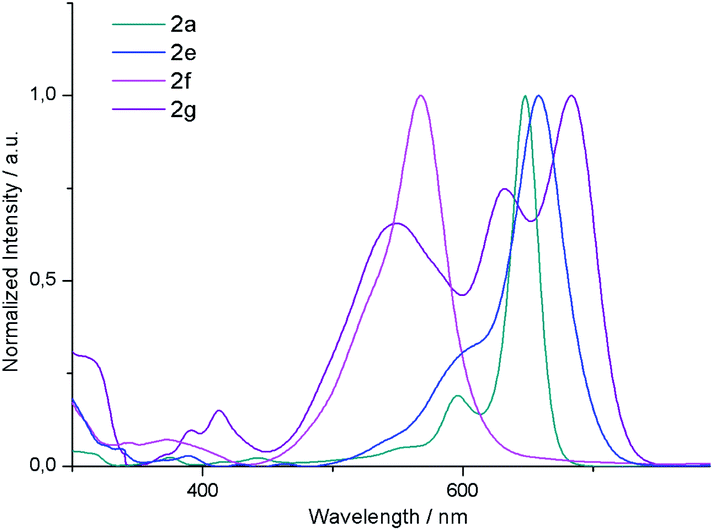 | ||
| Fig. 3 UV/Vis absorption spectra of 2a, 2e, 2f and 2g in dichloromethane. | ||
In order to gain further insight into the electronic structure, density functional theory (DFT) calculations were conducted at the B3LYP/6-31G(d) level (see the ESI† for more details). As can be seen from Fig. 4, the HOMO and LUMO of 2a, 2b and 2f are extended over the fused benzene rings. Consequently, these derivatives exhibit lower HOMO–LUMO band gaps compared to their unsubstituted counterpart arising from both the stabilization of the LUMO and the destabilization of the HOMO, as expected, given the α ring fusion.13,15 Remarkably, while the central benzene rings of 2a are involved in both the HOMO and the LUMO delocalization, the outer benzene ring only significantly affects the HOMO. This fact would explain the slight bathochromic shift of the maximum absorption observed in 2e relative to 2a.
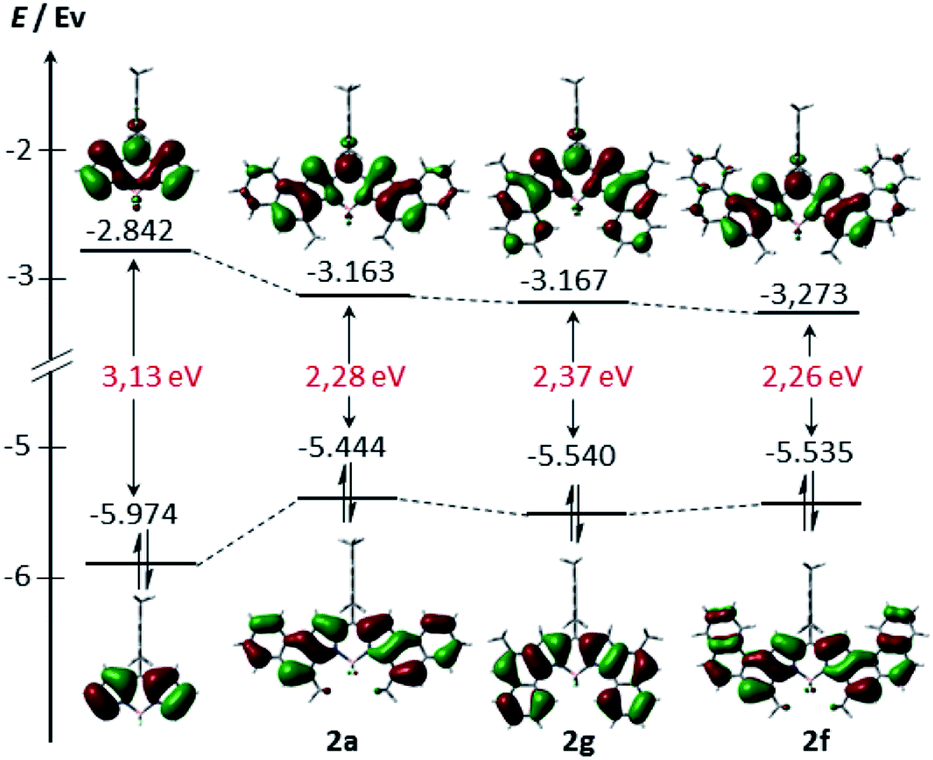 | ||
| Fig. 4 Energy diagrams and Kohn–Sham orbital representations of the HOMO and LUMO of unsubstituted BODIPY 2a, 2f and 2g. All calculations were performed at the B3LYP/6-31G level. | ||
In addition, nucleus-independent chemical shift (NICS(0)) values at the B3LYP/6-31G(d,p)//B3LYP/6-31G(d) level were calculated to evaluate the ring current effect of these systems (Fig. S6.2†). For the benzene rings directly linked to pyrroles (Scheme 2, blue benzene), NICS(0) values are −3.0, −3.1 and −2.5 in 2a, 2e and 2g, respectively. These values are substantially lower than those reached in the benzo[e]indole, naphtho[1,2-e]indole and benzo[g]indole (−8.5, −8.2 and −8.5, respectively), indicating a higher double-bond character of the K-region of 2a, 2e and 2g. Interestingly, all the NICS(0) values of the aromatic rings that contain the BODIPY core of 2g, are lower than those of 2a (see Fig. SX†). In line with the spectroscopic results, this result suggests different π-conjugation in 2a compared to its isomer 2g.
Time-dependent (TD) calculations at the PCM(CH2Cl2)-TD-BMK/6-31G(d,p)//B3LYP/6-31G(d) level were carried out to assign the nature of the absorptions observed in the UV-vis spectra of 2a, 2e, and 2g.25 Selected TD-DFT transition energies, oscillator strengths (f) and molecular orbital configurations are shown in Table 2. With regard to the calculated spectra of 2a and 2e, only one band corresponding to the HOMO–LUMO transition is observed. Moreover, the calculations predict a 10 nm bathochromic shift of 2e relative to 2a, which is in good agreement with the experimental result. In the case of 2g, the spectrum displays three intense bands with oscillator strength values of 0.59, 0.46 and 0.32, which well reproduced the shape of the experimental spectra (see S6.3†). The lowest-energy absorption band is mainly derived also from the HOMO–LUMO transition, while the second one arises from the HOMO−1–LUMO transition. Interestingly, the highest-energy band arises from a charge-transfer transition between the meso-mesityl ring (HOMO−5) and the BODIPY core (LUMO) orbitals, which are minimally overlapped through the mesityl moiety (see S6.4†). This unprecedented finding renders the [1,2]naphtho-fusion a novel and powerful strategy to achieve a wide absorption in the visible range with BODIPY derivatives.
Conclusions
In this work, we have reported the efficient synthesis of β-naphtho- and phenanthro-fused-BODIPYs by means of gold(I)-catalyzed cycloisomerization as a key step, being the first example of functionalization of a BODIPY using this kind of catalysis. We found that the catalytic system PPhF3AuCl/AgSbF6 enabled the synthesis of 2a in a quantitative yield at room temperature. The reaction is compatible with other substituents on the alkyne such as -H or –Ph; however, in the case of bulkier p-methoxyphenyl BF2 decomplexation takes place as a result of steric interactions. The reaction is totally regioselective toward the 6-endo-dig and α-cyclization product. The origin of the α-selectivity was studied by DFT calculations and it can be concluded that it arises from (a) the higher nucleophilicity of the α-carbon, which lowers the energy of the nucleophilic attack (rate-determining step), (b) the higher stability of all intermediates and transition states involved in the α-path and (c) in the particular case of Mes- as a substituent, a shifted equilibrium to the starting materials in the β-attack. The synthesis of other β-aryl-fused-BODIPYs, [3,4]phenanthro- (2e and 2f) and [1,2]naphtho-fused (2e), was also successfully achieved using this methodology just by changing the position or the design of the ortho-alkynyl-aryl moiety. Their molecular and electronic structures were compared by NMR, UV-vis and single-crystal X-ray diffraction analysis. With regard to the X-ray structures, 2a, 2e and 2g present significant crystallographic differences. While 2a and 2g display a quasi-planar π-skeleton, 2e shows a slightly curved π-skeleton owing to its [4]-helicene character. In addition, 2a and 2e exhibit a unidirectional packing based on π-stacking interactions, which is not favoured in the case of 2g. The UV/vis spectra of 2a–2e exhibit an intense peak (ε = 100000–200000 M−1 cm−1) at 650–660 nm. In contrast, 2g displays three intense absorption bands in the range of 450 to 750 nm. Time-dependent DFT calculations revealed that these transitions arise from the π–π* transition from the HOMO and the HOMO−1 to LUMO and a charge transfer transition from the meso-mesityl group (HOMO−5) and LUMO. NICS(0) values also indicate a different π-conjugation in 2a and 2g, as revealed by the less aromatic character of each benzene ring of 2g.
Given the reactivity of the pyrrole ring of the BODIPY towards Au-activated alkynes, this work serves as a proof of concept to explore other gold(I)-catalyzed reactions to functionalize BODIPY or other pyrrole-based porphyrinoids. In this sense, further transformations are being investigated in our laboratories.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from Spanish MINECO (CTQ2017-85393-P) is acknowledged. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686). J. L. acknowledges MECD, Spain, for a F.P.U. fellowship. The European Commission under the Marie Sklodowska-Curie Action Cofund 2015 (EU project 713366-InterTalentum) is acknowledged for the support for G.D.S. Generous allocation of computational time from the Centro de Computación Científica UAM is gratefully acknowledged.Notes and references
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed.
- (a) E. Gonzalez-Fernandez, L. D. M. Nicholls, L. D. Schaaf, C. Fares, C. W. Lehmann and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 1428 CrossRef CAS PubMed; (b) M. Satoh, Y. Shibata and K. Tanaka, Chem.–Eur. J., 2018, 24, 5434 CrossRef CAS PubMed.
- (a) R. Zuzak, R. Dorel, M. Kolmer, M. Szymonski, S. Godlewski and A. M. Echavarren, Angew. Chem., Int. Ed., 2018, 57, 10500 CrossRef CAS PubMed; (b) R. Dorel, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 55, 11120 CrossRef CAS PubMed.
- (a) M. Tanaka, Y. Shibata, K. Nakamura, K. Teraoka, H. Uekusa, K. Nakazono, T. Takata and K. Tanaka, Chem.–Eur. J., 2016, 22, 9537 CrossRef CAS PubMed; (b) B. Alcaide, P. Almendros, C. Aragoncillo, E. Busto, C. G. Lopez-Calixto, M. Liras, V. A. de la Pena O'Shea, A. Garcia-Sanchez and H. V. Stone, Chem.–Eur. J., 2018, 24, 7620 CrossRef CAS PubMed.
- (a) M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297 CrossRef CAS PubMed; (b) R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517 CrossRef CAS PubMed; (c) V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369 RSC; (d) M. Urbani, G. de la Torre, M. K. Nazeeruddin and T. Torres, Chem. Soc. Rev., 2019, 48, 2738 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- J. J. Chen, S. M. Conron, P. Erwin, M. Dimitriou, K. McAlahney and M. E. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 662 CrossRef CAS PubMed.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- B. M. Squeo and M. Pasini, Supramol. Chem., 2020, 32, 56–70 CrossRef CAS.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953 RSC.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- A. Wakamiya, T. Murakami and S. Yamaguchi, Chem. Sci., 2013, 4, 1002 RSC.
- (a) Y. Hayashi, N. Obata, M. Tamaru, S. Yamaguchi, Y. Matsuo, A. Saeki, S. Seki, Y. Kureishi, S. Saito, S. Yamaguchi and H. Shinokubo, Org. Lett., 2012, 14, 866 CrossRef CAS PubMed; (b) W. Miao, Y. Feng, Q. Wu, W. Sheng, M. Li, Q. Liu, E. Hao and L. Jiao, J. Org. Chem., 2019, 84, 9693–9704 CrossRef CAS PubMed; (c) H. Ito, H. Sakai, Y. Suzuki, J. Kawamata and T. Hasobe, Chem.–Eur. J., 2020, 26, 316–325 CrossRef CAS PubMed.
- Z. Zhou, J. Zhou, L. Gai, A. Yuan and Z. Shen, Chem. Commun., 2017, 53, 6621 RSC.
- X. Yang, L. Jiang, M. Yang, H. Zhang, J. Lan, F. Zhou, X. Chen, D. Wu and J. You, J. Org. Chem., 2018, 83, 9538 CrossRef CAS PubMed.
- (a) A. Fürstner and V. Mamane, J. Org. Chem., 2002, 67, 6264 CrossRef PubMed; (b) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556 CrossRef CAS PubMed; (c) A. Fürstner and V. Mamane, Chem. Commun., 2003, 2112 RSC; (d) A. Fürstner and J. W. J. Kennedy, Chem.–Eur. J., 2006, 12, 7398 CrossRef PubMed.
- (a) J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753 CrossRef CAS PubMed; (b) J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuskova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815 CrossRef CAS PubMed.
- A. Homs, C. Obradors, D. Leboeuf and A. M. Echavarren, Adv. Synth. Catal., 2014, 356, 221 CrossRef CAS PubMed.
- J. Chen, A. Burghart, A. Derecskei-Kovacs and K. Burgess, J. Org. Chem., 2000, 65, 2900 CrossRef CAS PubMed.
- M. H. Chua, K. Huang, J. Xu and J. Wu, Org. Lett., 2015, 17, 4168 CrossRef CAS PubMed.
- G. W. Gribble and W. J. Kelly, Tetrahedron Lett., 1985, 26, 3779 CrossRef CAS.
- Z. Yao, J. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7 CrossRef CAS.
- J. Karolin, L. B. A. Johansson, L. Strandberg and T. Ny, J. Am. Chem. Soc., 1994, 116, 7801 CrossRef CAS.
- It should be noted that there is an intrinsic error in the calculation of vertical absorption energy of BODIPY derivatives. See: M. R. Momeni and A. Brown, J. Chem. Theory Comput., 2015, 11, 2619 CrossRef CAS PubMed and references therein. However, the goal of this analysis is to understand the transitions observed in our derivatives (see Fig. S6.2†).
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for synthesis, characterization data, and supplementary figures. CCDC 1985328–1985338. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01054e |
| This journal is © The Royal Society of Chemistry 2020 |