
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in metal-free aerobic C–H activation
André
Shamsabadi
 and
Vijay
Chudasama
*
and
Vijay
Chudasama
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk
First published on 13th February 2019
Abstract
Herein we describe recent developments in selective, metal-free, dioxygen-induced C–H activation. This method of activating C–H bonds is an attractive alternative to traditional methodologies as it uses dioxygen, an inherently sustainable and widely accessible oxidant, in place of expensive or toxic metals and/or hazardous peroxides. Reactions developed on the basis of using aerobic C–H activation are also discussed.
 André Shamsabadi | André Shamsabadi was born in London, UK. He received his undergraduate education at University College London (UCL) during which he was awarded the Jackson-Lewis Scholarship (2015), C. K. Ingold Prize (2015), Franz Sondheimer Prize (2016) and was a Dean's List commendee. In 2016, he was awarded a UCL Graduate School Research Scholarship, which allowed him to continue in a postgraduate position at UCL under the guidance of Dr Vijay Chudasama. His research interests are based in radical organic synthesis and aerobic C–H activation. |
 Vijay Chudasama | Dr Vijay Chudasama obtained his MSci degree and PhD from University College London in 2008 and 2011, respectively. Following post-doctoral studies under the supervision of Prof. Stephen Caddick, Vijay obtained a Ramsay Memorial Fellowship. During this time, he was made Technical Director of a biotechnology spin-out (ThioLogics™). In April 2015, he was awarded a lectureship (Reader from October 2017) at UCL for him to focus on the research areas of aerobic C–H activation and various aspects of Chemical Biology. Vijay's research has recently been highlighted by Forbes, Scientific American, CNN News, Nature Chemistry and the Royal Society of Chemistry. |
C–H bond activation
Direct C–H bond activation has long been credited as the ‘holy grail’ of organic chemistry.1 The concept of discriminatively activating what is typically an inert C–H bond is a fundamentally powerful tool in synthesis for a number of reasons: (i) C–H bonds are ubiquitous in organic molecules, (ii) C–H bond functionalisation typically reduces the amount of transformations in multi-step syntheses which can consist of long and laborious protection–deprotection sequences, (iii) it establishes an ideal transformation regarding atom economy and waste minimisation when compared to classic cross-coupling reactions (e.g. Negishi coupling, Suzuki coupling, etc.). Over the past few decades, great strides have been made in the development of elegant C–H bond activation methodologies which has seen the process turn from being an aspirational idea to a commonplace reaction that is now heavily studied2 and used in both academia and industry.3,4 Modern C–H functionalisation reactions are carried out through (i) C–H bond homolysis via use of radical initiators followed by radical functionalisation, (ii) C–H insertion through use of carbenes or nitrenes, or (iii) metallic C–H activation via a C–metal intermediate by way of σ-bond metathesis, concerted metalation deprotonation, and oxidative addition. Unfortunately, each protocol commonly utilises harsh reagents or additives to activate what is otherwise an unreactive C–H bond. Owing to the ever-increasing popularity and widespread usage of C–H activation processes, there is significant pressure to improve the sustainability of C–H reactions from an environmental standpoint: radical initiators are generally toxic or shock sensitive, nitrenes and carbenes are high-energy materials that require intricate and heavily-monitored reaction conditions and organometallic C–H bond activation often requires the use of precious metals and stoichiometric amounts of oxidants (i.e. MnO2). Indeed, it has been highlighted recently by the ACS Green Chemistry Institute® that C–H activation processes utilising green oxidants whilst giving predictable site-selectivities is one of the top three research areas that would benefit from improvements in process ‘greenness’.5 To achieve this, chemists have created increasingly elegant and boundary-pushing ideas for organometallic C–H bond activation including catalysis by Earth-abundant metals (e.g. iron,6 copper7) or forgoing the use of metals altogether.8 In the goal for effectively using greener and milder oxidants, recent advances have been achieved in using arguably the most sustainable oxidant: dioxygen. Dioxygen is abundant; indeed, molecular dioxygen makes up 20.94% of the Earth's atmosphere, and one of its most fascinating and under-exploited uses is its ability to initiate auto-oxidation (alternatively referred to as autoxidation) processes. Whilst process chemists have previously utilised an essentially free reagent (air) in the homogenous catalytic oxidation of organic and inorganic compounds, the use of dioxygen has very rarely been used as the focal point for C–H bond activation.Dioxygen-initiated auto-oxidation
The auto-oxidation process describes the auto-catalytic oxidation of organic or inorganic systems. For organic molecules, it is usually used to describe the propagation reactions in a chain reaction cycle in which carbon radicals couple with dioxygen to form peroxy radical species, which in turn can then abstract H-atoms to form a peroxide product and regenerate further carbon radicals (Scheme 1). However, in these radical chain reactions, use of a radical initiator or UV light is commonly employed to initially activate a C–H bond. A rare but intriguing discussion regarding auto-oxidation is the observation that molecular dioxygen can directly interact with certain C–H bonds to generate carbon radicals without the apparent use of any additional initiator species. This allows for the possibility of C–H functionalisation through reaction of the transiently-formed radical species.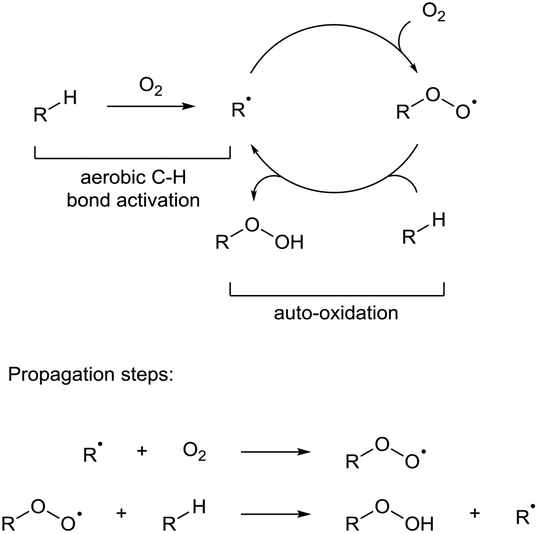 | ||
| Scheme 1 General dioxygen-initiated autoxidation pathway. | ||
This review highlights recent examples of ground-breaking research in which dioxygen is used to activate C–H bonds as a means to achieve green and sustainable C–H bond functionalisation (Scheme 2). Historically, aerobic C–H bond activation has been scarcely used in synthesis outside of simple C–H oxidation reactions,9 but its true potential in synthesis is starting to be uncovered with an increasing number of research groups utilising dioxygen as a radical initiator. It should be highlighted that different research groups have had differing logistic protocols of introducing dioxygen in their procedures: use of a pressurised environment of O2 through use of an autoclave (>1 atm), use of a dioxygen atmosphere via balloon (1 atm), bubbling compressed air into a reaction mixture under an inert environment, or by simple exposure of the reaction to the dioxygen in air (referred to as atmospheric dioxygen). We believe that, where possible, the strive should be made to use atmospheric dioxygen as the source of dioxygen as it represents the greenest and a freely accessible form of the oxidant to be utilised in synthesis.
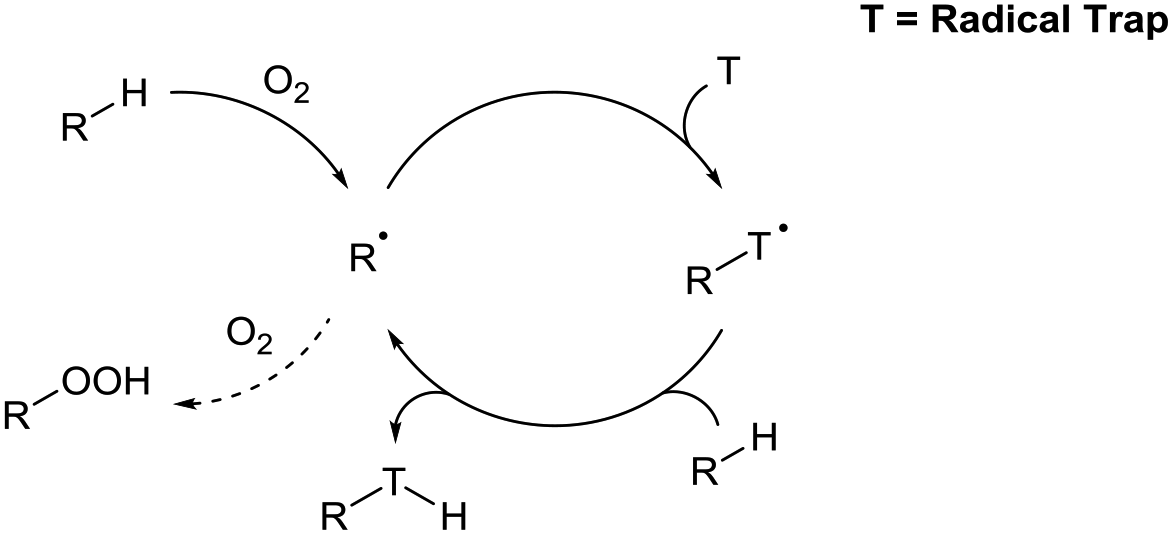 | ||
| Scheme 2 Model example of aerobic C–H bond activation via dioxygen-induced radical initiation through trapping of initial radical adduct species. | ||
Modern dioxygen-induced C–H bond functionalisation reactions have utilised aldehydes, ethers, benzylamines and glycine derivatives as precursors to aerobic activation, and reactions developed on this basis are described within.
Aldehydes
Aldehydes are amongst the most desirable substrates to achieve C–H functionalisation owing to ubiquity of the acyl group in molecular architecture. Radical C–H functionalisation is particularly useful for this purpose due to the susceptibility to hydrogen abstraction of aldehydic C–H bonds. Aldehydes are one of the most classical and studied examples of a reagent class that undergoes auto-oxidation.10,11 The mechanism for aldehyde auto-oxidation is identical to that shown above (Scheme 1) except that the peracyl product can undergo further reaction with another molecule of aldehyde to form an intermediate that breaks down into two molecules of carboxylic acid as the final product.11 The immediate species formed through aldehyde auto-oxidation is the acyl radical which is nucleophilic in nature and can efficiently participate in radical addition to electron-deficient unsaturated bonds.Since 2009, hydroacylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and NN bonds utilising acyl radicals formed through the exposure of aldehydes 1 to atmospheric dioxygen has allowed the green and efficient formation of various unsymmetrical ketones and acyl hydrazide moieties in good to excellent yields.12 Caddick and co-workers reported the reaction of aldehydes with several vinyl sulfonates 2 under atmospheric conditions to afford unsymmetrical ketones 3 in respectable yields (ca. 70%) and all was reported at room temperature (Scheme 3).13 Optimal yield was achieved when utilising 5 equivalents of aldehyde, which is very competitive with regards to existing hydroacylation methodologies. The reaction was completely inhibited when conducted in the presence of radical inhibitor BHT (2,6-di-tert-butyl-4-methylphenol), providing evidence of a radical mechanism. Whilst the initial account reported 1,4-dioxane as an optimal solvent, further research from the group found that the use of water as a solvent, the ideal “green” solvent, produced similar if not better yields of the ketones.14
C and NN bonds utilising acyl radicals formed through the exposure of aldehydes 1 to atmospheric dioxygen has allowed the green and efficient formation of various unsymmetrical ketones and acyl hydrazide moieties in good to excellent yields.12 Caddick and co-workers reported the reaction of aldehydes with several vinyl sulfonates 2 under atmospheric conditions to afford unsymmetrical ketones 3 in respectable yields (ca. 70%) and all was reported at room temperature (Scheme 3).13 Optimal yield was achieved when utilising 5 equivalents of aldehyde, which is very competitive with regards to existing hydroacylation methodologies. The reaction was completely inhibited when conducted in the presence of radical inhibitor BHT (2,6-di-tert-butyl-4-methylphenol), providing evidence of a radical mechanism. Whilst the initial account reported 1,4-dioxane as an optimal solvent, further research from the group found that the use of water as a solvent, the ideal “green” solvent, produced similar if not better yields of the ketones.14
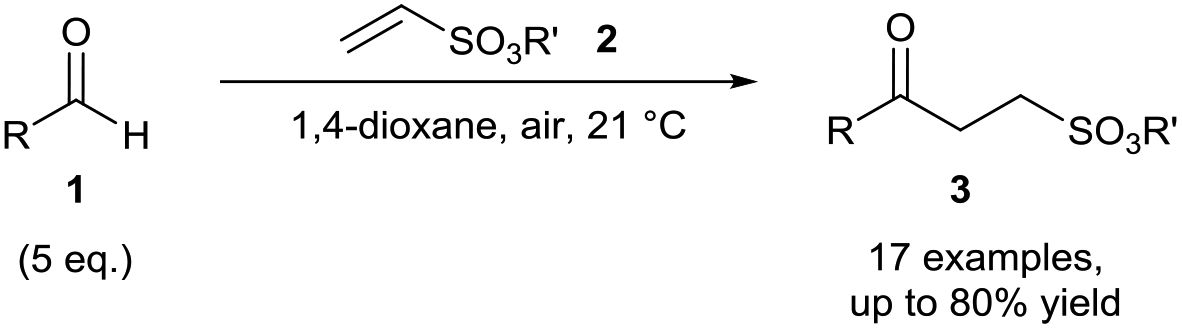 | ||
| Scheme 3 Hydroacylation of vinyl sulfonates 2via aerobic C–H activation. | ||
The Chudasama and Caddick groups then further utilised aerobically-generated acyl radicals to achieve hydroacylation of various vinyl sulfones 4. Here, aliphatic aldehydes were found to perform best in the reaction with vinyl sulfones to achieve consistent yields (ca. 60%) of the desired ketone product 5 (Scheme 4) at 21 °C using water as the reaction solvent.
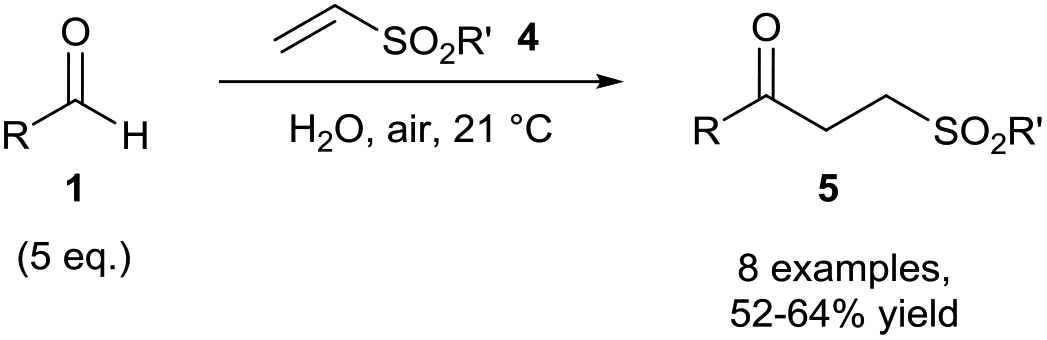 | ||
| Scheme 4 Hydroacylation of vinyl sulfones 4via aerobic C–H activation. | ||
Chudasama et al. then expanded the scope of this hydroacylation methodology to include various unsaturated esters 6–8 where aliphatic aldehydes were once again found to be the best performers, affording yields of 9–11 of up to 89% (Scheme 5). This work paved the way for expanding the scope of radical acceptors that were applicable for use in this methodology.15 It should be noted that efficient reaction conversion was achieved through raising the reaction temperature to 60 °C. It was postulated that the increase in temperature resulted in a lowering of dissolved oxygen in the reaction mixture, combating over-oxidation of the acyl radical (auto-oxidation) and increasing conversion of alkenes 6–8 to the desired ketone products 9–11.
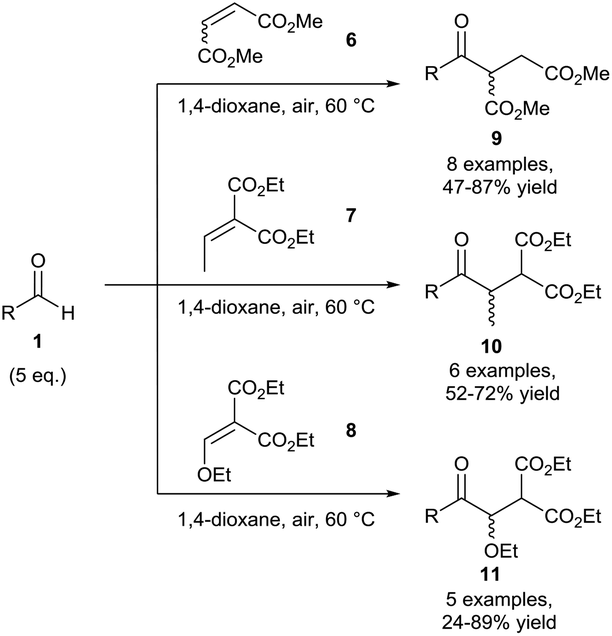 | ||
| Scheme 5 Hydroacylation of unsaturated esters 6–8via aerobic C–H activation. | ||
Chudasama et al. then extended the scope of the developed protocol to include vinyl phosphonates 12, which were successfully utilised as acyl radical acceptors (Scheme 6). A reaction temperature of 60 °C was again used to achieve efficient conversion of the alkene.16 The addition of a dioxane radical to a vinyl phosphonate was also observed when this protocol was used, providing supporting evidence of radicals being formed in the reaction mixture.
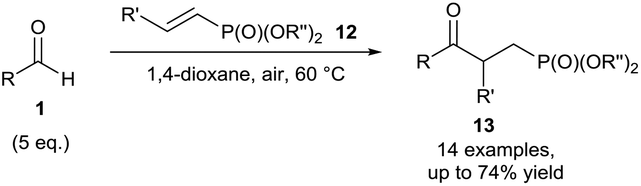 | ||
| Scheme 6 Hydroacylation of vinyl phosphonates 12via aerobic C–H activation. | ||
In 2011, using a similar aerobic activation protocol, acyl hydrazides 15 were synthesised in a highly efficient manner when using azodicarboxylates 14 as radical acceptors (Scheme 7). Acyl hydrazides are extremely useful synthetic intermediates which have found use in the formation of many medicinally desirable entities (amides, indazoles, hydroxamic acids, etc.).17–22 Both diethyl azodicarboxylate (DEAD) and diisopropyl azodicarboxylate (DIAD) were shown to be compatible with both aliphatic and aromatic aldehydes towards hydroacylation via aerobic C–H bond activation. The efficiency of the reaction was clearly demonstrated with excellent yields being attained even when utilising aldehydes as the limiting reagent.23 Chudasama et al. also showed that α-chiral aldehydes could undergo aerobic hydroacylation with azodicarboxylates and vinyl sulfonates with complete retention of optical purity. These transformations represented the first examples of hydroacylation achieved using enantio-enriched aldehydes with retention of enantiomeric excess.
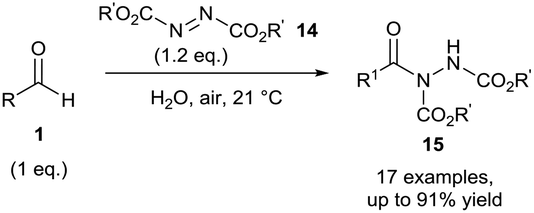 | ||
| Scheme 7 Hydroacylation of azodicarboxylates 14via aerobic C–H activation. | ||
In 2015, Guin and co-workers conducted reactions under a dioxygen environment through use of a dioxygen balloon as an alternative method to achieve aldehydic C–H bond activation. The group utilised a high reaction temperature (115 °C) as means of accessing alkyl radicals 17 through facile decarboxylation of initially formed secondary and tertiary acyl radicals 16. In effect, the group achieved efficient ortho-alkylation on a plethora of acidified heteroaromatic bases 18 (Scheme 8).24 Regrettably, a large excess of aldehyde had to be utilised for optimal yield (6–20 equivalents). It is important to note that a moderate yield† of desired product 19 was observed when the reaction mixture was exposed to air as opposed to use of an oxygen balloon.
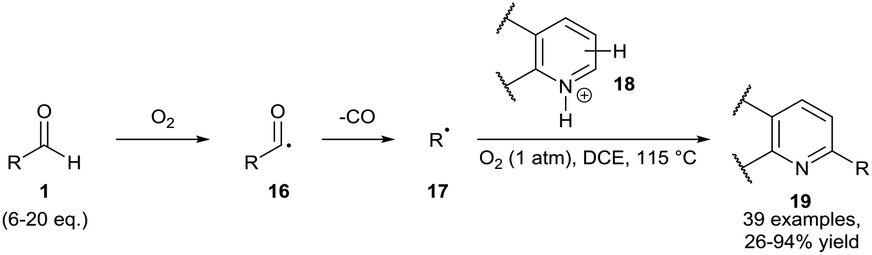 | ||
| Scheme 8 Dioxygen-mediated decarbonylative C–H alkylation of protonated heteroaromatic bases 18 with aldehydes 1. | ||
Guin and co-workers have since expanded their methodology to generate alkyl radicals 17 to achieve dicarbofunctionalisation of alkenes 18 (Scheme 9).25 As before, the methodology required a relatively high equivalence of aldehyde 1 (6–12 equivalents). Moderate yields where observed when secondary or tertiary aldehydes were utilised (ca. 60%) with poor yields only observed when a primary aldehyde was used (25%). When the reaction was repeated under an atmosphere of argon, only a trace amount of desired product 24 was observed, highlighting the importance of dioxygen for the reaction. Furthermore, no product formation was observed when the reaction was carried out in the presence of radical inhibitor TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl), with the alkyl-TEMPO product being detected by GCMS and HRMS, thereby corroborating the radical mechanism for the reaction. It is important to note that whilst the optimised methodology utilises a dioxygen atmosphere, it is stated that the reaction can be carried out in air albeit with a slightly lower yield of desired product.†
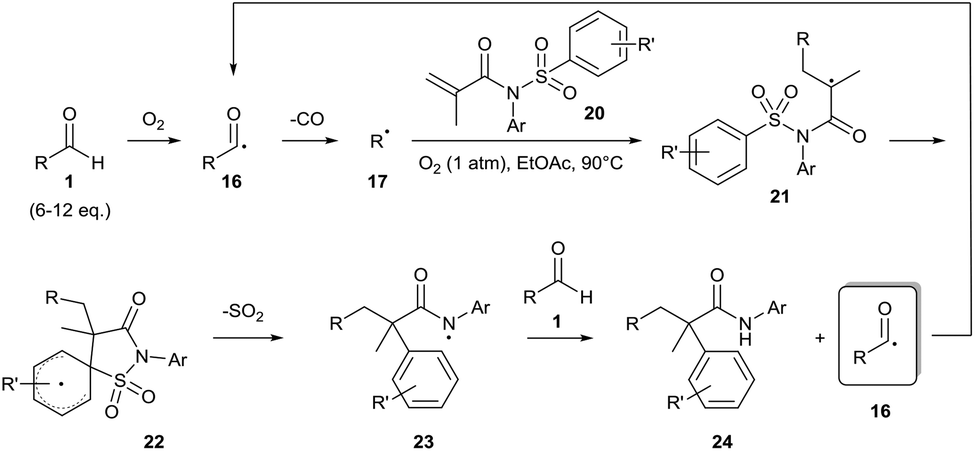 | ||
| Scheme 9 Dioxygen-mediated radical dicarbofunctionalisation of alkene 20 with aldehyde 1. R′ can be a variety of functional groups (e.g. alkyl, amide, nitro, halo, etc.). | ||
Guin and co-workers have also used their procedure to describe the oxidative 1,2-alkylarylation of α,β-unsaturated amides 25 for the synthesis of biologically-active oxindoles 28 (Scheme 10).26 A large excess of aldehyde had to be utilised (6–12 equivalents), which is perhaps unexpected as in the proposed radical chain process an acyl radical is generated as reaction byproduct. As before, complete radical inhibition was observed when conducted in the presence of TEMPO. Nonetheless, good to excellent yields of product 28 (44–81% yield) were achieved when utilising secondary or tertiary aldehydes, with the only poor yield (15%) observed when using butyraldehyde.
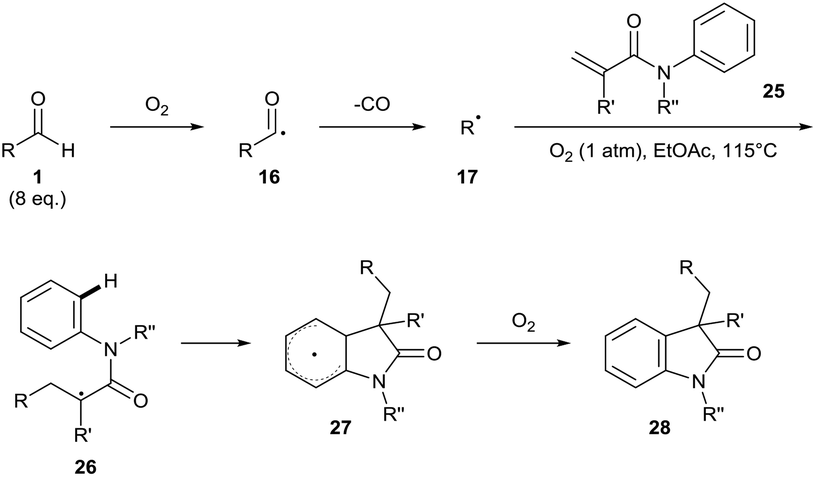 | ||
| Scheme 10 Oxidative radical 1,2-alkylarylation of α,β-unsaturated amides 25. | ||
In 2017, Miyamoto and co-workers initially reported the facile auto-oxidation of isobutyraldehyde 1a under a dioxygen atmosphere (use of balloon) in 1,2-dichloroethane at 40 °C in which perisobutyric acid 29 was the predominant species present after 1 h.27 Further reaction upon addition of iodobenzene 30 proceeded efficiently, granting a 75% yield of hypervalent iodine(III) species 31 after 5 h (Scheme 11). Miyamoto and co-workers then used this procedure of generating hypervalent iodine(III) intermediates to effectively carry out oxidative cleavage of 1,2-diols and Hofmann rearrangement of carboxamides. Whilst the isolation of hypervalent iodide(III) species 31 was never carried out when utilising air as the dioxygen source, it was reported that the reaction procedure for the oxidative cleavage of 1,2-diols was almost as effective when the reaction mixture was exposed to air (ca. 90% compared to quantitative yield when utilising a balloon).
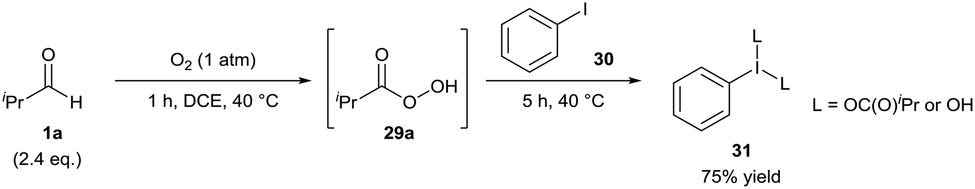 | ||
| Scheme 11 Aerobically-generated hypervalent iodine(III) species 31. | ||
Powers and co-workers carried out a similar experiment for the oxidation of iodobenzene 30 to iodobenzene diacetate 33 (Scheme 12).28 Their initial results highlighted the auto-oxidation of different aldehydes as a means to oxidise iodobenzene at 23 °C. Benzaldehyde, isobutyraldehyde and butyraldehyde all proved ineffective with minimal amount of product formed (0%, 2% and 6% respectively). However, use of acetaldehyde gave a somewhat capricious formation of desired product 33 with 42–91% yield observed over 5 repeats. The group speculated that the variability of oxidation efficiency was due to inconsistent initiation of radical auto-oxidation chemistry. Although the work published showed the possibility of the reaction being able to proceed through a dioxygen-mediated C–H activation process, the group eventually settled on using a cobalt-based initiator (1 mol%) and utilised the in situ formation of the iodine(III) diacetate species to oxygenate a wide range of substrates (alkenes, β-keto esters, etc.).
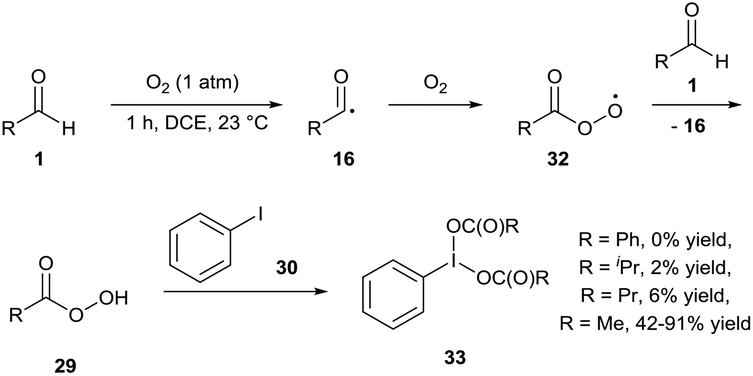 | ||
| Scheme 12 Aerobically-generated hypervalent iodine(III) dicarboxylate species 33. | ||
Ethers
Owing to their common usage as reaction solvents, ethereal auto-oxidation is the most studied and understood auto-oxidation process,29,30 however it should be noted that it is uncommonly utilised as a means for aerobic C–H bond activation. This is likely due to final oxidation product of ethers being relatively explosive peroxide species, meaning that historically, measures have been taken to prevent the activation of ethereal C–H bonds through dioxygen interaction (through use of an inert atmosphere or intentional doping with radical inhibitors). Nonetheless, a few research groups have shown the effective utilisation of (cyclic) ethers as precursors for aerobic C–H bond activation. All ethereal C–H bond activations have been carried out whilst utilising the ether substrate as the reaction solvent, which represents a great excess of the ether substrate. Owing to the observed sensitivity of ethers to auto-oxidation, all methods have utilised transient dioxygen in the atmosphere and forgo the need for a dioxygen balloon.An early example of ethereal aerobic C–H bond activation was achieved in 2000 by Parsons and co-workers who used bromotrichloromethane as a means of brominating the α-position on THF 34 (Scheme 13).31 The subsequent entity 36 was found to be unstable with regards to elimination and thereby resulted in oxocarbenium cation 37 formation. The group then used this cation as a means of tetrahydrofuranyl-protecting alcohol groups, a very desirable protecting group that can be selectively cleaved even in the presence of THP-ethers.32 It should be noted that owing to the use of THF as the reaction solvent, the substrate is used in a great excess (ca. 185 equivalents). It does not appear that efforts were made to utilise the ether in a smaller stoichiometry coupled with possibly a greener, non-nucleophilic solvent (i.e. esters).
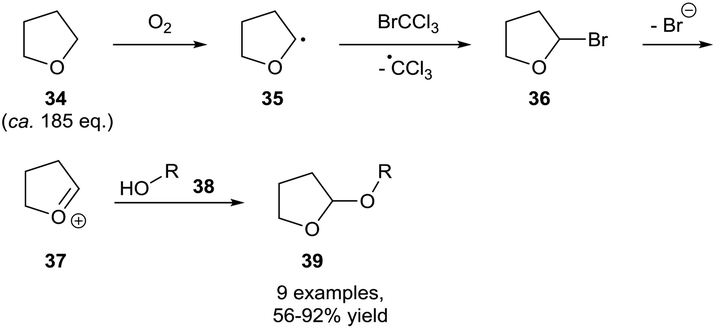 | ||
| Scheme 13 Aerobically-initiated protocol for the protection of alcohols 38 as 2-tetrahydrofuranyl ethers 39 utilising bromotrichloromethane. | ||
More recently, Troisi et al. extended the available reaction partners that can be coupled to THF following aerobic C–H bond activation.33 It was showed that allyl chloride 40 can be utilised as a chlorinating agent. Doing so formed an allyl radical capable of abstracting an ethereal H-atom, propagating the radical chain reaction. Although not explicitly stated, it was suspected that the resulting α-chloro product 41 had the same decomposition effect displayed by the α-bromo species 36 described by Parsons and co-workers in which release of halide resulted in oxocarbenium species 37, which can then be trapped using alcohols 38. Utilising this method, Troisi et al. showed the tetrahydrofuranylation of simple (phenol, benzylic) and complex (cholesterol) alcohols (Scheme 14). Further studies confirmed the importance of dioxygen in their protocol by observing no product formation when the reaction was conducted under a nitrogen atmosphere and using deaerated THF. Evidence supporting a radical mechanism was then provided by showing complete reaction inhibition when carried out in the presence of TEMPO. Interestingly, Troisi et al. then provided further evidence by observing no reaction when utilising commercially stabilised THF, and then rapid subsequent reaction upon addition of CrCl2 oxidant to the reaction mixture, which consumed the antioxidant stabiliser and allowed uninhibited aerobic C–H bond activation.
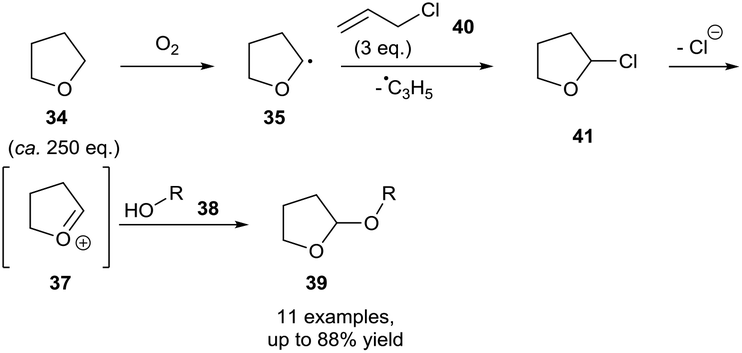 | ||
| Scheme 14 Aerobic protocol for the protection of alcohols 38 as 2-tetrahydrofuranyl ethers 39 utilising allyl chloride. | ||
Following this, Troisi then briefly showed the possibility of the tetrahydrofuranyl radical 35 participating in radical addition to imines and an alkyne 42 (Scheme 15). Troisi postulated that the reaction was likely to follow a similar mechanism. Although substrate scope was scarcely explored, and THF was again used as the reaction solvent, it was shown that the ethereal radical was able to add to CN bonds and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, both of which were unexplored with respect to aerobically-induced acyl radical addition.
C bonds, both of which were unexplored with respect to aerobically-induced acyl radical addition.
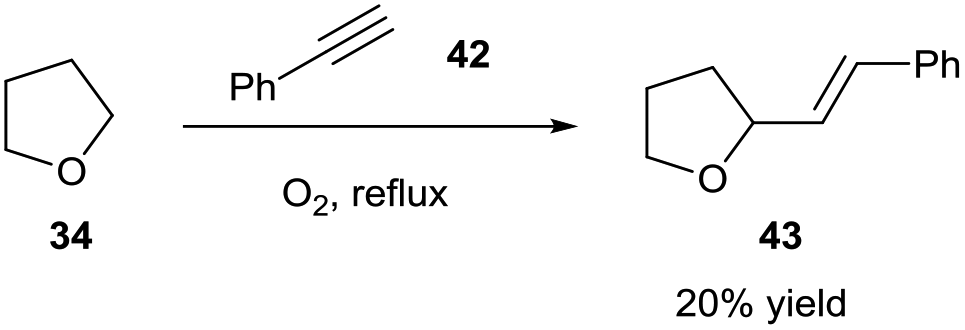 | ||
| Scheme 15 Ethereal radical addition to phenylacetylene 42 to afford alkene 43. | ||
Benzylamines
Benzylamines represent a class of molecules that have gathered recent attention due to observed imine formation in reactions that are heated in the presence of air. The Fu and Nguyen groups have both exploited this and have shown that aerobically-induced C–H bond activation of benzylamines 44 resulted in oxidative coupling to form imines 50 (Scheme 16).34,35 Fu and co-workers showed that by utilising a dioxygen balloon, metal-free aerobic oxidative coupling of amines can be achieved whilst utilising water as the reaction solvent, with good to excellent yields (51–85%) achieved in 12–64 h. Furthermore, there was no product formation observed when the reaction was conducted under a nitrogen atmosphere, highlighting the significance of dioxygen for the reaction. The group also declared an unchanging yield when the reaction was conducted in a Teflon-reactor, excluding the possibility of light- or glass-catalysed processes. Although aliphatic amines have low susceptibility to aerobic C–H bond activation, they were able to be effectively utilised (45–77% yield) in the cross-coupling with benzylamines when used in excess (1.8 equivalents). Whilst the exact mechanistic pathway was not discerned, it was clear dioxygen played a role in the reaction via aerobic C–H bond activation.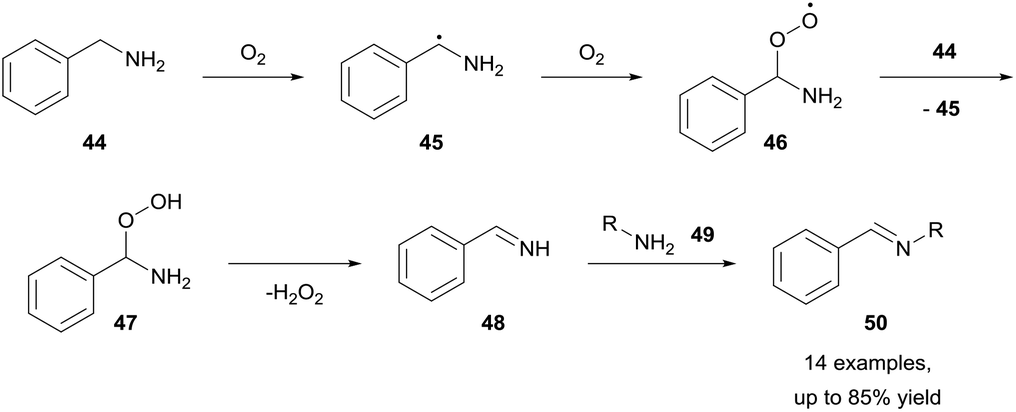 | ||
| Scheme 16 Aerobic oxidative coupling of benzylamines 44. | ||
Nguyen et al. then reported a similar reaction that forgoes the need for a solvent altogether.35 Nguyen reported that simply heating benzylamines under an oxygen environment was sufficient for achieving oxidative coupling of amines to imines in good to excellent yields (62–83%) in 24 h. Nguyen reported that whilst effective benzylamine auto-oxidation was observed utilising a flask exposed to air, evaporation of volatile benzylamines rendered the method inconvenient. Nonetheless, the reaction was proven to be effective, and Nguyen et al. used this process of generating imines to construct many medicinally relevant nitrogen heterocycles (i.e. benzimidazoles, Scheme 17).
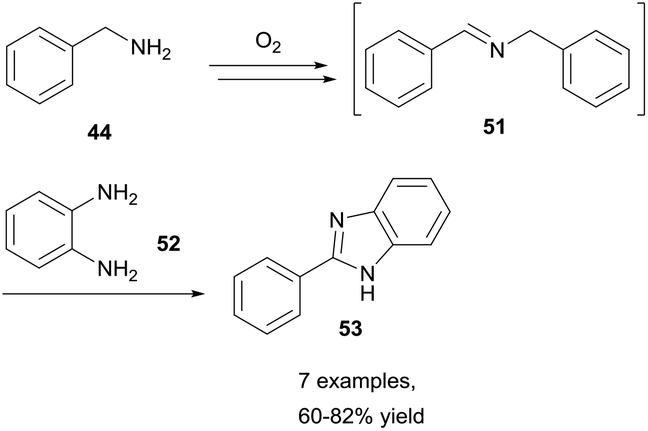 | ||
| Scheme 17 Synthesis of benzimidazoles 53 through aerobic C–H bond activation of benzylamines 44. | ||
Glycine derivatives
Pioneering research by Huo and co-workers has shown the functionalisation of the α-carbon in glycine derivatives 54.36,37 The modification of glycine is particularly attractive for medicinal chemistry owing to the possibility of simple diversification of unnatural amino acids. Since 2014, Huo and co-workers have exploited the auto-oxidation of glycine derivatives 54. The mechanism for glycine auto-oxidation is similar to that displayed by benzylamines in which an imine intermediate 58 is formed through the release of H2O2 (Scheme 18). The group have developed elegant methodologies for C–C formation via nucleophilic attack on the imine intermediate formed via aerobic activation; styrenes, indoles and arenes were used as nucleophilic partners for C–C bond formation.36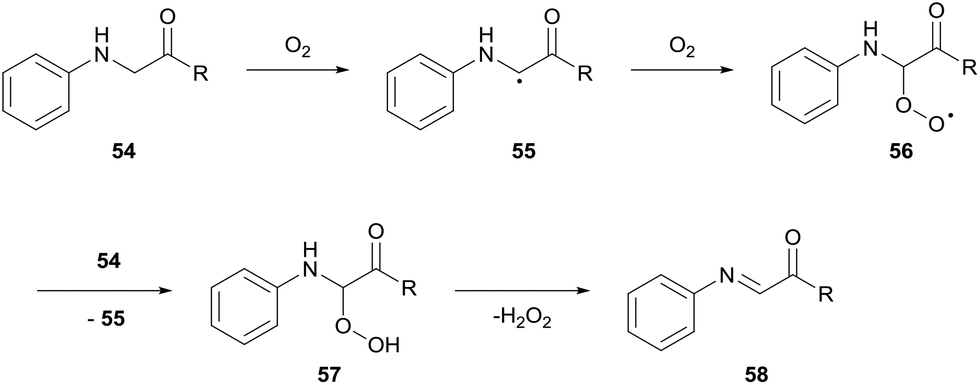 | ||
| Scheme 18 Aerobic C–H bond activation of glycine derivatives 54. | ||
For the reaction with indoles, a dioxygen balloon was utilised at 40 °C. Reaction times varied (4–60 h) with average to high yields reported (30–83%). Analogous reactions conducted under an argon atmosphere reported a significant drop in yield, thus indicating the significance of dioxygen for the reaction. Furthermore, no desired product was observed upon the addition of TEMPO, thereby providing further evidence of a radical mechanism.
For the coupling of the glycine imine to styrene moieties 59, an open flask with access to air was utilised (Scheme 19). However, yields were comparatively low (28–53%) owing to an alternative pathway providing formation of self-oxidation side product 61 (7–40% yield). Finally, the use of electron-rich arenes allowed for efficient nucleophilic addition to the protonated imine intermediate at room temperature and utilising atmospheric dioxygen. Moderate to good yields were observed (34–76%) when utilising a stoichiometric amount of arene to glycine ester.
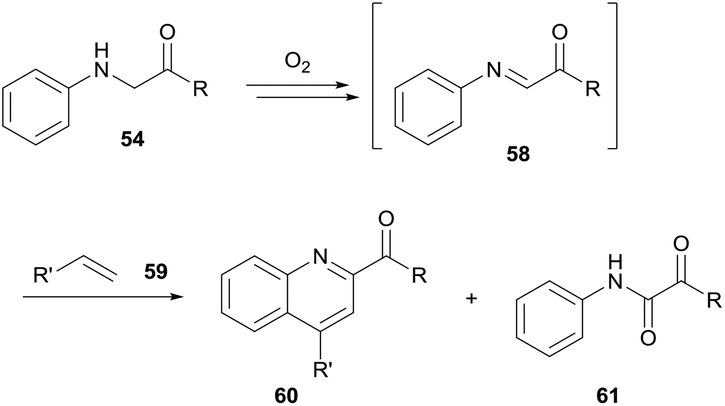 | ||
| Scheme 19 Utilising aerobic generation of 58 to facilitate Povarov/aromatisation tandem reactions with styrenes 59. | ||
To provide further proof of the mechanism for benzylamine auto-oxidation, the group showed that when 1 mol% of MnO2 was added at the end of a reaction, gas evolution was observed. This suggested the formation of H2O2 as an auto-oxidation by-product since MnO2 reacts with H2O2 to form dioxygen and water.
Conclusions
Aerobic C–H bond activation in which dioxygen interacts and activates a C–H bond is a fundamentally green progress with regards to atom economy and waste minimisation when compared with typical C–H activation reagents. Utilising dioxygen as the activating species ensures the use of an inherently green oxidant species (especially through the use of atmospheric dioxygen) which is of timely interest as scientists strive for more environmentally friendly protocols. In this review we highlight the realisation of new synthetic protocols that utilise aerobic C–H bond activation as a means of initiating radical reactions. Whilst these new reactions use relatively few functional groups as aerobic C–H activation precursors (e.g. aldehydes, ethers, benzylamines and glycine derivatives) it is hoped that these breakthroughs will lead to further efforts in exploring novel motifs that are amenable to this methodology. For example, by looking more closely at other molecules/functionalities that “degrade” aerobically and/or appraising various parameters (e.g. dissolved oxygen concentration of solvent, stirring rate, surface area to volume ratio, etc.) that promote the aerobic oxidation of alternative C–H bearing functional groups.Conflicts of interest
There are no conflicts to declare.References
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS.
- H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, Bioorg. Med. Chem. Lett., 2016, 26, 5378–5383 CrossRef CAS PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- M. A. Légaré, M. A. Courtemanche, É. Rochette and F. G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed.
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W. D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S. F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, Chem. Commun., 2010, 46, 145–147 RSC.
- L. Vanoye, A. Favre-Réguillon, A. Aloui, R. Philippe and C. de Bellefon, RSC Adv., 2013, 3, 18931–18937 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 RSC.
- R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Org. Biomol. Chem., 2009, 7, 235–237 RSC.
- V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133–135 RSC.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed.
- V. Chudasama, J. M. Ahern, R. J. Fitzmaurice and S. Caddick, Tetrahedron Lett., 2011, 52, 1067–1069 CrossRef CAS.
- A. Shamsabadi, J. Ren and V. Chudasama, RSC Adv., 2017, 7, 27608–27611 RSC.
- A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC.
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 RSC.
- A. Shamsabadi and V. Chudasama, Chem. Commun., 2018, 54, 11180–11183 RSC.
- A. R. Akhbar, V. Chudasama, R. J. Fitzmaurice, L. Powell and S. Caddick, Chem. Commun., 2014, 50, 743–746 RSC.
- G. N. Papadopoulos and C. G. Kokotos, Chem. – Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- S. Paul and J. Guin, Chem. – Eur. J., 2015, 21, 17618–17622 CrossRef CAS PubMed.
- P. Biswas and J. Guin, J. Org. Chem., 2018, 83, 5629–5638 CrossRef CAS PubMed.
- P. Biswas, S. Paul and J. Guin, Synlett, 2017, 28, 1244–1249 CrossRef CAS.
- K. Miyamoto, J. Yamashita, S. Narita, Y. Sakai, K. Hirano, T. Saito, C. Wang, M. Ochiai and M. Uchiyama, Chem. Commun., 2017, 53, 9781–9784 RSC.
- A. Maity, S. M. Hyun and D. C. Powers, Nat. Chem., 2018, 10, 200–204 CrossRef CAS PubMed.
- S. Di Tommaso, P. Rotureau, O. Crescenzi and C. Adamo, Phys. Chem. Chem. Phys., 2011, 13, 14636–14645 RSC.
- S. Di Tommaso, P. Rotureau, B. Sirjean, R. Fournet, W. Benaissa, P. Gruez and C. Adamo, Process Saf. Prog., 2014, 33, 64–69 CrossRef CAS.
- J. M. Barks, B. C. Gilbert, A. F. Parsons and B. Upeandran, Tetrahedron Lett., 2000, 41, 6249–6252 CrossRef CAS.
- C. G. Kruse, N. L. J. M. Broekhof and A. van der Gen, Tetrahedron Lett., 1976, 17, 1725–1728 CrossRef.
- L. Troisi, C. Granito, L. Ronzini, F. Rosato and V. Videtta, Tetrahedron Lett., 2010, 51, 5980–5983 CrossRef CAS.
- L. Liu, S. Zhang, X. Fu and C. H. Yan, Chem. Commun., 2011, 47, 10148–10150 RSC.
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Green Chem., 2013, 15, 2713–2717 RSC.
- C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 13544–13547 CrossRef CAS PubMed.
- Y. Wei, J. Wang, Y. Wang, X. Yao, C. Yang and C. Huo, Org. Biomol. Chem., 2018, 16, 4985–4989 RSC.
Footnote |
| † Precise yield not stated. |
| This journal is © The Royal Society of Chemistry 2019 |