DOI:
10.1039/C8CE02133C
(Paper)
CrystEngComm, 2019,
21, 1130-1136
Organic molecular tessellations and intertwined double helices assembled by halogen bonding†
Received
18th December 2018
, Accepted 9th January 2019
First published on 11th January 2019
Abstract
Isomeric 2,5- and 2,6-bis(iodoethynyl)pyrazine have been employed to assemble single-component polygonal molecular tessellations and intertwined double helices via intermolecular ethynyl C—I⋯N halogen bonding. X-ray crystallography elucidated the 3D supramolecular architectures of five ditopic heteroaromatic polymorphs obtainable via crystallization in various organic solvents.
Introduction
Tessellation or tiling on a plane is the highly-ordered arrangement of one or more planar shapes, called prototiles, to fill the surface without gaps and overlaps to generate various attractive patterns. Although molecular tessellations using organic precursors fabricated on a metal surface have been observed with a combination of scanning tunnelling microscopy, synchrotron radiation photoelectron spectroscopy and X-ray spectroscopy techniques,1 detailed three-dimensional structural information derived from X-ray crystallographic studies of tessellation materials is still lacking. Moreover, exploration in two-dimensional organic molecular tessellations remains limited to uniform pore patterns in a homogeneous environment owing to the formidable challenge in designing molecular building units that readily undergo covalent linkage.2 Therefore, construction of self-assembled molecular tessellations usually involves two-component systems. We surmise that the use of custom-designed multi-functionalized small planar organic molecules interconnected by self-complementary non-covalent interactions may lead to novel single-component tiling patterns upon crystallization.
In recent years, hydrogen bonding has been utilized in the designed construction of a variety of helical molecules exemplified by supramolecular helices3 and foldamers.4 Among various helical structures, a meso-helix is a subtype under the shadow of the celebrated chiral helix. In contrast to the single screw-sense (either left- or right-handed) along the helical axis, a meso-helix exhibits a reversal of helicity along the strand so that it is intrinsically achiral. Since the publication of the first inorganic meso-helical structure in 1994,5 the number of reports on this type of metal complexes consolidated by coordination bonding has grown rapidly.6 However, in the realm of organic compounds, since the first construction of a hydrogen-bonded supramolecular meso-helix with meta-substituted phenylene dioxamic acid diethyl ester monomers in 2003,7 only a handful of related literature reports have appeared.8 To the best of our knowledge, as yet, there is no known example of a supramolecular organic meso-helix consolidated by halogen bonding.9
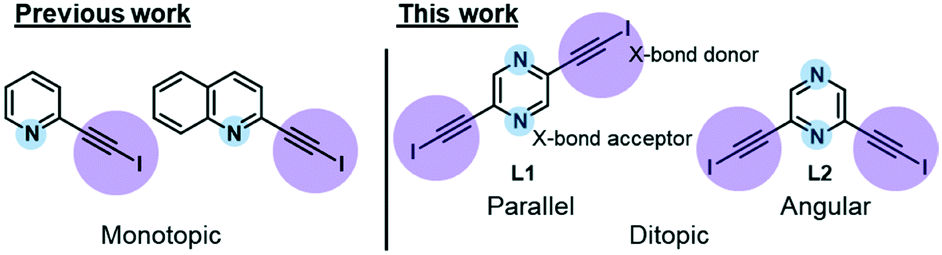
In our recent work, we reported the single-component self-assembly of 2-(iodoethynyl)pyridine and 2-(iodoethynyl)quinoline (Fig. 1, left side) each bearing one set of self-complementary halogen-bond donor and acceptor, which form a supramolecular triangle and an unprecedented pair of enantiomeric (31 and 32) double helices.10 The present study started with a simple pyrazine core possessing two para-nitrogen atoms and two outstretched iodoethynyl arms bearing a coaxial (∠ = 180°) or angular relationship (∠ = 120°) (Fig. 1, right side) that could facilitate the formation of diversified halogen-bonded networks and helical assemblies. Herein we report the construction and structural characterization of single-component molecular tessellations, a supramolecular network of double helices, and an unprecedented network of supramolecular meso-helices assembled by halogen-bonding interaction between isomeric bis(iodoethynyl)pyrazine molecules.
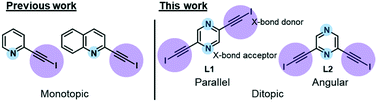 |
| | Fig. 1 Monotopic ligands employed in our previous study versus ditopic ligand 2,6-bis(iodoethynyl)pyrazine L1 and its 2,5-regioisomer L2 in this work. | |
New heteroaromatic 2,5-bis(iodoethynyl)pyrazine L1 and its 2,6-regioisomer L2 were readily synthesized by two-fold iodination11 of bisacetylenes, which were obtained from the commercially available 2,5- and 2,6-dichloropyrazine by Sonogashira reaction.12
Experimental
Synthesis
All reagents were purchased from commercial suppliers (Sigma-Aldrich, Acros and Dieckmann) and used without further purification. All reactions were carried out under a N2 atmosphere unless otherwise stated. All reactions were monitored by thin-layer chromatography on pre-coated silica gel plates, which were visualized by UV irradiation at 254 or 365 nm and/or stained using 5% (w/v) dodecamolybdophosphoric acid in ethanol followed by heating. Flash column chromatography was performed on a glass column of silica gel (230–400 mesh), and solvent ratios were expressed in volume to volume. The 1H and 13C NMR spectra were recorded on a Bruker Advance III HD 500 MHz nuclear magnetic resonance spectrometer (1H, 500 MHz and 13C, 125 MHz). Unless otherwise stated, all NMR measurements were conducted in (CD3)2SO at 25 °C. The chemical shifts were reported as parts per million in a δ scale using a solvent residual peak as the internal standard. Coupling constants (J) were reported in hertz. All the mass spectra were obtained on a Thermo Scientific Q Exactive Focus Orbitrap LC-MS/MS system. The reported molecular mass (m/z) values were monoisotopic unless otherwise stated. The melting points were measured on a digital melting point apparatus and were uncorrected.
General procedure for the iodination of terminal bisacetylenes
Modifying the literature procedure for the iodination11 of terminal acetylene, bisacetylene (1 mmol, 1 eq), tert-butyl hydroperoxide (3 eq) and potassium iodide (2.2 eq) were dissolved in methanol. The reaction mixture was stirred at room temperature overnight. The precipitate formed was then filtered and washed with ice-cold methanol and dichloromethane to afford the desired product.
2,5-Bis(iodoethynyl)pyrazine L1
Yellow solid (150 mg, 39%). Mp: 196 °C (dec.). Rf: 0.5 (hexane/ethyl acetate = 3/1). 1H NMR: 8.69 (s, 2 H). 13C NMR: 147.5, 137.3, 90.3, 28.7. HRMS (ESI) calcd for [M + H]+ = C8H3I2N2+, 380.83801; found 380.83862.
2,6-Bis(iodoethynyl)pyrazine L2
Yellow solid (200 mg, 53%). Mp: 180 °C (dec.). Rf: 0.5 (hexane/ethyl acetate = 3/1). 1H NMR: 8.69 (s, 2 H). 13C NMR: 146.6, 138.5, 89.7, 27.0. HRMS (ESI) calcd for [M + H]+ = C8H3I2N2+, 380.83801; found 380.83867.
X-ray crystallography
The unit-cell and intensity data of crystals L1a–L2c were collected at 298 or 173 K on a Bruker D8 VENTURE diffractometer with Mo Kα radiation (λ = 0.71073 Å) from a sealed tube generator. Data collection, reduction, and empirical absorption corrections were performed using APEX2 software.13 All five crystal structures were solved by direct methods with the SHELXS14 program, and all non-hydrogen atoms were subjected to anisotropic refinement against F2 with full-matrix least-squares techniques using the SHELXL-97 program. All hydrogen atoms were included in the structure factor calculation at idealized positions with fixed isotropic thermal displacement parameters relative to the attached atoms. The SQUEEZE15 process had been applied for crystal forms L1a, L2a and L2b to remove highly-disordered residual electron density in the void. The crystallographic data and structure refinement parameters for crystalline phases L1a–L3d are given in Table S1 in the ESI.†
Results and discussion
Crystal structure of L1a featuring two-dimensional monohedral rhombic tessellation
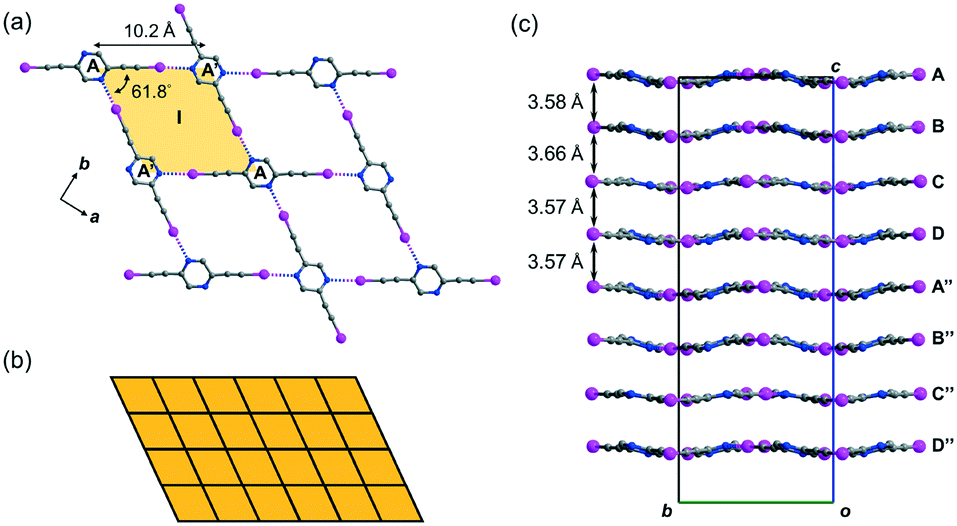
Crystal form L1a16 deposited from a solution of 2,5-regioisomer L1 in acetonitrile belongs to non-centrosymmetric orthorhombic space group Pca21 (No. 29) with Z = 16, so that there are four crystallographically independent L1 molecules (labelled A, B, C, and D)17 per unit cell. Its crystal structure exhibits stacked sinusoidal layers of monohedral rhombic tessellations formed by intermolecular C–I⋯N halogen bonding, such that a single independent L1 molecule is used to construct each successive layer. For instance, two molecules A and their glide-related counterparts A′ are connected to form rhombus I (length AA′ = 10.2, ∠A′AA′ = 61.8°) through intermolecular C–I⋯N halogen bonds of lengths 2.88 and 2.94 Å (relative distance18RIN = 0.82 and 0.83, respectively) (Fig. 2a). Rhombus I is replicated along both the crystallographic a- and b-axes to form a complete tessellation with wallpaper group cmm (Fig. 2b). The sinusoidal layers of the rhombic tessellation each composed of independent L1 molecules A, B, C and D and their 21-related counterparts A′′, B′′, C′′ and D′′ constitute a stacked layer structure with an interlayer spacing ranging from 3.57 to 3.66 Å along the c-axis (Fig. 2c).
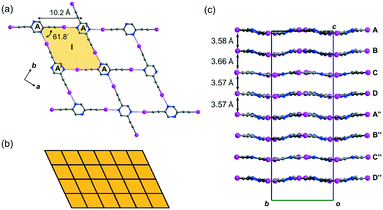 |
| | Fig. 2 (a) Crystal form L1a showing the monohedral rhombic tessellation stabilized by C–I⋯N halogen bonding generated from a single independent L1 molecule. (b) Schematic diagram showing the monohedral rhombic tessellation pattern of a single layer in crystal form L1a. (c) Projection diagram along the a-axis showing the stacking of wavy tessellated layers. All hydrogen atoms are omitted for clarity. | |
Crystal structure of L1b featuring a network of halogen-bonded double helices
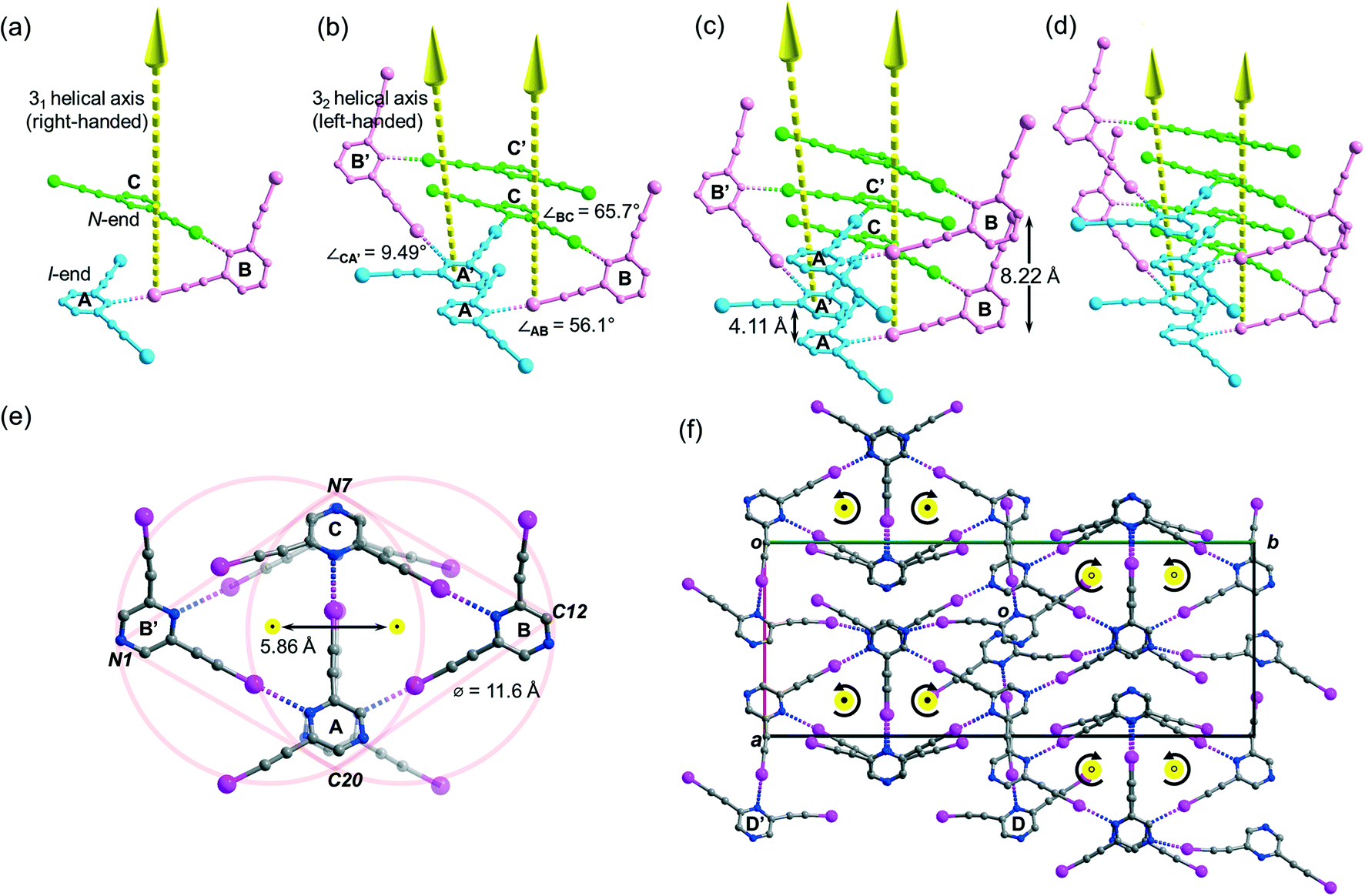
Besides acetonitrile, crystallization of L1 in other common organic solvents including ethyl acetate, methanol, benzene, toluene, and xylene failed to yield X-ray quality crystals. However, slow evaporation of a solution of L1 in chloroform gave crystal form L1b belonging to space group P21/c with Z = 4. X-ray structure analysis revealed two independent L1 molecules each located at a crystallographic inversion center while a chloroform molecule sits in a general position. The crystal structure of L1b features a network of supramolecular C–I⋯N halogen-bonded double helices directed along the a-axis. In each column of the supramolecular double helix, the backbone of the constituent single helical strands is formed by intermolecular C–I⋯N halogen bonds between successive L1 monomers with distances of 2.88 and 2.95 Å (RIN = 0.84 and 0.81). Interestingly, uncommon non-crystallographic 41 (right-handed) and 43 (left-handed) screw axes along the a-axis direction could be found in respective supM- and supP-helices19 within the network, such that each helix completes one turn in four molecules with a pitch distance 2a = 8.68 Å in the sequence …A⋯B⋯A′⋯B′… along the strand (Fig. 3a). In the subsequent discussion, the end of the strand terminating at a pyrazine nitrogen atom is designated as the “N-end”, whereas the iodoalkyne terminus is designated as the “I-end”. The right- and left-handed helices screw in the direction from N- to I-end in an anti-clockwise and clockwise sense, respectively. The diameter of the circular helical column is estimated to be 15.4 Å20 from the average of pairwise interatomic distances between the projections of atoms C4, C8, N1 and N2 in the peripheral square onto the bc-plane (Fig. 3b). Notably, chloroform solvate molecules are located inside the cavity of the helical channel along the screw axis. Tilting of pyrazine rings A, B, and A′ leads to the helical formation, and its extent can be measured by the pairwise inter-planar angles ∠AB = 19.4° and ∠BA′ = 56.5° (Fig. 3c). With stabilization from inter-helical π–π stacking between the pyrazine rings along the helices, two identical helical strands intertwine to give a double helix with a parallel orientation. In contrast to the major and minor grooves commonly found in DNA, the grooves of the double helices in crystal L1b are of the same length a = 4.34 Å. Each column of four-fold symmetrical double helices stacks along the orthogonal a- and b-axes so that a column of the double helix always shares its backbone with four neighboring columns in the crystallographic b, −b, c and −c directions (Fig. 3d). Furthermore, the supramolecular helicity of the double helices is retained along the b-axis while reversal of handedness is observed along the orthogonal c-axis. Despite the presence of supramolecular chiral supP- and supM-double helices in L1b, the overall chirality of the crystal vanishes because of internal helicity cancellation.
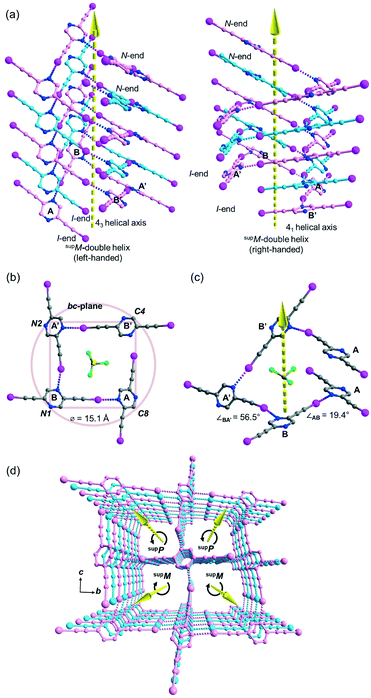 |
| | Fig. 3 (a) Perspective diagram showing the left- and right-handed supramolecular double helices in crystal form L1b, which are formed by C–I⋯N halogen bonds with two identical single strands (carbon skeleton colored in pink and pale blue) oriented in the same direction (arrow head pointing upward from I-end to N-end). The solvated chloroform molecules along the helix are omitted for clarity. (b) Projection of a section of the right-handed helix in L1b along the a-axis, showing four-fold rotational axis pointing towards the reader and the proposed diameter of the circular helix. (c) Perspective diagram showing a section of the single-stranded right-handed helix in L1b and the inter-planar angle between the pyrazine planes of A, B and A′ along the helix (symmetry transformation ′: x, 1/2 − y, 1/2 + z). (d) Perspective diagram showing a packing of supP- and supM-double helices parallel to the a-axis. All hydrogen atoms are omitted for clarity. | |
Buckled ribbon composed of halogen-bonded triangles and parallelograms in the crystal structure of L2a
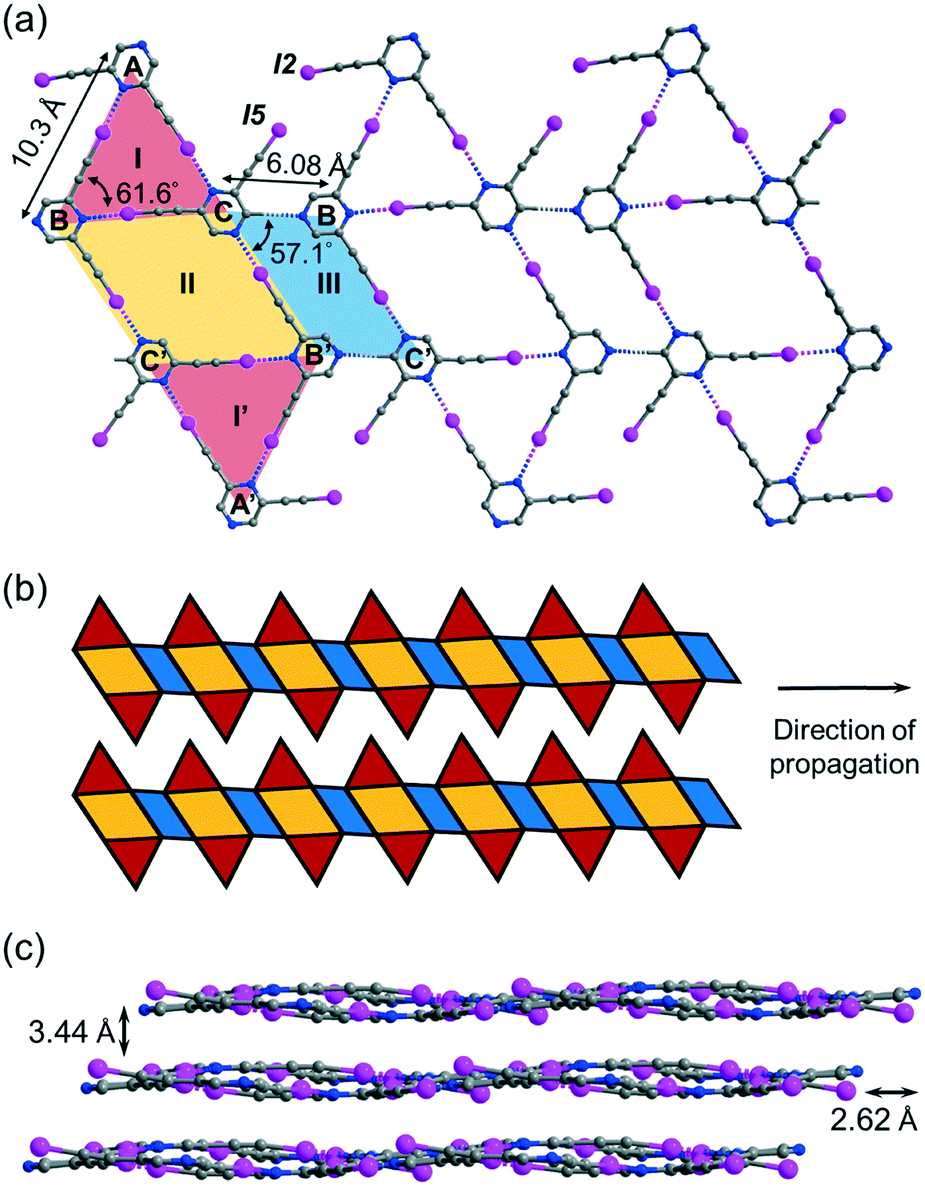
Crystallization of regioisomer 2,6-bis(iodoethynyl)pyrazine L2 in acetonitrile under the same condition gave crystal form L2a with three independent molecules per unit cell (Z′ = 3) in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The crystal structure contains layers of supramolecular C–I⋯N halogen-bonded buckled ribbons composed of equilateral triangles, parallelograms and rhombuses (Fig. 4a). Equilateral triangle I is composed of independent L2 molecules A, B and C connected by C–I⋯N halogen bonds ranging from 2.94 (RIN = 0.83) to 2.99 Å (RIN = 0.85). Moreover, molecules B and C and their inversion-related molecules B′ and C′ are halogen-bonded with a bond length of 2.86 Å (RIN = 0.81) to give rhombus II adjoining equilateral triangle I such that they share common edge BC of length 6.08 Å. Equilateral triangle I and its inversion I′ are congruent such that I′ shares common edge B′C′ with rhombus II. Interestingly, parallelogram III with vertices B′, C, B, and C′ is formed by two halogen bonds and two weak intermolecular C–H⋯N hydrogen bonds with a donor–acceptor distance DCHN = 3.36 Å and angle ∠CHN = 174.6°. The I-II-I′-III pattern propagates along one direction to give an infinite ribbon with saw-tooth edges that repeats alongside to fill up the entire plane (Fig. 4b). The ribbon layers are stacked together with an interlayer spacing of 3.44 Å (Fig. 4c).
. The crystal structure contains layers of supramolecular C–I⋯N halogen-bonded buckled ribbons composed of equilateral triangles, parallelograms and rhombuses (Fig. 4a). Equilateral triangle I is composed of independent L2 molecules A, B and C connected by C–I⋯N halogen bonds ranging from 2.94 (RIN = 0.83) to 2.99 Å (RIN = 0.85). Moreover, molecules B and C and their inversion-related molecules B′ and C′ are halogen-bonded with a bond length of 2.86 Å (RIN = 0.81) to give rhombus II adjoining equilateral triangle I such that they share common edge BC of length 6.08 Å. Equilateral triangle I and its inversion I′ are congruent such that I′ shares common edge B′C′ with rhombus II. Interestingly, parallelogram III with vertices B′, C, B, and C′ is formed by two halogen bonds and two weak intermolecular C–H⋯N hydrogen bonds with a donor–acceptor distance DCHN = 3.36 Å and angle ∠CHN = 174.6°. The I-II-I′-III pattern propagates along one direction to give an infinite ribbon with saw-tooth edges that repeats alongside to fill up the entire plane (Fig. 4b). The ribbon layers are stacked together with an interlayer spacing of 3.44 Å (Fig. 4c).
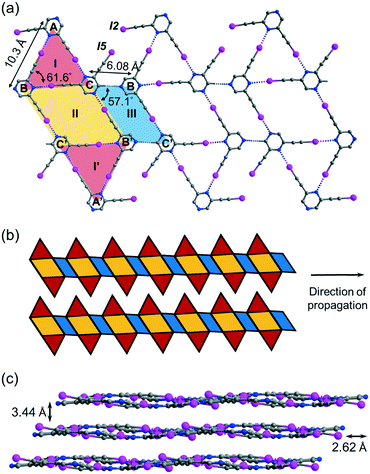 |
| | Fig. 4 (a) Projection diagram showing the triangle-rhombus-parallelogram ribbon constructed by C–I⋯N halogen bonds and C–H⋯N hydrogen bonds between three independent L2 molecules in crystal form L2a. (b) Schematic diagram showing two adjacent toothy-edge ribbons in a single layer in L2a. (c) Projection diagram along the a-axis showing the stacking of wavy tessellated layers. All hydrogen atoms are omitted for clarity. | |
Crystal structure of L2b featuring tacit trihedral snub trihexagonal tessellation
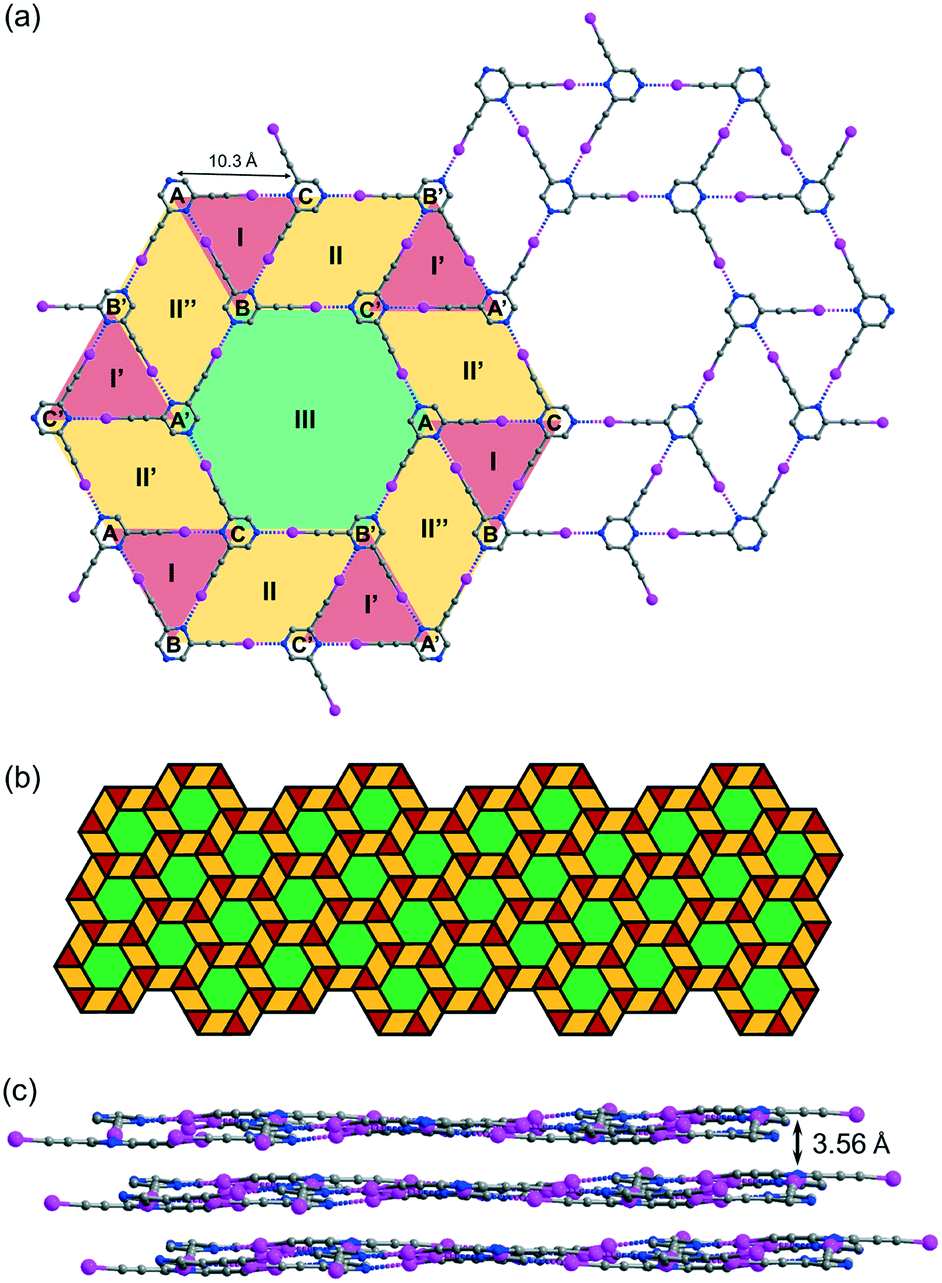
While crystallization of L2 in benzene or xylene only produced an amorphous solid residue, evaporation of the solution in toluene gave crystal form L2b, which belongs to space group P with three independent L2 molecules per unit cell. Remarkably, L2b features a supramolecular C–I⋯N halogen-bonded trihedral snub trihexagonal tessellation composed of equilateral triangles, rhombuses and regular hexagons (Fig. 5a). Intermolecular C–I⋯N halogen bonds in the range of 2.97 (RIN = 0.84) to 2.99 Å (RIN = 0.85) connecting three independent molecules A, B and C form triangle I with an average edge length of 10.3 Å. Furthermore, molecules B and C and their inversion-related molecules B′ and C′ are halogen-bonded with a distance of 2.92 (RIN = 0.83) to 2.99 Å (RIN = 0.85) to give rhombus II adjacent to triangle I and its inversion counterpart I′ at shared edges BC and B′C′. Also, three congruent symmetry-related rhombuses II, II′ and II′′ are each sandwiched between a pair of triangles labelled I and I′. Remarkably, regular hexagon III is formed by sharing six edges AB′, B′C, CA′, A′B, BC′ and C′A of rhombuses II, II′ and II′′ along with six vertices A, B′, C, A′, B and C′ from triangles I and I′. Hence, every hexagon III is surrounded by triangles and rhombuses in the sequence I, II, I′, II′, I, II′′, I′, II, I, II′, I′ and II′′ running in the clockwise sense. The entire tessellation pattern extends throughout the plane with wallpaper group p6 (Fig. 5b). Ordinary snub trihexagonal tessellation is a dihedral tiling pattern composed of two types of prototiles, namely triangles and hexagons. However, in addition to these two types, the trihedral pattern in L2b also contains rhombuses, each of which could be bisected by its shorter diagonal to give two triangles to fit the tiling definition. Therefore, the term “tacit trihedral snub trihexagonal tessellation” is coined to designate this subtle difference. Each tessellation layer is inclined to the crystallographic bc-plane with a tilt angle of 32.1°, and stacking along the a-axis occurs with an interlayer spacing of 3.56 Å (Fig. 5c).
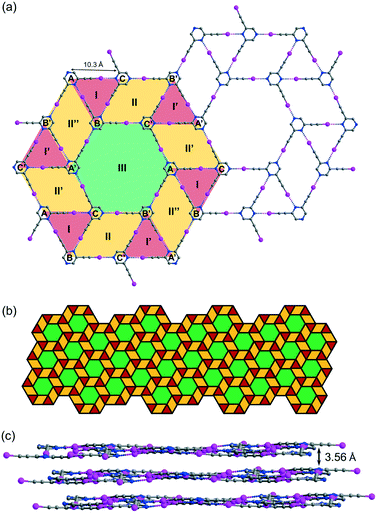 |
| | Fig. 5 (a) Projection diagram showing the tacit trihedral snub trihexagonal tessellation stabilized by C–I⋯N halogen bonds with three independent L2 molecules in L2b. (b) Schematic diagram showing the pattern of the complete tessellation of a single layer in L2b. (c) Projection diagram showing the stacking of the tiling layers in the crystal structure. All hydrogen atoms are omitted for clarity. | |
Halogen-bonded meso-helix in the crystal structure of L2c
Crystallization of L2 in chloroform yielded crystal form L2c belonging to centrosymmetric space group P21/c with Z = 16 and showed four independent L2 molecules (Z′ = 4) per unit cell. Its crystal structure features a supramolecular C–I⋯N halogen-bonded single-stranded meso-helix directed along the c-axis composed of three independent L2 molecules (labelled A, B and C and colored blue, pink and green, respectively), while the fourth one labelled D serves as an auxiliary bridge between columns of the meso-helices (Fig. 6c). The backbone of the meso-helical strand is formed by continuous linkage of short helical segments [A⋯B⋯C] that are connected by intermolecular C–I⋯N halogen bonds ranging from 2.82 (RIN = 0.80) to 3.00 Å (RIN = 0.85) with handedness supP (right-handed) (Fig. 5a). Similarly, their c-glide-related counterparts A′, B′ and C′ are connected to form a left-handed (supM) helical segment [A′⋯B′⋯C′] (Fig. 6b). These enantiomeric supP- and supM-helical segments possess respective non-crystallographic 31 and 32 screw axes, such that each segment completes one turn in three L2 molecules with a pitch distance c/2 = 4.11 Å. Therefore, the meso-helix in L2c may be considered as the propagation of independent L2 molecules in the sequence …[A⋯B⋯C], [A′⋯B′⋯C′], [A⋯B⋯C]… along the 31 and 32 screw axes. The handedness along the infinite helical strand alternates strictly (i.e. …supP, supM, supP, supM…) for every segment so that periodic helicity inversion at molecules C to A′ (or C′ to A) leads to the formation of a single-stranded supramolecular meso-helix with an inter-planar distance c = 8.22(3) Å between two consecutive pyrazine rings (same color) of the same handedness (Fig. 6c). Tilting of the pyrazine planes of L2 accounts for the formation of its meso-helical structure, and individual tilts can be measured by the inter-planar angles ∠AB and ∠BC (56.1° and 65.7°, respectively) while a small torsion angle ∠CA′ = 9.49° was observed for helicity inversion (Fig. 6b). The diameter of the meso-helix may be estimated with reference to two equal-sized circles each of diameter 11.6 Å21 whose centers (located on the parallel 31 and 32 axes) are separated by 5.86 Å (see Fig. 6e). Despite the Cs-symmetry of L2 as well as the achirality of the meso-helix, the meso-helical strand is directional (from N-end to I-end, following the same terminology as in crystal L1a) because of the unequal environment of two iodoethynyl side arms. The running direction of the halogen-bonded meso-helix is retained along the a-axis and reversed along the b-axis (closed black dot: from I- to N-ends toward the reader; open black dot: from I- to N-end away from the reader; see Fig. 6f). Moreover, the auxiliary bridging molecule D and its c-glide counterpart D′ form halogen bonds with molecules A and B (A′ and B′) within each column of the meso-helix, while one set of donor iodine and acceptor nitrogen atoms in molecule D or D′ does not partake in halogen bonding.
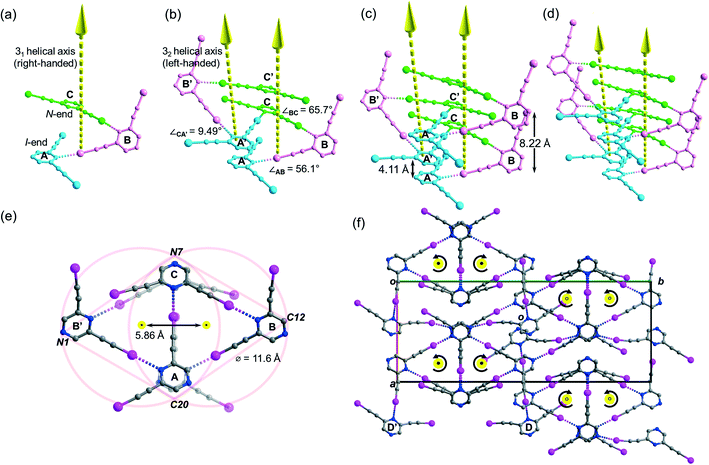 |
| | Fig. 6 Perspective diagram showing progressive propagation of the supramolecular meso-helix in L2c assembled by C–I⋯N halogen bonds. In part (a), three independent L2 molecules labelled A, B and C (shown in blue, pink and green colours, respectively) are arranged around the 31 helical axis in the c-direction. In part (b), the c-glide-related molecules labelled A′, B′ and C′ (symmetry transformation ′: x, 1/2 − y, 1/2 + z) are arranged around the 32 helical axis. In parts (c) and (d), the helical strand propagates along the 31 and 32 axes in the sequence … A, B, C, and A′, B′, C′, respectively. In part (e), projection of a section of the meso-helix in L2c along the c-axis showing the three-fold rotational symmetry and the proposed diameter of the circular helix. The yellow and closed black dots, respectively, indicated end-on projection of the 31 and 32 screw axes and their arrow tips pointing towards the reader. In part (f), projection diagram along the c-axis showing the running directions and the screw sense (labeled by the curved arrows) of the meso-helices, with auxiliary bridging molecules D and D′ located between the meso-helices in L2c. The open black dots indicate that the arrow tips of the 31 and 32 screw axes point away from the reader. All hydrogen atoms are omitted for clarity. | |
Tessellation or helical structure?
Despite the subtle difference in the arrangement of two iodoethynyl side arms in isomeric L1 and L2, they share two families of crystal structures, namely molecular tessellation (crystal forms L1a, L2a and L2b) and helical assembly (L1b and L2c). Apparently, tessellations as two-dimensional patterns are fundamentally dissimilar and unrelated to three-dimensional helices, yet they are found to be connected through the crystal forms of L1 and L2 in this study. A comparison of solvents used in crystallization and the resultant crystal structures revealed that tessellation formation is favored by acetonitrile whereas the use of chloroform leads to manifestation of helical assembly. The only solvated molecule found in these crystal structures is chloroform, which occupies the channels of the double helices in L1b. Based on these findings, both L1 and L2 molecular crystals exhibit polymorphism, and the choice of solvent plays a crucial role in the resulting structures. To some extent, a three-dimensional helical structure could be considered as a tessellation pattern upon projection along the helical axis onto the a plane. We propose that the formation of the helical structure is primarily determined by the tilt of the molecule in non-covalent supramolecular assembly during the initial stage of crystallization. When the molecules are packed with small or zero tilt along the two directions, potential tessellation could be formed.
Conclusions
The present study illustrates the potential of strategic planning in building complex halogen-bonded architectures using custom-designed multi-functionalized small organic molecules. While recent literature reports have focused on tiling of surfaces at the liquid/solid interface by regular polygons constructed by supramolecular assembly of organic building blocks bearing functional groups designed to form intermolecular linkages by hydrogen bonding, van der Waals interaction, or coordination to metal centers,22 the present work is the first one that deals with single-component molecular tessellation and meso-helices consolidated by halogen bonding.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Wei Lun Foundation and the University Grants Committee of Hong Kong (Project no: AoE/P-03/08).
Notes and references
-
(a) D. Écija, J. I. Urgel, A. C. Papageorgious, S. Joshi, W. Auwärter, A. P. Seitsonen, S. Klyatskaya, M. Ruben, S. Fischer, S. Vijayaraghavan, J. Reichert and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6678–6681 CrossRef PubMed;
(b) Z. Tao, T. Wang, D. Wu, L. Feng, J. Huang, X. Wu and J. Zhu, Chem. Commun., 2018, 54, 7010–7013 RSC;
(c) Y.-Q. Zhang, M. Paszkiewicz, P. Du, P. Zhang, T. Lin, Z. Chen, S. Klyatskaya, M. Ruben, A. P. Seitsonen, J. V. Barth and F. Klappenberger, Nat. Chem., 2018, 10, 296–304 CrossRef CAS PubMed.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef CAS.
-
(a) G. D. Pantoş, P. Pengo and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194–197 CrossRef PubMed;
(b) J. Kang, D. Miyajima, T. Mori, Y. Inoue and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed;
(c) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed;
(d) For review, see E. Yashima, N. Ousaka, D. Taura, K. Shimonmura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
-
(a) N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334–341 CrossRef CAS PubMed;
(b) G. W. Collies, R. Bailly, K. Pulka-Ziach, C. M. Lombardo, L. Mauran, N. Taib-Maamar, J. Dessolin, C. D. Mackereth and G. Guichard, J. Am. Chem. Soc., 2017, 139, 6128–6137 CrossRef PubMed;
(c) R. Wechsel, M. Žabka, J. W. Ward and J. Clayden, J. Am. Chem. Soc., 2018, 140, 3528–3531 CrossRef CAS PubMed;
(d) For review, see D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- G. Becker, B. Eschbach, O. Mundt and N. Seidler, Z. Anorg. Allg. Chem., 1994, 620, 1381–1390 CrossRef CAS.
-
(a) L. Plasseraud, H. Maid, F. Hampel and R. W. Saalfrank, Chem. – Eur. J., 2001, 7, 4007–4011 CrossRef CAS PubMed;
(b) C. Xu, Q. Guo, X. Wang, H. Hou and Y. Fan, Cryst. Growth Des., 2011, 11, 1869–1879 CrossRef CAS;
(c) S. Y. Moon, E. Kim, T. H. Noh, Y.-A. Lee and O.-S. Jung, Dalton Trans., 2013, 42, 13974–13980 RSC;
(d) K. M. Patil, S. A. Cameron, S. C. Moratti and L. R. Hanton, CrystEngComm, 2014, 16, 4587–4601 RSC;
(e) W. Cullen, C. A. Hunter and M. D. Ward, Inorg. Chem., 2015, 54, 2626–2637 CrossRef CAS PubMed;
(f) M. Li, L. Cong, J. Zhao, T. Zheng, R. Tian, J. Sha, Z. Su and X. Wang, J. Mater. Chem. A, 2017, 5, 3371–3376 RSC.
- G. Blay, I. Fernández, J. R. Pedro, R. Ruiz-García, M. C. Muñoz, J. Cano and R. Carrasco, Eur. J. Org. Chem., 2003, 2003, 1627–1630 CrossRef.
-
(a) J. S. González-González, F. J. Martínez-Martínez, A. L. P. Campos, M. de J. Rosales-Hoz, E. V. García-Báez and I. I. Padilla-Martínez, CrystEngComm, 2011, 13, 4748–4761 RSC;
(b) R. Wechsel, J. Raftery, D. Cavagnat, G. Guichard and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 9657–9661 CrossRef CAS PubMed.
-
(a) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS;
(b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed;
(c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- C. F. Ng, H.-F. Chow and T. C. W. Mak, Angew. Chem., Int. Ed., 2018, 57, 4986–4990 CrossRef CAS PubMed.
- K. R. Reddy, M. Venkateshwar, C. U. Maheswari and P. S. Kumar, Tetrahedron Lett., 2010, 51, 2170–2173 CrossRef.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
-
APEX2 Data Collection Software, version 2012.4, Bruker AXS, Delft, The Netherlands, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- The letters a, b, and c are consistently used to denote different crystalline forms.
- Capital letters A, B, and C and Roman numerals I, II, and III are consistently used to denote independent molecules and polygons, respectively. The symbol ′ is used to designate symmetry transformation. Translation-equivalent molecules share the same symbol.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- For terminology of supramolecular helicity, see T. Sasaki, I. Hisaki, S. Tsuzuki, N. Tohnai and M. Miyata, CrystEngComm, 2012, 14, 5749–5752 RSC.
- By elementary geometry, the diameter d of the circle and the length b of its inscribed square are related by the equation: d = √2b.
- The diameter d of the circle and the length b of its inscribed equilateral triangle are related by the equation: d = 2b/√3. As the three interatomic distances are not identical, the average value for pairwise interatomic distances between the projections of peripheral atoms N1(C12), N7 and C20 onto the ab-plane is used for the calculation.
-
(a) J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7332 CrossRef CAS PubMed;
(b) K. Iritani, M. Ikeda, A. Yang, K. Tahara, K. Hirose, J. S. Moore and Y. Tobe, Langmuir, 2017, 33, 12453–12462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and X-ray crystallographic structural analyses of five crystal forms with CCDC 1861306–1861310 (L1a–L2c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce02133c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Hak-Fun
Chow
a,
Hak-Fun
Chow