
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeoxygenation of amine N-oxides using gold nanoparticles supported on carbon nanotubes†
Simon
Donck
ab,
Edmond
Gravel
b,
Nimesh
Shah
a,
Dhanaji V.
Jawale
b,
Eric
Doris
*b and
Irishi N. N.
Namboothiri
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Mumbai 400 076, India. E-mail: irishi@chem.iitb.ac.in
bCEA, iBiTecS, Service de Chimie Bioorganique et de Marquage, 91191 Gif-sur-Yvette, France. E-mail: eric.doris@cea.fr
First published on 2nd June 2015
Abstract
Deoxygenation of a variety of aromatic and aliphatic amine N-oxides has been carried out in excellent yield using dimethylphenylsilane as the reducing agent under the catalytic influence of a carbon nanotube–gold nanohybrid at room temperature. Low catalyst loading, good TON and TOF values, and recyclability of the catalyst are some of the salient features of our methodology.
Introduction
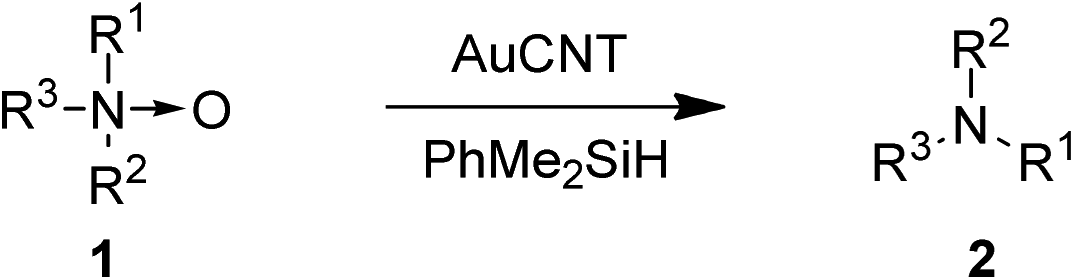
Deoxygenation of amine N-oxides to corresponding amines is a useful transformation in organic synthesis.1 Execution of this transformation in a mild and highly selective fashion is of significance not only in synthetic chemistry but also in biology, for instance, for drug metabolism studies.2 Although numerous stoichiometric and catalytic methods are reported in the literature for the reduction of amine N-oxides to amines,1,3–6 most of them require harsh reaction conditions and/or suffer from low activity or selectivity. Recently, Kaneda and co-workers have reported the deoxygenation of amine N-oxides using hydroxyapatite-supported gold nanoparticles (AuHAP)7 but the latter method is limited to pyridine N-oxides and requires rather high catalytic loadings.In recent years, we have exploited the remarkable catalytic activity of nanoparticles of various metals such as Au, Ru, Rh and Pd supported on carbon nanotube (CNT)–organic bilayer assembly in diverse organic transformations.8,9 Carbon nanotubes were chosen as support due to their chemical inertness, high surface area, and ability to stabilize transient higher oxidation states of metals.10 The activity of our gold–carbon nanotube assembly (AuCNT) has already been demonstrated for the oxidation of silanes and alcohols, the reductive amination and N-formylation of aldehydes, as well as in the one-pot alcohol oxidation–imine formation for the synthesis of quinoxalines.8 In a similar fashion, RuCNT was successfully employed for the selective reduction of nitroarenes to anilines and arylhydroxylamines, PdCNT for the room temperature Suzuki coupling of aryl halides and Tsuji–Wacker oxidation of terminal olefins, and RhCNT for the dehydrogenation of N-heterocycles.9 Herein we report for the first time the use of the AuCNT nanohybrid in the catalytic deoxygenation of amine N-oxides (Scheme 1).
 | ||
| Scheme 1 General scheme for the deoxygenation of amine N-oxides over AuCNT. | ||
Results and discussion
The AuCNT nanohybrid was assembled as reported elsewhere.8a Briefly, carbon nanotubes were sonicated in the presence of a polymerizable anionic amphiphilic surfactant in water, resulting in the formation of nanoring-like structures at the surface of the CNTs. The rings were then polymerized by UV irradiation which permitted to reinforce the cohesion of the supramolecular assembly. In a second step, a polycationic polymer layer was deposited on the primary coating and gold nanoparticles were finally added to afford the final hybrid (AuCNT). The nanohybrid was obtained as an aqueous suspension ([Au] = 3 mM, as measured by ICP-MS). The deoxygenation of quinoline N-oxide 1a to quinoline 2a was chosen as the model reaction for our studies (Table 1). Two different silanes (e.g. iPr3SiH and PhMe2SiH) were screened as hydride source in dry THF, at room temperature, and under N2 atmosphere (entries 1–2). Silanes were selected as hydride source because of their mildness and easy handling and the use of an anhydrous solvent in the above transformations was governed by the fact that water classically acts as oxygen atom source in the AuCNT-mediated oxidation of silanes into silanols.8a While there was no reaction in the presence of iPr3SiH (entry 1), which was attributed to the high steric demand of the reagent, quinoline 2a was isolated in 84% yield in the presence PhMe2SiH as the reducing agent (entry 2). Other solvents such as dichloromethane and toluene were less effective than THF affording quinoline 2a in lower yields (76% and 78%, respectively, entries 3–4). As expected, there was no reaction in the absence of silane or catalyst confirming the requirement of both the reducing agent and the catalyst in this transformation (entries 5–6). The reaction also did not proceed when the multilayer CNT-assembly alone (without AuNP) or HAuCl4 was used as the catalyst (entries 7–8).|
|
||||
|---|---|---|---|---|
| Entry | Silane | Catalyst | Solvent | Yieldb (%) |
| a Conditions: amine N-oxide (0.1 mmol), catalyst (0.4 mol%), THF (1 mL), PhMe2SiH (1.1 equiv.), N2, RT, 24 h. b Isolated yield after silica gel column chromatography. c No reaction. | ||||
| 1 | iPr3SiH | AuCNT | THF | NRc |
| 2 | PhMe 2 SiH | AuCNT | THF | 84 |
| 3 | PhMe2SiH | AuCNT | CH2Cl2 | 76 |
| 4 | PhMe2SiH | AuCNT | Toluene | 78 |
| 5 | — | AuCNT | THF | NRc |
| 6 | PhMe2SiH | — | THF | NRc |
| 7 | PhMe2SiH | CNT | THF | NRc |
| 8 | PhMe2SiH | HAuCl4 | THF | NRc |

Having confirmed the key role of AuCNT in catalyzing the silane-mediated deoxygenation of N-oxides 1, we proceeded to investigate the scope of the reaction using various aromatic and aliphatic amine N-oxides 1a–l (Table 2). Besides quinoline N-oxide 1a which afforded quinoline 2a in 84% yield (entry 1), isoquinoline N-oxide 1b and several pyridine N-oxides 1c–h were subjected to deoxygenation under our optimized conditions (entries 2–8). Thus, parent pyridine N-oxide 1c and alkyl and aryl substituted pyridine N-oxides 1d and 1e, respectively, underwent smooth deoxygenation to provide corresponding pyridines 2c–e in excellent yield (92–94%, entries 3–5). The remarkable functional group tolerance of our experimental conditions is evident from facile deoxygenation of pyridine N-oxides 1f–h, bearing substituents such as bromine, thiol, and carboxylic acid, to pyridines 2f–h in good to excellent yield (79–96%, entries 6–8). The deoxygenation of adenosine N-oxide 1i to adenosine 2i also took place in excellent yield (91%, entry 9), highlighting the wide applicability of our methodology to complex molecules including natural products. Representative examples for the deoxygenation of aliphatic, including alicyclic and benzylic, N-oxides 1j–l to amines 2j–l in high yield (88–91%, entries 10–12) further exemplified the scope of the AuCNT nanocatalyst, although two equivalents of silane were needed for the latter substrates.
|
|
|||||
|---|---|---|---|---|---|
| Entry | N-Oxide | Amine | Yieldb (%) | ||
| a Conditions: amine N-oxide (0.1 mmol), AuCNT (0.4 mol%), THF (1 mL), PhMe2SiH (1.1 equiv.), N2, RT, 24 h. b Isolated yield after silica gel column chromatography. c DMSO was used instead of THF. d 2 equiv. of PhMe2SiH. | |||||
| 1 | 1a |

|
2a |

|
84 |
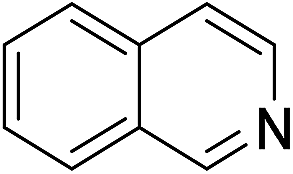
| 2 | 1b |

|
2b |
|
94 |
| 3 | 1c |

|
2c |

|
94 |
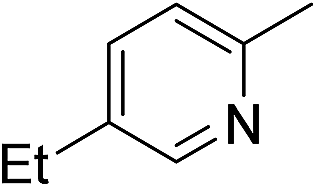
| 4 | 1d |

|
2d |
|
92 |
| 5 | 1e |

|
2e |

|
92 |
| 6 | 1f |
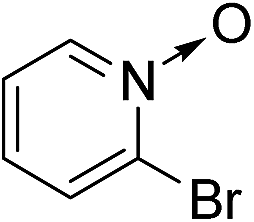
|
2f |

|
96 |
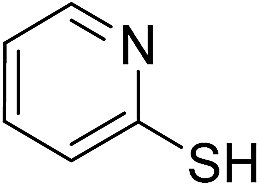
| 7 | 1g |

|
2g |
|
88 |
| 8 | 1h |

|
2h |

|
79 |
| 9c | 1i |

|
2i |

|
91 |
| 10d | 1j |

|
2j |
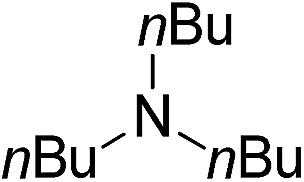
|
88 |
| 11d | 1k |

|
2k |
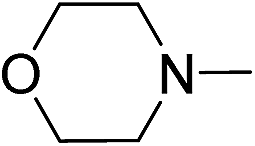
|
89 |
| 12d | 1l |

|
2l |

|
91 |
In order to understand the nature of the catalysis, we carried out a partial deoxygenation of quinoline N-oxide 1a under the optimized conditions (vide supra). After stirring for 7 h, the AuCNT catalyst was removed by centrifugation. At that point, only 22% conversion was reached, as determined by 1H-NMR. The catalyst-free supernatant was then stirred for another 16 h, but no further conversion was detected, thus confirming the heterogeneous nature of the catalyst.
The efficacy of the AuCNT catalyst was further evaluated by performing the deoxygenation of quinoline N-oxide 1a using only 0.03 mol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) % of the catalyst. After 24 h of reaction, quinoline 2a was formed in 39% yield, which corresponded to a turn-over number (TON) of 1300 and a turn-over frequency (TOF) of 54 h−1.
% of the catalyst. After 24 h of reaction, quinoline 2a was formed in 39% yield, which corresponded to a turn-over number (TON) of 1300 and a turn-over frequency (TOF) of 54 h−1.
Recyclability of the AuCNT catalyst was assessed in the deoxygenation of quinoline N-oxide 1a to quinoline 2a as the model reaction (Table 3). At the end of the reaction, the mixture was centrifuged and the supernatant was subjected to usual work up to isolate quinoline 2a. The recovered AuCNT catalyst was reused 4 times without any appreciable drop in the yield or the reaction rate.

The mechanism of the AuCNT/PhMe2SiH-mediated deoxygenation of amine N-oxides was thereafter investigated using an isotopic labelling experiment. The classical silane source was replaced by deuterated PhMe2SiD11 and a reaction was set starting from quinoline N-oxide 1a (Scheme 2). After the usual work-up, 2H-NMR showed the incorporation of deuterium at the C2-position of quinoline. By analogy to what occurs with other noble metals, it is likely that phenyldimethyl silane undergoes oxidative Si-D insertion on the surface of the supported gold nanoparticles. This insertion produces an activated silane species that reacts with the N-oxide substrate with simultaneous transfer of a deuteride at C2. The transient intermediate then rearomatizes with concomitant elimination of silanol, affording deuterated 2a′ in 83% yield and with ca. 50% isotope incorporation.
 | ||
| Scheme 2 Proposed mechanism for the deoxygenation of amine N-oxides. | ||
Conclusions
Carbon nanotube–gold nanohybrid was found to be an efficient catalytic system to perform the silane-mediated reduction of a variety of aromatic and aliphatic amine N-oxides. Some attractive features of our methodology are: (i) the mildness of the reaction conditions as the deoxygenated amines were formed at room temperature, (ii) the very low catalytic loading (0.4 mol%), (iii) the possible recyclability of the catalyst, and (iv) the scope of the process that was also applied to aliphatic amines N-oxides. The results obtained in the context of this study thus compare favourably to previous reports in terms of overall efficiency.Acknowledgements
Support from the Indo-French Centre for the Promotion of Advanced Research (IFCPAR)/Centre Franco-Indien pour la Promotion de la Recherche Avancée (CEFIPRA) is gratefully acknowledged (Project no. 4705-1). S.D. thanks the Department of Science and Technology (Government of India) and the Science & Technology Department (French Embassy in India) for the award of a Raman-Charpak fellowship. The TEM-team platform (CEA, iBiTec-S) is acknowledged for help with TEM images. The “Service de Chimie Bioorganique et de Marquage” belongs to the Laboratory of Excellence in Research on Medication and Innovative Therapeutics (ANR-10-LABX-0033-LERMIT).Notes and references
- (a) D. P. Sebesta, Handbook of Reagents for Organic Synthesis: Reagents for Silicon-Mediated Organic Synthesis, ed. P. L. Fuchs, 2011, p. 309 Search PubMed; (b) S. Kano, Ventron Alembic, 1980, 19, 1 CAS; (c) T. Cohen, U. S. C. F. S. T. I., AD Rep., 1965, 625414, 14 Search PubMed; (d) F. Devinsky, Acta Fac. Pharm. Univ. Comenianae, 1986, 40, 63 CAS; (e) E. Ochiai, in Aromatic Amine Oxides, Elsevier, Amsterdam, 1967, pp. 184–209 Search PubMed; (f) S. Oae, Lect. Heterocycl. Chem., 1978, 4, 69 CAS.
- P. Kulanthaivel, R. J. Barbuch, R. S. Davidson, P. Y. Gregory, A. Rener, E. L. Mattiuz, C. E. Hadden, L. A. Goodwin and W. J. Ehlhardt, Drug Metab. Dispos., 2004, 32, 966 CAS.
- CeCl3·7H2O–Zn: (a) B. W. Yoo, H. I. Jung, S. H. Kim, Y. S. Ahn and J. Y. Choi, Bull. Korean Chem. Soc., 2013, 34, 359 CrossRef CASBiCl3/In: (b) B. W. Yoo and J. W. Choi, Synth. Commun., 2009, 39, 3550 CrossRef CAS PubMedNbCl5/Zn: (c) K. Oh and W. E. Knabe, Tetrahedron, 2009, 65, 2966 CrossRef CAS PubMedRuCl3·xH2O: (d) S. Kumar, A. Saini and J. S. Sandhu, Tetrahedron Lett., 2005, 46, 8737 CrossRef CAS PubMedNaBr: (e) A. H. Khuthier, T. Y. Ahmed and R. S. Al-Taan, Iraqi J. Sci., 1986, 27, 313 CrossRef CASNiCl2·6H2O/In: (f) J. H. Han, J. W. Choi, K. I. Choi, J. H. Kim, C. M. Yoon and B. W. Yoo, Bull. Korean Chem. Soc., 2005, 26, 1921 CrossRef CASCu(I) salts: (g) S. K. Singh, M. S. Reddy, M. Mangle and K. R. Ganesh, Tetrahedron, 2007, 63, 126 CrossRef CAS PubMedMoO2Cl2: (h) P. M. Reis and B. Royo, Tetrahedron Lett., 2009, 50, 949 CrossRef CAS PubMedDichlorodioxomolybdenum(VI): (i) R. Sanz, J. Escribano, Y. Fernandez, R. Aguado, M. R. Pedrosa and F. J. Arnaiz, Synlett, 2005, 1389 CrossRef CAS.
- Pd/C: (a) J.-P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781 CrossRef CAS PubMed; (b) W. Sun, M. Wang, Y. Zhang and L. Wang, Org. Lett., 2015, 17, 426 CrossRef CAS PubMed, [Pd(OAc)2]/dppf: (c) J. A. Fuentes and M. L. Clarke, Synlett, 2008, 2579 CAS.
- Metal clusters/complexes: (a) Rh-carbonyl cluster: K. Kaneda, T. Takemoto, K. Kitaoka and T. Imanaka, Organometallics, 1991, 10, 846 CrossRef CASN-fused porphyrin-rhenium complexes: (b) M. Toganoh, K. Fujino, S. Ikeda and H. Furuta, Tetrahedron Lett., 2008, 49, 1488 CrossRef CAS PubMedOxorhenium(v): (c) Y. Wang and J. H. Espenson, Org. Lett., 2000, 2, 3525 CrossRef CAS PubMedMo(CO)6: (d) K. W. Yoo, J. W. Choi and C. M. Yoon, Tetrahedron Lett., 2006, 47, 125 CrossRef PubMedFe clusters: (e) T. Itoh, T. Nagano and M. Hirobe, Chem. Pharm. Bull., 1986, 34, 2013 CrossRef CAS.
- Fe powder: (a) R. Ma, A.-H. Liu, C.-B. Huang, X.-D. Li and L.-N. He, Green Chem., 2013, 15, 1274 RSC Borohydride exchange resin-copper sulphate; (b) T. B. Sim, J. H. Ahn and N. M. Yoon, Synthesis, 1996, 324 CrossRef CAS Sodium borohydride-RANEY® nickel:; (c) N. B. Gowda, G. K. Rao and R. A. Ramakrishna, Tetrahedron Lett., 2010, 51, 5690 CrossRef CAS PubMed; (d) S. Lindsay, R. John, J. M. Linford, L. C. McKeer and P. M. Morris, J. Chem. Soc., Perkin Trans. 2, 1984, 1099 Search PubMed.
- Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem.–Eur. J., 2011, 17, 1768 CrossRef CAS PubMed.
- AuCNT: (a) J. John, E. Gravel, A. Hagège, H. Li, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533 CrossRef CAS PubMed; (b) R. Kumar, E. Gravel, A. Hagège, H. Li, D. V. Jawale, D. Verma, I. N. N. Namboothiri and E. Doris, Nanoscale, 2013, 5, 6491 RSC; (c) D. V. Jawale, E. Gravel, V. Geertsen, H. Li, N. Shah, I. N. N. Namboothiri and E. Doris, ChemCatChem, 2014, 6, 719 CrossRef CAS PubMed; (d) R. Kumar, E. Gravel, A. Hagège, H. Li, D. Verma, I. N. N. Namboothiri and E. Doris, ChemCatChem, 2013, 5, 3571 CrossRef CAS PubMed; (e) N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2014, 6, 2201 CrossRef CAS PubMed; (f) N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2015, 7, 57 CrossRef CAS PubMed; (g) D. V. Jawale, E. Gravel, V. Geertsen, H. Li, N. Shah, R. Kumar, J. John, I. N. N. Namboothiri and E. Doris, Tetrahedron, 2014, 70, 6140 CrossRef CAS PubMed.
- RuCNT: (a) D. V. Jawale, E. Gravel, C. Boudet, N. Shah, V. Geertsen, H. Li, I. N. N. Namboothiri and E. Doris, Chem. Commun., 2015, 51, 1739 RSCPdCNT: (b) D. V. Jawale, E. Gravel, C. Boudet, N. Shah, V. Geertsen, H. Li, I. N. N. Namboothiri and E. Doris, Catal. Sci. Technol., 2015, 5, 2388 RSC; (c) S. Donck, E. Gravel, N. Shah, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2015 DOI:10.1002/cctc.201500241RhCNT: (d) D. V. Jawale, E. Gravel, N. Shah, V. Dauvois, H. Li, I. N. N. Namboothiri and E. Doris, Chem.–Eur. J., 2015, 21, 7039 CrossRef CAS PubMed.
- Reviews: (a) J. John, E. Gravel, I. N. N. Namboothiri and E. Doris, Nanotechnol. Rev., 2012, 1, 515 CAS; (b) E. Gravel, D. Bernard, I. N. N. Namboothiri and E. Doris, Actual. Chim., 2015, 393–394, 82 Search PubMed.
- S. B. Takale, S. M. Tao, X. Q. Yu, X. J. Feng, T. Jin, M. Bao and Y. Yamamoto, Org. Lett., 2014, 16, 2558 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H, 2H, 13C spectra, a typical experimental procedure for amine N-oxide deoxygenation, and spectral data. See DOI: 10.1039/c5ra08845c |
| This journal is © The Royal Society of Chemistry 2015 |