
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli responsive behavior in a D–A–D organic fluorophore: the role of cold crystallization†
Mohammad Afsar
Uddin‡
 *a,
Sergio
Gámez-Valenzuela‡
b,
M. Carmen
Ruiz Delgado
*b and
Berta
Gómez-Lor
*a
*a,
Sergio
Gámez-Valenzuela‡
b,
M. Carmen
Ruiz Delgado
*b and
Berta
Gómez-Lor
*a
aInstituto de Ciencia de Materiales de Madrid, CSIC, Cantoblanco, 28049, Madrid, Spain. E-mail: m.auddin@csic.es; bgl@icmm.csic.es
bDepartment of Physical Chemistry, University of Málaga, Campus de Teatinos s/n, 29071 Málaga, Spain. E-mail: carmenrd@uma.es
First published on 28th April 2025
Abstract
Herein, dimethyl-4,7-di(2-thienyl)-2,1,3-benzothiadiazole-5,6-dicarboxylate (DTBTE) is introduced as a highly efficient fluorophore with multi-stimuli responsive properties. Its molecular structure, consisting of a benzothiadiazole (BTD) core functionalized with ester groups and thiophene rings, results in a highly flexible framework with dynamic conformational behavior and low rotational barriers. These characteristics allow DTBTE to exhibit cold crystallization at 80 °C, accompanied by a pronounced and reversible color change. This transition can be reversed, restoring the amorphous state via external stimuli such as solvent fuming or mechanical shearing. Density functional theory (DFT) calculations, combined with advanced characterization techniques such as single-crystal X-ray analysis and IR and Raman spectroscopy, provide detailed insight into the mechanisms driving the phase transformations and their associated color changes. In particular, the flexibility of the ester groups plays a crucial role in enabling cold crystallization, as it facilitates the formation of a complex hydrogen bond network. Additionally, the switchable emission colours are attributed to cis/trans conformational changes in the thiophene groups during the transition between the crystalline and the amorphous phases. These findings highlight DTBTE as a promising platform for designing efficient stimuli-responsive materials and provide valuable guidelines for the development of new fluorophores exhibiting a dynamic behavior.
Introduction
Luminescent solids that exhibit changes in their structural arrangement and emission properties in response to external stimuli such as pressure, temperature or solvent vapours represent a fascinating area of research with a wide range of promising applications, such as sensors,1 security features,2–4 optoelectronics,5,6 optical data storage and recording,7,8 biomedical applications,9,10etc.In the pursuit of developing stimuli-responsive light-emitting materials, organic compounds offer clear advantages.11 Their luminescence arises from delocalized π-electrons along the molecular backbone, where the extent of π-conjugation plays a critical role in determining their optical properties.12 As a result, conjugated molecules with easily accessible conformers are particularly appealing for designing stimuli-responsive fluorophores.13 Additionally, the light-emission characteristics of π-conjugated molecular solids are profoundly influenced by the supramolecular organization of their molecular components.14 This strong dependence between emission characteristics and molecular or supramolecular arrangement offers enormous versatility in designing fluorescent materials capable of responding to external stimuli through phase transformations. Furthermore, their inherent flexibility, solution processability and compatibility with lightweight, large-area substrates make them particularly attractive for most of the intended applications.
To effectively optimize these functionalities, significant efforts have been dedicated to understanding the underlying mechanisms that govern the structural transformations and resultant luminescence modulation in these materials. Although many stimuli-responsive fluorophores are still discovered serendipitously, recent advances in material synthesis, state-of-the-art characterization techniques, and theoretical modelling have significantly deepened our understanding of the dynamic processes operating in these materials, unlocking their potential for practical applications.15,16
For instance, in the search for stimuli-responsive fluorophores, molecules with a donor–acceptor (D–A) electronic structure are of particular interest.17–19 Small modifications in their intramolecular configuration or intermolecular arrangement can lead to substantial changes in their electronic delocalization, giving rise to pronounced spectral shifts in their absorption or emission properties. In this context, benzothiadiazole (BTD) is a preferred moiety for designing responsive D–A light-emitting compounds.16,19–22 This electron-deficient heterocycle exhibits high fluorescence quantum yields and a strong propensity to adopt quinoid conformations, which promotes charge transfer between substituents and allows for tunable optical properties.23 Moreover, the presence of N and S atoms in its structure, capable of forming weak interactions,24,25 significantly enhances the propensity of BTD derivatives for crystallization and polymorphism.16,22,26 The inherent weakness of these interactions facilitates solid–solid phase transformations, leading to distinct supramolecular arrangements, altered electronic delocalization, and, consequently, diverse optical properties.16,27 Particularly interesting are molecules containing BTD moieties flanked by thiophene rings, owing to the low inter-ring rotational barriers afforded by their molecular structure. This structural feature facilitates access to both stable and metastable states with varying degrees of electronic delocalization, enabling the modulation of their light-emitting properties by external stimuli such as grinding, thermal annealing, and solvent vapor annealing.22,28–30
Cold crystallization is an intriguing and somewhat counterintuitive phase transition observed in certain organic materials. Unlike conventional crystallization processes, which typically occur as substances cool from a hot solution or melt, cold crystallization involves crystallization during heating. This phenomenon has generated significant interest as a mechanism for developing heat storage materials31,32 or more active pharmaceutical compounds.33,34 However, its potential has only been recently investigated in the context of responsive fluorochromic materials.17,35
While cold crystallization behavior is relatively common in semicrystalline polymers,36 it is far less frequent in small molecules, although it can still be observed in certain glass-forming compounds. This phenomenon typically requires functional groups capable of forming different rotamers to inhibit crystallization and facilitate supercooling, as well as long molecular chains that enhance molecular mobility.37,38 Upon reheating, these molecules gain the necessary thermal energy and increased molecular mobility, allowing them to reorganize and form a crystalline structure.
The connection between cold-crystallization temperature (Tcc) and mechanochromic luminescence in organic materials has been recently highlighted.39 In fact, the Tcc has been proposed as a crucial parameter for reversion from the force-induced amorphous metastable states.40 However, controlling Tcc based on the chemical structure of small molecules can be complex. The length and branching of the attached alkyl groups can effectively influence Tcc,41,42 but also the position of alkyl substituents has been found to influence the steric barrier that affects the cold-crystallization process and polymorphism.43
Building on this concept, we present dimethyl-4,7-di(2-thienyl)-2,1,3-benzothiadiazole-5,6-dicarboxylate (DTBTE) as a highly emissive multi-stimuli responsive fluorophore (see Fig. 1). In this compound, cold crystallization effectively triggers changes in both color and photoluminescence, inducing a phase transition between amorphous and crystalline states, as confirmed by differential scanning calorimetry (DSC) and X-ray diffraction (XRD). Notably, both shearing and solvent fuming produce the amorphization of this material, thereby reversing the color change and further highlighting the dynamic and reversible nature of its thermoresponsive behavior. Note that although this compound has previously been reported as a monomer in the synthesis of D–A polymers for photovoltaics, its detailed photophysical properties remain unexplored.44–46
 | ||
| Fig. 1 Chemical structure of DTBTE. | ||
Results and discussion
DTBTE was synthesized by a Diels–Alder reaction between 4,6-di(thien-2-yl)-thieno[3,4-c][1,2,5]-thiadiazole and dimethyl acetylenedicarboxylate in anhydrous xylene under reflux, following a previously reported procedure45 (see Scheme S1 for the detailed synthetic route, ESI†). After purification by column chromatography using CH2Cl2, DTBTE was obtained as a powdery solid in good yield. The compound was subsequently characterized by NMR spectroscopy, which confirmed its purity. Interestingly, despite this confirmation, the solid exhibited a non-homogeneous color, with distinct orange and yellow regions. These differences were more pronounced when the powder was illuminated under a UV lamp, revealing highly emissive orange and green areas. Curiously, heating the sample to 80 °C induced a transformation into the green-emitting form (see Fig. 2a inset). Further heating of the green-emitting solid resulted in melting, which takes place at around 140 °C. Interestingly, upon heating the compound above its melting temperature and cooling down to room temperature, it regains the orange emission. | ||
| Fig. 2 (a) DSC curves of DTBTE during heating (red) and cooling (blue) at 10 °C min−1, with exothermic events directed upward. Inset: Color transformation observed under UV-light. (b) Evolution of the PXRD patterns upon annealing at 80 °C. (c) PXRD patterns showing the interconversion from amorphous to crystalline upon heating and from crystalline to amorphous upon shearing. | ||
To shed light on this thermochromic behavior, the thermal properties of DTBTE were investigated by DSC. As outlined in Fig. 2a, the first heating cycle revealed a glass transition near room temperature (26 °C), followed by an exothermic cold crystallization transition at 86 °C and an endothermic peak corresponding to the melting point at 136 °C. During the cooling cycle, only the glass transition process was observed, suggesting the formation of a supercooled amorphous state. As shown in Fig. 2, powder X-ray diffraction (PXRD) analysis further elucidated the cold crystallization process of DTBTE. The as-prepared solid showed only a low intensity peak at low angles and a broad diffuse halo at medium angles, indicating that it predominantly corresponded to an amorphous phase. However, heating the sample for different periods of time showed the emergence of a larger number of additional peaks, clearly indicating that crystallization was taking place (Fig. 2b). Scanning electron microscopy (SEM) images clearly demonstrate the textural changes undergone by DTBTE films during the transition from the amorphous to the crystalline phase after annealing (Fig. S23, ESI†), further supporting the conclusions reached by PXRD analysis. Interestingly, simply shearing the crystallized material with a spatula induces its amorphization once again, as confirmed by X-ray diffraction, resulting in the reversal of the color change (Fig. 2c and Fig. S24, ESI†). Alternatively, amorphization can be induced by solvent fuming, probably involving a solution/supercooling process upon solvent evaporation. Such reversibility of the thermochromic behavior is of critical importance for its practical applications.
The photophysical properties of DTBTE were initially studied in solution using UV-vis and fluorescence spectroscopy (Fig. S5, ESI†). The UV-vis absorption spectrum of a 10−4 M solution of DTBTE in CH2Cl2 shows four predominant absorption bands at 266, 312, 325 and 406 nm. According to time-dependent DFT (TD-DFT) calculations, the bands localized at higher energies (266, 312 and 325 nm) are assigned to π–π* electronic excitations, whereas the lowest-energy band (S0 → S1) centered at 406 nm corresponds to an intramolecular charge transfer (ICT) transition from the HOMO, delocalized on the thiophene rings and the phenyl group of the BTD unit, to the LUMO, which is mainly localized on the central BTD unit but also delocalizes along the flanking thiophene rings (see Fig. S9 and S10, ESI†). Calculations reveal that the position of this absorption band is notably influenced by the relative orientation of the flanking thiophene rings with respect to the central BTD unit. In fact, a progressive redshift is observed when going from a cis–cis to a cis–trans or trans–trans conformation (see Fig. S11, ESI†), attributed to enhanced intramolecular electronic communication between the electron-deficient BTD unit and the electron-rich thienyl rings.
Interestingly, the dichloromethane solution of DTBTE exhibits strong fluorescence when irradiated at 400 nm, with an emission band centered at 577 nm and a high photoluminescence quantum yield of ΦF = 66.9%. As shown in Fig. 3a, the fluorescence of DTBTE undergoes a redshift as the solvent polarity increases from cyclohexane to DMSO. This solvatochromic behavior suggests the presence of a highly polar excited state. In polar solvents like DMSO, the excited state is more stabilized than the ground state due to stronger solvent–solute interactions, likely involving dipole–dipole interactions and hydrogen bonding. This stabilization reduces the energy gap between the excited and ground states, resulting in emission at longer wavelengths as the solvent polarity increases. Conversely, in aqueous environments, DTBTE exhibits a distinctly different behavior characterized by a marked blueshift in fluorescence. This opposite trend suggests the involvement of unique intermolecular interactions in this solvent, and in fact visualization of the aqueous solution under a microscope reveals the presence of nanocrystals in suspension. The stability of the crystalline phase in water may lead to distinct packing arrangements that alter intermolecular interactions, thereby altering the photophysical properties of DTBTE compared to its behavior in other solvents.
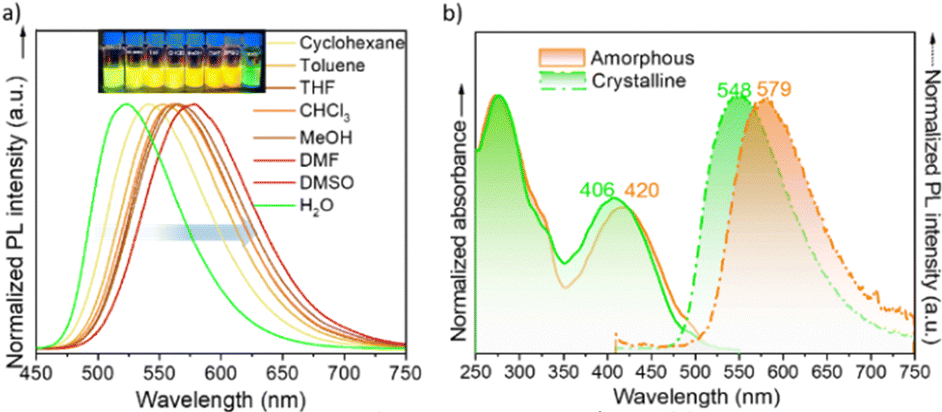 | ||
| Fig. 3 (a) Fluorescence spectra of DTBTE in various solvents. (b) UV-vis absorption (solid line) and fluorescence (dashed line) spectra for both the amorphous (orange) and crystalline (green) states of DTBTE. The fluorescence spectra were recorded using an excitation wavelength of 400 nm. | ||
Curiously, in the solid state, the as-prepared amorphous phase shows optical properties remarkably similar to those observed in solution, with an emission maximum (λmax) at 579 nm and a quantum yield of ΦF = 68.4% (see Fig. 3b). This behavior is likely attributed to the random conformational distribution and lack of molecular ordering in the amorphous phase, which in some ways resembles the dynamic nature of the solution state. Importantly, heating the sample for 60 minutes leads to a crystallization-induced emission enhancement (CIEE) effect, with the crystalline state achieving a quantum yield of ΦF = 77.4% and notable blueshifts of 14 nm in the lowest-energy absorption band and 31 nm in the emission maximum (λmax = 548 nm). Interestingly, similar lifetimes (τF ≈ 7–8 ns) are observed in solution, crystalline, and amorphous phases (Fig. S7–S9, ESI†), thus suggesting that the radiative decay is an intrinsic property of the molecule itself, and is not heavily influenced by the surrounding environment.
To shed light on the origin of the color changes accompanying the phase transformation, we attempted to grow single crystals suitable for X-ray analysis. Green-emitting crystals were obtained by the slow evaporation of a solution of DTBTE in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1). Single crystal structure determination revealed that DTBTE crystallizes in the orthorhombic space group Pna21, with two independent molecules per unit cell. These molecules differ primarily in the dihedral angles between the flanking thiophene rings and the BTD moiety, which are situated one above and one below the BTD plane (60.9° and 44.5° in one molecule vs. 56.5° and 43.5° in the other; Fig. S3, ESI†). In both independent molecules, the sulphur atoms of the thiophene rings align in the same direction as the sulphur atom of the BTD core, thereby assuming a cis–cis conformation with respect to the BTD unit. The two independent molecules crystallize with BTD groups oriented oppositely, resulting in the formation of dimers via 2S–2N square chalcogen interactions, a common motif in the crystallization of BTD moieties.24 The dimers further organize into slipped stacks, stabilized by CH–π interactions involving the six-membered ring of the BTD moiety and one of the flanking thiophene rings of a neighboring molecule in the stack (Fig. 4a). These stacks subsequently arrange into layers (Fig. 4b), where the peripheral ester groups participate in additional interactions, creating a complex network of non-covalent bonds (Fig. 4c). It should be noted that a different polymorph of this compound was previously reported;46 however, in our hands, only this form could be obtained. In fact, the powder X-ray diffractogram simulated from the single crystal data matches perfectly with that obtained through cold crystallization (see Fig. S4, ESI†).
:MeOH (1:1). Single crystal structure determination revealed that DTBTE crystallizes in the orthorhombic space group Pna21, with two independent molecules per unit cell. These molecules differ primarily in the dihedral angles between the flanking thiophene rings and the BTD moiety, which are situated one above and one below the BTD plane (60.9° and 44.5° in one molecule vs. 56.5° and 43.5° in the other; Fig. S3, ESI†). In both independent molecules, the sulphur atoms of the thiophene rings align in the same direction as the sulphur atom of the BTD core, thereby assuming a cis–cis conformation with respect to the BTD unit. The two independent molecules crystallize with BTD groups oriented oppositely, resulting in the formation of dimers via 2S–2N square chalcogen interactions, a common motif in the crystallization of BTD moieties.24 The dimers further organize into slipped stacks, stabilized by CH–π interactions involving the six-membered ring of the BTD moiety and one of the flanking thiophene rings of a neighboring molecule in the stack (Fig. 4a). These stacks subsequently arrange into layers (Fig. 4b), where the peripheral ester groups participate in additional interactions, creating a complex network of non-covalent bonds (Fig. 4c). It should be noted that a different polymorph of this compound was previously reported;46 however, in our hands, only this form could be obtained. In fact, the powder X-ray diffractogram simulated from the single crystal data matches perfectly with that obtained through cold crystallization (see Fig. S4, ESI†).
 | ||
| Fig. 4 (a) View of the 2S–2N square interactions responsible for the formation of the dimers and the close contacts involved in the formation of slipped stacks. (b) Layered arrangement of the molecules, viewed along the a-axis. (c) Close contacts between the peripheral ester groups. | ||
It should be noted that although DFT calculations for an isolated DTBTE molecule (at both B3LYP/6-31G** and ωB97X-D/6-31G** levels of theory) predict an asymmetric cis–cis conformation as the most stable structure, in agreement with the conformation observed in the crystal, the calculated potential energy surface probes the flexibility of DTBTE around the C–C inter-ring between the BTD and thienyl rings (see Fig. 5a and Fig. S12, S13, S16–S18, ESI†). In fact, a very low potential energy barrier of 1.86 kcal mol−1 is predicted for the rotation of one external thiophene ring relative to the central BTD unit to reach the cis–trans conformation, which is subsequently doubled (3.34 kcal mol−1) when the trans–trans disposition is achieved (Fig. 5b).
 | ||
| Fig. 5 (a) Dihedral potential energy hypersurface E = E(ϕ1, ϕ2) for an isolated DTBTE molecule, calculated at the ωB97X-D/6-31G** level of theory. The most stable conformation is shown as an inset. (b) Schematic potential energy profile for the three possible conformations of DTBTE, highlighting their relative energies with respect to the most stable conformation and the energy barriers for interconversion. The optimized molecular structures of each conformer are also shown. | ||
Considering the high rotational flexibility between the BTD and thienyl rings, and the significant influence of their conformation on the photophysical properties of DTBTE, it seems reasonable to consider that the red-shifting observed in the absorption and emission bands during the crystalline to amorphous phase transformation is the result of cis/trans conformational changes.
Interestingly, the conformational flexibility of the carbonyl ester groups with respect to the BTD unit has also been explored, resulting in energy barriers of ∼4 kcal mol−1 (Fig. S14 and S15, ESI†). These results highlight that the molecular motion of the ester groups might also play a relevant role in the responsive transformation.
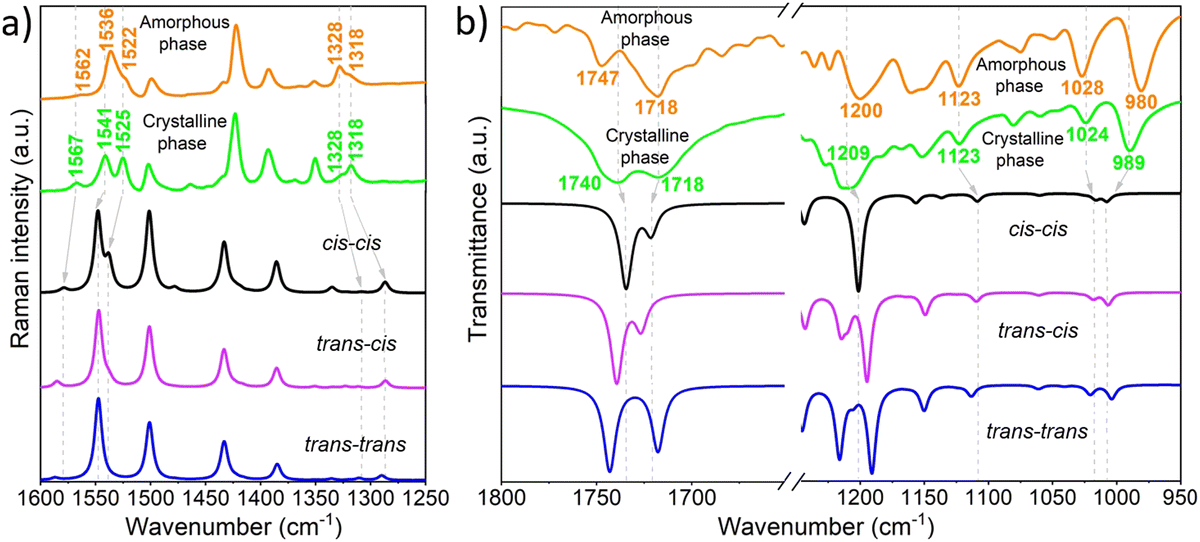
Raman spectroscopy has proven to be a powerful tool for providing valuable molecular structural information on BTD derivatives, including insights into π-conjugation efficiency and backbone planarity,20,47 while indirectly revealing intermolecular changes during phase transitions.16,26,48,49 In this study, Raman spectroscopy was utilized to investigate conformational changes occurring during the interconversion between crystalline and amorphous states. As shown in Fig. 7a, notable differences are observed in the Raman spectra of the two phases. The Raman band located at 1567 cm−1 in the crystal, ascribed to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the phenyl ring of BTD, downshifts 5 cm−1 when transitioning from the crystalline to the amorphous phase. A similar downshift is observed in the bands localized at 1541 and 1525 cm−1, which correspond to C–C/CC stretching vibrations mainly delocalized on the thiophene rings with contribution from the phenyl ring of BTD (see Fig. S19 and S20, ESI†), together with an increase in the I1541/I1525 intensity ratio in the amorphous phase. These frequency shifts point to alterations in the electronic communication between the central BTD unit and the external thiophene rings, leading to an increased π-electron delocalization in the amorphous phase. These spectral changes are nicely reproduced by DFT-calculations when rotating from cis to trans conformation (see Fig. 6a), which also predict a decrease in the intensity of the low frequency band at ∼1525 cm−1 for the trans conformers. This supports the hypothesis that the increased I1541/I1525 intensity ratio in the amorphous phase is due to a higher ratio of trans-conformers compared to the crystalline state, where molecules predominantly adopt the cis conformation.
C stretching vibrations of the phenyl ring of BTD, downshifts 5 cm−1 when transitioning from the crystalline to the amorphous phase. A similar downshift is observed in the bands localized at 1541 and 1525 cm−1, which correspond to C–C/CC stretching vibrations mainly delocalized on the thiophene rings with contribution from the phenyl ring of BTD (see Fig. S19 and S20, ESI†), together with an increase in the I1541/I1525 intensity ratio in the amorphous phase. These frequency shifts point to alterations in the electronic communication between the central BTD unit and the external thiophene rings, leading to an increased π-electron delocalization in the amorphous phase. These spectral changes are nicely reproduced by DFT-calculations when rotating from cis to trans conformation (see Fig. 6a), which also predict a decrease in the intensity of the low frequency band at ∼1525 cm−1 for the trans conformers. This supports the hypothesis that the increased I1541/I1525 intensity ratio in the amorphous phase is due to a higher ratio of trans-conformers compared to the crystalline state, where molecules predominantly adopt the cis conformation.
 | ||
| Fig. 6 Comparison of experimental (a) Raman and (b) IR spectra for the amorphous (orange line) and crystalline (green line) phases of DTBTE, as well as the DFT-predicted spectra (ωB97X-D/6-31G**) considering the three possible conformations. | ||
Furthermore, the crystalline-to-amorphous transformation induces an interconversion in the relative intensity ratio of the bands at 1328 and 1318 cm−1. These bands correspond to CC and CO stretching vibrations localized on the thiophene rings and ester groups, and CC and CN vibrations of the BTD unit, respectively (see Fig. S20, ESI†). Notably, these vibrations are highly sensitive to the surrounding chemical environment. In fact, different intramolecular close contacts (i.e., S–N vs. S–O and N–H vs. O–H) are expected depending on whether the thiophene rings align in the same or opposite orientation relative to the ester groups. This observation further supports the idea of increased conformational changes involving the thiophene groups during the phase transformation.
The low rotation barrier predicted for the carbonyl groups in DTBTE (see Fig. S14 and S15, ESI†), along with the complex network of hydrogen bonds observed involving the ester groups at positions 4 and 5, suggests that the molecular motion of these moieties may also play a fundamental role during the amorphous-to-crystal transition. Heating is expected to facilitate their mobility, allowing them to reorganize and establish interactions among themselves, which could induce crystallization. It is important to note that, even though the carbonyl groups in esters are not typically considered strong hydrogen bond acceptors, their involvement in the induction of cold crystallization in polymers via hydrogen bonding has been previously reported.50
With the aim of deepening the knowledge of the role played by ester groups in the molecular arrangement of these phases, infrared spectroscopy (IR) was used. Interestingly, remarkable differences are observed in the carbonyl ν(CO) stretching vibrations of the ester groups when comparing the two phases (see Fig. 7b and Fig. S21, ESI†). Specifically, upon amorphization, a substantial narrowing of these bands is observed together with an upshifting of the higher-energy peak located at 1740 cm−1 in the crystal, suggesting a distinct chemical environment surrounding the ester groups in the two phases. This effect is likely attributed to the disappearance of CO⋯H bonds between the ester groups present in the DTBTE crystal. Note that these hydrogen bonds might facilitate partial electron density delocalization from CO, weakening the bond and reducing the ν(CO) frequency.51,52
 | ||
| Fig. 7 Messages written and decorated on various substrates using a DTBTE solution (0.5% wt) in THF as ink, illustrating emission colour changes upon heating. Photographs were taken under irradiation with a 365 nm UV lamp. Top row: Images of paper, al foil, glass, wood, and flexible polypropylene film (PP) at 25 °C; Bottom row: Images obtained after heating at 80 °C. | ||
Interestingly, IR spectral features, whose trends are well reproduced by molecular modelling (see Fig. 7b and Fig. S21, S22, ESI†), also support a predominant cis–trans distribution in the amorphous phase (see ESI† for more details). These include the 9 cm−1 downshift observed in the band at 1209 cm−1 and the increased separation between the two bands at 1024 and 989 cm−1 going from the crystalline to the amorphous phase, all of them strongly associated with C–H bending of the thiophene rings and C–O stretching vibrations of the ester groups. It is important to highlight that, unlike the typical crystallization observed during solvent vapor exposure, DTBTE experiences a transition from a crystalline to an amorphous phase. This atypical behavior is likely due to a solvent-induced disruption of the hydrogen bond network, particularly involving the ester groups,53,54 as well as conformational changes around the BTD-thiophene dihedral angle triggered by solvent interactions during the transition from crystalline to amorphous phases55,56 as supported by Raman and IR spectroscopy studies, along with DFT calculations. This study clearly underscores the critical role of the ester moieties in facilitating cold crystallization and driving the thermo/mechanochromic behavior of DTBTE.
DTBTE, with its multi-stimuli responsiveness and reversible fluorochromic properties, shows significant potential as a security ink due to its dynamic features. Security inks that enable the creation of thermally activated, color-changing messages, arouse much interest as they are difficult to reproduce yet easily recognizable. Notably, messages written on paper using a damp cotton swab soaked in a THF solution of DTBTE (0.5% wt) changed their emission color when heated to 80 °C (Fig. 7a and b) demonstrating the material's responsiveness and potential applications in security ink technologies.
To further investigate the versatility of highly emissive DTBTE, we evaluated whether its stimuli-responsive properties were preserved across different substrates. Orange-emitting amorphous films were cast on various materials, including glass, wood, and flexible polypropylene sheets. Upon controlled heating, these films transformed into crystalline green-emitting phases (Fig. 7c–j). Interestingly, vapor fuming using a mask allowed us to reverse the color changes in the uncovered areas, enabling the creation of messages or motifs that could subsequently be erased by reheating the films (Fig. S27, ESI†). The high compatibility of this material with various substrates, while maintaining its switchable emission, demonstrates the adaptability and potential of DTBTE for applications such as responsive security labels and erasable recording or sensing technologies.
Conclusion
In conclusion, this study establishes DTBTE, a highly flexible compound consisting of a BTD core functionalized at the 4,7-positions with thiophene rings and at the 5,6-positions with ester groups, as a highly efficient multi-stimuli responsive fluorophore. The combination of low rotational barriers of groups able to establish hydrogen-bond networks, and dynamic conformational behavior enables DTBTE to exhibit cold crystallization at 80 °C, which can be easily reversed by solvent fuming and mechanical shearing. Interestingly, this reversible amorphous/crystalline phase is accompanied by pronounced emission color changes. Through a combined study involving DFT and TD-DFT calculations, single crystal X-ray analysis and advanced spectroscopic techniques, valuable insights on the molecular dynamics responsible for these reversible fluorochromic transformations have been achieved. Particularly, this study has allowed us to identify the molecular motion of ester groups as a key driver of these phase transformations, while the accompanying switchable emission color is attributed to cis/trans conformational changes involving the thiophene groups. Interestingly, the ability of DTBTE to maintain its multi-stimuli responsive properties across a variety of substrates, from paper to glass, wood, and flexible polymers, highlights its potential for practical applications in fields ranging from security inks, sensing, security labels, and erasable recording. These findings position DTBTE as a versatile and innovative material for next-generation luminescent technologies.Author contributions
MAU and SGV were contributed equally. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this study's findings are available within the article and its ESI.† All relevant experimental data, including spectral analyses (IR, Raman), X-ray single crystal data, and density functional theory (DFT) computational results, can be accessed upon reasonable request from the corresponding author. Additionally, detailed characterization results relevant to the synthesis and properties of dimethyl-4,7-di(2-thienyl)-2,1,3-benzothiadiazole-5,6-dicarboxylate (DTBTE) are available at DOI: https://doi.org/10.1002/advs.201600032Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by MCIN/AEI/10.13039/501100011033 (project PID2022-139548NB-I00, PID2023-150022NB-I00), and by Junta de Andalucía (P09FQM-4708 and P18-FR-4559). MAU thanks to Spanish Ministry of Science, Innovation and Universities for RAMÓN Y CAJAL 2021 fellowship (Project RYC2021-034211-I) and CSIC for ATRACCIÓN DE TALENTO RAMÓN Y CAJAL 2021 (Project 20236AT033). The authors would like to thank the computer resources, technical expertise and assistance provided by the SCBI (Supercomputing and Bioinformatics) center and the vibrational spectroscopy (EVI) lab of the Research Central Services (SCAI) of the University of Málaga. The authors acknowledge the support of SCXRD Laboratory at SIdI - Universidad Autónoma de Madrid. The equipment used was funded by MCINN/AEI/10.13039/501100011033 NextGenerationEU/PRTR (EQC2021-007518-P). The authors also acknowledge the MICIN for the REDES project “RED2022-134503-T”.Notes and references
- V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed.
- N. Sardari, A. Abdollahi and M. Farokhi Yaychi, ACS Appl. Mater. Interfaces, 2023, 15(49), 57656–57678 CAS.
- R. Martín, M. Echeverri, F. González, A. de Andrés, M. C. Ruiz Delgado, A. Concellón, J. L. Serrano and B. Gómez-Lor, Adv. Opt. Mater., 2024, 2400965 Search PubMed.
- H. Mardani, H. Roghani-Mamaqani, S. Shahi and D. Roustanavi, ACS Appl. Polym. Mater., 2023, 5, 1092–1102 CrossRef CAS.
- T. Pan, S. Liu, L. Zhang, W. Xie and C. Yu, Light: Sci. Appl., 2022, 11, 59 CrossRef CAS PubMed.
- R. Zheng, Y. Wei, Z.-C. Zhang, Z.-Y. Wang, L.-L. Ma, Y. Wang, L. Huang and Y.-Q. Lu, Responsive Mater., 2023, 1, e20230017 CrossRef.
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef.
- S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS.
- M. Bril, S. Fredrich and N. A. Kurniawan, Smart Mater. Med., 2022, 3, 257–273 CrossRef.
- W. Ma, D. Hua, R. Xiong and C. Huang, Mater. Adv., 2023, 4, 458–475 RSC.
- Y. Huang, L. Ning, X. Zhang, Q. Zhou, Q. Gong and Q. Zhang, Chem. Soc. Rev., 2024, 53, 1090–1166 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, NY, 3rd edn, corrected at 4, printing., 2010 Search PubMed.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499 RSC.
- B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592–3606 RSC.
- M. Echeverri, C. Ruiz, S. Gámez-Valenzuela, I. Martín, M. C. Ruiz Delgado, E. Gutiérrez-Puebla, M. Á. Monge, L. M. Aguirre-Díaz and B. Gómez-Lor, J. Am. Chem. Soc., 2020, 142, 17147–17155 CrossRef CAS PubMed.
- R. Martín, A. Sánchez-Oliva, A. Benito, I. Torres-Moya, A. M. Garcia, J. Álvarez-Conde, J. Cabanillas-González, P. Prieto and B. Gómez-Lor, J. Mater. Chem. C, 2024, 12, 2903–2910 RSC.
- Y. Wang, Z. He, G. Chen, T. Shan, W. Yuan, P. Lu and Y. Zhang, Chin. Chem. Lett., 2017, 28, 2133–2138 CrossRef CAS.
- T. Jadhav, B. Dhokale, Y. Patil, S. M. Mobin and R. Misra, J. Phys. Chem. C, 2016, 120, 24030–24040 CrossRef CAS.
- M. Echeverri, C. Ruiz, S. Gámez-Valenzuela, M. Alonso-Navarro, E. Gutierrez-Puebla, J. L. Serrano, M. C. Ruiz Delgado and B. Gómez-Lor, ACS Appl. Mater. Interfaces, 2020, 12, 10929–10937 CrossRef CAS.
- A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2018, 6, 10888–10901 RSC.
- S. Ito, S. Nagai, T. Ubukata and T. Tachikawa, CrystEngComm, 2021, 23, 5899–5907 RSC.
- P. Rietsch, S. Sobottka, K. Hoffmann, A. A. Popov, P. Hildebrandt, B. Sarkar, U. Resch-Genger and S. Eigler, Chem. – Eur. J., 2020, 26, 17361–17365 CrossRef CAS PubMed.
- M. R. Ams, N. Trapp, A. Schwab, J. V. Milić and F. Diederich, Chem. – Eur. J., 2019, 25, 323–333 CrossRef CAS PubMed.
- M. Echeverri, I. Martín, A. Concellón, C. Ruiz, M. S. Anselmo, E. Gutiérrez-Puebla, J. L. Serrano and B. Gómez-Lor, ACS Omega, 2018, 3, 11857–11864 CrossRef CAS PubMed.
- M. A. Uddin, R. Martín, S. Gámez-Valenzuela, M. Echeverri, M. C. Ruiz Delgado, E. Gutiérrez Puebla, A. Monge and B. Gómez-Lor, J. Am. Chem. Soc., 2024, 146, 27690–27700 CrossRef CAS PubMed.
- M. Echeverri, C. Ruiz and B. Gómez-Lor, CrystEngComm, 2021, 23, 5925–5930 RSC.
- G. He, L. Du, Y. Gong, Y. Liu, C. Yu, C. Wei and W. Z. Yuan, ACS Omega, 2019, 4, 344–351 CrossRef CAS PubMed.
- R. Yoshida, T. Tachikawa and S. Ito, Cryst. Growth Des., 2022, 22, 547–558 CrossRef CAS.
- R. Yoshida, T. Tachikawa and S. Ito, Chem. Commun., 2022, 58, 6781 RSC.
- K. Turunen, M. R. Yazdani, S. Puupponen, A. Santasalo-Aarnio and A. Seppälä, Appl. Energy, 2020, 266, 114890 CrossRef CAS.
- M. R. Yazdani, J. Etula, J. B. Zimmerman and A. Seppälä, Green Chem., 2020, 22, 5447–5462 RSC.
- R. Chang, Q. Fu, Y. Li, M. Wang, W. Du, C. Chang and A. Zeng, CrystEngComm, 2017, 19, 335–345 RSC.
- S. E. Lapuk, T. A. Mukhametzyanov, C. Schick and A. V. Gerasimov, Int. J. Pharm., 2021, 599, 120427 CrossRef CAS.
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS.
- B. Wunderlich, J. Chem. Phys., 1958, 29, 1395–1404 CrossRef CAS.
- A. Honda, S. Kakihara, M. Kawai, T. Takahashi and K. Miyamura, Cryst. Growth Des., 2021, 21, 6223–6229 CrossRef CAS.
- T. Rozwadowski, M. Massalska-Arodź, Ł. Kolek, K. Grzybowska, A. Bąk and K. Chłędowska, Cryst. Growth Des., 2015, 15, 2891–2900 CrossRef CAS.
- S. Ito, J. Photochem. Photobiol., C, 2022, 51, 100481 CrossRef CAS.
- S. A. Sharber, K.-C. Shih, A. Mann, F. Frausto, T. E. Haas, M.-P. Nieh and S. W. Thomas, Chem. Sci., 2018, 9, 5415–5426 RSC.
- S. Nakamura, K. Okubo, Y. Nishii, K. Hirano, N. Tohnai and M. Miur, J. Mater. Chem. C, 2024, 12, 2370–2378 RSC.
- A. Honda, S. Kakihara, M. Kawai, T. Takahashi and K. Miyamura, Cryst. Growth Des., 2021, 21, 6223–6229 CrossRef CAS.
- A. Honda, Y. Hibi, K. Matsumoto, M. Kawai and K. Miyamura, RSC Adv., 2022, 12, 7229–7236 RSC.
- L. Lan, Z. Chen, Q. Hu, L. Ying, R. Zhu, F. Liu, T. P. Russell, F. Huang and Y. Cao, Adv. Sci., 2016, 3, 1600032 CrossRef.
- B. A. Abdulahi, X. Li, M. Mone, B. Kiros, Z. Genene, S. Qiao, R. Yang, E. Wang and W. Mammo, J. Mater. Chem. A, 2019, 7, 19522–19530 RSC.
- C. B. Nielsen, R. S. Ashraf, N. D. Treat, B. C. Schroeder, J. E. Donaghey, A. J. P. White, N. Stingelin and I. McCulloch, Adv. Mater., 2015, 27, 948–953 CrossRef CAS PubMed.
- S. Gámez-Valenzuela, M. Comí, S. R. González, M. C. R. Delgado, M. Al-Hashimi and R. Ponce Ortiz, J. Mater. Chem. C, 2023, 11, 8027–8036 RSC.
- H. Chung, D. Dudenko, F. Zhang, G. D’Avino, C. Ruzié, A. Richard, G. Schweicher, J. Cornil, D. Beljonne, Y. Geerts and Y. Diao, Nat. Commun., 2018, 9, 278 CrossRef PubMed.
- D. W. Davies, B. Seo, S. K. Park, S. B. Shiring, H. Chung, P. Kafle, D. Yuan, J. W. Strzalka, R. Weber, X. Zhu, B. M. Savoie and Y. Diao, Nat. Commun., 2023, 14, 1304 CrossRef CAS.
- O. Toledano, O. Gálvez, M. Sanz, C. Garcia Arcos, E. Rebollar, A. Nogales, M. C. García-Gutiérrez, G. Santoro, I. Irska, S. Paszkiewicz, A. Szymczyk and T. A. Ezquerra, Macromolecules, 2024, 57, 2218–2229 CrossRef CAS PubMed.
- H. Samuel, U. NwekeMaraizu and E. E. Etim, J. Chem. Rev., 2023, 5(4), 439–465 CAS.
- S. C. Edington, J. C. Flanagan and C. R. Baiz, J. Phys. Chem. A, 2016, 120, 3888–3896 CrossRef CAS.
- T.-F. Zhang, C. Jiang, Y.-B. Wang, J.-X. Wang, X. Zhang, G. Qian and B. Li, J. Mater. Chem. A, 2025, 13, 12492–12499 RSC.
- P. Sudhakar and T. P. Radhakrishnan, J. Mater. Chem. C, 2019, 7, 7083–7089 RSC.
- K. Dyk, V. Kinzhybalo, G. Czernel, W. Grudziński, Y. Horak, S. Butenko and D. M. Kamiński, CrystEngComm, 2023, 25, 971–980 RSC.
- Z. Li, G. Jin, W. Yuan, B. Huang, X. Liang and Y. Tao, Dyes Pigm., 2022, 206, 110605 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, single crystal X-ray structure determination, photophysical characterization, computational details and DFT and TD-DFT calculations, Raman analysis, IR analysis, stimuli-responsive luminescence experiments etc. CCDC 2415534. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00852b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |