
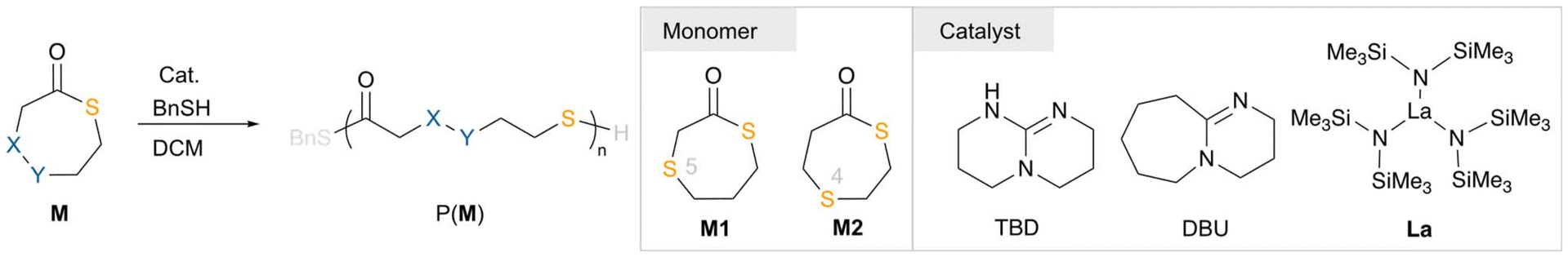
Chemically recyclable poly(thioether-thioester)s via ring-opening polymerization of seven-membered thiolactones†
Long-Hai
Liu
,
Si-Qi
Wang
,
Hua-Zhong
Fan
,
Qing
Cao
,
Zhongzheng
Cai
 * and
Jian-Bo
Zhu
*
* and
Jian-Bo
Zhu
*
National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), College of Chemistry, Sichuan University, Chengdu 610064, People's Republic of China. E-mail: zzcai@scu.edu.cn; jbzhu@scu.edu.cn
First published on 16th January 2025
Abstract
Developing new sulfur-containing polymers with chemical recyclability as next-generation high-performance sustainable polymeric materials is in high demand. Herein, we prepared two seven-membered thiolactone monomers (M1 and M2) with sulfur incorporated at different positions. Both monomers displayed excellent reactivity towards ring-opening polymerization with >90% monomer conversion at room temperature. The resulting poly(thioether-thioester) products P(M)s exhibited high thermal stability and comparable mechanical properties (σB = ∼30 MPa, εB = ∼660%) to commercial low-density polyethylene. Chemical recycling of P(M1) to its monomer M1 could be accomplished with excellent yield and purity. Intriguingly, P(M1) could be applied for selective absorption and recovery of Au3+ with >99% efficiency by exploiting the chemical recyclability of P(M1).
Introduction
The accumulation of petroleum-based plastic waste has caused serious environmental problems.1–4 Currently, chemically recyclable polymers that are able to depolymerize back to their monomers under mild conditions have been developed as promising alternatives to traditional petroleum-based polymers.5–10 The recent decade has witnessed significant breakthroughs in chemically recyclable polymers from polyesters to polyamides.11–24 Monomer design represented an important strategy to manipulating the thermodynamics of the reversible ring-opening polymerization (ROP) and ring-closing depolymerization (RCD).25–28 The development of new chemically recyclable polymers as next-generation sustainable polymeric materials is in high demand.Sulfur-containing polymers have gained growing interest owing to their distinct merits such as outstanding optical and electrical characteristics and excellent adhesion to heavy metal ions.29–44 As a result, sulfur-containing polymeric materials provide potential as engineering plastic, optical, photoelectronic, and heavy metal capture materials.43,45–47 Consequently, developing a sulfur-containing polymer system with chemical recyclability and outstanding properties would be significant and highly desired. To date, polythioesters and polydisulfides have been investigated as chemically recyclable polymers (Fig. 1).48 These polythioesters illustrated stronger depolymerizability in comparison with their polyester analogues due to their elongated thioester bonds.49–57 In addition, polydisulfides have been reported as elegant systems for chemical recycling to monomers.58–63 Gutekunst and our group have reported a DTO monomer (1,4-dithian-2-one) as a facile platform to construct chemically recyclable poly(thioether-thioester)s.64,65 However, this monomer suffered from low polymerization conversions at room temperature due to the low ring strain and its polymer product displayed poor thermal stability. To overcome these shortcomings, we proposed to construct seven-membered thiolactones owing to their enhanced ring strain. Herein, we have prepared two seven-membered-ring based monomers 1,4-dithiepan-2-one (M1) and 1,4-dithiepan-5-one (M2) and investigated their ROP performance with organic bases or La complexes as catalysts. The produced poly(thioether-thioester) P(M)s exhibited high air and thermal stability and distinct thermal and mechanical properties. P(M2) with sulfur incorporated at the 4 position exhibited a melting temperature (Tm) of 95 °C and demonstrated high-performance mechanical properties (σB = 29.86 ± 2.02 MPa and εB = 663 ± 60%). Notably, P(M1) with sulfur at the 5 position behaved as a soft material, which could be applied for selective absorption and recovery of Au3+ (>99% efficiency). Intriguingly, chemically recycling of P(M1) to the monomer could be carried out in solution or bulk with excellent yields, featuring a highly efficient recycling system.
 | ||
| Fig. 1 Representative chemically recyclable sulfur-containing polymers. Access to chemically recyclable seven-membered-based poly(thioether-thioester)s. | ||
Results and discussion
Ring-opening polymerization
1,4-Dithiepan-2-one (M1) and 1,4-dithiepan-5-one (M2) were prepared according to a modified synthetic route.66 2.1 g of M1 as colourless crystals was obtained via cyclization of 1,3-dimercaptopropane and chloroacetyl chloride in the presence of Et3N. Further scaling up of this synthetic procedure led to a significant decrease in monomer yield. M2 was synthesized using a similar procedure except that 1,2-ethanedithiol and 3-chloropropionyl chloride were used as the starting materials. The 1H NMR spectrum of M1 was in a good agreement with a literature report.66 For M2, its 1H NMR spectrum was inconsistent with a previous report.67 However, the accurate chemical structure of M2 was confirmed by single-crystal X-ray diffraction analysis (Fig. S4†).With these monomers in hand, the polymerization study of M1 and M2 was next carried out in DCM solution at room temperature with organic bases TBD, DBU, and La as catalysts and benzyl mercaptan (BnSH) as an initiator (Table 1). Among these catalysts, TBD acted as the most effective catalyst and promoted robust polymerization of M1, which reached >90% conversions within 10 min (Table 1, entries 1–4). Size exclusion chromatography (SEC) analysis revealed that increasing the [M1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TBD]:[BnSH] ratios from 100:1:1 to 1000:1:1 led to a gradual increase in Mn from 8.8 to 62.1 kDa. These Mn values were lower than the theoretical values (Mn,Calcd) calculated from [M1]:[BnSH] ratios and monomer conversions. We believe that this is due to the inevitable chain transfer or the formation of a cyclic polymer. To verify our hypothesis, a P(M1) sample prepared with [M1]:[TBD]:[I] = 50:1:1 was examined by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. It displayed only one set of molecular ion peaks with the space between two neighbouring ion peaks being ∼148, which is in good agreement with the molar mass of M1. Upon a linear fitting of the data, it showed an intercept of 22.98 which matched the molecular weight of Na+(23.0), confirming the presence of the cyclic structure of P(M1) (Fig. S32†). At a [M1]:[DBU]:[BnSH] ratio of 500:1:1, the polymerization approached 73% conversion within 60 min, delivering P(M1) with an Mn of 33.9 and a dispersity (Đ) of 1.95 (Table 1, entry 5). Catalyst La displayed diminished activity with or without an alcohol initiator (Table 1, entries 6 and 7), presumably due to the stable coordination of thioether groups to the metal center, which further inhibited monomer insertion.
:[TBD]:[BnSH] ratios from 100:1:1 to 1000:1:1 led to a gradual increase in Mn from 8.8 to 62.1 kDa. These Mn values were lower than the theoretical values (Mn,Calcd) calculated from [M1]:[BnSH] ratios and monomer conversions. We believe that this is due to the inevitable chain transfer or the formation of a cyclic polymer. To verify our hypothesis, a P(M1) sample prepared with [M1]:[TBD]:[I] = 50:1:1 was examined by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. It displayed only one set of molecular ion peaks with the space between two neighbouring ion peaks being ∼148, which is in good agreement with the molar mass of M1. Upon a linear fitting of the data, it showed an intercept of 22.98 which matched the molecular weight of Na+(23.0), confirming the presence of the cyclic structure of P(M1) (Fig. S32†). At a [M1]:[DBU]:[BnSH] ratio of 500:1:1, the polymerization approached 73% conversion within 60 min, delivering P(M1) with an Mn of 33.9 and a dispersity (Đ) of 1.95 (Table 1, entry 5). Catalyst La displayed diminished activity with or without an alcohol initiator (Table 1, entries 6 and 7), presumably due to the stable coordination of thioether groups to the metal center, which further inhibited monomer insertion.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Monomer | Cat. | [M]/[Cat.]/[I] | Time (min) | Conv.b (%) | M n,Calcd (kg mol−1) | M n (kDa) | Đ |
| a Conditions: [M] = 1.1 M, initiator (I) = benzyl mercaptan, RT. b Monomer conversion measured by 1H NMR of the quenched solution. c Calculated from [M]0/[BnSH]0 × Conv. × MWM + MWBnSH. d Number-average molecular weight (Mn) and dispersity index (Đ = Mw/Mn), determined by size exclusion chromatography (SEC) at 40 °C in THF. e Number-average molecular weight (Mn) and dispersity index (Đ = Mw/Mn), determined by size exclusion chromatography (SEC) in DCM. | ||||||||
| 1 | M1 | TBD | 100:1:1 |
1 | 93 | 13.8 | 8.8 | 1.40 |
| 2 | M1 | TBD | 200:1:1 |
1 | 93 | 27.5 | 13.7 | 1.41 |
| 3 | M1 | TBD | 500:1:1 |
5 | 94 | 69.6 | 44.0 | 1.43 |
| 4 | M1 | TBD | 1000:1:1 |
10 | 95 | 140.6 | 62.1 | 1.65 |
| 5 | M1 | DBU | 500:1:1 |
60 | 73 | 54.0 | 33.9 | 1.92 |
| 6 | M1 | La | 1000:1:0 |
180 | 17 | — | 18.7 | 1.84 |
| 7 | M1 | La | 1000:1:3 |
180 | 12 | 5.9 | 24.7 | 1.51 |
| 8 | M2 | TBD | 200:1:1 |
1 | 97 | 28.7 | 47.2e | 1.51e |
| 9 | M2 | TBD | 500:1:1 |
10 | 98 | 72.5 | 101e | 1.55e |
| 10 | M2 | TBD | 1000:1:1 |
60 | 97 | 144 | 220e | 1.56e |
| 11 | M2 | TBD | 2000:1:1 |
120 | 77 | 228 | 256e | 1.62e |
M2 showed a similar reactivity pattern to M1 and polymerized rapidly to achieve >95% conversions with TBD as the catalyst (Table 1, entries 8–11). It should be noted that the resulting P(M2) products showed poor solubility in THF and their Mn values were then determined by SEC in a DCM eluent. With an increasing [M2]:[TBD]:[BnSH] ratio from 200:1:1 to 1000:1:1, the produced P(M2) exhibited a gradual growth in Mn from 47.2 to 220 kDa that was 1-fold higher than its corresponding theoretical value. We hypothesized that a poor correlation between relative molecular weights using polystyrene calibration and absolute molecular weights in these polar eluents could be responsible for the increased Mn. Notably, the polymerization of M2 with an [M2]/[TBD]/[I] ratio of 2000/1/1 afforded P(M2) with an Mn of 256 kDa and a Đ of 1.62.
Polymer characterization
To evaluate the thermal stability of these synthesized P(M) products, thermal gravimetric analysis (TGA) was first conducted. P(M1) and P(M2) demonstrated outstanding thermal stability with decomposition temperatures (Td, defined as the temperature at 5% weight loss) of 293 and 275 °C, respectively (Fig. 2a). These values were significantly higher than those of previously reported chemically recyclable poly(thioether-thioester) P(DTO) (Td = 227 °C).65 Next, the air stability was tested by placing P(M1) samples (Mn = 68.6 kDa, Đ = 1.67) inside an incubator at 25 °C, 40% relative humidity (RH) or 35 °C, 50% RH to monitor their weight and Mn changes over 5 weeks. As shown in Fig. 2b and c, the P(M1) sample exhibited negligible changes in Mn and Đ over 5 weeks. Moreover, these samples showed identical 1H NMR spectra to those of the pristine material after 5 weeks at 25 °C and 40% RH and 35 °C and 50% RH (Fig. S24 and S25†). These experiments underscored the excellent air and moisture stability of P(M1). | ||
| Fig. 2 Polymer characterization. (a) TGA curves of P(M)s. (b) Humidity stability test of P(M1) at 25 °C, 40% RH. (c) Humidity stability test of P(M1) at 35 °C, 50% RH. (d) DSC curves of P(M)s on the first heating scan. (e) DSC curves of P(M)s on the second heating scan. (f) Strain–stress curves of P(M)s. | ||
Differential scanning calorimetry (DSC) was utilized to study the thermal properties of the P(M) product. P(M1) with sulfur incorporated at the 5 position displayed a near-room-temperature Tm of 47 °C on the first heating scan. This melting transition disappeared on the second heating scan and only a clear glass transition with a glass-transition temperature (Tg) of −49 °C was observed (Fig. 2d and e), suggesting a slow crystallization rate. In contrast, sulfur-incorporation at the 4 position contributed to a Tm of ∼95 °C on the first and second heating scans for P(M2). The distinct crystallization property between P(M1) and P(M2) illustrated the influence of the heteroatom incorporation position. Specifically, P(M2) with sulfur-incorporation at the 4 position adopted a more rigid and organized backbone structure, which resulted in an increased Tm and crystallization rate in comparison with those of P(M1). Their semi-crystallinity was also confirmed by powder X-ray diffraction (PXRD) (Fig. S17 and S18†).
We next investigated the mechanical properties of P(M1) and P(M2) by tensile testing experiments. The dog-bone-shaped opaque P(M1) and P(M2) specimens were prepared by melt pressing at 50 °C and 110 °C, respectively. The formed P(M) films displayed similar Mn and Đ values with the original solid samples (Table S4†), indicative of no obvious degradation by hot pressing and further confirming the excellent thermal stability of P(M1) and P(M2). Both P(M1) and P(M2) were subjected to uniaxial extension experiments at a strain rate of 10 mm min−1 and exhibited distinct mechanical performance (Fig. 2f). P(M1) (Mn = 94.9 kDa, Đ = 1.56) containing an S atom at the 5 position showed a tensile strength at break (σB) of 13.19 ± 1.45 MPa, an elongation at break (εB) of 572 ± 45%, and a Young's modulus (E) of 165 ± 8 MPa. In comparison with the soft P(M1), P(M2) (Mn = 131.5 kDa, Đ = 2.08) exhibited both improved tensile strength and ductility, demonstrating εB = 29.86 ± 2.02 MPa, εB = 663 ± 60%, and E = 247 ± 28 MPa. These values were comparable to those of commercial low-density polyethylene (LDPE).21
Chemical recycling to the monomer
To gain better understanding of the influence of the sulfur incorporation position on the chemical recyclability of the resulting polymers, we set out to calculate the thermodynamics of these two polymerization systems. Their ceiling temperature (Tc) was determined by monitoring the polymerization equilibrium changes over a temperature range of 40 to 130 °C. Their standard-state thermodynamic parameters of and
and  were calculated according to van't Hoff plots (Tables S7 and 8†). The thermodynamic parameters were calculated to be
were calculated according to van't Hoff plots (Tables S7 and 8†). The thermodynamic parameters were calculated to be  and
and  for P(M1). P(M2) demonstrated
for P(M1). P(M2) demonstrated  and
and  . Their relative Tc values at [M]0 = 1.0 M for P(M1) and P(M2) were calculated to be 512 and 658 °C, respectively, manifesting the feasibility for depolymerizability. We inferred that the sulfur at the 5 position of M1 could contribute to a more stable ring conformation, thereby delivering a lower Tc in comparison with M2 having sulfur at the 4 position.
. Their relative Tc values at [M]0 = 1.0 M for P(M1) and P(M2) were calculated to be 512 and 658 °C, respectively, manifesting the feasibility for depolymerizability. We inferred that the sulfur at the 5 position of M1 could contribute to a more stable ring conformation, thereby delivering a lower Tc in comparison with M2 having sulfur at the 4 position.
In line with our thermodynamic study, P(M1) underwent efficient chemical recycling to M1 in the presence of 1 mol% of Sn(Oct)2 at 150 °C, producing pure M1 in 93% yield (Table S9†). To establish a close-loop economy of P(M1), the recycled M1 was treated with TBD and BnSH for repolymerization. To our delight, the recycled M1 underwent repolymerization at [M1]:[TBD]:[I] ratios of 100:1:1 and 500:1:1 without a decrease in polymerization reactivity, approaching 95% and 92% conversion within 10 min, respectively (Table S12†). These results highlighted the feasibility and efficiency of chemical recycling of P(M1) (Fig. 3).
 | ||
| Fig. 3 Chemical recycling of P(M)s. (a) Scheme for depolymerization. (b) The sublimation setup for the recycling of P(M1). (c) 1H NMR spectra of starting M1, P(M1), recycled M1, and recycled P(M1). | ||
Surprisingly, although various catalytic systems including t-BuOK, PhSNa, Sn(Oct)2, Sn(OAc)2, La, and Sb2O3 were screened for thermal bulk depolymerization of P(M2) at 100–175 °C (Table S10†), it was found that only 60% yield of M2 could be collected from the depolymerization of P(M2) with PhSNa in 11 h at 170 °C. We speculated that it could be attributed to the poor polymer–catalyst contact effectivity. Next, we examined the depolymerization of P(M2) in dilute solution (Table S11†). A 0.01 M solution of P(M2) at 160 °C with 5 mol% Sn(OAc)2 converted back to M2 in 62% yield in 11 h. Extending the depolymerization time led to no change in monomer recovery. Collectively, we inferred that the depolymerization of P(M2) was limited to the depolymerization kinetics.
Application of P(M1) for Au3+ absorption and recovery
Motivated by the excellent chemical recyclability of P(M1) and the presence of thioether groups in P(M1), we speculated that it could be applied for Au3+ absorption and recovery. The sulphur atoms in the thioether groups readily form coordinate bonds with the empty d orbitals of metal ions because of their lone pair electrons, creating a strong electrostatic attraction. In a typical test, P(M1) (4 mg) dissolved in 100 μL DCM was directly added into an aqueous solution containing ∼60 ppm Au3+ (4 mL). After stirring for 2 h, the precipitated solid was separated by filtration. The atomic absorption spectroscopy (AAS) analysis of the remaining solution after filtration showed a >99% removal efficiency of Au3+ (Table S13† and Fig. 4a). In comparison, the adsorption of other metal ions such as Co2+, Mn2+, and Cu2+ was almost negligible, which underlined the excellent adsorption selectivity of P(M1) for Au3+ over other metal ions. More importantly, P(M1) was able to accomplish the absorption and subsequent recovery process of Au3+ by the exploitation of its chemical recyclability. The P(M1) adsorbent after Au3+ adsorption was dried under vacuum and then transferred into a sublimation equipment. Sublimation was carried out at 140 °C under vacuum in the presence of Sn(OAc)2 as the catalyst. M1 could be collected as the sublimate in 70–73% yields (Table S16 and Fig. S31†). 1H NMR analysis revealed the high purity of the recovered M1. Meanwhile, the left solid at the bottom of the sublimate contained the recovered Au3+. This process revealed that P(M1) could serve as a promising chemically recyclable material for Au3+ adsorption and recovery. | ||
| Fig. 4 Application of P(M1) for Au3+ absorption and recovery. (a) Metal ion removal efficiency of P(M1). (b) The diagram of the Au3+ absorption and recovery process. | ||
Experimental
Materials and methods
All synthesis and manipulations of air- and moisture-sensitive materials were carried out in an argon-filled glovebox. All reagents from Adamas-beta and Energy Chemical were used as received unless otherwise stated.1H NMR and 13C NMR spectroscopy
1H and 13C NMR spectra were recorded on an Agilent 400-MR DD2 or a Bruker Advance 400 spectrometer (1H: 400 MHz, 13C: 100 MHz). Chemical shifts (δ) for 1H and 13C NMR spectra are given in ppm relative to TMS. The residual solvent signals were used as references for 1H and 13C NMR spectra and the chemical shifts converted to the TMS scale (CDCl3: δH = 7.26 ppm, δC = 77.00 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. All spectra were processed with MestReNova 14.1.1 software, and coupling constants were reported as observed.Size exclusion chromatography (SEC)
Measurements of polymer absolute weight-average molecular weight (Mw), number average molecular weight (Mn), and molecular weight distributions or dispersity indices (Đ = Mw/Mn) were performed via size exclusion chromatography (SEC). The SEC instrument consisted of an Agilent LC system equipped with one guard column and two PL gel 5 μm mixed-C gel permeation columns and coupled with an Agilent G7162A 1260 Infinity II RI detector. The analysis was performed at 40 °C using THF as the eluent at a flow rate of 1.0 mL min−1. The instrument was calibrated with nine polystyrene standards, and chromatograms were processed with Agilent OpenLab CDS Acquisition 2.5 molecular weight characterization software.Differential scanning calorimetry (DSC)
The melting-transition temperature (Tm) and glass-transition temperature (Tg) of purified and thoroughly dried polymer samples were measured by differential scanning calorimetry (DSC) on a TRIOS DSC25, TA Instrument. All Tg values were obtained from a second scan after the thermal history was removed from the first scan.Thermo-gravimetric analysis (TGA)
The decomposition temperatures (Td) and maximum rate decomposition temperatures (Tmax) of the polymers were measured by thermo-gravimetric analysis (TGA) on a TGA55 Analyzer, TA Instrument. The polymer samples were heated from ambient temperature to 500 °C at a heating rate of 10 °C min−1. The values of Tmax were obtained from derivative (wt%/°C) vs. temperature (°C) plots and defined by the peak values, while Td values were obtained from wt% vs. temperature (°C) plots and defined as the temperature at 5% weight loss.X-ray crystallographic analysis
Single crystal data were obtained using a Bruker D8 Venture single crystal X-ray diffractometer.Wide angle X-ray diffraction (WXRD)
Powder X-ray diffraction data were obtained using a Bruker D2 Phaser diffractometer with Cu-Kα radiation (λ = 1.5416 Å) at 30 kV and 10 mA (scan of 2θ = 1.5–60° with a speed of 1° min−1).Mechanical analysis
Tensile stress/strain testing was performed using an Instron 5967 universal testing system. Samples were made by melt press in a steel mold (50 × 4 × 0.4 mM2) and were stretched at a strain rate of 10 mm min−1 at ambient temperature until break. The measurements were performed 4 times for each test, and the values reported are averaged from the measured data.Atomic absorption spectrometer (AAS)
AAS data were obtained using a VARIAN SpectrAA 220FS/220Z.Conclusions
In conclusion, we have prepared two seven-membered thiolactone monomers M1 and M2 with sulfur incorporated at 4 and 5 positions. The robust polymerization of these thiolactone monomers approached >90% conversion at room temperature, affording semicrystalline P(M) products with Tm values up to 95 °C. These P(M) products exhibited high thermal stability (Td up to 293 °C) and excellent air stability. P(M1) and P(M2) exhibited distinct mechanical properties. P(M1) represented a soft material while P(M2) illustrated superior tensile strength and ductility to LDPE. More importantly, P(M1) could efficiently and selectively convert back to M1, which underwent repolymerization to establish a closed-loop lifecycle. By exploiting the chemical recyclability, P(M1) showcased high efficiency capability for selective adsorption and recovery of Au3+. Collectively, these poly(thioether-thioester)s based on the seven-membered thiolactones paved the pathway towards the development of next-generation chemically recyclable sulfur-containing polymers.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Data are available upon reasonable request from the authors.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1501700), the National Natural Science Foundation of China (22071163, 22301197, and 22371194), and the Fundamental Research Funds from Sichuan University (2023SCUNL103 and 2024SCUQJTX005). We would like to thank Peng-Chi Deng from the Analytical & Testing Center of Sichuan University as well as Dongyan Deng and Jing Li from the College of Chemistry at Sichuan University for compound testing.References
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51–55 CrossRef CAS PubMed.
- G. Hole and A. S. Hole, Sustainable Prod. Consum., 2020, 23, 42–51 CrossRef.
- F. Zhang, F. Wang, X. Wei, Y. Yang, S. Xu, D. Deng and Y.-Z. Wang, J. Energy Chem., 2022, 69, 369–388 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- X.-L. Li, K. Ma, F. Xu and T.-Q. Xu, Chem. – Asian J., 2023, 18, e202201167 CrossRef CAS PubMed.
- C. M. Plummer, L. Li and Y. Chen, Macromolecules, 2023, 56, 731–750 CrossRef CAS PubMed.
- Y. Sun, Z. An, Y. Gao, R. Hu, Y. Liu, H. Lu, X.-B. Lu, X. Pang, A. Qin, Y. Shen, Y. Tao, Y.-Z. Wang, J. Wang, G. Wu, G.-P. Wu, T.-Q. Xu, X.-H. Zhang, Y. Zhang, Z. Zhang, J.-B. Zhu, M. Hong and Z. Li, Sci. China: Chem., 2024, 67, 2803–2841 CrossRef CAS.
- S. Yang, S. Du, J. Zhu and S. Ma, Chem. Soc. Rev., 2024, 53, 9609–9651 RSC.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y.-X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS.
- R. M. Rapagnani, R. J. Dunscomb, A. A. Fresh and I. A. Tonks, Nat. Chem., 2022, 14, 877–883 CrossRef CAS.
- Y.-T. Yan, G. Wu, S.-C. Chen and Y.-Z. Wang, Sci. China: Chem., 2022, 65, 943–953 CrossRef CAS.
- C. Li, L. Wang, Q. Yan, F. Liu, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202201407 CrossRef CAS PubMed.
- Y.-M. Tu, X.-M. Wang, X. Yang, H.-Z. Fan, F.-L. Gong, Z. Cai and J.-B. Zhu, J. Am. Chem. Soc., 2021, 143, 20591–20597 CrossRef CAS.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2023, 62, e202212543 CrossRef CAS PubMed.
- L. Ye, X. Liu, K. B. Beckett, J. O. Rothbaum, C. Lincoln, L. J. Broadbelt, Y. Kratish and T. J. Marks, Chem, 2024, 10, 172–189 CAS.
- W. Zhang, J. Dai, Y.-C. Wu, J.-X. Chen, S.-Y. Shan, Z. Cai and J.-B. Zhu, ACS Macro Lett., 2022, 11, 173–178 CrossRef CAS.
- H. Chen, Z. Shi, T.-G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 25493–25498 CrossRef CAS.
- B. A. Abel, R. L. Snyder and G. W. Coates, Science, 2021, 373, 783–789 CrossRef CAS PubMed.
- C. Shi, W. Diment and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2024, e202405083 CAS.
- Q. Cao, Y.-M. Tu, H.-Z. Fan, S.-Y. Shan, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202400196 CrossRef CAS.
- D. Sathe, S. Yoon, Z. Wang, H. Chen and J. Wang, Chem. Rev., 2024, 124, 7007–7044 CrossRef CAS PubMed.
- C. Shi, Z.-C. Li, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Chem, 2021, 7, 670–685 CAS.
- Y.-M. Tu, F.-L. Gong, Y.-C. Wu, Z. Cai and J.-B. Zhu, Nat. Commun., 2023, 14, 3198 CrossRef CAS.
- J. Zhou, D. Sathe and J. Wang, J. Am. Chem. Soc., 2022, 144, 928–934 CrossRef CAS.
- Z. Cai, Y. Liu, Y. Tao and J.-B. Zhu, Acta Chim. Sin., 2022, 80, 1165–1182 CrossRef CAS.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185–267 CrossRef CAS.
- H. Li, S. M. Guillaume and J.-F. Carpentier, Chem. – Asian J., 2022, 17, e202200641 CrossRef CAS.
- C.-J. Zhang, T.-C. Zhu, X.-H. Cao, X. Hong and X.-H. Zhang, J. Am. Chem. Soc., 2019, 141, 5490–5496 CrossRef CAS PubMed.
- W. Xiong, J. Dai, Z. Cai and J.-B. Zhu, Polymer, 2024, 290, 126515 CrossRef CAS.
- K. Li, J.-L. Cheng, M.-Y. Wang, W. Xiong, H.-Y. Huang, L.-W. Feng, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202405382 CrossRef CAS PubMed.
- H.-Z. Fan, X. Yang, J.-H. Chen, Y.-M. Tu, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202117639 CrossRef CAS PubMed.
- J. M. M. Pople, T. P. Nicholls, L. N. Pham, W. M. Bloch, L. S. Lisboa, M. V. Perkins, C. T. Gibson, M. L. Coote, Z. Jia and J. M. Chalker, J. Am. Chem. Soc., 2023, 145, 11798–11810 CrossRef CAS.
- S. Wang, Z.-Y. Tian and H. Lu, Angew. Chem., Int. Ed., 2024, 63, e202411630 CrossRef CAS PubMed.
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. Lavilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156 CrossRef CAS PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS.
- T.-J. Yue, L.-Y. Wang and W.-M. Ren, Polym. Chem., 2021, 12, 6650–6666 RSC.
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CrossRef CAS.
- A. L. Speelman, I. Čorić, C. Van Stappen, S. DeBeer, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2019, 141, 13148–13157 CrossRef CAS.
- Y. Sun, C. Zhang and X. Zhang, Chem. – Eur. J., 2024, 30, e202401684 CrossRef CAS PubMed.
- T.-J. Yue, W.-M. Ren and X.-B. Lu, Chem. Rev., 2023, 123, 14038–14083 CrossRef CAS.
- J.-Z. Zhao, T.-J. Yue, B.-H. Ren, X.-B. Lu and W.-M. Ren, Nat. Commun., 2024, 15, 3002 CrossRef CAS PubMed.
- W. Cao, F. Dai, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 978–986 CrossRef CAS.
- T. Tian, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS.
- W. Xiong and H. Lu, Sci. China: Chem., 2023, 66, 725–738 CAS.
- C. Shi, M. L. McGraw, Z. C. Li, L. Cavallo, L. Falivene and E. Y. X. Chen, Sci. Adv., 2020, 6, 1–12 Search PubMed.
- Y.-T. Guo, C. Shi, T.-Y. Du, X.-Y. Cheng, F.-S. Du and Z.-C. Li, Macromolecules, 2022, 55, 4000–4010 CrossRef CAS.
- M. Wang, Z. Ding, X. Shi, Z. Ma, B. Wang and Y. Li, Macromolecules, 2024, 57, 869–879 CrossRef CAS.
- Y. Wang, M. Li, J. Chen, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 22547–22553 CrossRef CAS PubMed.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, J. Am. Chem. Soc., 2019, 141, 4928–4935 CrossRef CAS PubMed.
- W. Xiong, W. Chang, D. Shi, L. Yang, Z. Tian, H. Wang, Z. Zhang, X. Zhou, E.-Q. Chen and H. Lu, Chem, 2020, 6, 1831–1843 CAS.
- D. Zhang, X. Wang, Z. Zhang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2024, 63, e202402233 CrossRef CAS PubMed.
- L. Zhou, L. T. Reilly, C. Shi, E. C. Quinn and E. Y.-X. Chen, Nat. Chem., 2024, 16, 1357–1365 Search PubMed.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303 CrossRef CAS PubMed.
- Q. Zhang, Y. Deng, C.-Y. Shi, B. L. Feringa, H. Tian and D.-H. Qu, Matter, 2021, 4, 1352–1364 CrossRef CAS.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2022, 4, eaat8192 CrossRef.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed.
- S. Pal, A. Sommerfeldt, M. B. Davidsen, M. Hinge, S. U. Pedersen and K. Daasbjerg, Macromolecules, 2020, 53, 4685–4691 CrossRef CAS.
- K. A. Stellmach, M. K. Paul, M. Xu, Y.-L. Su, L. Fu, A. R. Toland, H. Tran, L. Chen, R. Ramprasad and W. R. Gutekunst, ACS Macro Lett., 2022, 11, 895–901 CrossRef CAS.
- J. Dai, W. Xiong, M.-R. Du, G. Wu, Z. Cai and J.-B. Zhu, Sci. China: Chem., 2023, 66, 251–258 CrossRef CAS.
- C. M. Archer, J. R. Dilworth, D. V. Griffiths, M. J. Al-Jeboori, J. D. Kelly, C. Lu, M. J. Rosser and Y. Zheng, J. Chem. Soc., Dalton Trans., 1997, 1403–1410 RSC.
- Z. Xiao, Y. Wang, X. Chen, J. Long and Z. Wei, J. Chem. Sci., 2017, 129, 1595–1601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2354510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py01442a |
| This journal is © The Royal Society of Chemistry 2025 |