DOI:
10.1039/D4PY01169D
(Communication)
Polym. Chem., 2025,
16, 409-414
Synthesis of cyclic peptide-based [2]rotaxanes via copper-catalyzed azide–alkyne cycloaddition†
Received
19th October 2024
, Accepted 23rd December 2024
First published on 28th December 2024
Abstract
Cyclic peptide-based [2]rotaxanes were synthesized from cyclo(PG)4 and monocationic ammonium threads via copper-catalyzed azide–alkyne cycloaddition (CuAAC), achieving relatively high yields of up to 36%. Cyclic peptides that do not contain bulky side chains or amino acids that induce the formation of cis-amide bonds were found to be unsuitable for rotaxane synthesis. This innovative synthetic approach advances the development of multifunctional rotaxanes and opens new avenues for their applications in various fields.
Peptide-based materials are of great interest due to their exceptional biodegradability and biocompatibility because their degradation products are readily metabolized.1–6 In particular, cyclic peptides have attracted substantial attention owing to their unique cyclic topology, which offers greater stability than their linear counterparts.7–10 This distinctive cyclic structure enables the formation of complex structures, leading to the ability to develop advanced functionalities that are not achievable with linear or monocyclic peptides.11–16 Preorganized cyclic peptides serve as superior host compounds owing to their minimalized entropy loss during complex formation.17–22 Thus, interlocked structures such as rotaxanes can be formed by utilizing the receptor ability of cyclic peptides.23 Peptides and proteins with mechanically interlocked structures, whether naturally occurring or artificially synthesized, exhibit unique properties,24 featuring attributes such as enhanced thermal, mechanical, and chemical stability,25,26 proteolytic resistance27 and dynamic behaviors such as sliding and switching.28,29 Furthermore, peptide-based rotaxanes have the potential to act as cross-linking points, effectively enhancing the toughness of the resulting polymer materials without compromising their biodegradability or compatibility with polypeptides and proteins.30,31 Thus, peptide-based rotaxanes enable the development of peptide- or protein-based materials with unprecedented functions, and these peptide- and protein-based materials are anticipated to be applied in a variety of fields. As a first step in this direction, we successfully synthesized an all-peptide-based rotaxane in 1.2% yield via the active template method.32 Enhancing the yield of peptide-based rotaxanes is essential for establishing them as practical tools for a wide range of applications. However, synthesizing rotaxanes from peptides is challenging because the hydrogen bonds between cyclic and axial peptides are too weak for the axial peptide to penetrate the cyclic peptide. To address this issue, we propose that the yield of rotaxanes can be improved by using stronger interactions, such as electrostatic interactions, to drive rotaxane formation. A cyclic octapeptide with an alternating sequence of proline and glycine, cyclo(PG)4, was reported to efficiently bind cations because this peptide adopts a γ-turn structure with a glycine carbonyl pointing toward the center of the cyclic peptide.22 By leveraging this receptor ability, rotaxanes were formed from cyclo(PG)4 and diammonium threads in good yields (56–63%).23 However, this process took 7 days because of the poor reactivity of capping with cationic diol threads and bulky acid chlorides during rotaxane formation. Furthermore, when a monocationic ammonium thread was used without detailed molecular design, only trace amounts of rotaxane were obtained.
In this study, we employed copper-catalyzed azide–alkyne cycloaddition (CuAAC),33–36 a rapid and bioorthogonal reaction, to cap the pseudorotaxane synthesized in situ by the complexation of a monoammonium thread and cyclo(PG)4 to form a rotaxane structure. This approach minimized the dissociation of the preformed pseudorotaxane structure, enabling rotaxanes to be synthesized in moderate yields, even when a monoammonium thread was used.
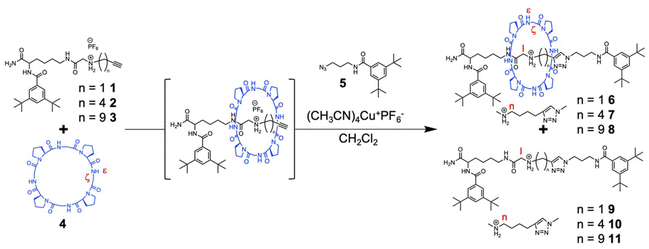
Cyclo(PG)4 and a sec-ammonium thread with both bulky and alkyne termini (Scheme 1, thread 1) were synthesized, which was confirmed by 1H NMR (Fig. S11 and S15†). The CuAAC-mediated strategy for the synthesis of peptide-based [2]rotaxanes is shown in Scheme 1. Cyclo(PG)4 (3 equiv.) and thread 1 (1 equiv.) were mixed in dichloromethane (DCM). The resulting mixture was then subjected to sonication for 1 min. Sonication enhanced the solubility of the monocationic ammonium thread in DCM and promoted its threading through the cyclic peptide, which is essential for the formation of the pseudorotaxane structure. To this solution, an azide compound with bulky termini (Scheme 1, compound 5 and Fig. S14,† 3 equiv.), a copper catalyst (1.2 equiv.), and lutidine (0.05 equiv.) were added, and the mixture was left to stand for 1 h at 25 °C. Rotaxane formation was confirmed by high-performance liquid chromatography (HPLC). The molecular weights of the eluates corresponding to each peak were determined, and the peak at 31.5 min was identified as the rotaxane. The rotaxane yield was determined to be 4.1% based on the HPLC peak areas (Fig. S1† and Table 1, entry 1).
 |
| | Scheme 1 Scheme of cyclic peptide-based [2]rotaxane synthesis. | |
Table 1 Effect of the molecular structure of sec-ammonium thread on rotaxane formation
| Entry |
Cyclic peptide |
Ammonium thread |
Yield/% |
| 1 |
Cyclo(PG)4 |
1 (n = 1) |
4.1 |
| 2 |
Cyclo(PG)4 |
2 (n = 4) |
36.1 |
| 3 |
Cyclo(PG)4 |
3 (n = 9) |
3.0 |
We investigated the effect of the molecular structure of the sec-ammonium thread with both bulky and alkyne termini on rotaxane formation. Given that the close proximity between the alkyne and NH2 groups results in reduced rotaxane yields due to steric hindrance in the CuAAC reaction and the tendency of the complex to dissociate, we synthesized a new monoammonium thread, thread 2 (Scheme 1), in which the alkyne and NH2 groups were positioned further apart (Scheme S1, Fig. S9, and S12†). As anticipated, the rotaxane yield increased to 36.1% when monoammonium thread 2 was used (Fig. 1 and Table 1, entry 2). To further enhance the stability of the pseudorotaxane structure, we synthesized monocationic ammonium thread 3 (Scheme 1), which contains a longer CH2 linkage than that of thread 2 (Scheme S1, Fig. S10, and S13†). This modification was expected to render the pseudorotaxane structure more resistant to dissociation, thereby improving the rotaxane yield. However, the yield of rotaxane was 3.0%, which was lower than that obtained with thread 2 (Fig. S2† and Table 1, entry 3). This is likely due to the increased distance between the alkyne and NH2 moiety, which hindered the formation of the pseudorotaxane structure. To further optimize the reaction conditions, we investigated the effect of reaction temperature. As the temperature increases, the pseudorotaxane becomes more susceptible to dissociation, leading to a decrease in rotaxane yield. Conversely, at lower temperatures, pseudorotaxane is more stable; nevertheless, the solubility of the starting materials and the efficiency of the capping reaction are decreased, meaning that the rotaxane yield is system-dependent.37 To investigate the effect of lower temperature on our rotaxane synthesis method, we conducted the rotaxane-forming reaction at 0 °C. The rotaxane yield was 30%, which was slightly lower than that obtained at 25 °C (Fig. S3† and Table 2, entry 4). Thus, in our rotaxane synthesis method, the decreased solubility and reduced efficiency of the capping reaction at lower temperatures have a greater impact than the stabilization of the pseudorotaxane. Consequently, synthesizing rotaxanes at lower temperatures is unsuitable for our synthetic system.
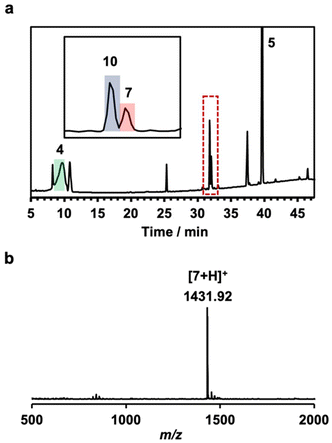 |
| | Fig. 1 (a) HPLC chromatogram of the crude reaction mixture. The inner box shows an enlarged view of the area within the red dashes. Red peak: rotaxane; blue peak: free thread; and green peak: free cyclic peptide. (b) MALDI-TOF MS spectrum of the fraction containing the red peak from the chromatogram in (a). | |
Table 2 Effect of reaction temperature on rotaxane formation
| Entry |
Cyclic peptide |
Reaction temperature/°C |
Yield/% |
| 2 |
Cyclo(PG)4 |
25 |
36.1 |
| 4 |
Cyclo(PG)4 |
0 |
30.1 |
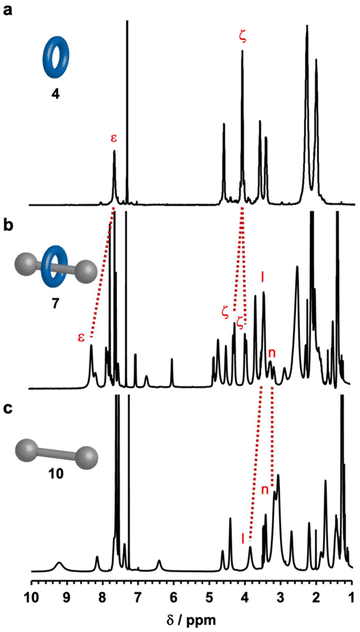
The chemical structure of the rotaxane was determined by 1H NMR spectroscopy (Fig. 2). Eight glycine Cα proton resonances were split, with four shifting upfield and the other four shifting downfield. This phenomenon occurred because two of the four carbonyl oxygens on the glycine residues within cyclo(PG)4 formed hydrogen bonds with –H2N+–, whereas the remaining two carbonyl groups experienced disruption of their internal amide–amide hydrogen bonds owing to the formation of an interlocked structure. Although the internal hydrogen bonds in cyclo(PG)4 were lost within the rotaxane, the amide proton resonances shifted downfield, which was attributed to inductive effects resulting from strong electrostatic interactions between the glycine carbonyl groups and the ammonium cation. This result is consistent with previous research on cyclo(PG)4-ammonium thread rotaxanes.23 Additionally, the formation of hydrogen bonds between the carbonyl oxygens in the thread and the amide protons in cyclo(PG)4 further enhanced this shift. Compared with that in the free thread, the proton Hl in the rotaxane was shielded, whereas the proton Hn remained unchanged, indicating that cyclo(PG)4 was positioned over Hl on the thread. The 2D diffusion ordered spectroscopy (DOSY) NMR spectrum of rotaxane 7 revealed that every peak had the same diffusion coefficient, which also indicated the formation of a rotaxane structure (Fig. 3).
 |
| | Fig. 2
1H NMR spectra (400 MHz, CDCl3, 298.15 K) of (a) cyclo(PG)4, (b) [2]rotaxane 7, and (c) monocationic ammonium thread 10. The peaks are labeled according to Scheme 1. The complete attribution of each NMR spectrum is detailed in the ESI (Fig. S15–S17†). | |
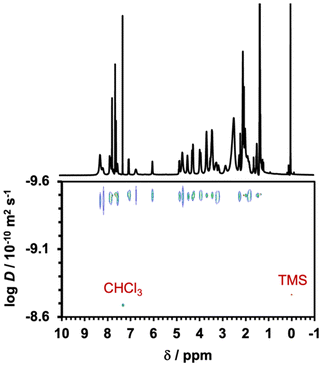 |
| | Fig. 3 DOSY NMR spectrum of [2]rotaxane 7. Compound 7 had a single diffusion coefficient, indicating the formation of a rotaxane. | |
Cyclo(PG)4 does not contain any residues with reactive side chains, which significantly limits its functionality owing to its inability to bind to other molecules. Incorporating reactive side chains into cyclic peptides is crucial to enhance the versatility of peptide-based rotaxanes for diverse applications. To assess the applicability of this rotaxane synthesis method to other cyclic peptides, we designed and synthesized cyclo[(PG)3PC(StBu)], in which one glycine residue was substituted with S-tert-butylthio-cysteine (C(StBu)). Compared with cyclo(PG)4, cyclo[(PG)3PC(StBu)] afforded rotaxanes in lower yield (Fig. S4† and Table 3, entry 5). As demonstrated in a previous study, cyclo[(PG)3PC(StBu)] adopts a planar structure with a relatively large inner cavity in two of its five metastable conformations.32 Several structures in which the cysteine side chain was present near the center of the cyclic peptide were identified. This steric hindrance caused the yield of the rotaxanes to be lower than the rotaxanes prepared with cyclo(PG)4.
Table 3 Effects of the cyclic peptide amino acid sequence on rotaxane formation
| Entry |
Cyclic peptide |
Ammonium thread |
Yield (%) |
| 2 |
Cyclo(PG)4 |
2 (n = 4) |
36.1 |
| 5 |
Cyclo[(PG)3PC(StBu)] |
2 (n = 4) |
16.0 |
| 6 |
Cyclo[GC(ΨMe,MePro)(GP)3] |
2 (n = 4) |
0.0 |
Glycine is a flexible amino acid that lacks a side chain; therefore, its impact on the structure of cyclo(PG)4 is minimal. As a result, proline may have a major effect on the conformation of cyclo(PG)4, which in turn affects the formation of rotaxanes. To examine the influence of proline, we synthesized a proline derivative, C(ΨMe,MePro) (Fig. S8†), to use in the synthesis of the analogous cyclic peptide cyclo[GC(ΨMe,MePro)(GP)3] (Fig. S7†). Unfortunately, cyclo[GC(ΨMe,MePro)(GP)3] did not afford a rotaxane (Fig. S5† and Table 3, entry 6). To determine why the rotaxane was not formed, we analyzed the solution structure of cyclo[GC(ΨMe,MePro)(GP)3]. The secondary structure of cyclo[GC(ΨMe,MePro)(GP)3] was analyzed by circular dichroism (CD) in DCM. A negative Cotton effect was observed at 227 nm, which is characteristic of peptides with γ-turn structures (Fig. S6†).22 Moreover, a γ-turn structure with 1 ← 3 intramolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N hydrogen bonds gives rise to a relatively planar structure in which the glycine carbonyls point toward the center. Compared with that of cyclo(PG)4,32 the CD signal of cyclo[GC(ΨMe,MePro)(GP)3] was less intense, indicating a reduced propensity to form γ-turn structures. We further investigated the effect of C(ΨMe,MePro) on the solution structures of cyclic peptides via replica exchange molecular dynamics (REMD) simulations (Fig. 4).38 The characteristic feature of C(ΨMe,MePro) is its tendency to adopt a cis configuration at the amide bond preceding the C(ΨMe,MePro) residue.39–41 During the simulations, 55.2% of the amide bonds preceding the C(ΨMe,MePro) residue in cyclo[GC(ΨMe,MePro)(GP)3] adopted a cis configuration (Table 4), which was more than two times that observed for the amide bond preceding the proline in cyclo(PG)4 (25.1%). When a cyclic peptide interacts with a cation to form a rotaxane, the carbonyl oxygen at the point of interaction is typically directed toward the center of the cyclic peptide. With this in mind, if the amide bond before C(ΨMe,MePro) in cyclo[GC(ΨMe,MePro)(GP)3] remains in a cis configuration during rotaxane formation, the carbonyl oxygen of glycine and the Cδ atom of C(ΨMe,MePro) both face the same direction. This means that the five-membered ring of C(ΨMe,MePro) is oriented toward the center of cyclo[GC(ΨMe,MePro)(GP)3], which increases steric hindrance. Transitioning from the cis conformation to the trans conformation would avoid such steric hindrance, but this conversion requires energy. In other words, the entropy loss of cyclo[GC(ΨMe,MePro)(GP)3] during complex formation is expected to be even greater than that of cyclic peptides that do not adopt a cis configuration. Owing to the steric hindrance or increased entropy loss induced by C(ΨMe,MePro), cyclo[GC(ΨMe,MePro)(GP)3] would not have formed a rotaxane.
O⋯H–N hydrogen bonds gives rise to a relatively planar structure in which the glycine carbonyls point toward the center. Compared with that of cyclo(PG)4,32 the CD signal of cyclo[GC(ΨMe,MePro)(GP)3] was less intense, indicating a reduced propensity to form γ-turn structures. We further investigated the effect of C(ΨMe,MePro) on the solution structures of cyclic peptides via replica exchange molecular dynamics (REMD) simulations (Fig. 4).38 The characteristic feature of C(ΨMe,MePro) is its tendency to adopt a cis configuration at the amide bond preceding the C(ΨMe,MePro) residue.39–41 During the simulations, 55.2% of the amide bonds preceding the C(ΨMe,MePro) residue in cyclo[GC(ΨMe,MePro)(GP)3] adopted a cis configuration (Table 4), which was more than two times that observed for the amide bond preceding the proline in cyclo(PG)4 (25.1%). When a cyclic peptide interacts with a cation to form a rotaxane, the carbonyl oxygen at the point of interaction is typically directed toward the center of the cyclic peptide. With this in mind, if the amide bond before C(ΨMe,MePro) in cyclo[GC(ΨMe,MePro)(GP)3] remains in a cis configuration during rotaxane formation, the carbonyl oxygen of glycine and the Cδ atom of C(ΨMe,MePro) both face the same direction. This means that the five-membered ring of C(ΨMe,MePro) is oriented toward the center of cyclo[GC(ΨMe,MePro)(GP)3], which increases steric hindrance. Transitioning from the cis conformation to the trans conformation would avoid such steric hindrance, but this conversion requires energy. In other words, the entropy loss of cyclo[GC(ΨMe,MePro)(GP)3] during complex formation is expected to be even greater than that of cyclic peptides that do not adopt a cis configuration. Owing to the steric hindrance or increased entropy loss induced by C(ΨMe,MePro), cyclo[GC(ΨMe,MePro)(GP)3] would not have formed a rotaxane.
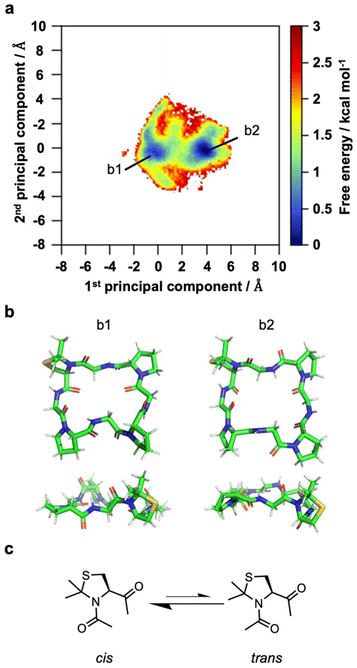 |
| | Fig. 4 (a) Free energy landscape of cyclo[GC(ΨMe,MePro)(GP)3] determined by PCA. (b) Molecular structures of the two main representative metastable states of cyclo[GC(ΨMe,MePro)(GP)3]. (c) Schematic of proline cis–trans isomerization. | |
Table 4 Percentages of cis amide bonds preceding the Pro residue in cyclo(PG)4 or the C(ΨMe,MePro) residue in cyclo[GC(ΨMe,MePro)(GP)3]
| Cyclic peptide |
Percentages of cis amide bonds preceding the Pro or C(ΨMe,MePro) residue |
| Cyclo(PG)4 |
25.1 |
| Cyclo[GC(ΨMe,MePro)(GP)3] |
55.2 |
In summary, we herein successfully synthesized peptide-based [2]rotaxanes via a CuAAC capping reaction following the pseudorotaxane formation of cyclo(PG)4 with monocationic ammonium threads. Rapid execution of the capping reaction was found to prevent dissociation of the pseudorotaxane intermediate, resulting in the synthesis of rotaxanes in up to 36% yield. We further explored how the cyclic peptide sequence affects rotaxane formation. Rotaxane formation was inhibited by specific amino acid residues in the cyclic peptide sequence, highlighting the critical roles of the peptide primary structure on the molecular structure and rotaxane formation. Although numerous studies have explored the interactions between cyclic peptides and cations, comprehensive design guidelines for these systems have not yet been established.17–22 Consequently, we continue to focus on identifying the optimal cyclic peptides for enhanced rotaxane synthesis (Fig. 5). This study bridges the gap between peptide chemistry and supramolecular chemistry, paving the way for the development of multifunctional rotaxanes by exploiting the broad chemical diversity of natural and nonnatural amino acids. The development of peptide-based rotaxanes has enabled the creation of innovative materials for a broad range of applications. This research lays the groundwork for the advancement of these innovative materials.
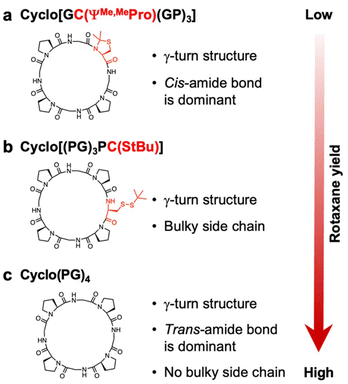 |
| | Fig. 5 Summary diagram of the effects of the cyclic peptide amino acid sequence on rotaxane formation in this study. (a) Cyclo[GC(ΨMe,MePro)(GP)3], which adopts a γ-turn structure with predominantly cis-amide bonds (Table 2), was unable to form a rotaxane. (b) Cyclo[(PG)3PC(StBu)], which adopts γ-turn structure with bulky side chains, formed a rotaxane in a low yield. (c) Cyclo(PG)4, which adopts a γ-turn structure with predominantly trans-amide bonds (Table 2) and lacks bulky side chains, demonstrated the highest produced a rotaxane in the highest yield to date. This figure underscores how strategically modifying the amino acid sequence modifications in of a cyclic peptide can significantly enhance rotaxane yield. Moreover, continued optimization of these cyclic peptides has the potential to achieve even greater improvements in could lead to further improvements in rotaxane yield. | |
Author contributions
T. K. and K. N. conceived and designed the research. T. K. performed all the experiments and analyzed the data. T. K. and K. N. wrote the manuscript.
Data availability
All the data in the current study is included in the main text and ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge the RIKEN Advanced Center for Computing and Communication (ACCC) for providing access to the HOKUSAI BigWaterfall Supercomputer resources. This research was financially supported by JST ERATO grant no. JPMJER1602, Japan (K. N.); a Grant-in-Aid for Transformative Research Areas (B) (grant no. JP20H05735 (K. N.)); the MEXT Data Creation and Utilization-type MaTerial R&D project (K. N.); JST CREST (grant no. JPMJCR2091 (K. N.)) and a JSPS Research Fellowships for Young Scientists (grant no. 23KJ1250 (K. T.)).
References
- B. J. Pepe-Mooney and R. Fairman, Curr. Opin. Struct. Biol., 2009, 19, 483–494 CrossRef CAS.
- M. J. Harrington and P. Fratzl, Prog. Mater. Sci., 2021, 120, 1–40 CrossRef.
- B. E. Ramakers, J. C. van Hest and D. W. Lowik, Chem. Soc. Rev., 2014, 43, 2743–2756 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 17, 1700177 CrossRef.
- T. Kurita and K. Numata, Phys. Chem. Chem. Phys., 2024, 26, 28776–28792 RSC.
- G. Abbenante, D. R. March, D. A. Bergman, P. A. Hunt, B. Garnham, R. J. Dancer, J. L. Martin and D. P. Fairlie, J. Am. Chem. Soc., 1995, 117, 10220–10226 CrossRef.
- H. L. Sham, C. A. Rempel, H. Stein and J. Cohen, J. Chem. Soc., Chem. Commun., 1990, 666–667 RSC.
- R. M. Schultz, J. P. Huff, P. Anagnostaras, U. Olsher and E. R. Blout, Int. J. Pept. Protein Res., 1982, 19, 454–469 CrossRef.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef PubMed.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef.
- M. Amorin, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef PubMed.
- T. Kurita, T. Terabayashi, S. Kimura, K. Numata and H. Uji, Biomacromolecules, 2021, 22, 2815–2821 CrossRef.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef PubMed.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- L. R. Gahan and R. M. Cusack, Polyhedron, 2018, 153, 1–23 CrossRef.
- S. Kubik and R. Goddard, Eur. J. Org. Chem., 2001, 311–322 CrossRef.
- S. Kubik and R. Goddard, J. Org. Chem., 1999, 64, 9475–9486 CrossRef.
- S. Kubik, J. Am. Chem. Soc., 1999, 121, 5846–5855 CrossRef.
- V. Madison, M. Atreyi, C. M. Deber and E. R. Blout, J. Am. Chem. Soc., 1974, 96, 6725–6734 CrossRef PubMed.
- V. Madison, C. M. Deber and E. R. Blout, J. Am. Chem. Soc., 1977, 99, 4788–4798 CrossRef PubMed.
- V. Aucagne, D. A. Leigh, J. S. Lock and A. R. Thomson, J. Am. Chem. Soc., 2006, 128, 1784–1785 CrossRef PubMed.
- X. W. Wang and W. B. Zhang, Trends Biochem. Sci., 2018, 43, 806–817 CrossRef.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef PubMed.
- X. W. Wang and W. B. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13985–13989 CrossRef PubMed.
- F. Saito and J. W. Bode, Chem. Sci., 2017, 8, 2878–2884 RSC.
- H. V. Schroder, M. Stadlmeier, M. Wuhr and A. J. Link, Chem. – Eur. J., 2022, 28, e202103615 CrossRef.
- Y. Liu, W. H. Wu, S. Hong, J. Fang, F. Zhang, G. X. Liu, J. Seo and W. B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19153–19161 CrossRef PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef.
- J. Sawada, D. Aoki, S. Uchida, H. Otsuka and T. Takata, ACS Macro Lett., 2015, 4, 598–601 CrossRef PubMed.
- T. Kurita, M. Higashi, J. Gimenez-Dejoz, S. Fujita, H. Uji, H. Sato and K. Numata, Biomacromolecules, 2024, 25, 3661–3670 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- N. I. Hassan, V. del Amo, E. Calder and D. Philp, Org. Lett., 2011, 13, 458–461 CrossRef PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef.
- P. Dumy, M. Keller, D. E. Ryan, B. Rohwedder, T. Wöhr and M. Mutter, J. Am. Chem. Soc., 1997, 119, 918–925 CrossRef.
- M. Keller, C. Sager, P. Dumy, M. Schutkowski, G. S. Fischer and M. Mutter, J. Am. Chem. Soc., 1998, 120, 2714–2720 CrossRef.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Keiji
Numata
a and
Keiji
Numata





