DOI:
10.1039/D4PY01086H
(Paper)
Polym. Chem., 2025,
16, 1272-1284
Redox-responsive micellar nanoparticles using benzothiazole-disulfide terminated polymers: employing host–guest complexation for targeted delivery of curcumin†
Received
28th September 2024
, Accepted 15th January 2025
First published on 11th February 2025
Abstract
A host–guest interaction-based redox-responsive polymeric nanoparticle system for targeted delivery of curcumin is fabricated. Host–guest interaction between a β-cyclodextrin (β-CD) terminated hydrophilic polymer and curcumin is employed to formulate stable nanosized aggregates. A redox-sensitive disulfide linkage is used to tether the CD moiety onto the polymer chain ends to obtain efficient release inside cancerous cells. For the synthesis of the β-CD terminated redox-responsive polymer, a polyethylene glycol (PEG)-based polymer containing a benzothiazole-disulfide (BDS) group at the chain end was utilized. The BDS-terminated polymers were synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization by using a BDS-containing chain-transfer agent (CTA). Initially, the post-polymerization modification abilities of these polymers were demonstrated by successful conjugation of model thiol functional compounds such as furfuryl thiol and Bodipy-SH, a fluorescent hydrophobic dye. To prepare a nanoaggregate forming polymer precursor, a thiol functional β-CD host moiety was conjugated to the BDS end groups of the reactive polymer through redox-responsive disulfide linkage formation. The other end of the β-CD-containing polymer was capped with an integrin-targeting cyclic peptide (cRGDfK). While no aggregates were observed in the aqueous medium for the BDS-containing parent polymer, the β-CD attached polymers gave nanosized aggregates with average sizes below 150 nm. Curcumin-loaded nanoaggregates were obtained through the inclusion complexation of curcumin molecules with β-CD moieties. It was shown that enhanced drug release occurred upon exposure of the nanoaggregates to the glutathione (GSH) environment. While the parent BDS functional polymers and β-CD-containing polymeric aggregates were non-toxic to healthy fibroblast cells, curcumin-loaded aggregates showed dose-dependent toxicity against the U-87 glioblastoma cancer cell line. The peptide-targeting group containing nanoparticles showed slightly higher toxicity and enhanced cellular internalization. The modular nanoparticle system disclosed here could be employed to address challenges in the delivery of hydrophobic drugs to cancer cells.
Introduction
The utilization of nanosized polymeric drug delivery agents such as micelles, nanoparticles, and nanogels continues to play an ever-increasing role in providing a viable solution to challenges in the delivery of hydrophobic anti-cancer drugs.1–5 Apart from solubilizing the drug, the polymeric constructs provide a biocompatible platform for protecting drugs from premature degradation and improving their circulation time in vivo. Furthermore, polymer-based delivery systems allow for targeted drug delivery through appropriate functionalization with specific ligands such as peptides, antibodies or bioactive small molecules to target disease cells, thus minimizing off target effects and enhancing therapeutic outcomes. The efficient solubilization of hydrophobic drugs plays an important role since many effective anticancer compounds will be ruled out because of their low bioavailability. One such hydrophobic anticancer compound is curcumin, a naturally occurring polyphenol derived from turmeric, which exhibits remarkable anti-inflammatory, antioxidant, and anticancer properties.6–10 Despite its therapeutic potential, curcumin's clinical utility is limited by its poor aqueous solubility, compromising its bioavailability and effectiveness.
Commonly used approaches for fabricating polymer-based micellar carriers utilize block copolymers, which assemble into nanosized aggregates in aqueous media and thus encapsulate the hydrophobic drugs in their interior. Although the past years have seen remarkable advances in the synthesis of block copolymers, their synthesis is still not trivial. Instead of using a hydrophobic second block, we envisioned employing a host moiety to encapsulate the hydrophobic drug and increase hydrophobic interactions leading to micellar aggregates. For this purpose, beta-cyclodextrin (β-CD), a cyclic oligosaccharide composed of glucose units, emerges as a viable candidate. It is well established that β-CD is a valuable excipient for enhancing the solubility and bioavailability of hydrophobic drugs.11–14 Previous studies have demonstrated β-CD's utility in enhancing curcumin's solubility and stability, making it an ideal candidate for formulating curcumin-based therapeutics.14–21 However, most of these studies solubilize curcumin in molecular form. Thus, the size of the complex usually falls much below the domain of nanosize necessary for drug delivery with passive targeting mediated by the enhanced permeability and retention (EPR) effect.22 Apart from using CDs in molecular form, a variety of macromolecular constructs with CDs have been reported,23–25 To date, the CD moiety has been conjugated to the chain end,26,27 as side chain residues,28 or utilized as a core motif in star-like multiarm polymers.29 In most cases, the CD moiety is non-reversibly conjugated to the polymeric scaffolds, and thus, the encapsulated guest molecule is released predominantly by reversible host–guest interactions.30,31 In this study, we aimed for conjugation with a disulfide-based reversible linkage so that the encapsulated hydrophobic guest molecule remains soluble upon release from the polymer-based aggregate.
In recent years, it has been established that the incorporation of redox-responsive linkage in the drug delivery platform further enhances the cytotoxicity in cancerous cells with increased levels of glutathione (GSH).32–39 Thus, we targeted constructing a hydrophilic polymer containing a chain end β-CD unit linked with a redox-responsive linker. To obtain such a polymer in a modular fashion, we envisioned that a thiol–disulfide exchange using a benzothiazole disulfide (BDS) chain-terminated polymer could be utilized. The BDS group is known to undergo the thiol–disulfide exchange reaction with high efficiency, and we recently employed it for the functionalization of side chain residues of polymers, polymer-coated surfaces and hydrogels.40–44 Until now, the usage of the BDS group as a thiol-reactive group on a polymer chain end has remained unexplored. The present work demonstrates the versatility of polymers containing the BDS group as a chain end moiety, along with demonstrating their utilization for obtaining β-CD-terminated polymers.
Herein, we aimed to fabricate curcumin-loaded micellar nanoparticles using a redox-responsive polymeric construct in which the CD moiety is linked to the end of the polymer chain through a redox-responsive disulfide linkage (Scheme 1). We employed a BDS-based linker at the chain end of a PEG-based hydrophilic polymer to obtain such constructs in a modular fashion. We report here the synthesis and functionalization of BDS group terminated polymers and show that the CD moiety could be efficiently added to the polymer chain end through a redox-responsive disulfide linkage. Thus the obtained β-CD-containing polymers are used to fabricate drug-loaded aggregates upon complexation with curcumin. It is also demonstrated that peptide-based cell targeting groups can be incorporated at the other chain end of the β-CD-containing polymer. Drug loading and release studies, as well as cytotoxicity and cellular internalization studies, suggest these polymers as promising candidates for the delivery of curcumin. Although the study here is undertaken with curcumin, one could expect that the modular approach reported here could be adapted for the delivery of other hydrophobic drugs to cancerous cells in a targeted fashion.
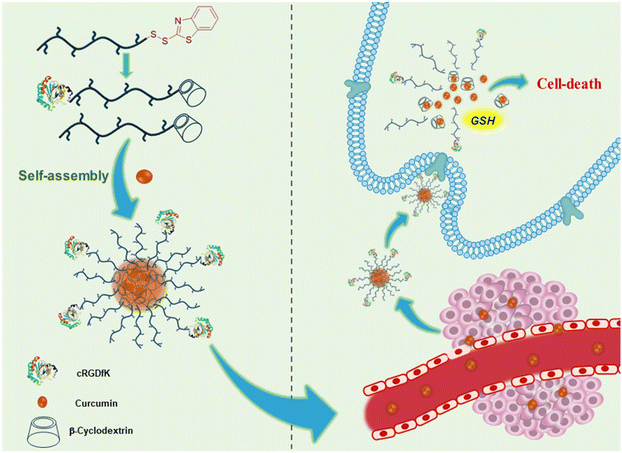 |
| | Scheme 1 Illustration of the fabrication of a CD-based redox-responsive targeted delivery system using a BDS-terminated polymer. | |
Experimental section
Materials
Poly(ethylene glycol) methyl ether methacrylate (PEGMEMA, Mn = 300 gmol−1) was obtained from Sigma-Aldrich and used after purification through activated aluminum oxide filtration. N,N′-Diisopropylcarbodiimide (DIPC), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB), 4-dimethylaminopyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC), triethylamine (TEA), 2,2′-azobis(2-methylpropionitrile) (AIBN), 2-furfurylthiol, glutathione, 4,4′-azobis(4-cyanovaleric acid) (ACVA) and N-hydroxysuccinimide were purchased from Sigma-Aldrich. The cyclic arginine–glycine–aspartic acid–phenylalanine–lysine (c-RGDfK),45 benzothiazole disulfide alcohol (BDSOH),46 and a thiol-bearing dye (Bodipy-SH)47 were synthesized according to the literature. Other solvents were obtained from Merck, and ultrapure water was obtained using a Milli-Q water purification system (Milli-Q system, Millipore, Billerica, MA, USA). The L929 mouse fibroblast (American Type Culture Collection, ATCC) and U-87 cancer cell lines (American Type Culture Collection, ATCC) were used for cell studies.
Instrumentation
1H and 13C NMR spectroscopy (Bruker Avance Ultrashield 400 (400 MHz) spectrometer), dynamic light scattering (DLS, Malvern), scanning transmission electron microscopy (STEM, Thermo Scientific, Quattro S), and UV-vis spectroscopy (Shimadzu, UV 1800 spectrophotometer) were used to characterize the synthesized monomers, polymers, and polymeric nanoparticles. The polymer molecular weights were determined by size exclusion chromatography (SEC, Shimadzu, Japan) equipped with an RID-10A RI- detector and PSS SDV Linear M column, calibrated with poly(methyl methacrylate) standards. A mobile phase solution of 0.05 M LiBr in dimethylacetamide (DMAc) was used as an eluent at a flow rate of 1 mL min−1. Cytotoxicity assay experiments were carried out using a plate reader (Multiskan FC, Thermo Scientific, USA) using a Cell Counting Kit-8 (CCK-8, Fluka) obtained from Sigma-Aldrich, and internalization experiments were performed using a laser scanning confocal microscope (Leica Stellaris 5, Germany). The imaging was performed with an HC PL APO CS2 63x/1.40 OIL objective. For excitation, Diode 405 and WLL lasers were used to excite the nuclear stain DAPI and curcumin, respectively. Emission signals were detected within the ranges of 425–480 nm for DAPI and 503–650 nm for curcumin.
Synthesis of BDS-CTA
BDSOH (0.2 g, 0.821 mmol), CPADB (0.25 g, 0.896 mmol), and DMAP (0.04 g, 0.327 mmol) were added to a round-bottom flask and dissolved in anhydrous CH2Cl2 (3 mL). DIPC (0.114 g, 0.904 mmol) was dissolved in another flask in anhydrous CH2Cl2 (3 mL). The content of the second flask was slowly added to the ice-cold mixture in the first flask. The combined reaction mixture was stirred at room temperature for 4 h. Purification using column chromatography on silica gel using CH2Cl2 and hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluents yielded the pure product (256 mg, 61%). 1H NMR (CDCl3, δ, ppm) δ 7.89 (t, 3H), 7.82 (d, 1H), 7.56 (t, 1H), 7.39 (m, 4H), 4.46 (t, 2H), 3.22 (t, 2H), 2.79–2.31 (m, 4H), 1.92 (s, 3H). 13C NMR (CDCl3, δ ppm): 222.3, 171.5, 154.7, 144.6, 135.8, 133.2, 128.6, 126.7, 125.0, 122.3, 121.40, 118.5, 62.6, 45.81, 37.9, 33.4, 29.8, 24.3.
:1) as eluents yielded the pure product (256 mg, 61%). 1H NMR (CDCl3, δ, ppm) δ 7.89 (t, 3H), 7.82 (d, 1H), 7.56 (t, 1H), 7.39 (m, 4H), 4.46 (t, 2H), 3.22 (t, 2H), 2.79–2.31 (m, 4H), 1.92 (s, 3H). 13C NMR (CDCl3, δ ppm): 222.3, 171.5, 154.7, 144.6, 135.8, 133.2, 128.6, 126.7, 125.0, 122.3, 121.40, 118.5, 62.6, 45.81, 37.9, 33.4, 29.8, 24.3.
Synthesis of BDS-containing polymer P1
PEGMEMA (0.500 g, 1.60 mmol), AIBN (0.00273 g, 0.016 mmol), and BDS-CTA (0.042 g, 0.083 mmol) were added to a round bottom flask and dissolved in anhydrous dioxane (0.9 mL). The solution was purged with N2 for 20 min. The reaction vessel was immersed in a preheated oil bath at 70 °C. After 16 h of polymerization, the reaction solution was concentrated by rotary evaporation. The residue was taken up with a minimum amount of CH2Cl2 and precipitated into cold diethyl ether. The precipitated polymer was collected and dried under high vacuum. The obtained polymer was characterized by SEC and 1H NMR analyses. Mn,SEC = 7100 g mol−1, and Mw/Mn = 1.25. 1H NMR (CDCl3, δ, ppm) δ 7.86 (m, 3H), 7.64–7.31 (m, 6H), 4.42 (t, 2H), 4.08 (bs, 48H), 3.37 (s, 68H), 3.19 (t, 2H), 2.08–0.60 (m).
Post-polymerization modification of P1 with 2-furfurylthiol
Polymer P1 (58 mg, 0.0082 mmol) and 2-furfurylthiol (1.12 mg, 0.0098 mmol) were dissolved in anhydrous CH2Cl2 (0.5 mL). To the reaction mixture, a catalytic amount of acetic acid (AcOH) was added, and the reaction was continued for 16 h. After the reaction, the functionalized polymer was purified by precipitation into cold diethyl ether. 1H NMR (CDCl3, δ, ppm) δ 7.88 (m, 2H), 7.58–7.31 (m, 4H), 6.34 (s, 1H), 6.28 (s, 1H), 4.45–3.96 (m, 75H), 3.37 (s, 99H), 2.20–0.60 (m).
Post-polymerization modification of P1 with Bodipy-SH
Polymer P1 (11 mg, 0.0015 mmol), Bodipy-SH (3.5 mg, 0.0083 mmol) and TEA (8 μL) were dissolved in anhydrous DMF (0.2 mL) and reacted at 25 °C for 18 h. After the reaction, the purified polymer was obtained through dialysis (Mw cut-off: 3.5 kDa) using acetonitrile. 1H NMR (CDCl3, δ, ppm) δ 7.98 (d, 2H), 7.57–7.46 (m, 1H), 7.38 (dd, 2H), 6.04 (s, 2H), 4.08 (s, 51H), 3.38 (s, 81H), 2.92 (m, 2H), 2.76–2.67 (m, 2H), 2.50 (s, 6H), 2.37 (d, 6H), 2.15–0.68 (m).
Conjugation of β-CD to the α-chain end of P1
Polymer P1 (0.1 g, 0.014 mmol) and β-CD-SH (0.024 g, 0.021 mmol) were dissolved in anhydrous DMF (2 mL) and degassed for 5 min. After adding a catalytic amount of AcOH, the reaction time was continued for 16 h at 25 °C. After the reaction, the purified polymer was obtained through dialysis (Mw cut-off: 3.5 kDa) using H2O:MeOH (80:20). 1H NMR (CDCl3, δ, ppm) δ 7.86 (m, 2H), 7.60–7.31 (m, 3H), 5.01 (bs, 7H), 4.08 (s, 74H), 3.38 (s, 128H), 2.19–0.70 (m).
Conjugation of the cRGDfK peptide to the ω-chain end of P1
Initially, a radical cross-coupling reaction was performed to remove the benzodithioate end group of the polymer and install a carboxylic acid functionality. The polymer CD-PEG (10 mg, 0.0013 mmol) and ACVA (9.34 mg, 0.033 mmol) were dissolved in anhydrous DMF (0.4 mL) and the solution was purged with N2 for 15 min. The reaction vessel was immersed in a preheated oil bath at 70 °C and the reaction was continued for 4 h. The reaction solution was concentrated using rotary evaporation, and the yellow residue was washed with cold diethyl ether twice. 1H NMR (CDCl3, δ, ppm) δ 4.67 (s, 1H), 4.08 (s, 30H), 3.38 (s, 49H), 1.88–0.71 (m). The obtained polymer CD-PEG-COOH (10 mg, 0.00125 mmol) was reacted with N-hydroxysuccinimide (0.43 mg, 0.0037 mmol) in the presence of DCC (0.77 mg, 0.0037 mmol) and CH2Cl2 (0.5 mL). The reaction mixture was incubated at room temperature for 16 h. After completion of the reaction time, CH2Cl2 was evaporated, and the cRGDfK peptide (1.5 mg, 0.0025 mmol) was added to the reaction vessel dissolved in anhydrous DMF (0.5 mL). After purging the reaction mixture for 5 min, TEA (10 μL) was added, and the reaction was continued for 16 h at 25 °C. Thereafter, the purification of the polymer was conducted through dialysis (Mw cut-off: 3.5 kDa) using deionized water. 1H NMR (D2O, δ, ppm) δ 7.40–7.17 (m, 2H), 4.99 (bs, 7H), 4.15 (bs, 56H), 3.36 (s, 97H), 2.77–0.68 (m).
Curcumin loading into the β-CD terminated polymer and release profiles
The polymer CD-PEG-RGD (3 mg, 0.0004 mmol) and curcumin (1.2 mg, 0.320 mmol) were added into an Eppendorf tube, followed by deionized water (1.5 mL). The reaction mixture was stirred for two days at 25 °C. After two days, the reaction solution was filtered with a syringe filter (0.45 μm, PTFE) to remove unreacted curcumin. To obtain a curcumin-loaded mixture with a β-CD-containing polymer and β-CD-cRGDfK-containing polymer, the reaction was repeated by adding polymer P6 and polymer P7 (30% of P6) in the same equivalent of curcumin and deionized water for two days. The curcumin loading efficiency and loading level were calculated to be 50% and 2 wt%, respectively. A dialysis bag method was employed to assess the release kinetics of curcumin from the nanoparticles. Curcumin-loaded nanoparticles containing 20 ppm curcumin were enclosed within a 3.5 kDa dialysis bag and immersed in three different environments (10 mL): neutral (pH 7.4, PBS), neutral-reducing (pH 7.4, 5 mM GSH), and acidic-reducing (pH 5.5, 5 mM GSH). The incubation was conducted at 37 °C with constant shaking. Samples (2 mL) were collected from the external medium at predetermined intervals, and fresh medium was replenished to maintain consistent conditions. The concentration of the released curcumin in the samples was quantified using UV spectroscopy, with a calibration curve generated using known concentrations of curcumin as a reference.
Cytotoxicity experiments
The cytotoxicity of the polymers was investigated via a CCK-8 viability assay on L929 mouse fibroblast cells. Cells (6000 cells per well) were seeded in a 96-well plate in quadruplicate with 100 μL of the culture medium (DMEM) and incubated at 37 °C overnight to grow and adhere completely. After removing the culture media, cells were treated with polymers (0.1, 0.01, and 0.001 mg) dissolved in culture media for 48 h. After incubation, the polymer solutions were removed, and the cells were treated with a 10% CCK-8 solution for two hours. The absorbance values at 450 nm were measured using a microplate reader. Viability results were obtained using GraphPad Prism software in the nonlinear regression mode. The cytotoxicity of the curcumin-loaded polymers was investigated via a CCK-8 viability assay on the U-87 cancer cell line using a similar procedure. To evaluate the efficiency of cRGDfK-mediated uptake of curcumin-loaded nanoparticles, an in vitro cellular internalization experiment was conducted using U-87 cancer cells. First, U-87 cells were seeded at a density of 50000 cells per well in a 6-well plate. Duplicate wells were prepared for each treatment group to ensure data reproducibility. The cells were then incubated overnight at 37 °C in a humidified incubator with 5% CO2 to allow for proper attachment and growth. Following overnight incubation, the cells were treated with either non-targeted or cRGDfK-conjugated nanoparticles or both excess cRGDfV and cRGDfK-conjugated nanoparticles containing an identical amount of curcumin. The cells were incubated with the nanoparticles for 24 hours for sufficient internalization. After incubation, the culture medium containing nanoparticles was removed, and the cells were washed twice with PBS to remove any residual media components. Subsequently, the cells were fixed, stained, and analyzed using a confocal microscope. For the flow cytometry experiment, U-87 cells (60000 cells per well) were seeded into a 24-well plate as duplicates in 1 mL of culture media and incubated at 37 °C overnight. The samples’ properties were identical to those used in the cellular internalization experiment. Cell media were removed after 4 and 24 h, and the cells were trypsinized with 0.05% trypsin solution for 2 min at 37 °C. After 2 min, the culture medium (700 μL) was added to the wells, and curcumin intensities were determined by flow cytometry (Guava easyCyte).
Results and discussion
Synthesis of thiol-reactive BDS-terminated polymers
Polymers containing the thiol-reactive BDS group at chain ends are synthesized using RAFT polymerization. We chose RAFT polymerization since it is well established for obtaining polymers with control over molecular weight and dispersity, is metal catalyst-free, and is tolerant to a wide variety of functional groups.48,49 To synthesize polymers containing the thiol-reactive BDS unit at the chain end, a BDS-functionalized chain transfer agent (CTA) was synthesized. The synthesis involved the esterification of BDS-group containing alcohol (BDSOH) with a carboxylic acid functional CTA (Fig. 1a). The successful formation of BDS-CTA was confirmed by 1H and 13C NMR spectroscopy (Fig. 1b and Fig S1†). The 1H NMR spectrum displayed characteristic peaks at 7.80 ppm and 4.48 ppm, corresponding to the aromatic and methylene protons of the BDS moiety, respectively, along with the expected peaks at 7.56 ppm and 1.93 ppm attributed to the protons from the CPADB component.
 |
| | Fig. 1 (a) Synthesis and (b) 1H NMR spectrum of the BDS-bearing CTA in CDCl3. | |
After obtaining BDS-CTA, a PEGMEMA-based polymer was synthesized using RAFT polymerization. The polymerization was conducted in dioxane at 70 °C using AIBN as the initiator (Fig. 2a). The successful formation of the polymer was confirmed by 1H-NMR spectroscopy (Fig. 2b). A peak at 7.52 ppm was attributed to the protons in the CPADB moiety at one end of the polymer chain. Another peak at 7.45 ppm was assigned to the aromatic protons of the BDS group at the other end of the chain. Additionally, a singlet peak at 3.34 ppm confirmed the presence of the methoxy (–OCH3) groups from the PEG-based side chains.
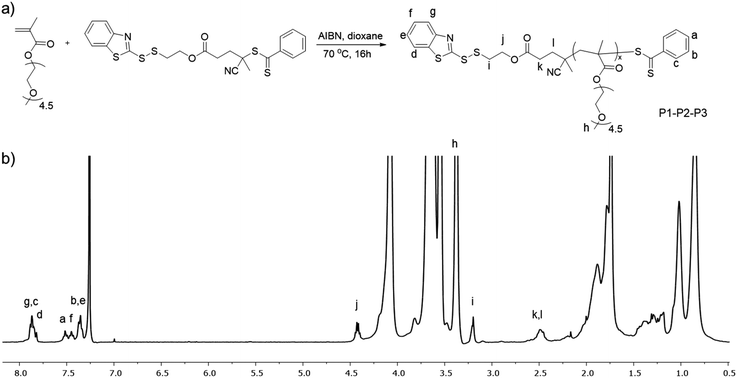 |
| | Fig. 2 (a) Synthesis of the BDS-containing polymer and (b) 1H NMR spectrum of the polymer (P1) in CDCl3. | |
The molar ratio of BDS-CTA to AIBN was kept constant while the PEGMEMA content was varied to investigate the effect of the monomer feed ratio on the polymer properties. The detailed polymerization conditions and the corresponding results for different PEGMEMA/BDS-CTA ratios are summarized in Table 1. As expected, the polymer's average molecular weight increased with a higher feed ratio of the PEGMEMA monomer. Polymerization was conducted with moderately high monomer conversions and narrow dispersities. The SEC results of the obtained polymers are shown in the ESI figures (Fig. S2†).
Table 1 Synthesis conditions and characterization of poly(PEGMEMA) polymers
| Polymer |
Reactant ratios [M]:[CTA]:[AIBN] |
M
n,theo (g mol−1) |
Conv. % |
M
n,cal. (g mol−1) |
M
n,NMRa (g mol−1) |
M
n,SECb (gmol−1) |
DPa |
M
w/Mnb |
| [M]: PEGMEMA and [CTA]: BDS-CTA. Calculated using 1H NMR spectroscopy. Determined by SEC and eluted with DMAC, relative to linear PMMA standards. |
| P1 |
20:1:0.2 |
6000 |
96 |
5760 |
6900 |
7100 |
22 |
1.25 |
| P2 |
50:1:0.2 |
15000 |
84 |
12600 |
13000 |
15000 |
49 |
1.21 |
| P3 |
100:1: 0.2 |
30000 |
76 |
22800 |
30500 |
29000 |
133 |
1.22 |
Post-polymerization functionalization of BDS-containing polymers with model compounds
Polymer P1 was utilized to evaluate the post-polymerization modification efficiencies of the obtained reactive polymers. Thiol-containing compounds furfuryl mercaptan and Bodipy-SH were employed as model molecules for the modification of the polymer's α-chain end via a thiol–disulfide exchange reaction. First, the polymer was treated with furfuryl mercaptan in the presence of a catalytic amount of acetic acid (Fig. 3a). After the reaction, polymer P4 was purified by precipitation in cold diethyl ether. The 1H NMR spectrum revealed the complete disappearance of the characteristic peaks at 7.45 and 7.80 ppm belonging to the terminal BDS group on the polymer, and new peaks emerged at 7.36, 6.36, and 6.30 ppm, indicating the successful quantitative incorporation of furfuryl mercaptan (Fig. 3b).
 |
| | Fig. 3 (a) Synthesis of the furfuryl mercaptan conjugated polymer and (b) 1H NMR spectrum of the polymer (P4) in CDCl3. | |
To further demonstrate the versatility of this approach for post-polymerization functionalization, the polymer was reacted with Bodipy-SH, a hydrophobic fluorophore commonly utilized for imaging and drug delivery applications.50–53 The reaction was conducted in the presence of TEA, and the product was dialyzed in acetonitrile to remove any unreacted dye (Fig. 4a). 1H NMR analysis confirmed the successful conjugation of Bodipy-SH to yield polymer P5. The disappearance of the aromatic proton peaks of the BDS group at 7.80 and 7.45 ppm was observed, along with the emergence of new peaks at 6.04, 2.50, and 2.40 ppm. These new peaks are attributed to the Bodipy dye; the peak at 6.04 ppm corresponds to the proton on the pyrrole moiety, and the peaks at 2.50 and 2.40 ppm belong to the methyl groups (Fig. 4b). Fig. 4c highlights the dramatic shift in the solubility behavior of the dye upon conjugation to the polymer. Bodipy-SH, due to its hydrophobic character, dissolves in the nonpolar ether phase, whereas the polymer-conjugated dye gains water solubility.
 |
| | Fig. 4 (a) Synthesis of the Bodipy thiol conjugated polymer, (b) 1H NMR spectrum of the polymer (P5) in CDCl3, and (c) dye conjugation via the exchange reaction; images upon UV illumination of (c1) free dye, and (c2) dye-attached polymer, partitioned into Et2O and H2O phases. | |
Conjugation of β-CD and the cRGDfK peptide at polymer chain-ends
After establishing the synthesis and post-polymerization functionalization of the BDS-terminated polymer, we investigated if the approach could be extended to obtain CD-conjugated polymers. To this end, the BDS-terminated polymer P1 was treated with β-CD-SH to obtain the CD-PEG polymer (Fig. 5a). After the reaction, the excess β-CD-SH was removed by dialysis, and the purified product was characterized by 1H NMR and SEC analyses. The SEC result indicated a slight increase in the polymer molecular weight (see Fig. S3†), as evident from the shift in retention time. The 1H NMR spectrum of the CD-PEG polymer confirmed the chemical composition; the aromatic peaks corresponding to the cleavable disulfide group (BDS) disappeared, while a new peak at 5.01 ppm emerged, characteristic of the β-CD unit (Fig. 5b). Interestingly, although the expected disappearance of the proton resonances from the BDS group and the appearance of anomeric protons were observed, peaks adjacent to the disulfide bonds were not discernable and seemed close to the baseline presumably due to the proximity to the bulky CD group. The successful place exchange reaction was also indicated by the appearance of yellow color upon addition of CDSH, as expected from the release of the benzothiazolethione group.
 |
| | Fig. 5 (a) Synthesis of a β-CD conjugated polymer and (b) 1H NMR spectrum of the polymer CD-PEG in CDCl3. | |
The β-CD conjugated polymer CD-PEG was further modified with a cell-targeting peptide to achieve a dual-functional polymeric construct CD-PEG-RGD. The peptide cRGDfK is known to interact with specific receptors on the surface of certain cells, potentially enabling the nanoparticles to accumulate at these targeted sites preferentially.54–57 The first step involved exchanging the benzodithioate end group of the polymer with a carboxylic acid group using an azo-based radical exchange reaction. In the 1H NMR spectrum of the CD-PEG-COOH polymer, complete loss of the aromatic proton peaks of the CTA end group was observed, suggesting successful end-group transformation (Fig. S4†). The carboxylic acid end group was activated using NHS and then coupled with the amine group on the cRGDfK peptide. The final product was purified by dialysis in water, yielding a polymer with β-CD at one end and cRGDfK at the other end. The construct composition was validated using 1H NMR analysis, where the peaks between 7 and 8 ppm are attributed to the phenyl proton on the peptide, the peak at 5.01 ppm is attributed to the protons from β-CD, and the peak at 3.34 ppm is attributed to the terminal methoxy group on the PEG-based side chains (Fig. 6b). Also, according to the SEC analysis, the molecular weight of the polymers remained quite similar during the end group transformations (Fig. S5†).
 |
| | Fig. 6 (a) Synthesis pathway and (b) 1H NMR spectrum of cRGDfK and the β-CD conjugated polymer in D2O. | |
Micellar nanoparticle formulation, curcumin loading, and release
To assess the changes in size resulting from the conjugation process, the hydrodynamic diameters of the polymers were determined using DLS. From the DLS analysis, the Z-average value, representing the particles’ average hydrodynamic diameter, was determined as 6.5 nm for unconjugated polymer P1. This value increased significantly to 134 nm with β-CD conjugated polymers, indicating the successful formation of nanoparticles. Furthermore, the size of the polymer further increased to 149 nm upon conjugation with cRGDfK. Importantly, the subsequent conjugation of the hydrophilic cRGDfK peptide does not disrupt the nanoparticle structure, resulting in only a slight increase in size (Fig. 7).
 |
| | Fig. 7 DLS-based size analysis of (a) BDS-polymer, (b) β-CD-conjugated polymer, and (c) cRGDfK, and the β-CD conjugated polymer. | |
In the subsequent step, curcumin was loaded into the CD-containing nanoparticles during the self-assembly step (Fig. 8a). To achieve this, the β-CD-SH/cRGDfK-conjugated polymer was dissolved in water and subsequently mixed with curcumin. The initially insoluble drug slowly solubilized, presumably through the interaction with the hydrophobic cavity of the β-CD moiety. Following incubation, the reaction mixture was filtered to remove any excess curcumin that remained insoluble in water. As shown in Fig. 8b, the resulting solution was yellow and clear, signifying the successful encapsulation of curcumin within the nanoparticles. DLS analysis was performed to assess the size of the curcumin-loaded nanoparticles (Fig. 8c). Interestingly, the average hydrodynamic diameter slightly decreased compared to the unloaded nanoparticles, which could be due to the compactness of the structure due to enhanced hydrophobic interactions. Furthermore, the size of the curcumin-loaded nanoparticles was determined using STEM, which revealed an average size of approximately 90 nm (Fig. 8d and Fig. S6†). Such reduction in size obtained using STEM in comparison with DLS analysis is expected due to the dry state of particles as opposed to their solvated state in DLS analysis, and a similar trend has been observed in earlier studies.58–60
 |
| | Fig. 8 (a) Schematic illustration of curcumin-loaded nanoparticles, (b) the solutions of curcumin and curcumin-loaded nanoparticles after sonication (b1) before filtration and (b2) after filtration, (c) DLS analysis of the curcumin-loaded nanoparticles, (d) STEM image of curcumin-loaded nanoparticles (scale bar: 500 nm), and (e) curcumin release in non-reducing, reducing, acidic, and neutral environments for 72 h at 37 °C. | |
To investigate the potential of these redox-responsive nanoparticles for hydrophobic drug delivery, curcumin was released under different conditions. Curcumin-loaded nanoparticles in a dialysis bag were incubated in three distinct environments: neutral (pH 7.4, PBS), neutral-reducing (pH 7.4, 5 mM GSH), and acidic-reducing (pH 5.5, 5 mM GSH). The neutral environment mimics physiological conditions, while the neutral-reducing environment introduces the reducing agent GSH commonly over-expressed in cancerous cells. The acidic-reducing environment combines a slightly acidic pH, potentially encountered in tumors, along with the reducing agent GSH. At predetermined time intervals, samples were withdrawn from the sink and analyzed using UV spectroscopy to quantify the amount of released curcumin (Fig. 8e and Fig. S7†). The results revealed a significant difference in curcumin release between environments with and without GSH, suggesting that the presence of GSH plays a crucial role in curcumin release. Also, a higher amount of curcumin release is observed at 24 and 48 h when a combination of reducing and acidic environments is used, while almost complete release was observed at 72 h. Presumably, the large difference between neutral and reducing environments arises from the fact that curcumin, being a very hydrophobic drug, remains entrapped in the nanoparticle structure under neutral conditions, whereas it is released in solubilized form as a curcumin–CD complex upon cleavage of the disulfide bond in the presence of GSH. Size analysis through DLS indicated that the curcumin loaded nanoparticles upon exposure to GSH (10 mM) degraded over time as the single monomodal size distribution in PBS became multimodal (Fig. S8†).
Cytotoxicity and cellular uptake
The fabricated polymers were evaluated using cell viability assays to assess their safety and efficacy. First, the cytotoxicity of the conjugated polymers was evaluated using the healthy L929 cell line. The results showed no significant toxicity, even at various concentrations, indicating good biocompatibility (Fig. 9a). Next, the anti-cancer potential of curcumin-loaded nanoparticles was investigated using the U-87 cancer cell line. Notably, nanoparticles conjugated with cRGDfK displayed significantly higher cytotoxicity at concentrations of 45 μg mL−1 and 4.5 μg mL−1 compared to non-targeted nanoparticles (Fig. 9b). This enhanced cell death suggests that the cRGDfK moiety effectively targets the nanoparticles to U-87 cancer cells, facilitating greater curcumin internalization. An in vitro cellular internalization experiment on U-87 cells was conducted to compare both targeting group-containing and non-targeted nanoparticles. Confocal microscopy and flow cytometry analyses confirmed that targeted nanoparticles were internalized more than non-targeted ones, highlighting the important role of the cRGDfK peptide for enhanced cellular uptake (Fig. 10). To confirm receptor-mediated internalization of the targeted nanoparticles, a competitive inhibition assay was performed using the cRGDfV peptide, which exhibits a higher affinity for integrin receptors. The addition of cRGDfV reduced nanoparticle internalization compared to the targeted nanoparticles alone, as evidenced by confocal microscopy and flow cytometry, supporting the hypothesis of receptor-mediated uptake. To further elucidate the internalization mechanism, experiments were conducted at 37 °C and 4 °C to investigate the involvement of energy-dependent processes. At 37 °C, a substantially higher fluorescence signal was observed in the cytoplasm, indicative of active endocytosis. In contrast, nanoparticle internalization was significantly compromised at 4 °C, a temperature that inhibits energy-dependent cellular processes. Flow cytometry analysis also suggested similar trends. This temperature-dependent uptake strongly suggests that the internalization of targeted nanoparticles is an energy-dependent process driven by active transport mechanisms (Fig. S10†).
 |
| | Fig. 9 (a) Cell viabilities of L929 healthy cells treated with BDS-polymer (blue) and β-CD-conjugated polymer (green), and (b) cytotoxicity assay of non-targeted (orange) and targeted (red) curcumin-loaded nanoparticles with U-87 cells after 24 hours. All data N ≥ 3 and p < 0.0001. | |
 |
| | Fig. 10 (a) Confocal microscopy images, and (b, c) flow cytometry data of non-targeted, targeted, and cRGDfV-added curcumin-loaded nanoparticles with U-87 cells after 24 hours. Scale bar: 20 μm, all data N ≥ 3 and p < 0.003. | |
Conclusion
In summary, polymers that undergo facile chain-end functionalization through a redox-responsive disulfide linkage using the BDS group are reported. A CTA containing the BDS group is synthesized and utilized to obtain thiol-reactive polymers using RAFT polymerization. Facile chain-end functionalization through various thiol-containing molecules is demonstrated. Polymers containing CD units at chain ends linked through disulfide linkages are obtained. The other chain end is modified to install a peptide-based cancer cell targeting unit. The self-assembly of these polymers in the presence of curcumin yields drug-loaded micellar nanoparticles. Drug loading and release studies, as well as cytotoxicity and cellular internalization studies, suggest that these CD-containing polymers are promising candidates for the delivery of curcumin. Importantly, the CD-containing redox-responsive polymeric system here is demonstrated for solubilization and targeted cellular delivery of curcumin; other hydrophobic drugs and desired targeting motifs can be incorporated due to the modular nature of the construct.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
References
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef CAS PubMed.
- K. Manna, A. Roy, S. Dey, S. Ghosh, P. Pradhan and S. Pal, J. Macromol. Sci., Part A, 2024, 61, 349–375 CrossRef CAS.
- I. Altinbasak, Y. Alp, R. Sanyal and A. Sanyal, Nanoscale, 2024, 16, 14033–14056 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- J. Liu, H. Cabral and P. Mi, Adv. Drug Delivery Rev., 2024, 207, 115239 CrossRef CAS PubMed.
- M. M. Yallapu, P. K. B. Nagesh, M. Jaggi and S. C. Chauhan, AAPS J., 2015, 6, 1341–1356 CrossRef PubMed.
- T. Jiang, W. Liao and C. Charcosset, Food Res. Int., 2020, 132, 109035 CrossRef CAS PubMed.
- A. Amaroli, I. Panfoli, M. Bozzo, S. Ferrando, S. Candiani and S. Ravera, Cancers, 2024, 16, 2580 CrossRef CAS PubMed.
- S. Liu, J. Liu, L. He, L. Liu, B. Cheng, F. Zhou, D. Cao and Y. He, Molecules, 2022, 27, 4400 CrossRef CAS.
- F. L. Yen, T. H. Wu, C. W. Tzeng, L. T. Lin and C. C. Lin, J. Agric. Food Chem., 2010, 58, 7376–7382 CrossRef CAS PubMed.
- Á. Sarabia-Vallejo, M. d. M. Caja, A. I. Olives, M. A. Martín and J. C. Menéndez, Pharmaceutics, 2023, 15, 2345 CrossRef.
- C. Scavone, A. C. Bonagura, S. Fiorentino, D. Cimmaruta, R. Cenami, M. Torella, T. Fossati and F. Rossi, Drugs R D, 2016, 16, 129–140 CrossRef CAS.
- P. Jansook, M. D. Moya-Ortega and T. Loftsson, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 229–236 CrossRef CAS.
- Y. Bai, N. An, D. Chen, Y.-z. Liu, C.-p. Liu, H. Yao, C. Wang, X. Song and W. Tian, Carbohydr. Polym., 2020, 231, 115714 CrossRef CAS.
- C. S. Mangolim, C. Moriwaki, A. C. Nogueira, F. Sato, M. L. Baesso, A. M. Neto and G. Matioli, Food Chem., 2014, 153, 361–370 CrossRef CAS.
- C. H. Liu, G. W. Lee, W. C. Wu and C. C. Wang, Colloids Surf., B, 2020, 186, 110726 CrossRef CAS.
- Y. Zhou, C. Zhou, Y. Zou, Y. Jin, S. Han, Q. Liu, X. Hu, L. Wang, Y. Ma and Y. Liu, Biomater. Sci., 2020, 8, 5029–5046 RSC.
- G. Fan, Z. Yu, J. Tang, R. Dai and Z. Xu, Mater. Exp., 2021, 11, 655–662 CrossRef CAS.
- P. Arya and N. Raghav, J. Mol. Struct., 2021, 1228, 129774 CrossRef CAS.
- B. Khatun, P. Baishya, A. Ramteke and T. K. Maji, New J. Chem., 2020, 44, 4887–4897 RSC.
- S. Orbay, R. Sanyal and A. Sanyal, Micromachines, 2023, 14, 1969 CrossRef.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
- Z. Liu, L. Ye, J. Xi, J. Wang and Z.-G. Feng, Prog. Polym. Sci., 2021, 118, 101408 CrossRef CAS.
- M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 615–629 RSC.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef CAS PubMed.
- J. Stadermann, H. Komber, M. Erber, F. Däbritz, H. Ritter and B. Voit, Macromolecules, 2011, 44, 3250–3259 CrossRef CAS.
- J. Willenbacher, B. V. K. J. Schmidt, D. Schulze-Suenninghausen, O. Altintas, B. Luy, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 7056–7059 RSC.
- D. Yoshida, J. Park, N. Yamashita, R. Ikura, N. Kato, M. Kamei, K. Ogura, M. Igarashi, H. Nakagawa and Y. Takashima, Polym. Chem., 2023, 14, 3277–3285 RSC.
- R. Hoogenboom, B. C. Moorea and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- M. Arslan, T. N. Gevrek, A. Sanyal and R. Sanyal, RSC Adv., 2014, 4, 57834–57841 RSC.
- A. Degirmenci, H. Ipek, R. Sanyal and A. Sanyal, Eur. Polym. J., 2022, 181, 111645 CrossRef CAS.
- B. Zhu, T. Zong, R. Zheng, X. Chen, Y. Zhou, Y. Liu, J. Yan, B. Zhao and J. Yin, Biomacromolecules, 2024, 25, 1838–1849 CrossRef CAS.
- V. K. Kannaujiya, Y. Qiao, R. H. Sheikh, J. Xue, T. R. Dargaville, K. Liang and P. R. Wich, Biomacromolecules, 2024, 25, 1775–1789 CrossRef CAS PubMed.
- X. Wang, Y. Wu, H. Shang, X. Sun, K. An, Q. Zhang and N. Qiao, J. Drug Delivery Sci. Technol., 2023, 86, 104652 CrossRef CAS.
- T. Liu and M. Lang, J. Macromol. Sci., Part A, 2022, 59, 513–525 CrossRef CAS.
- D. Basak, R. Bej and S. Ghosh, Polym. Chem., 2015, 6, 6465–6474 RSC.
- F. Calik, A. Degirmenci, R. Sanyal and A. Sanyal, Eur. Polym. J., 2023, 201, 112548 CrossRef CAS.
- B. Saha, S. Bhattacharyya, S. Mete, A. Mukherjee and P. De, ACS Appl. Polym. Mater., 2019, 1, 2503–2515 CrossRef CAS.
- I. Altinbasak, S. Kocak, R. Sanyal and A. Sanyal, Polym. Chem., 2023, 14, 3897–3905 RSC.
- A. Degirmenci, R. Sanyal, M. Arslan and A. Sanyal, Polym. Chem., 2022, 13, 2595–2607 RSC.
- J. Li, C. Tian, Y. Yuan, Z. Yang, C. Yin, R. Jiang, W. Song, X. Li, X. Lu, L. Zhang, Q. Fan and W. Huang, Macromolecules, 2015, 48, 1017–1025 CrossRef CAS.
- N. M. Matsumoto, D. C. González-Toro, R. T. Chacko, H. D. Maynard and S. Thayumanavan, Polym. Chem., 2013, 4, 2464–2469 RSC.
- Y. L. Lo, M. F. Tsai, Y. Soorni, C. Hsu, Z. X. Liao and L. F. Wang, Biomacromolecules, 2020, 21, 3342–3352 CrossRef CAS PubMed.
- M. Arslan, Eur. Polym. J., 2020, 126, 109543 CrossRef CAS.
- Z. Hu, R. L. Arrowsmith, J. A. Tyson, V. Mirabello, H. Ge, I. M. Eggleston, S. W. Botchway, G. D. Pantos and S. I. Pascu, Chem. Commun., 2015, 51, 6901–6904 RSC.
- E. Brzezinska and A. L. Ternay, J. Org. Chem., 1994, 59, 8239–8244 CrossRef CAS.
- J. L. Shepherd, A. Kell, E. Chung, C. W. Sinclar, M. S. Workentin and D. Bizzotto, J. Am. Chem. Soc., 2004, 126, 8329–8335 CrossRef CAS.
- B. Tilottama, K. Manojkumar, P. M. Haribabu and K. Vijayakrishna, J. Macromol. Sci., Part A, 2022, 59, 180–201 CrossRef CAS.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- O. Gok, I. Kosif, T. Dispinar, T. N. Gevrek, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2015, 26, 1550–1560 CrossRef CAS.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- X. Dai, X. Chen, Y. Zhao, Y. Yu, X. Wei, X. Zhang and C. Li, Biomacromolecules, 2018, 19, 141–149 CrossRef CAS.
- B. Aktan, L. Chambre, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 490–497 CrossRef CAS PubMed.
- F. Li, Y. Zhao, C. Mao, Y. Kong and X. Ming, Mol. Pharm., 2017, 14, 2793–2804 CrossRef CAS PubMed.
- L. Deng, F. Zhang, Y. Wu, J. Luo, X. Mao, L. Long, M. Gou, L. Yang and D. Y. B. Deng, ACS Biomater. Sci. Eng., 2019, 5, 6254–6264 CrossRef CAS.
- D. Wang, T. Zhang, Y. Hu, Y. Luo, Y. Li, D. Duan, L. Zhang and Y. Zhu, ACS Appl. Nano Mater., 2024, 7, 11432–11444 CrossRef CAS.
- C. L. Waite and C. M. Roth, Bioconjugate Chem., 2009, 20, 1908–1916 CrossRef CAS PubMed.
- E. Buhr, N. Senftleben, T. Klein, D. Bergmann, D. Gnieser, C. G. Frase and H. Bosse, Meas. Sci. Technol., 2009, 20, 084025 CrossRef.
- F. Calik, A. Degirmenci, M. Eceoglu, A. Sanyal and R. Sanyal, Bioconjugate Chem., 2019, 30, 1087–1097 CrossRef CAS.
- B. S. Bolu, E. M. Gecici and R. Sanyal, Mol. Pharm., 2016, 13, 1482–1490 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Amitav
Sanyal
*c and
Amitav
Sanyal










