
Green solvents in hydroformylation-based processes and other carbonylation reactions
Fábio G.
Delolo†
 *a,
Leandro D.
Almeida†
b,
Gabriel M.
Vieira
a,
Eduardo N.
dos Santos
*a and
Elena V.
Gusevskaya
*a
*a,
Leandro D.
Almeida†
b,
Gabriel M.
Vieira
a,
Eduardo N.
dos Santos
*a and
Elena V.
Gusevskaya
*a
aDepartamento de Química, Universidade Federal de Minas Gerais, 31270-901, Belo Horizonte, MG, Brazil. E-mail: fabiodelolo@ufmg.br; nicolau@ufmg.br; elena@ufmg.br
bAdvanced Catalytic Materials (ACM), KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
First published on 27th March 2025
Abstract
Carbonylations are an important class of reactions that involve the addition of carbon monoxide (CO) to organic substrates in the presence of nucleophiles, leading to the formation of carbonyl-containing compounds. Despite its security issues, CO is a cheap feedstock, widely used in the chemical industry, that can be produced by sustainable processes. Usually, carbonylations are catalysed by transition metal complexes in solution, and solvents are employed. As solvents have a major impact on the sustainability of industrial processes, the focus of this review is the use of greener or more sustainable solvents for reactions involving carbonylations. The recent literature on hydroformylation, tandem reactions involving hydroformylation, hydroxycarbonylations, alkoxycarbonylations, aminocarbonylations, and other miscellaneous carbonylations was broadly covered. Aspects regarding renewable feedstocks, more efficient synthetic protocols (one-pot and tandem processes), milder reaction conditions, and easier catalyst recovery were also highlighted.
Green foundation1. This article gives a broad view of state-of-the art, transition metal catalysed carbonylations with an emphasis on their sustainable and green aspects, particularly concerning the solvents, since they play a major role in the overall sustainability of the processes.2. Catalytic carbonylations, especially hydroformylation, represent a synthetically efficient, diverse, and atom-economical pathway to carbonyl-containing compounds and are widely used to produce bulk and fine chemicals in industry. 3. We expect this review will inspire further developments of greener and more sustainable carbonylation processes by the replacement of fossil-based and hazardous solvents with greener alternatives and encourage researchers to design new reactions employing more recommended solvents from the outset. |
1. Introduction
1.1. Scope and applications of carbonylation reactions
Carbonylation reactions involve the addition of CO to organic substrates in the presence of nucleophiles, leading to the formation of carbonyl-containing compounds. The versatility of substrates that can be used in combination with several possibilities of their nucleophilic counterparts makes the carbonylation reaction a crucial synthetic and industrial tool, which offers convenient access to a wide range of valuable chemical products.1–8In particular, since its discovery 86 years ago, hydroformylation has been a remarkable strategy in the chemical industry for producing aliphatic aldehydes (Fig. 1).9 As mentioned above, this process consists of the addition of CO and H2 (syngas) to alkenes, enabling the conversion of alkenes into valuable aldehydes that can be used directly or as key intermediates for further chemical transformations. It is estimated that over 10 million tons of aldehydes are produced annually through hydroformylation.10,11
 | ||
| Fig. 1 The scope of catalytic carbonylation: examples of hydroformylation-based processes and other carbonylation reactions. | ||
A second example that must be highlighted is the carbonylation of methanol to produce acetic acid.12–16 In 2019, global acetic acid production reached 9.1 million tons, with around 85% derived from methanol carbonylation.12,17 Another significant industrial application of carbonylation reactions is in the production of methyl propionate (MP), a key intermediate for manufacturing methyl methacrylate polymers and plastics.18 The leading commercial method for MP synthesis, known as the Lucite Alpha process, consists of the reaction of CO and alcohol with ethylene (this transformation is called alkoxycarbonylation).19 Alkoxycarbonylation (Fig. 1) is a valuable synthetic method for producing esters, which are important intermediates for pharmaceuticals, agrochemicals, and fine chemicals.
Despite the great success and significance of established industrial carbonylation processes, many other chemical transformations based on the insertion of CO into organic substrates show significant potential for large-scale industrial applications. Some examples are shown in Fig. 1. Hydroaminomethylation, a tandem process that combines hydroformylation with reductive amination, directly produces amines from alkenes, offering an efficient route to the synthesis of nitrogen-containing compounds. Similarly, hydroformylation–acetalization couples hydroformylation with the formation of acetals, streamlining the production of aldehydes protected as acetals for further chemical transformations. The hydroformylation–hydrogenation reaction sequence is another tandem process in which hydroformylation is followed by hydrogenation, allowing for the efficient conversion of alkenes into homologous alcohols. Hydroxycarbonylation, a reaction that produces carboxylic acids via the addition of CO and water to alkenes, holds promise for the sustainable production of biorenewable acids. Aminocarbonylation enables the direct synthesis of amides from aryl halides, representing an especially valuable tool for the pharmaceutical and agrochemical sectors. Furthermore, strategies based on the cross-coupling reaction in the presence of CO enable the production of ketones.1,20 Lastly, thiocarbonylation offers a pathway for producing sulphur-containing compounds, which have important applications in the pharmaceutical and rubber industries.
1.2. Sustainability aspects – the role of solvents
Further development of this chemistry could lead to new sustainable and efficient processes that complement or even surpass current carbonylation methods, offering routes to various key intermediates for a wide range of industries. The exploration and industrial application of carbonylation reactions and tandem processes based on carbonylation could greatly enhance the efficiency of chemical manufacturing, especially if sustainability aspects are considered.The development of sustainable processes aims to minimize energy consumption, waste generation, and the use of hazardous reagents. Furthermore, as sustainability becomes a key concern in chemical processes, the choice of solvent is increasingly important not only for its effect on the reaction outcome but also for its environmental and toxicological impacts. In the pharmaceutical sector, for example, it is estimated that solvents contribute to more than 70% of the total mass used in a production sequence.21 Beyond their sheer volume, life cycle assessments reveal that around 50% of the energy consumed during the production of active pharmaceutical ingredients (APIs) is linked to solvent usage. Moreover, solvents are responsible for nearly 60% of the greenhouse gas emissions associated with the industrial process.22
Solvents play a fundamental role in daily life, laboratories, and industrial applications.22,23 Their main function is to increase the mobility of the reactants and allow collisions that lead to the reaction products. However, the effect of solvents on chemical reactions and reactivity is profound, influencing multiple aspects, such as the reaction rate, yield, and selectivity, by stabilizing intermediates or transition states. Solvents can impact the solubility of reactants, facilitating their interaction and promoting more efficient reactions. They also play a critical role in heat and mass transfer, ensuring that reactions occur under optimal conditions.24
However, despite this well-established understanding, solvents are often overlooked in reaction design and optimization, and more emphasis is placed on catalysts and ligands.25 Solvents are frequently chosen based on their ability to dissolve reactants, without much consideration of their broader impact on the reaction mechanism, intermediates, and transition states. This oversight has led to a lack of in-depth studies on the critical role that solvents can play in shaping reaction outcomes. However, several scholars recognize the significant influence of the solvent on the reaction medium. In 2010, Kou and co-workers reviewed the importance of green solvents in transition metal nanoparticle catalysis.26 The authors claimed that selected green solvents could enhance the efficiency of nanoparticle catalysis by providing a more favourable environment for reactions, which could lead to improved selectivity and catalytic activity. Particularly, in challenging processes like the partial hydrogenation of arenes, certain green solvents can provide better outcomes compared to traditional solvents. However, the authors recognized that environmental benefits alone might not be sufficient for the widespread adoption of alternative solvents. Efficiency and cost-effectiveness are also critical factors that need to be addressed.
Recently, Resasco and colleagues reviewed the effects of solvent on catalytic reactions and related phenomena at liquid–solid interfaces.27 The authors recognized that the role of solvents in competitive adsorption was well documented, but quantification of their impact on reactant–catalyst binding was still at an early stage. However, understanding these interactions is essential for optimizing catalytic processes.
Homogeneous catalysts are more affected by solvents due to their intimate interaction with the catalyst molecules. In 2016, Dyson and Jessop reviewed the critical role of solvents in homogeneous catalysis based on a mechanistic approach.28 In catalytic systems, solvents interact with both catalysts and substrates, affecting the catalytic activity and selectivity by altering the energy profiles of intermediates and transition states. These interactions can shift the reaction pathway, leading to different products or enhancing the desired selectivity. The choice of solvent can thus make a significant difference to both the chemical efficiency and economic feasibility of a process.28 An acceleration effect can be related to the direct participation of the solvent in coordination–dissociation of ligands and reactants, the stabilization of transition states in relation to ground states, the change in the solubility of reactive gases, as well as the change in the rate of the mass transfer of reactive gases into the liquid phase in different reaction media. In 2019, Slattery and collaborators reviewed the effects of solvents in popular palladium-catalysed cross-coupling methodologies, such as Suzuki, Stille, Kumada, Negishi, Hiyama, Heck, Sonogashira, and Buchwald–Hartwig reactions.29 The paper emphasizes that the solvent plays a crucial role, affecting reaction efficiency and selectivity. For instance, in the Buchwald–Hartwig and Sonogashira reactions, an appropriate solvent can facilitate various reaction steps, such as oxidative addition and reductive elimination. In some cases, polar coordinating solvents can enhance isomerization and reductive elimination processes, while low polarity solvents may hinder these steps. The authors advocate for further studies on solvent effects to optimize cross-coupling methodologies, which could lead to greener and more sustainable synthetic practices.
1.3. Solvent selection guides
To assist in solvent selection, solvent selection guides (SSGs) have been developed as essential resources for researchers. These guides provide structured frameworks for evaluating and choosing solvents based on multiple criteria, including chemical properties, environmental impact, and safety considerations. By incorporating parameters such as polarity, miscibility, and toxicity, SSGs enable informed decision-making that aligns with green chemistry and sustainability principles.The effective use of SSGs not only promotes the selection of suitable solvents but also advances the broader goals of sustainability and safety in chemical research and industrial applications. Over the years, several companies, such as Sanofi,30 Syngenta,31 Pfizer,32 AstraZeneca,33 and GlaxoSmithKline (GSK),34,35 developed SSGs, some in collaboration with the public sector.36
In 2014, Prat and co-authors published a survey of solvent selection guides.37 Researchers have been developing special tools and techniques to assist in the selection of more sustainable solvent alternatives, which were reviewed in 2016 by Clark and coauthors.38 In 2015, Pena-Pereira and collaborators proposed a more quantitative and informative approach for solvent selection.39 The authors ranked more than 150 solvents according to their toxicological endpoints, hazards, the potential to be appropriately disposed of after utilization, and the potential to be obtained from renewable feedstock. In 2016, Isoni and colleagues developed Q-SA√ESS (quick sustainability assessment via experimental solvent selection) to perform an initial solvent selection based on environmental, health and safety (EHS) factors and their suitability for specific chemical reactions.40
1.4. Green and sustainable solvents
Green chemistry is a transformative approach aimed at designing chemical processes and products that reduce or eliminate the use of hazardous substances, thus minimizing environmental impact and enhancing safety.41–47 This discipline is guided by 12 principles, which serve as foundational tenets for sustainable practices in chemical research and industry.48 These principles emphasize waste prevention, the design of safer chemicals, energy efficiency, and the use of renewable feedstocks, among others. A particularly critical aspect of green chemistry is the selection of solvents, which significantly influences the sustainability and safety of chemical processes.A green solvent is one that minimizes environmental and health impacts throughout its life cycle. The key characteristics of green solvents include low toxicity, biodegradability, and reduced volatility, which helps to lower exposure risks and minimize air pollution. Green solvents are often derived from renewable sources, such as biomass, and have a reduced carbon footprint compared to traditional petroleum-based solvents. They should be safe for humans, environmentally benign, and efficient in terms of energy required during their recycling or disposal. It is important to include in the environmental scoring of solvents a life-cycle impact analysis of their synthesis and end-of-life recovery; in particular, their carbon footprint and total energy demand. The use of green solvents is part of the green chemistry principles that enables the development of safer and more sustainable chemical processes, i.e., the processes aligned with environmental regulations and the reduced generation of hazardous waste.
Analyzing the most common solvents in the SSGs, it can be observed that some classes are more common, such as alcohols and esters. Solvents like water, ethanol, isopropanol, n-butanol, ethyl acetate, isopropyl acetate, n-butyl acetate, anisole, and ethylene glycol are generally recommended solvents in most of the guides. The main parameters to evaluate the greenness of a solvent are health, safety, and environmental impact. In terms of safety, flammability is the main concern and to assess this last issue, the combination of flash point and boiling point is considered. This is why some low boiling substances like ethanol or ethyl acetate are less recommended, although they present low toxicity. However, high boiling point solvents might result in energy waste during solvent recycling. To be sustainable, the solvent must be, at least in a long-term analysis, produced from biobased feedstocks. Examples of solvents that can be obtained from renewable sources are ethanol, n-butanol, acetone, isobutanol, isoamyl alcohol (from sugar fermentation), triglycerides, their methyl esters, and glycerol (from the biodiesel industry), pinenes (turpentine, from pine resin), limonene (from the citrus industry) dipentene (from the cellulose industry), and the ones produced from lignocellulosic biomass (γ-valerolactone, 2-methyltetrahydrofuran, tetrahydrofurfuryl alcohol, cyrene). Organic carbonates have also attracted attention as green solvent alternatives due to their safety and production process, which involves CO2 capture. Besides, they have a wide range of polarity and are indicated to be good replacements for ketone and glycol ether solvents. Cyclic carbonates could replace hazardous aprotic polar solvents, such as DMF (Fig. 2).
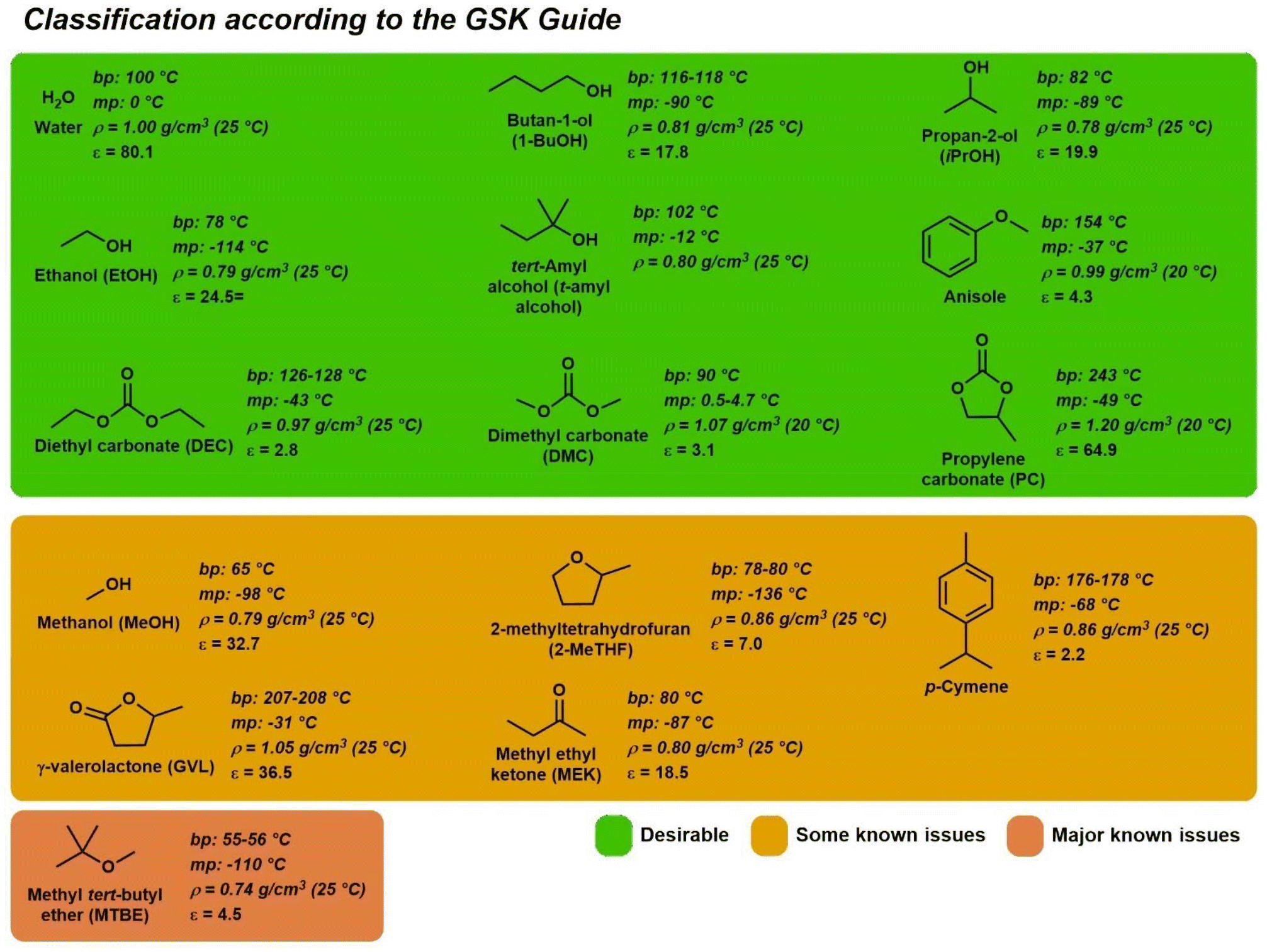 | ||
| Fig. 2 Chemical structures of selected solvents. | ||
Of course, the replacement of traditional fossil-based solvents by greener or more sustainable alternatives is not an easy task, particularly in continuous, large-scale processes. Beyond the chemical performance, factors such as local price and availability, costs for recycling and disposal, among others, should be considered. Nevertheless, the above-mentioned green and sustainable solvents can be “drop-in” replacements, meaning that they can be used without significant changes to existing chemical plants. The implementation of biphasic, thermomorphic or supercritical carbon dioxide systems discussed hereafter involves significant changes to infrastructure and these systems are more likely to be implemented in new production plants.
1.5. Scope of the review
The replacement of fossil-based and hazardous solvents with environmentally friendly alternatives can lead to the development of greener and more sustainable synthetic practices in catalytic carbonylation. Numerous research works have been published on this challenging topic, most of them in the last ten years, and the present article will give an overview of this information. Recently, the use of renewable solvents in palladium-catalysed carbonylation reactions was discussed in excellent review articles by Bayer49 and Schwab.50 In 2024, Árvai and Mika published a more general review highlighting recent advances in catalytic carbonylation reactions in alternative reaction media, in which the use of other metals besides palladium was also addressed.51 Hydroformylation – a central point of the present contribution – was reviewed by Árvai and Mika for aqueous biphasic systems, with only a few examples in other alternative reaction media being included. The present review article is mainly focused on the use of green solvents in hydroformylation and hydroformylation-based tandem processes, but an overview of the literature on other carbonylative transformations, such as alkoxycarbonylation, hydroxycarbonylation, aminocarbonylation, thiocarbonylation, carbonylative cross-coupling, and ring-expansion reactions in environmentally friendly media, is also provided. Differently from the previous reviews mentioned above, the information is classified according to the type of chemical transformation rather than the nature of the solvent. Not surprisingly, since in homogeneous catalysis the solvent plays multiple roles, the optimal green solvent strongly depends on the system, even considering the same class of reactions. Nonetheless, to guide the reader, at the end of each subsection, we recommend green solvents that present good results in different systems.2. Hydroformylation
Hydroformylation, often referred to as the oxo process, is a fundamental chemical process in which an olefin (unsaturated hydrocarbon) reacts with carbon monoxide (CO) and hydrogen (H2) in the presence of a catalyst, typically a transition metal complex, to produce aldehydes.11,52–60 The choice of catalyst and reaction conditions can influence the regioselectivity of hydroformylation, a crucial factor in industrial applications. The industrial catalysts for hydroformylation are based on cobalt and rhodium complexes in solution. However, other metals have also shown hydroformylation activity: e.g., Ir, Ru, Os, Pt, Pd, Fe, and Ni.59,61–63A key advantage of the hydroformylation reaction is its atom economy. This reaction incorporates all atoms of starting materials into the final product, minimizing waste. Hydroformylation is the main process for the production of aliphatic aldehydes and plays a vital role in the synthesis of many fine chemicals, pharmaceuticals, and agrochemicals.64,65 This reaction is of immense importance in industrial chemistry, enabling the synthesis of key intermediates for the production of various chemicals, including plasticizers, detergents, and synthetic lubricants. In addition to the hydroformylation of olefins, there are reports in the literature on the hydroformylation of epoxides.66 However, despite the various patents filed on the subject, there is no record of industrial plants that carry out the hydroformylation of epoxides.67–77
The hydroformylation reaction can be, in principle, performed without solvent, if one of the reactants is liquid.78 However, this approach introduces several limitations on the process, so that in industrial hydroformylation solvents are usually employed. In some industrial processes for the production of aldehydes from alkenes, a high boiling point solvent is used to ensure the catalyst remains dissolved during product distillation, but this can cause thermal stress to the catalyst.79 A significant advancement was achieved by Ruhrchemie/Rhône Poulenc, which developed a biphasic process for the hydroformylation of propene, in which a rhodium catalyst was maintained in an aqueous solution through the use a water-soluble phosphorus ligand. The catalyst is recovered by separating the water phase before distillation, but the process is limited to olefins with at least a moderate solubility in water, usually limited to five carbons. This approach has inspired the development of other biphasic systems, including fluorous phases80,81 and ionic liquid systems.82–84 Furthermore, alternative solvents such as supercritical carbon dioxide85 and thermomorphic solvent86–90 systems have been extensively researched in recent years.
All the above-mentioned approaches have limitations, and the search for greener solvents and catalytic systems for hydroformylation can enhance the sustainability and environmental friendliness of this important chemical transformation, making it an area of active research within the realm of green chemistry.
2.1. Solvent-free hydroformylation
Solvent-free reactions are considered the best situation according to green chemistry principles. Solvent-free hydroformylation, also known as neat hydroformylation, would represent a significant advancement in the field, eliminating the need for traditional organic solvents, and, thereby, reducing waste generation, minimizing environmental impact, and streamlining the purification process. In solvent-free hydroformylation, the substrate is a liquid phase that dissolves the gas phase and the catalyst. This method offers several advantages, e.g., enhanced selectivity towards desired products and simplified procedures for product isolation. Solvent-free hydroformylation can be particularly advantageous for industrial processes, as it eliminates the challenges associated with solvent removal and reduces the overall energy consumption.91 However, it should be considered that even when the reaction is performed under solvent-free conditions, product purification requires the use of a solvent. In addition, mass-transfer limitations can be a problem in these processes as the reactions are carried out in a gas–liquid phase system. Moreover, solvent-free conditions may be detrimental to the activity of the system.92 Only a few examples of solvent-free hydroformylation have been reported in the literature, even for substrates that could, in principle, act as solvents.Monnereau, Sémeril, and Matt investigated solvent-free hydroformylation using rhodium complexes with hemispherical 1,3-calix[4]arene-diphosphites as catalysts (Fig. 3).93 For the hydroformylation of 1-octene, an impressive turnover frequency (TOF) of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 290 h−1 was achieved under optimized conditions. The authors disclosed that the reaction rates were seven times higher in comparison with the reactions performed in toluene. In addition, the use of the reported rhodium complexes favoured the regioselectivity for linear aldehydes (l/b = 84.5 for 1-octene and 4.3 for styrene) due to the strong steric hindrance imposed by the bulky ligand.
290 h−1 was achieved under optimized conditions. The authors disclosed that the reaction rates were seven times higher in comparison with the reactions performed in toluene. In addition, the use of the reported rhodium complexes favoured the regioselectivity for linear aldehydes (l/b = 84.5 for 1-octene and 4.3 for styrene) due to the strong steric hindrance imposed by the bulky ligand.
 | ||
| Fig. 3 Solvent-free hydroformylation of olefins.78,93 | ||
In 2015, Alsalahi and Trzeciak reported the solventless hydroformylation of olefins using Rh(acac)(CO)2 and PPh3 as the auxiliary ligand (Fig. 3).78 They observed fast complete conversion of 1-hexene with a TOF of 970 h−1, whereas the reaction performed in toluene showed a TOF of only 438 h−1. Catalyst recycling was carried out by “vacuum transfer” followed by the addition of a new portion of the olefin, which was converted even faster than in the first cycle (TOF = 1330 h−1). In the optimization study, they found an optimum ratio between the substrate and the catalyst (S/C = 3000) and obtained an impressive TOF of 4353 h−1 at 80 °C, and 10 bar of syngas. Then, the authors performed the hydroformylation of 1-octene with a special focus on catalyst recovery. Remarkably, from the first to the fifth cycle, the conversion only decreased from 100% to 80% in 1 h, without the loss of selectivity for the aldehydes.
In 2016, Shaikh and collaborators developed a magnetic nanoparticle-supported ferrocenylphosphine material to anchor metals like Pd or Rh.94 The authors compared the catalytic activity of the rhodium-based material in styrene hydroformylation in DCM and THF, as well as under solvent-free conditions. Although the material performed well under solvent-free conditions, the reactions with solvents displayed higher rates and regioselectivities.
Using a heterogeneous catalyst, which consisted of a porous organic polymer containing rhodium species, Wang and Yang performed the solvent-free hydroformylation of olefins.95 Remarkably, l/b regioselectivity of up to 158.5 was achieved with 1-hexene as the substrate at almost complete conversion. The catalyst could be recycled 5 times without significant activity changes, highlighting the environmental aspects of this work.
In 2023, the use of triethylamine as an additive in the [Rh(cod)Cl2]/PPh3 catalytic system, which operated under solvent-free conditions, was reported (Fig. 4).96 The TOFs obtained in the solvent-free experiments (62 h−1) were higher than the ones obtained in DMF, toluene, DMAc, DMA, and DIPEA solutions (up to 8 h−1). The authors speculated that the role of triethylamine was to act as an HCl scavenger to form catalytically active hydride species. Various aldehydes were obtained by this methodology; nonetheless, no functional group tolerance was disclosed. To demonstrate the greenness of the process, recycling experiments were performed. To accomplish this, the final mixture was distilled under vacuum at 25 and 50 °C, and then fresh substrate and triethylamine were added for subsequent runs. The catalytic system could be recycled up to 7 times. From the fifth to seventh cycle, a conversion decrease was observed due to the formation of insoluble rhodium black powder.
 | ||
| Fig. 4 Rhodium catalysed solvent-free hydroformylation of olefins.96 | ||
In 2024, Zhang and collaborators studied hydroformylation reactions using a heterogeneous single-atom rhodium catalyst.97 The study demonstrated that leaching of rhodium species was lower at higher substrate concentrations. For this reason, the hydroformylation of 1-hexene was performed under solvent-free conditions. An increase in the formation of aldehydes during the 50 h experiment was observed, with a linear l/b ratio close to 3.
Recently, an effective catalytic system composed of tetradentate P-ligands and Rh(acac)(CO)2 was disclosed.98 The presence of these ligands suppressed the formation of hydrogenation and isomerization products. In the solvent-free system, higher regioselectivity was achieved than in toluene solutions under similar conditions (l/b ratio of 14.9 vs. 11.0). Unfortunately, product and catalyst recovery were not addressed by the authors.
2.2. Hydroformylation in green solvents
Behr and collaborators conducted the hydroformylation of piperylene utilizing a Rh(acac)(CO)2/Xantphos catalytic system and propylene carbonate (PC) as the solvent (Fig. 5).99 PC is known as an eco-friendly alternative to traditional organic solvents due to its low toxicity, biodegradability, and low corrosivity. During the hydroformylation of piperylene, regioselectivity control can be a challenging task due to the possibilities of 1,2- and 1,4-additions as well as substrate isomerization. Interestingly, the main product obtained in this study was an E/Z mixture of α,β-unsaturated aldehydes. After an exhaustive optimization, with a focus on recycling the catalytic system, PC was chosen as the optimal solvent, allowing α,β-unsaturated aldehydes to be obtained in 51% yield (l/b ratio of 1:3.6). The use of other solvents, like methanol, dimethylformamide, acetonitrile, toluene, and heptane, resulted in yields of 36%, 15%, 8%, 58%, and 29%, respectively. In recycling experiments, the reactor was directly connected to vacuum distillation apparatus and heated at 40 °C to remove the reactants and the products. A decreasing performance was achieved in five consecutive runs due to the ligand decomposing; however, the addition of a new portion of the ligand enabled the recovery of the catalytic activity.
 | ||
| Fig. 5 Hydroformylation in propylene carbonate (PC) (top) and γ-valerolactone (GVL) (bottom) solutions.99,101 | ||
In 2016, Mika and collaborators reported an enantioselective catalytic system for the hydroformylation of styrene in γ-valerolactone (GVL) solutions, which consisted of in situ generated platinum–chiral diphosphine–tin(II) chloride complexes.100 GVL is a biomass-based and non-toxic renewable compound considered to be a platform molecule for renewable chemicals. In the ligand screening study, all the results obtained in GVL were compared with the benchmark solvent – toluene. Optically active versions of BDPP, BINAP, SEGPHOS, and JOSIPHOS ligands were evaluated, resulting in moderate enantiomeric excess (ee) for the desired product. In general, lower activities were obtained in GVL solutions under the same reaction conditions as those in toluene. However, the GVL-based systems showed improved chemoselectivity towards the aldehydes and ee values comparable to those obtained in toluene, especially at a relatively low temperature of 80 °C. Catalyst recycling was performed by vacuum distillation of the reaction mixture followed by redissolution in GVL. Nonetheless, a continuous decrease in catalyst performance was observed from the first to the fourth cycle (from 76% to 25% conversion in the same reaction time). 31P NMR analysis of the spent catalyst suggested the dissociation of tin(II) chloride from the catalyst, which was suggested as a reason for the decrease in catalytic activity.
In 2017, GVL was efficiently applied as a solvent for the hydroformylation of various functionalized olefins, such as α-methylstyrene, dimethyl itaconate, and limonene, in the presence of rhodium–chiral diphosphine catalytic systems.101 The rhodium–BDPP combination showed lower catalytic activity than that observed in toluene, but with higher selectivity for the desired aldehydes, which was attributed to the coordinating nature of GVL. The system performed excellently in three consecutive recycling runs (Fig. 5).
In 2016, the group of Gusevskaya and dos Santos reported the selective hydroformylation of β-caryophyllene in ethanol solutions using a [Rh(cod)(OMe)]2/PPh3 catalytic system (Fig. 6).102 The strained bicyclic structure of this sesquiterpene substrate poses a challenge for selective transformation due to its possible isomerization. Interestingly, the obtained hydroformylation product was derived from the apparently less reactive endocyclic olefinic bond rather than the terminal one. A systematic study was performed to optimize the reaction parameters and achieve high selectivity. Remarkably, the reaction rates were similar to those observed in toluene. Despite the possibility of the formation of acetals in ethanol solutions (tandem sequence of hydroformylation–acetalization reactions), 71% yield of the corresponding monoaldehyde product was achieved, along with small amounts of di-hydroformylation products and acetals. Later, the same group performed the hydroformylation of caryophyllene oxide in various green, biorenewable solvents: ethanol, dimethyl carbonate (DMC), diethyl carbonate (DEC), p-cymene, and 2-methyltetrahydrofuran (2-MeTHF) (Fig. 6).103 Excellent yields were obtained in DMC, DEC, and p-cymene (90–95%). Decreased catalytic activity in 2-MeTHF was ascribed to the possible coordination of the solvent on rhodium, whereas reduced selectivity for the aldehyde in ethanol solutions was ascribed to the formation of acetals due to the relatively high reaction temperature required for the hydroformylation of the sterically encumbered olefinic bond in caryophyllene oxide.
 | ||
| Fig. 6 Hydroformylation of β-caryophyllene (top) and caryophyllene oxide (bottom) in green renewable solvents.102,103 | ||
In 2019, the group of Gusevskaya and dos Santos described another example of hydroformylation focused on the synthesis of fragrance molecules (Fig. 7).104 The hydroformylation of (−)-myrtenol and nopol was performed in solutions of ethanol, DEC, and 2-MeTHF. In the case of myrtenol, besides the desired aldehydes resulting from hydroformylation, the substrate isomerization product – myrtanal – was produced in significant amounts. The best selectivity for hydroformylation was obtained in DEC solutions.
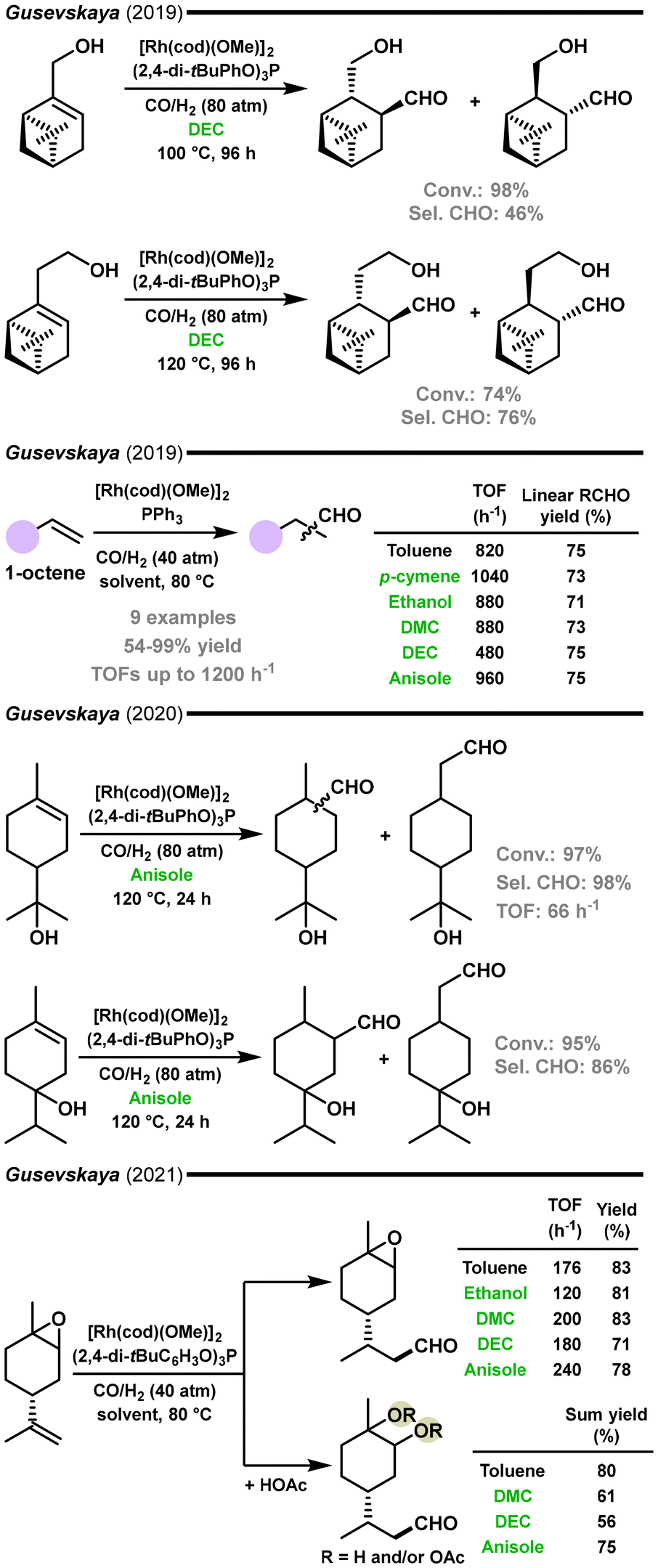 | ||
| Fig. 7 Hydroformylation of fragrance compounds: nopol, myrtenol and limonene oxide.104–107 | ||
Also in 2019, the same group demonstrated that anisole could be used as an efficient green solvent for the hydroformylation of olefins, including terpene-derived olefins (Fig. 7).105 The performance of various green solvents, such as p-cymene, DEC, DMC, ethanol, and anisole, was compared with that of toluene, as a benchmark reaction medium. For the hydroformylation of 1-octene, regioselectivity for the linear aldehyde was nearly the same in all solvents tested; however, the reaction rates (TOFs) in green solvents (except DEC) were higher than that in toluene. Anisole showed the best results and was applied in the hydroformylation of various olefins, including commercially relevant molecules like estragole, limonene, carveol, α-pinene, myrtenol, nopol, and perillyl alcohol. The obtained yields of the corresponding fragrance aldehydes varied from 54 to 99%. Remarkably, the improvement in selectivity was particularly important for substrates with a hydroxyl group in their structure, such as perillyl alcohol, carveol, myrtenol, and nopol. It was suggested that the beneficial effect of anisole was related to the protection of the hydroxyl group from coordination on rhodium due to hydrogen bond formation with anisole (the solvent). In other words, anisole helped to prevent the chelation of the substrate on rhodium to form less active hydroformylation chelate complexes, thus accelerating the whole hydroformylation process.
In 2020, the group of Gusevskaya and dos Santos reported the successful hydroformylation of recalcitrating olefinic bonds in a series of terpenes, such as α-terpineol, terpinene-4-ol, limonene, and α-ionone (Fig. 7).106 The accomplishment of this work was achieved due to the use of the [Rh(COD)(OMe)]2/(2,4-di-tBuC6H3O)3P catalytic system. A solvent screening performed for α-terpineol in solutions of toluene, p-cymene, DEC, and anisole resulted in total aldehyde yields of 91%, 97%, 97%, and 95%, respectively. Interestingly, the reaction in anisole showed higher TOFs than in toluene (66 vs. 56 h−1). The double hydroformylation of limonene resulted in 91% yield of the aldehydes. In the case of α-ionone, the aldehyde product was derived from the hydroformylation of the less hindered olefinic bond (91% yield).
The same group recently reported the hydroformylation of limonene oxide and a one-pot process involving simultaneous hydroformylation of this substrate and epoxy ring cleavage using various green solvents as the reaction medium (Fig. 7).107 With the [Rh(cod)(OMe)]2/(2,4-di-tBuC6H3O)3P catalytic system, the rate of limonene oxide hydroformylation was 176, 120, 200, 180, and 240 h−1 in toluene, ethanol, DMC, DEC, and anisole solutions, respectively. The selectivity for the aldehyde was high in all the solvents. In the presence of an acylating agent (acetic anhydride or acetic acid), epoxy ring cleavage occurred simultaneously with hydroformylation to give diols, molecules with multiple functionalities and new olfactory characteristics.
In 2021, the one-pot rhodium-catalysed hydroformylation/O-acylation of propenylbenzenes using anisole as the solvent was reported by the group of Gusevskaya and dos Santos (Fig. 8).108 Anisole performed better than the benchmark solvent toluene. The process combined the hydroformylation reaction and parallel O-acylation of the phenyl group in the substrate molecule with acetic anhydride resulting in difunctionalized compounds. The single-step procedure developed allowed for the simultaneous incorporation, in a synthetically efficient manner, of two different organic groups into the substrate molecule: the acetate and the aldehyde, known for their interesting olfactory properties. The desired products were obtained in up to quantitative yields with high regioselectivity. Later, the methodology was extended to biomass-based hydroxyolefins (i.e., perillyl alcohol, carveol, myrtenol, nopol, isopulegol, and isoprenol) (Fig. 8).109 In 2024, the same group reported the use of formic acid and acetic anhydride as a syngas (CO/H2) surrogate to carry out the simultaneous incorporation of the acetate and aldehyde groups (Fig. 8).110 THF showed the best performance as the solvent (91% yield); however, its greener alternative 2-MeTHF allowed for a similar outcome (86% yield). The substrate screening was performed in 2-MeTHF, with a series of propenylbenzenes and terpene derivatives being obtained high yields.
 | ||
| Fig. 8 Hydroformylation of renewable propenyl benzenes and terpene-based hydroxyolefins in anisole solutions.108–110 | ||
In 2021, Beller's group presented the first example of the cobalt-catalysed hydroformylation of olefins using phosphine oxides as ligands and anisole as a green solvent (Fig. 9).111 The hydroformylation of 1-octene occurred similarly in toluene and anisole; however, in DMC, DEC, and ethanol, catalytic activities were lower. After process optimization, the aldehydes from 1-octene were obtained in 91% yield with an l/b ratio of 69:31. A wide scope of olefinic substrates were successfully tested, providing the desired aldehydes in good to excellent yields and high regioselectivity.
 | ||
| Fig. 9 Cobalt-catalysed hydroformylation of olefins.111 | ||
After considering the presented examples and recent trends in the application of green solvents in hydroformylation, anisole stands as a good alternative to the benchmark toluene. Key factors, such as similar chemical structures, polarity, boiling point, and density, can facilitate the replacement of toluene in current industrial processes. In addition, the use of organic carbonates such as propylene carbonate deserves to be highlighted. The advantage that can be underscored in this case is the possibility of selecting carbonates with different polarity and boiling points that will better suit the desired catalytic system.
2.3. Hydroformylation in supercritical CO2
Supercritical carbon dioxide (scCO2) has emerged as a promising green solvent for carbonylation reactions, offering a sustainable alternative to traditional organic solvents. In its supercritical state, CO2 exhibits unique properties that combine the characteristics of both a gas and a liquid, such as low viscosity and high diffusivity, which enhance mass transfer and reaction rates.112,113 The use of this non-toxic and non-flammable solvent not only minimizes environmental impact but also facilitates the selective extraction and purification of products. Furthermore, the ability to easily tune the density and solvent properties of scCO2 through pressure and temperature adjustments allows for improved control of the reaction conditions.The solubility of transition metal catalysts is rather limited in scCO2 due to its low polarity. To overcome this limitation, Jin and Subramaniam introduced the application of CO2-expanded liquids in hydroformylation, combining the advantages of scCO2 and organic solvents.114 The hydroformylation of 1-octene was performed in the presence of the unmodified Rh(acac)(CO)2 catalyst in CO2-expanded acetone in comparison with pure acetone and pure scCO2. The turnover number (TON) obtained in CO2-expanded acetone was higher than that in pure acetone (147 vs. 35 in 6 h); this was attributed to the higher solubility of syngas in CO2-expanded acetone due to the lower dielectric constant of this medium. The best results were obtained using a mixture of CO2 and acetone in a volume ratio of 75/25. Alternatively, the reaction in pure scCO2 resulted in an even higher TON (209); however, it required a higher pressure (210 vs. 103 bar).
More recent advances in hydroformylation in scCO2 was reviewed previously.115
2.4. Hydroformylation in thermomorphic solvent systems
Thermomorphic solvent systems (TMS) represent an eco-friendly option to replace common organic solvents. The use of TMS allows for a decrease in energy usage, more precise reaction control, and a simplification of product recovery, thus contributing to process sustainability. Within this approach, the reaction medium consists of at least two solvents, which have a highly temperature-dependent miscibility. A catalytic reaction occurs under homogeneous conditions in a single liquid phase, whereas the catalyst is recovered by liquid–liquid separation at lower temperature, when the solvents are not miscible and form at least two liquid phases.86Behr and co-workers applied a TMS composed of propylene carbonate (PC), dodecane, and p-xylene as the reaction medium for the isomerizing hydroformylation of 4-octene catalysed by the Rh(acac)(CO)2/BIPHEPHOS system (Fig. 10).116 Outstandingly, the addition of p-xylene to the PC/dodecane mixture favoured selectivity to n-nonanal, with the effect being dependent on the relative amounts of PC in the mixture. p-Xylene, as a third component of the solvent, was supposed to play the role of a solvation mediator between the other two solvents. Under optimized conditions, 99% conversion was obtained with ca. 90% selectivity for the desired linear aldehyde. However, significant rhodium leaching (47%) into the nonpolar dodecane phase was observed, thus limiting future industrial applications until this drawback is overcome.
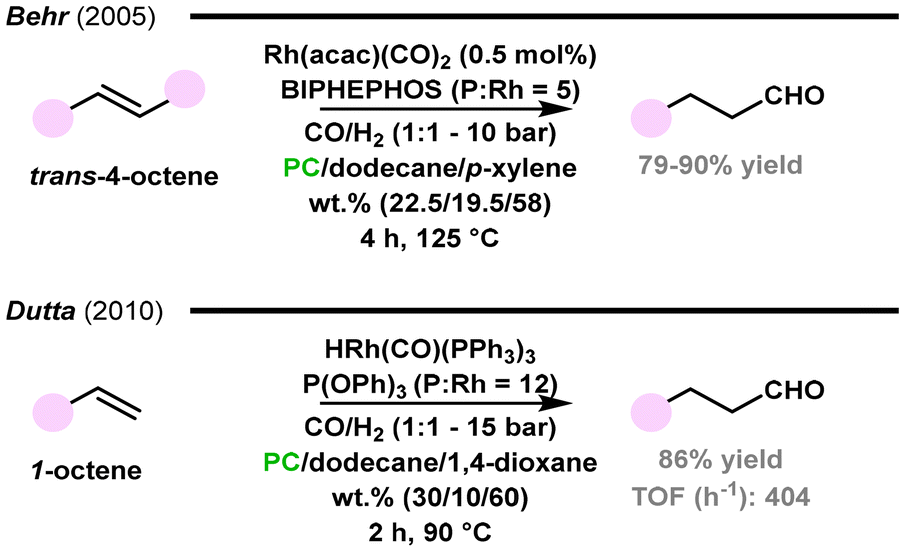 | ||
| Fig. 10 Hydroformylation of olefins in thermomorphic solvent systems.116,117 | ||
Later on, Dutta and collaborators, in an attempt to reduce rhodium leaching, evaluated the TMS composed of PC, dodecane, and 1,4-dioxane, using the latter as the mediator solvent, given its higher polarity compared to p-xylene (Fig. 10).117 In the TMS of the optimized composition, the rhodium loss was minimized to 2.8%. Nearly complete conversions and 89% selectivity for the linear aldehyde were achieved for the hydroformylation of 1-octene in the presence of the HRhCO(PPh3)3/P(OPh)3 catalytic system.
Recently, Vossen, Leitner, and Vorholt presented an atypical example of a TMS using ethylene carbonate (EC) as a solvent. This compound has a melting point of 36 °C, which makes it solid at room temperature, thus enabling liquid product separation and retaining the catalyst in the polar solid phase (EC) due to crystallization (Fig. 11).118 At a certain product yield, the formation of a solid–liquid–gas system was observed under the reaction conditions. This was ascribed to the precipitation of the catalyst, which limited substrate conversion. To circumvent the problem, two different methodologies were adopted: the addition of either water (to increase the polarity of the catalyst phase) or n-decane (to decrease the polarity of the organic phase). Water addition resulted in 93% yield with a TOF of 1215 h−1, while n-decane addition allowed for 89% yield with a TOF of 579 h−1. Both systems demonstrated good recyclability, activity, and catalyst stability.
 | ||
| Fig. 11 Schematic representation of the phase behaviour during the hydroformylation of 1-octene in ethylene carbonate (EC). UCST – upper critical solution temperature; AT – ambient temperature.118 This figure has been adapted/reproduced from ref. 118 with permission from the American Chemical Society, copyright 2023. | ||
Despite the progress achieved in the application of TMS in hydroformylation, the use of fossil-based solvents as part of the system still represents a challenge to be overcome in the field.
2.5. Hydroformylation in biphasic systems
The tremendous success of the Ruhrchemie/Rhone-Poulenc biphasic hydroformylation process fostered intense research activity in this field.119,120 In this process, the rhodium catalyst is kept in a water phase through the use of tris(sulfonatophenyl)phosphine trisodium salt (TPPTS) as a ligand and the products can be separated from the catalyst after the reaction by decantation before distillation, whereas the water phase containing the catalyst is fed again into the reactor for a new pass. Facilitating catalyst recycling is the main goal of the use of biphasic transition metal catalysed systems. In the scientific literature, hydroformylation in biphasic systems is usually performed using an immiscible organic solvent, which will dissolve the products. By employing water-soluble ligands, such as sulfonated phosphines, the metal catalyst can be effectively dissolved in the aqueous phase, facilitating its recovery after the reaction. On the other hand, the reaction products are dissolved in the organic phase, and therefore can be easily separated from the catalyst. The aldehydes can then be extracted from the organic layer, while the aqueous phase containing the catalyst can be treated to recover the catalyst for reuse in subsequent reactions. Catalyst recycling can involve various methods, such as precipitation, membrane filtration, or solvent exchange. An efficient separation and recycling strategy not only minimizes catalyst loss but can also enhance selectivity toward the desired aldehyde products. While the use of biphasic systems for hydroformylation offers numerous advantages, it presents certain disadvantages, particularly those concerning the types of substrates that can be effectively utilized. A significant limitation is the requirement for the substrate to be compatible with both phases. Specifically, the substrate must possess sufficient solubility in the organic phase to participate in the reaction while remaining stable in the presence of water. This requirement can exclude certain polar or highly functionalized substrates that cannot be distributed favourably between the two phases. Additionally, the presence of water can lead to the hydrolysis of sensitive functional groups or the formation of undesired by-products, further restricting the range of viable substrates. Moreover, the choice of solvent and ligand can significantly influence substrate compatibility, often necessitating extensive optimization and limiting the general applicability of the method. Biphasic hydroformylation processes were recently reviewed by Baricelli and co-authors.121Vorholt and co-authors recently studied the role of cyclodextrins in the acceleration of biphasic hydroformylation (Fig. 12).122 In this work, in situ image-based boroscopic technology was used for the first time to observe a liquid–liquid interfacial area during the reaction. A kinetic study showed that RAME-β cyclodextrins reduced the rate-limiting effect of the aldehyde product concentration on the interface. Cyclodextrins removed the product from the interface thus facilitating the availability of the substrate (1-octene) for the surface bound reaction. The use of cyclodextrins in aqueous biphasic hydroformylation and hydroaminomethylation was recently reviewed by Monflier and collaborators.123
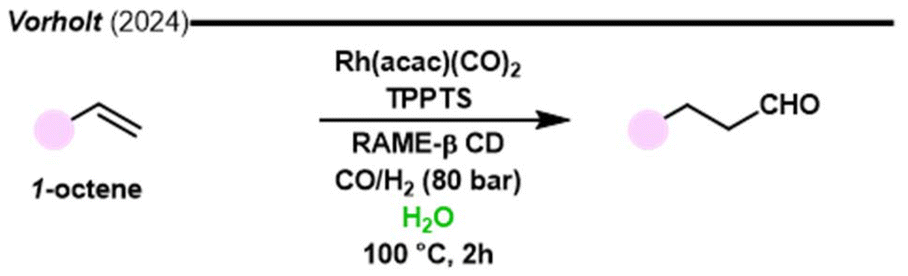 | ||
| Fig. 12 Aqueous biphasic hydroformylation assisted by cyclodextrins.122 | ||
In 2015, the group of Gusevskaya and dos Santos developed an aqueous biphasic water/toluene system for the synthesis of fragrance candidates. The hydroformylation of various terpene compounds (nerolidol, linalool, and β-citronellene) was performed using the Rh/TPPTS [tris(3-sulfonatophenyl)phosphine, trisodium salt] catalytic system combined with cetyltrimethylammonium chloride (CTAC) as a surfactant (Fig. 13).124 It was shown that the presence of CTAC had a positive impact on the hydroformylation activity, facilitating contact between liquid phases. The hydroformylation of β-citronellene occurred with excellent selectivity (100%) for the aldehydes and high regioselectivity for the linear product (90%). In the case of linalool and nerolidol, due to the proximity of the formyl group formed and the hydroxyl moiety, spontaneous intramolecular cyclization occurred, resulting in the hemiacetal products with 90% and 85% yields, respectively. The system could be reused for five cycles without loss of catalytic activity and with negligible leaching of rhodium into the organic phase.
 | ||
| Fig. 13 Aqueous biphasic hydroformylation of terpenes.124 | ||
In 2019, the Seidensticker group reported the aqueous biphasic hydroformylation of methyl oleate, known as an important renewable raw material for the chemical industry.125 The reaction was performed with the water-soluble Rh/TPPTS catalytic system in the absence of any phase-transfer agent (Fig. 14). To overcome the challenge, the authors used alcohols (methanol, butanol, and isopropanol) to enhance the substrate solubility in the polar phase. The system with isopropanol allowed for high yields of the aldehyde products (up to 98%) with low rhodium and phosphine leaching (0.13%) into the product phase. However, other solvents showed inferior performance with significant rhodium leaching. The authors performed recycling experiments by transferring the final mixture to a separation funnel under inert conditions. The aqueous phase was returned to the reactor for sequential experiments. Recycling experiments using 2.5 mol% of the catalyst gave 76% yield of the aldehydes, with a gradual decrease in activity in the second and third cycles due to the formation of rhodium nanoparticles. Subsequent cycles (from the third to the tenth) gave a constant yield of ca. 50%, achieving a total TON of up to 20000.
 | ||
| Fig. 14 Top: (a) reaction mixtures after aqueous biphasic hydroformylation of methyl oleate with full conversion at catalyst concentrations of 2.5 mol% (left) and 0.01 mol% (right). (b) Hydroformylation of methyl oleate.125 Bottom: hydroformylation of castor oil-derived methyl 10-undecanoate in an aqueous biphasic system.126 This figure has been adapted/reproduced from ref 125 with permission from the Royal Society of Chemistry, copyright 2019. | ||
In the following year, the same group reported the efficient aqueous biphasic hydroformylation of castor oil-derived methyl 10-undecanoate (Fig. 14).126 1-Butanol was used as a green solvent, which facilitated the mixing of the catalyst-containing phase and the substrate. Remarkably high space–time yields of ca. 120 g L−1 h−1 with high TOFs of 5000 h−1 were obtained in this work.
To showcase the enhanced performance of the aqueous biphasic rhodium-catalysed hydroformylation of previously described fatty-acid-derived molecules, namely methyl 10-undecenoate and methyl oleate, a jet-loop reactor was utilized (Fig. 15).127 The jet-loop reactor offers improved mixing among the gas–liquid–liquid phases, thus generating large interfacial areas without any moving or rotating parts. As depicted in Fig. 15, a liquid reaction stream from the bottom is withdrawn and reintroduced at the top of the reactor, creating a well-dispersed mixture of the immiscible components. This allowed for a significant reduction in the amounts of the previously mentioned co-solvent, facilitating the straightforward recovery of the product mixture. An impressive 5-fold increase in the aldehyde yield was achieved in the jet-loop reactor compared to a stirred tank reactor.
 | ||
| Fig. 15 Schematic drawing of a jet-loop reactor for intensified mixing used for the biphasic hydroformylation of fatty-acid-derived substrates (methyl 10-undecenoate and methyl oleate).127 This figure has been adapted/reproduced from ref. 127 with permission from Wiley-VCH Verlag GmbH & Co. KGAA, copyright 2019. | ||
Chemical reactions performed in an aqueous medium with reactants insoluble in water were termed “on water” reactions.128 In this situation, the reaction takes place at the organic/water interphase. The “on-water” methodology was applied by Alsalahi and Trzeciak for the hydroformylation of 1-hexene using a rhodium catalyst immobilized on polyacrylic acid (PAA) and a hydrophobic phosphine (triphenylphosphine, tri-p-tolylphosphine, or diphenyl(2-methoxyphenyl) phosphine).129 The method, which gave excellent results for 1-hexene in terms of aldehyde yield and regioselectivity, was tested for various other olefins, such as 2-hexene, 1-octene, and styrene. Recycling experiments showed the possibility of catalyst recovery without the loss of its catalytic activity. However, significant leaching of rhodium (ca. 50%) from water to the organic phase was observed. For this reason, product separation from the catalyst was complicated and required rhodium recovery not only from the aqueous phase but also from the organic phase by the vacuum distillation of all liquid components.
In addition to rhodium-based aqueous biphasic systems, a few works describe the use of cobalt catalysts in aqueous biphasic systems. One of the first reports was published by Beller and Krauter in 1999 (Fig. 16).130 In this work, the hydroformylation of 2-pentene was performed in the water/anisole (1:1) two-phase system using Co2(CO)6(TPPTS)2 as the catalyst and TPPTS in excess. The corresponding aldehydes were obtained in up to 75% yield with high regioselectivity. Importantly, the cobalt catalyst was recovered within the aqueous phase under inert conditions and reused for four cycles, giving even higher aldehyde yields and the same regioselectivity (l/b = 64:36). Low cobalt leaching from the aqueous to the organic phase (0.9–6.0%) was attributed to the excess of ligand employed in the experiments.
 | ||
| Fig. 16 Cobalt-catalysed hydroformylation of olefins in aqueous biphasic and micellar systems.130–132 | ||
In 2008, Bajaj, Jasra, and co-workers performed the aqueous biphasic hydroformylation of 1-octene and 1-decene using CoCl2(TPPTS)2 as the catalyst, CTAB as the surfactant, and ethanol or methanol as co-solvents (Fig. 16).131 The presence of the surfactant improved the olefin conversion and aldehyde selectivity, which was explained by the increased interlayer area between the catalyst solution and the substrate facilitating their contact. The addition of ethanol also improved the reaction rate and the aldehyde yield. Notably, the formation of acetals was not observed despite the relatively high reaction temperature (100 °C). The catalytic system was recovered and reused for four cycles without a significant loss in performance, achieving 96% conversion and 93% selectivity in the fourth cycle.
An important contribution to sustainable hydroformylation, by taking advantage of both micellar catalysis and microwave irradiation, was made by Petricci's group (Fig. 16).132 In their study, the rhodium/Xantphos catalytic system was employed in water, using the commercially available surfactant DL-α-tocopherol methoxypolyethylene glycol succinate (TPGS-750M), under microwave irradiation. This approach proved to be highly effective for the hydroformylation of terminal olefins and the tandem hydroformylation/hemiacetalization of homoallylic alcohols. Interestingly, the product could be precipitated as a salt by the addition of NaHSO3. Subsequently, the aqueous phase containing the catalytic system could be recovered for reuse, with no appreciable loss in catalytic activity being observed in three additional cycles. The E-factor for a single run was determined to be 1.08 in this protocol, which was considered suitable for scaled up process development. A high tolerance for various functional groups, such as ester, amide, acetal, nitrile, and ether, was demonstrated.
Several green solvents are used as the organic phase of biphasic systems, but no general use solvent can be pointed out. Typically, alcohols (ethanol and 1-butanol) and anisole displayed promising results, but the range of examples was limited, which suggested the need for a further search for efficient systems using green solvents. The application of biphasic systems for hydroformylation contributes to overcoming one of the main problems of homogeneous catalysis: the separation of the catalyst from the products. However, the products should be isolated from the organic layer and this step can require an additional solvent. It should be considered that a solvent-based approach to product isolation, such as extraction, also affects process sustainability.
3. Hydroformylation-based tandem processes
Tandem or sequential reactions involving hydroformylation have been developed to further diversify the range of products and enhance process efficiency. These processes integrate hydroformylation with subsequent transformations, enabling the synthesis of complex molecules such as amines, acetals, and alcohols in a single procedure.133 In addition to the use of sustainable solvents, the development of one-pot methodologies also contributes significantly to reducing the environmental impact of chemical production. A search for appropriate green solvent alternatives is an especially challenging and important task in the case of tandem processing, because it combines several chemical reactions, several reagents, and sometimes several catalytic systems, which could be affected by the nature of the solvent.3.1. Hydroformylation–acetalization
The realization of the hydroformylation reaction in the presence of alcohols opens an opportunity for the synthesis of acetals through a tandem olefin hydroformylation–aldehyde acetalization reaction sequence. The transformation of aldehydes into acetals could be necessary for either protection or synthetic purposes, as acetals have found many commercial applications as, e.g., solvents, additives in fuels, and intermediates in the fine chemicals industry. The synthesis of acetals under hydroformylation conditions is not straightforward due to the possible conflict between the steps: the presence of basic phosphorus ligands is usually required for selective hydroformylation, whereas acidic catalytic sites can be needed for the acetalization step. In this reaction, an olefin is hydroformylated to produce an aldehyde, which then reacts with an alcohol to form an acetal. The acetalization step typically requires an acid catalyst, such as p-toluenesulfonic acid (PTSA) or sulphuric acid; however, there are some published examples without the use of additional acid co-catalysts. The process can be performed at moderate temperatures, and the choice of alcohol affects the type of acetal formed. This tandem process simplifies the synthesis by avoiding the isolation of the intermediate aldehyde, directly producing the acetal. Besides the traditional use of rhodium catalysts in these processes, there are some reports on iridium,134,135 ruthenium,136–138 and cobalt139 based systems.Several contributions to the synthesis of acetals by tandem hydroformylation–acetalization were made by the group of Gusevskaya and dos Santos (Fig. 17). The method was applied to a wide scope of biorenewable substrates, such as terpenes and propenylbenzenes. In the first work by the group on this topic in 2010, acetals were synthesized from a series of p-menthenic terpenes.140 The transformation was achieved in ethanol solutions using the rhodium/P(O-o-tBuPh)3 catalytic system in the absence of acid co-catalysts. The corresponding acetals were obtained from α-terpinene, γ-terpinene, terpinolene, and limonene in 85, 60, 67, and 100% yield, respectively (Fig. 17). Later, the group applied this tandem transformation to bicyclic (3-carene, 2-carene, α-pinene, and β-pinene)141 and acyclic (linalool and β-citronellene) monoterpenes using a different rhodium precursor but the same ligand142 and to natural allyl benzenes eugenol and estragole, using a heterogeneous rhodium catalyst anchored in a commercial anion exchange resin – IRA900/TPPMS/Rh, TPPMS – 3-sulfonatophenyldiphenyl phosphine monosodium salt143 (Fig. 17). All these tandem hydroformylation–acetalization processes were performed in the absence of extra acid co-catalysts.
 | ||
| Fig. 17 Rhodium-catalysed hydroformylation–acetalization of renewable substrates in ethanol solutions.140–143 | ||
Bhanage and colleagues reported the synthesis of various acetals employing a rhodium phosphinite complex in methanol, ethanol, and 1-butanol solutions also without the addition of an acid cocatalyst.144 Acetals derived from 1-hexene were obtained with moderate regioselectivity in 81–99% yield, depending on the nature of the alcohol. Styrene derivatives and cyclic olefins were also satisfactorily employed as substrates; the reactions in methanol solutions yielded the corresponding acetals in excellent, nearly quantitative, yields.
The efficiency of the acetalization step depends significantly on the presence of acidic sites, making the ligand design crucial for process efficiency. With this in mind, a bifunctional ligand containing both a phosphine and a Lewis acidic phosphonium site was developed and tested for the synthesis of various acetals in solutions of different alcohols (Fig. 18).145 For the systems containing Rh(acac)(CO)2 as the catalyst precursor, a synergetic effect was observed with the charged bifunctional ligand in comparison with the analogous neutral ligand (TON of 1710 vs. 1130 for 1-octene). The enhanced performance was attributed to the stabilization of the rhodium–acyl intermediate by the phosphonium and the phosphine site, cooperatively. The method was applied to various substrates, with excellent TONs ranging from 1228 to 1960 being obtained. The formation of linear products was favoured in the case of alkyl olefins, while styrene gave a branched product. The authors also demonstrated the system's recyclability by using a biphasic ionic liquid solvent, [Bmim]BF4. Excellent yields of acetals were obtained in this work despite a small decrease in TON over the first seven cycles (1960 vs. 1700).
 | ||
| Fig. 18 Rhodium catalysed hydroformylation–acetalization of olefins using phosphonium-based ligands.145,146 | ||
Also in 2016, the same group reported a similar approach for a different ligand design. Instead of phosphines, they used phosphonium-based aminophosphine ligands for the same transformation in methanol solutions (Fig. 18).146 The authors noted that the accessibility of the phosphonium sites played a crucial role in acetal formation. Interestingly, the use of the analogous carbon-based ligand (instead of the nitrogen-based one) resulted in similar yields of acetals; however, with a lower TON (700 vs. 960). Various cyclic, acyclic, and aromatic olefins were successfully tested. The reactions with methanol, ethanol, isopropanol, ethylene glycol, and 1,2-propylene glycol, gave the corresponding acetals in high yields.
In 2021, Karakhanov and collaborators demonstrated the use of a biphasic system for easy rhodium catalyst recovery after the tandem process of the hydroformylation–acetalization of ethylene, in which polyols were used in the acetalization step to produce cyclic acetals.147 The reaction was performed in the presence of the RhCl3/TPPTS catalytic system in a mixture of toluene/water (2:1) as the reaction medium, with the addition of 5 vol% of H2SO4. The final product dissolved in the organic phase could be easily separated from the aqueous phase containing the catalytic system. A pH of the reaction medium of 3.5 was found to be the optimal value. In more acidic solutions with a pH of 1.5, excellent selectivity was obtained but the rate of the hydroformylation step was slower. On the other hand, in neutral and basic solutions, the formation of propanal was favoured, whereas the yield of the acetals was poorer. In recycling runs, slower gas consumption was observed at the beginning, but after 100 min similar values were achieved. Moreover, decreased selectivity for the acetals was observed in six recycling runs at full ethylene conversion.
Jia and co-workers reported a highly effective system for tandem hydroformylation–acetalization employing the Rh/phosphoramidite catalytic system in the presence of ZSM-35(10) molecular sieves, which were used to promote the acetalization step (Fig. 19).148 Interestingly, DFT calculations revealed that rhodium-catalysed hydroformylation occurred outside the structure of ZSM-35(10), whereas acetalization took place inside the molecular sieves. To demonstrate the importance of the channel pore size, zeolites with different porosity were tested. Good regioselectivity was observed only for microporous zeolites, such as ZSM-5, β-zeolite, and ZSM-35. Also, the higher acidity of ZSM-35(10) was beneficial for achieving great TONs of up to 10000 in the reactions of a variety of olefins with several alcohols: methanol, ethanol, n-propanol, n-butanol, iso-butanol, etc. In subsequent work by the same group, the heterogeneous composite catalyst Rh/POP-BINAPa&PPh3@ZSM-35(10) was prepared for these reactions (Fig. 19).149 Similarly to the previous work, the authors claimed that the Rh/POP-BINAPa&PPh3 motif was responsible for the hydroformylation step, whereas the acetalization step was promoted by ZSM-35(10) and the rhodium species. The composite material presented excellent activity resulting in TONs of 9905–10000 for various linear and cyclic olefins and various alcohols. Remarkably, the catalyst could be recycled five times with no significant loss in catalytic activity.
 | ||
| Fig. 19 Zeolites as co-catalysts in the hydroformylation–acetalization of olefins.148,149 | ||
Ruthenium catalysts were also reported to be active in tandem hydroformylation–acetalization processes. In 2014, Börner's group performed the immobilization of Ru3(CO)12 in ionic liquids for application in hydroformylation–acetalization of olefins with 1,2- or 1,3-diols (Fig. 20).136 To achieve high yields, NEt4Cl was added to enhance the efficiency of the ruthenium catalyst in hydroformylation and acetic acid to improve acetal production. Regarding the nature of the solvent, both [Bmin]NTf2 (88% yield) and [Bmin]BF4 (87% yield) exhibited superior performance in the formation of the cyclic acetal from 1-octene and ethylene glycol than conventional solvents, such as THF (66% yield), EtOAc (65% yield), NMP (53% yield), PBu4Br (27% yield), and propylene carbonate (7% yield). Various diols and olefins were tested to give the corresponding cyclic acetals in good to excellent yields. The reuse of the catalytic system was performed by product extraction from the polar ionic liquid with organic solvents followed by bulb-to-bulb distillation to purify the ionic liquid containing the ruthenium catalyst and NEt4Cl. The recovered mixture was used for two additional cycles, resulting in a total TON of 410. The immobilization of this system in the ionic liquid [Bmim]PF6 enabled its reuse in six consecutive cycles without loss in catalytic activity or metal leaching; however, a prolonged reaction time was necessary in reuse experiments because of mass transfer limitations due to the formation of a biphasic system.
 | ||
| Fig. 20 Ruthenium and iridium catalysed hydroformylation–acetalization of olefins.134,150,136,137 | ||
Batista et al. employed various ruthenium(II) and ruthenium(III) pre-catalysts to promote the hydroformylation of 1-decene in alcohol solutions with the aim of obtaining acetals (Fig. 20).137 The corresponding acetals were obtained in 50–65% yield in methanol, 24% in ethylene glycol, and 59% in 1,3-propanediol solutions.
Iridium catalysts were also reported to be active in tandem hydroformylation–acetalization processes. In 2019, Liu and coauthors investigated the utilization of an Ir(III)-complex ligated with phosphine as a bi-functional catalyst for tandem hydroformylation–acetalization reactions of olefins (Fig. 20).134 The Ir(III)-complex acts as both a transition metal complex and a Lewis acid, leading to high conversion rates of 1-hexene and excellent selectivity towards acetals without requiring additional additives. The research underscores the promising potential of this catalyst system, particularly the L1-based one, and highlights its recyclability and stability in [Bmim]PF6 ionic liquid.
More recently, Yang, Tian, and co-workers reported a catalytic system consisting of an iridium(I) catalyst, triphenylphosphine, and aluminium chloride (Fig. 20).150 Several acetals were obtained from various substrates in moderate to good yields (up to 85%) with high regioselectivity. Interestingly, water served as a hydrogen source in this system, avoiding the use of hydrogen at high pressures. AlCl3 hydrolysis was suggested to be a driving force for proton generation, successfully promoting the tandem reaction. The resulting pH, due to the addition of the acid, was measured as 1.6, which the authors described as a beneficial “acidic-buffer system”. The use of water allowed the undesired olefin hydrogenation to be suppressed.
The obvious choice of green solvents in this case are primary alcohols such as ethanol, which will also act as a reactant.
3.2. Hydroformylation–hydrogenation
Another example of hydroformylation-based tandem processes is the hydroformylation–hydrogenation reaction sequence in which a hydroformylation product (aldehyde) is immediately subjected to hydrogenation, resulting in the formation of the corresponding alcohol.151 This process is also known as hydrohydroxymethylation. The process can be fine-tuned to favour the production of either primary or secondary alcohols, depending on the substrate and the catalyst used. The resulting alcohols are widely used in various industries as solvents, plasticizers, detergents, and intermediates in the synthesis of pharmaceuticals and other fine chemicals.There are various methods for producing alcohols through hydroformylation. The first approach consists of a two-step process conducted in separate vessels, utilizing different catalytic conditions. Alternatively, a one-pot procedure can be employed in which the hydroformylation and hydrogenation steps are performed in the same reactor. A one-pot process can involve the use of two different catalysts operating under different or the same reaction conditions. Another option is a one-pot process with the use of a single catalyst that operates under different reaction conditions. Lastly, there is an option of using a single catalyst operating under consistent reaction conditions throughout the whole process. Each method offers distinct advantages depending on the desired outcome and efficiency of alcohol production. In the literature, the one-pot procedure using a single catalyst is the most common strategy applied.152 Reports on the application of rhodium,9,153–155 cobalt,156–160 ruthenium,161–165 palladium,166 and iridium167 systems in tandem hydroformylation–hydrogenation can be found in the literature as well as some examples with the use of dual metal systems.168,169 Several studies were devoted to searching for appropriate ligands and cooperative ligand systems to perform this one-pot transformation.153,167,170
In 2012, Diebolt, Müller, and Vogt reported a one-pot hydroformylation/hydrogenation process to prepare linear alcohols from olefins in a biphasic “on water” system (Fig. 21).171 The use of water as a co-solvent improved both the reaction rate and selectivity towards primary alcohols. The beneficial effect of water was attributed to the higher polarity of the solvent mixture, which favoured the repulsion of the hydrophobic carbon chain, and to the increase in the protic character of the reaction medium, which favoured the formation of cationic rhodium species. A catalytic system based on Rh(CO)2(acac) and a phosphine ligand (Xantphos or PBu3) was initially studied in different solvent/water mixtures aiming to improve the aldehyde reduction step. Alcoholic solvents (methanol, ethanol, t-amyl alcohol, isopropanol) showed good results, with the best performance being obtained with the isopropanol/water (1:9) mixture (90% yield for the desired alcohol). Five non-aromatic alcohols were obtained in good to excellent yields and excellent regioselectivity to the linear product.
 | ||
| Fig. 21 Tandem hydroformylation/hydrogenation of olefins.171,172 | ||
In 2017, Behr, Vorholt, and co-workers reported the tandem reductive hydroformylation of castor oil-derived renewable substrates (Fig. 21).172 This straightforward approach enabled the preparation of potential polymer precursors for polycondensates. The aldehydes were obtained in up to 88% yield with regioselectivities to linear products of up to 95%. This atom economical transformation was performed in isopropyl alcohol solutions using a combination of two distinct metal pre-catalysts and ligands: Rh(CO)2(acac) with Biphephos for the hydroformylation step and Ru3(CO)12 with tetracyclone (tetraphenylcyclopentadienone) for the aldehyde reduction step. Methyl 10-undecenoate, 10-undecenol, and 10-undecenoic acid were also tested to give the corresponding linear alcohols in 82, 67, and 74% yield, respectively. The rhodium/ruthenium catalytic system was successfully recycled through temperature-induced selective product crystallization and used in two consecutive runs; however, the activity decreased in the third run, which was attributed to ligand loss during the recycling treatment.
The study reported by Tilloy and co-workers was also focused on upgrading castor oil and its derivatives (Fig. 22).173 The process of the hydrohydroxymethylation of castor oil and ethyl ricinoleate, which involved the hydroformylation reaction followed by the hydrogenation of the resulting aldehydes to give polyhydroxylated derivatives, was reported. The authors evaluated the performance of the Rh(acac)(CO)2/trialkylamine catalytic system in various renewable solvents, i.e., isopropanol, anisole, 2-MeTHF, and γ-valerolactone, in comparison with toluene; the best results were obtained in anisole.
 | ||
| Fig. 22 Hydrohydroxymethylation of ethyl ricinoleate.173 | ||
In 2019, Alexanian and Shenouda developed a manganese-catalysed stereospecific hydroxymethylation of alkyl tosylates under mild conditions with low CO pressure to obtain homologated alcohols (Fig. 23).174 Several chiral, nonracemic β-branched primary alcohols were obtained in this work with high enantiospecificity starting from easily accessible secondary alkyl substrates. In terms of solvents, the best results were obtained in a mixture of t-amyl-alcohol and dioxane (4:1).
 | ||
| Fig. 23 Manganese-catalysed hydroxymethylation of alkyl tosylates.174 | ||
Recently, Monflier, Tilloy and collaborators reported the rhodium catalysed hydroformylation–hydrogenation of methyl 10-undecenoate using an ionic liquid/heptane biphasic system (Fig. 24).175 Among the various tri(n-alkyl)amines and branched or cyclic tertiary amines tested under different reaction conditions, the optimal results were achieved with triethylamine (TEA), which playing a dual role as both a ligand and a Brønsted base. Using the TEA/BMIM(NTf2)/heptane combination as a reaction medium allowed not only for the high yields of the alcohol but also for the efficient immobilization of rhodium within the ionic liquid. Under optimized conditions, the ionic liquid layer containing the catalyst could be recycled nine times with alcohol yields above 50% and low rhodium leaching of ca. 1.5% per cycle. To further investigate the long-term stability, the batch system was transposed to a mini-pilot plant for continuous flow operation. The mini-pilot plant ran for a total of 45 h, achieving a TON of 232 for alcohol production and 780 for the hydroformylation step, with 7.4% loss of the initial rhodium amount over 25 h of continuous operation.
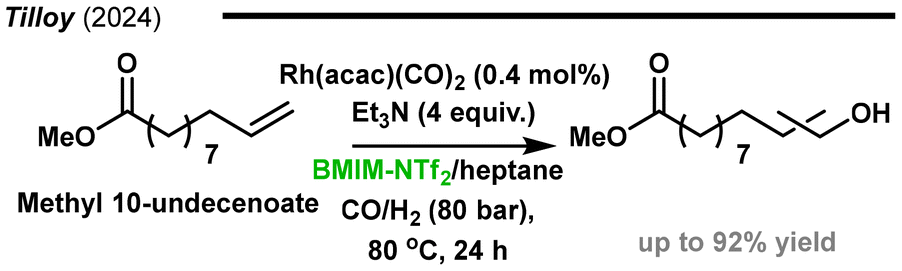 | ||
| Fig. 24 Rhodium/trialkylamine catalysed reductive hydroformylation in a mixture of ionic liquid and heptane.175 | ||
In the case of the hydroformylation–hydrogenation tandem transformation, there is an inclination to use polar solvents, especially bulky alcohols to decrease the possibility of acetal formation. A possible rationale for this can be the polarization or even protonation of the carbonyl group of the aldehyde by the protic solvent, which will favour hydrogenation of the aldehyde moiety to the desired alcohol product.
3.3. Hydroformylation–amination–hydrogenation (hydroaminomethylation)
Amines are an important class of chemicals both for the commodity and fine chemicals industrial fields. Hydroaminomethylation (HAM) is a powerful method for the synthesis of amines from olefins in a single synthetic procedure avoiding multistep methods.176–187 Formally, it corresponds to the addition of a hydrogen and an aminomethyl group to a C–C double-bond. This process involves three consecutive reactions occurring in tandem: hydroformylation, condensation of the aldehyde with its primary or secondary amine counterpart added from the outset, and the in situ hydrogenation of the enamine or imine formed in the previous step to give the desired final amine.184,187,188 Rhodium catalysts, often modified with phosphine ligands, are commonly used due to their high selectivity for the linear amine product. However, examples of ruthenium,189–197 cobalt,198,199 and copper200 were also reported. The reaction can be performed under mild conditions, but the choice of the amine and the solvent can significantly influence the reaction outcome.In 2003, Beller and collaborators presented a general and practical protocol for the HAM of olefins in toluene/methanol solutions (Fig. 25).201 The authors used a catalytic system based on [Rh(cod)2]BF4 and a diphosphine based on the Xantphos scaffold, which was tolerant of a variety of functional groups in the substrates and allowed for the successful synthesis of 21 amines in 59–99% yield with high regioselectivity (l/b ratio of up to 99:1). The authors noted that the combination of non-polar aromatic solvents with methanol favoured the hydrogenation step and suppressed the formation of N-formylpiperidine, a side product when piperidine was employed as its amine counterpart.
 | ||
| Fig. 25 General methods for the hydroaminomethylation of olefins.190,198,201 | ||
In 2013, the Beller group demonstrated an efficient and regioselective catalytic system based on trirutheniumdodecacarbonyl and a specific 2-phosphino-substituted imidazole ligand (L1) (Fig. 25) for the HAM of various substrates.190 Although the toluene/methanol mixture had been previously established as a suitable reaction medium for HAM, a comprehensive solvent screening was performed that included toluene, methanol, ethanol, THF, PC, and NMP. Excellent results were obtained for 1-octene in the toluene/methanol mixture (93% yield of the desired amine) and ethanol solutions (83% yield), while in PC the system showed only a moderate performance (40% yield). In PC, the yield for the desired product was compromised due to the extensive isomerization of 1-octene. Under optimized conditions, a broad scope of amines were successfully synthesized from olefins and piperidine as its amine counterpart.
Later on, the same group reported the cobalt catalysed HAM of olefins using tert-butyl-pyridine-Xantphos (L2) as the ligand and methyl tert-butyl ether (MTBE), which is a green solvent (Fig. 25).198 The process resulted in linear amines in good to excellent regioselectivity under mild conditions. Remarkably, the system demonstrated an improved selectivity compared to traditional cobalt(I) and rhodium catalytic systems. The ligand structure played a crucial role in promoting the desired reactions and suppressing the undesired ones, thus ensuring the successful synthesis of the target amines. A wide range of traditional solvents and their greener alternatives were tested, such as toluene, THF, dioxane, MTBE, heptane, ethyl acetate, triglyme, acetone, ethanol, etc. The best results for 1-octene – under non-optimized conditions – were achieved in MTBE (26% yield), followed by toluene (24% yield) and heptane (17% yield). The system showed excellent functional group tolerance under optimized conditions with various olefins and various anilines as their amine counterparts allowing for good yields of the final amines and excellent regioselectivity. The potential for scale-up was evaluated with the diisobutene mixture as the substrate (an important bulk industrial feedstock), achieving the corresponding amine in 73% yield on a 100 g scale.
In 2011, Zhang and co-workers described the use of rhodium complexes with the 2,2′,6,6′-tetrakis((diphenylphosphino)methyl)-1,1′-biphenyl (Tetrabi) ligand for the HAM of terminal olefins (Fig. 26).202 The system was successfully applied to several different olefins and secondary amines allowing for excellent regioselectivity, yields, and TONs at a high substrate/rhodium precursor ratio. A broad scope of green solvents was tested in the HAM of 1-hexene with piperidine, i.e., methanol, ethanol, isopropanol, and ethyl acetate, in comparison with traditional solvents, such as toluene, THF, and dioxane. The alcohols showed a superior performance (77% yield in isopropanol, 91% in ethanol, and 86% in methanol), compared to THF: 7% yield, the best result among the traditional solvents. The authors also explored a mixture of alcoholic solvents and chose the combination of isopropanol and ethanol (2:1) as an optimal reaction medium in terms of the product yield, l/b ratio, and efficiency in suppressing N-formylpiperidine formation.
 | ||
| Fig. 26 Rhodium-catalysed hydroaminomethylation of olefins using tetraphosphorus ligands in alcohol solutions.202,203 | ||
In a subsequent report, Zhang et al. presented a catalytic system consisting of [Rh(nbd)2]SbF6 and a pyrrole-based tetraphosphorus ligand to produce linear amines from styrene derivatives (Fig. 26).203 The authors noticed a considerable impact of the solvent nature on the system performance. The HAM products were highly favoured in solutions of alcohols, i.e., methanol, ethanol, isopropanol, and t-amyl alcohol; however, only t-amyl alcohol enabled a linear product to be obtained with high regioselectivity. A broad scope of styrenes and amines were tested as reactants, with the corresponding amines being obtained in moderate to excellent yields and high l/b ratios (up to 99/1).
In 2019, Zheng and Wang reported the HAM of 2,5-dihydrofuran with different anilines using rhodium/phosphine catalytic systems as a green and atom-economical method to prepare important pharmaceutical intermediates (Fig. 27).204 A systematic screening of solvents was performed to evaluate their impact on the process efficiency. Besides conventional problematic and hazardous solvents, greener alternatives were tested, such as methanol, ethanol, and isopropanol. Polar solvents were found to be more effective at producing the desired product, with methanol showing the best results. A broad scope of products containing the aniline moiety were successfully prepared in up to 92% isolated yield in methanol solutions from 2,5-dihydrofuran. Several other cyclic olefins were also tested to give the corresponding amines in up to 99% yield.
 | ||
| Fig. 27 Hydroaminomethylation of 2,5-dihydrofuran.204 | ||
In 2017, Behr, Vorholt, and collaborators showcased the bis-HAM of industrial cyclic dienes to obtain secondary diamines using [Rh(octanoate)2]2 as the catalyst under phosphine-free conditions (Fig. 28).205 In the initial screening, the model reaction involving dicyclopentadiene and n-butyl amine was performed in various solvents. Good results were obtained in toluene solutions: 88% yield of the desired secondary diamine. Green solvent alternatives, such as methanol and isopropanol, were evaluated due to their high polarity. However, the system's efficiency toward diamine production decreased, with significant amounts of intermediate being detected in the final mixture of products (monoimine and monoamine). Through tests with strongly coordinating solvents, the authors suggested that the coordination of the solvent to rhodium played a crucial role in controlling the hydrogenation step as well as hindering hydroformylation of the second C–C double bond.
 | ||
| Fig. 28 Rhodium-catalysed hydroaminomethylation of cyclic dienes.205 | ||
Focused on obtaining more diverse functionalized bio-based platform molecules, Vanbésien, Monflier, and Hapiot reported a novel HAM–hydrohydroxymethylation sequential route to obtain aminohydroxytriglycerides from triglycerides (Fig. 29).206 In this pioneering one-pot transformation, the amino-grafted product of HAM served as the ligand for rhodium to form an active species, which catalysed the hydrogenation step. Solvent effects were explored by comparing toluene, isopropanol, and THF. Despite the similar selectivity for the amine obtained in the greener solvent isopropanol, the substrate conversion was inferior compared to toluene and THF. In 2020, the same group reported the ruthenium-catalysed HAM of unsaturated oleochemicals using Ru3CO12 as the catalyst in the absence of auxiliary ligands.189 Isopropanol was again used as an environmentally friendly solvent alternative; however, a poor conversion of 6% was achieved vs. 45% in toluene under the same conditions.
 | ||
| Fig. 29 Hydroaminomethylation of biorenewable molecules.206,207 | ||
In 2019, the group of Gusevskaya and dos Santos reported a successful synthesis of various biomass-based amines starting from renewable olefins, such as estragole, limonene, camphene, and β-pinene (Fig. 29).207 With this methodology, based on [Rh(COD)(OMe)]2 and a phosphite ligand, various environmentally friendly solvents, i.e., p-cymene, anisole, and ethanol, were evaluated in comparison with the benchmark solvent, toluene. In most of the cases, the reactions performed in p-cymene and anisole gave results similar to those obtained in toluene. However, in ethanol solutions, considerable improvements in selectivity for the final amines were achieved due to the suppression of undesirable parallel reactions, such as the decomposition of the original amine.
In 2020, Shang, Lin and collaborators published the results of a study on the electronic and steric effects of neutral and ionic phosphines on the performance of iridium catalysts in HAM (Fig. 30).208 The behaviour of toluene, THF, and N-methyl-2-pyrrolidone (NMP) in the reaction of 1-octene with N-methylaniline in the presence of the [Ir(cod)Cl]2/neutral imidazolyl-phosphine system was compared with that of methanol, a less hazardous solvent. The best result was achieved in NMP (51% yield, l/b = 61:38), whereas methanol showed a similar second-best performance (46% yield, l/b = 50:50). Notably, the amounts of imine intermediates when the reaction was interrupted were lower in methanol than in other solvents.
 | ||
| Fig. 30 Hydroaminomethylation promoted by Ir(I) catalysts.208 | ||
In 1999, Beller's group reported an efficient protocol for HAM using ammonia to obtain primary amines (Fig. 31).209 To accomplish this transformation effectively in an aqueous biphasic medium, the use of a dual catalyst system composed of rhodium and iridium precursors and TPPTS (a water-soluble ligand) was necessary. MTBE, anisole, and toluene were tested as organic phases. An increase in selectivity for the desired primary amine was observed with a decrease in the polarity of the organic solvent. The correlation was attributed to the migration of the primary amine from the aqueous ammonia solution to the organic phase. The reactions in anisole and MTBE resulted in nearly the same yields; however, the ratio between the primary and secondary amines was higher in anisole.
 | ||
| Fig. 31 Rhodium-catalysed hydroaminomethylation of olefins.209–211 | ||
In 2014, the same group reported the direct synthesis of tertiary amines by the HAM reaction of olefins with aqueous ammonia using Ru3(CO)12 as the catalyst precursor and 2-phosphino-substituted imidazoles as auxiliary ligands (Fig. 31).210 A range of different solvents and solvent mixtures were tested, including methanol, ethanol, and isopropanol and their mixtures with THF and toluene. Tertiary amines were obtained in moderate to good yields (45–76%) with excellent regioselectivities (n/iso ratio of up to 99:1).
A remarkable study was performed by Faßbach, Sommer, and Vorholt in 2018, in which they envisaged the HAM reactions of olefins with highly functionalized hydrosoluble amines – rarely reported in the literature – in an aqueous biphasic system (Fig. 31).211 Highly polar diethanolamine and 1-octene were chosen as model substrates. The reactions were performed in aqueous/alcohol solvent mixtures using a catalytic system consisting of [Rh(cod)Cl]2 and a sulfonated diphosphine ligand Sulfoxantphos. Through the systematic variation of alcohols, 2-butanol was found to be a more effective co-solvent allowing for 79% yield of the amine. Notably, in the absence of the organic co-solvent, no reaction took place due to inefficient phase mixing.
Interesting work by Elsevier and collaborators showed the synthesis of cyclic amines by the intramolecular reaction of ethyl methallyl amine under HAM conditions using scCO2 as a green alternative to conventional organic solvents (Fig. 32).212 This approach allowed for the unexpected shift in selectivity from a cyclic amide (a major product in conventional solvents) to cyclic amines, as shown in Fig. 32, which was not observed in conventional solvents. The selectivity for the five-membered cyclic amines was less than 1% in dioxane solutions, whereas it was 76% in scCO2. Multinuclear high-pressure NMR spectroscopy revealed that in scCO2 the intramolecular cyclization of the acyl rhodium intermediate to give the amide was less favourable than its hydrogenolysis to give the amine products mainly due to the reversible formation of the carbamic acid. The authors suggested that scCO2 acted as a protective medium for amines, favouring their selectivity.
 | ||
| Fig. 32 Hydroaminomethylation of ethyl methallyl amine in supercritical CO2.212 | ||
In 2008, the group of Vogt reported the HAM of n-alkenes in a biphasic ionic liquid system (Fig. 33).213 High chemo- and regioselectivity to linear amines were obtained in the reactions with piperidine performed in 1-butyl-3-methyl-imidazolium tetrafluoroborate ([Pmin][BF4]), using a rhodium/Sulfoxantphos catalytic system. This approach led to the formation of a biphasic system that facilitated the efficient recovery of the product. After product recovery, the catalytic phase was reused four times in the reaction with 1-octene, demonstrating great stability. In comparison with the reaction performed in a mixture of toluene and methanol, the ionic liquid system showed increased chemoselectivity but slightly lower regioselectivity. In the next year, the same group extended this work by showcasing the use of xanthene-based amino-functionalized ligands in the HAM of 1-octene with piperidine (Fig. 33).214 The aim was to modulate the π-accepting character of the xanthene backbone with dipyrrolylphosphine, thus affecting both the electronic and steric hindrance properties of the ligand, and to study the ligand effect on the catalyst's performance. In addition, it was observed that the solvent choice significantly influenced the reaction selectivity. The screening of different mixtures composed of toluene and alcohols (1:1) revealed an important role of the alcohol's acidity: more acidic media allowed for higher activity, whereas less acidic media favoured regio- and chemoselectivity.
 | ||
| Fig. 33 Hydroaminomethylation in the presence of Xantphos-derived ligands in ionic liquids and alcohols.213,214 | ||
Behr and Roll performed the HAM of 1-octene with morpholine using temperature-dependent solvent systems (TMS).215 As commented on above, within this approach, the reaction medium consists of at least two solvents, which have a temperature-dependent miscibility, enabling the performance of a single-phase reaction at higher temperature and catalyst/product separation at lower temperature due to the formation of two liquid phases. Typically, the catalyst is dissolved in one phase, while the reaction products migrate to the other one. Interphase mixing can be facilitated by the presence of a third solvent, acting as a mediator. In this example, the green solvent PC was used as a polar phase to dissolve the catalyst, alkanes as a non-polar phase to dissolve the products and a semi-polar mediator solvent. With N-octylpyrrolidone as the mediator, high yields and high TONs were achieved. After the reaction, the catalyst was easily recovered by a simple phase separation with only a minimal loss of the rhodium catalyst. However, the authors acknowledged a limitation of the system due to the side reaction between morpholine and PC.
Later on, Behr and collaborators performed a systematic analysis of solvents to integrate a TMS for the HAM of 1-octene with morpholine in order to minimize catalyst leaching and side reactions.216 The study of liquid–liquid equilibria allowed for the selection of the three most suitable solvents, i.e., ethanol, NMP, and acetonitrile, for use as mediators in the TMS with 1-octene and water.
In 2020, Seidensticker and co-workers performed the HAM of 1-decene with diethylamine as a continuous-flow process in a thermomorphic multiphase system using a rhodium/Sulfoxantphos catalyst combination (Fig. 34).217 In the selection of the polar solvent, methanol exhibited superior results compared to DMF and acetonitrile, although the underlying reason for this behaviour remained unclear to the authors. To obtain the miscibility gap necessary for the application, n-dodecane was employed as the non-polar solvent. After optimization during batch recycling experiments, a continuous operation process was performed in a miniplant (Fig. 34a). An average yield of the linear amine of 61% was achieved after 60 h of stable process operation.
 | ||
| Fig. 34 (a) Flow scheme of the miniplant for continuous HAM and (b) continuous operation of HAM of 1-decene with diethylamine in TMS. Conditions: p = 36 bar CO/H2, CO/H2 = 1/2, Treactor = 125 °C, τ = 4 h, mn-dodecane/mmethanol = 1/1, mRh/mmethanol = 0.82 mg g−1, Rh/ligand = 1/3.5, n = 800 min−1, ṁdiethylamine = 4.8 g h−1, ṁn-dodecane = 26.6 g h−1, ṁ1-decene = 6.2 g h−1, ṁrecycle = 26.6 g h−1. Conversion (X) and yield (Y) of linear aldehyde l-2, enamine l-3 and amine l-4 were determined via GC-FID. Regioselectivity (l/b-selectivity) of the hydroformylation is calculated as the sum of l-2, l-3, and l-4 divided by the sum of 2, 3, and 4. The gray area shows the start-up procedure.217 This figure has been adapted/reproduced from ref. 217 with permission from the American Chemical Society, copyright 2020. | ||
In 2023, the group of Petricci demonstrated a micellar catalysis approach for HAM in water with full recovery of rhodium (Fig. 35).218 The authors started their study with the reaction of allylbenzene with aniline hydrochloride, using the surfactant TPGS-750-M to create micelles and Rh(CO)2(acac) with Xantphos as the catalytic system. Microwave irradiation was also used to perform the reactions in this study. Interestingly, an efficient work-up process was developed by filtering the final mixture through a strong cation exchange column with ethanol flushing. The water-phase was directly inserted into the reactor for the next reaction cycles. The product was obtained after washing the column with an ammonia solution in ethanol, followed by evaporation. With this protocol, nine amines were prepared in 55–99% yield. Remarkably, a complete life cycle assessment was made by the authors to demonstrate the sustainability of the overall process.
 | ||
| Fig. 35 Micellar catalysis for the hydroaminomethylation process in water.218 | ||
In 2014, Nairoukh and Blum reported a heterogeneous HAM process using a rhodium catalyst immobilized in a hydrophobic silica sol–gel matrix.219 The reactions of vinylarenes with aniline or nitrobenzene derivatives were performed in aqueous microemulsions using CTAB as the surfactant. The efficiency and regioselectivity of HAM were found to depend on the hydrophobicity of the sol–gel matrix and reaction conditions. A variety of amines were obtained in excellent yields and regioselectivity. The catalyst could be recycled at least 4 times, after that a decrease in activity and regioselectivity was observed due to morphological changes to the sol−gel matrix in water (Si–O bond breaking).
Due to the relevance of hydroaminomethylation, many examples of the use of green solvents have been reported in the past few years. A clear preference for alcoholic solvents, such as isopropanol and ethanol, can be observed. These protic solvents are believed to favour the imine/enamine hydrogenation step by protonating the nitrogen atom. It is also worthwhile highlighting the utility of anisole for this type of reaction.
4. Alkoxycarbonylation and hydroxycarbonylation
Differently from previously discussed examples, alkoxycarbonylation and hydroxycarbonylation reactions involve the incorporation of the alcoholic solvent or water in the final product, along with CO, resulting in the formation of carboxylic acids and their derivatives.220Hydroxycarbonylation is a specific type of carbonylation that involves the addition of CO and water to an unsaturated substrate, such as an alkene or alkyne, in the presence of a catalyst, usually a transition metal complex.221–223 Palladium-based catalysts are frequently used for hydroxycarbonylation, often in combination with phosphine ligands, which improve the catalyst performance. Other metals, such as rhodium and ruthenium224 can also serve as catalysts in this reaction. Hydroxycarbonylation is a versatile synthetic method widely employed in the preparation of carboxylic acids, which are known as fundamental building blocks in organic synthesis found in various natural products, pharmaceuticals, and polymers.
Alkoxycarbonylation consists of the addition of CO and an alcohol to an unsaturated compound, such as an alkene or alkyne, in the presence of a catalyst, usually, a transition metal complex (Fig. 1).220,225 The most common catalysts include palladium,226–228 nickel,229–231 and rhodium complexes;232–234 however, examples of ruthenium,235–238 cobalt,239 copper,240 and iron241 catalysts have also been reported. Catalytic systems often contain phosphine ligands to enhance the metal's activity and reaction selectivity. For example, palladium–phosphine complexes are widely used in alkoxycarbonylation due to their high efficiency and ability to operate under mild conditions.242–250
In 2021, Seidensticker and Cole-Hamilton reviewed the literature dealing with 1,2-bis(di-tert-butylphosphinomethyl)benzene (1,2-DTBPMB), one of the best ligands for performing alkoxycarbonylation and used in the commercial production of methyl methacrylate from ethylene (Alpha process).249 However, despite the success of 1,2-DTBPMB in the methoxycarbonylation of ethylene, scaling up other reactions using this ligand has faced challenges, such as catalyst deactivation and product separation issues. The modifications of 1,2-DTBPMB were explored, including the replacement of some tert-butyl groups with 2-pyridyl moieties performed by Beller's group, which enabled the catalyst's performance to be improved in carbonylation reactions at low temperatures. In 2021, Beller and colleagues reviewed state-of-the-art palladium-catalysed alkoxycarbonylation with special attention paid to the incorporation of 2-pyridyl moieties into the ligand.220
In an early report, Mortreux et al. disclosed a method for alkoxycarbonylation of natural allylic terpenic olefins (Fig. 36).251 This methodology enables the synthesis of β- and γ-unsaturated esters, which serve as building blocks for fine chemicals. The reactions with allylic carbonates, such as perillyl, myrtenyl, carvyl, and geranyl carbonates, resulted in esters in 67–80% yield in ethanol solutions. However, the reaction with verbenyl carbonate gave a poor yield of only 4%. The authors also used chlorides with the same terpenic skeletons as substrates instead of carbonates and obtained the esters in even higher yields, except for carvyl chloride (46% yield vs. 74% from carvyl carbonate). The lower reactivity of allylic carbonates compared to the corresponding chlorides was explained by the lower reactivity of the carbonates toward palladium(0) species and/or by the difficulty of their decarboxylation to give a key (π-allyl)(alkoxy)Pd intermediate. Interestingly, the C–C double bond in the substrate molecules remained untouched under the conditions applied.
 | ||
| Fig. 36 Alkoxycarbonylation of allylic terpenic compounds in ethanol solutions.251 | ||
In 2008, the group of Buchwald reported a system based on Xantphos for the palladium-catalysed methoxycarbonylation of aryl bromides under atmospheric pressure (Fig. 37).252 Several methyl esters were obtained in 80–92% yield in the presence of Et3N in methanol solutions. The mild conditions applied in these reactions allowed the presence of various functional groups in the substrate structure, including an ethyl ester (with no observable transesterification), aryl fluoride, nitrile, and tert-butyl carbamate.
 | ||
| Fig. 37 Palladium-catalysed methoxycarbonylation of aryl bromides.252 | ||
In 2010, Yamamoto revealed a general and selective alkoxycarbonylation of arylboronates in solutions of different alcohols (Fig. 38).253 The author evaluated the steric hindrance around the boron centre, which played a pivotal role in the overall yield of the ester. Despite the lower rates observed for bulkier p-chlorophenylboron substrates, higher yields for the desired products were obtained due to the suppression of side reactions. In addition to an in-depth mechanistic investigation, a broad scope of substituted arylboronates containing different functional groups were successfully tested to give the desired products in 48–94% yield. Nevertheless, for tertiary alcohols, a limitation was found: the reaction with tert-butanol resulted in only 6% yield.
 | ||
| Fig. 38 Palladium-catalysed alkoxycarbonylation of phenylboronic acid pinacol esters.253 | ||
A remarkable contribution was published by Clarke and collaborators in 2010, in which the palladium-catalysed enantioselective hydroxycarbonylation and alkoxycarbonylation of olefins were accomplished (Fig. 39).254 In particular, the hydroxycarbonylation of styrene was performed in a solution of methyl ethyl ketone (MEK), a solvent recommended by Pfizer's and Sanofi's solvent sustainability guides.30,255 Under optimized conditions, 71% yield of the corresponding carboxylic acid was obtained with a b/l ratio of 1.1 and ee of 80%. Furthermore, methoxycarbonylation of styrene in methanol solution resulted in 62% isolated yield of the methyl ester with a b/l ratio of 0.95 and an excellent ee of 91%.
 | ||
| Fig. 39 Hydroxycarbonylation and alkoxycarbonylation of olefins.235,254,256,260 | ||
In 2014, Beller's group demonstrated a significant breakthrough with the first alkene alkoxycarbonylation using paraformaldehyde as the CO source, thus avoiding the use of high-pressure CO (Fig. 39).235 Employing a combination of a ruthenium catalyst, tricyclohexylphosphine (PCy3) ligand, and an imidazolium chloride in an alcohol/NMP (1:3) mixture as the solvent, the authors obtained various esters in 40–90% yield. The catalytic system could be recycled by removing the unreacted substrate, and adding new portions of the substrate, paraformaldehyde, and methanol. Despite the decrease in efficiency after each run, 63% yield was achieved in the fourth cycle.
Gehrtz, Hirschbeck, and Fleischer introduced a novel methodology for alkoxycarbonylation of alkenes under ambient conditions with the indirect utilization of CO2 as a C1 source (Fig. 39).256N-Formylsaccharin, a recyclable product of CO2 reduction, was used as a CO transfer reagent. This transformation was achieved using a two-chamber pressure reactor. The first chamber produced N-formylsaccharin from saccharin and CO2 followed by the base-mediated decarbonylation of N-formylsaccharin to regenerate saccharin and produce CO, which was then used for alkoxycarbonylation. The second chamber contained a solvent mixture of alcohol and DCM (1:3), palladium catalyst, ligand, co-catalyst, and vinyl arenes as substrates. Differently from previously commented on reports, the catalytic system was efficient at synthesizing branched esters. A broad scope of substituted arenes and different alcohols were tested to give the corresponding esters in high yields (up to 97%) and high regioselectivity (b/l up to 93:7). More information on the use of N-formylsaccharin as a CO surrogate can be found in a recent review by Mi and Zheng.257
Inspired by these achievements, in 2017 Bhanage and collaborators developed a similar approach using N-formylsaccharin as a CO surrogate and a heterogeneous Pd/C catalyst, employing PC as a green solvent in a single high-pressure tube (Fig. 40).258 They successfully performed the phenoxycarbonylation of aryl iodides for 32 substrates with yields of 46–90% using a ligand- and co-catalyst-free system. It is worth mentioning that the catalyst could be recycled for four cycles without loss in performance, which contributed to process sustainability.
 | ||
| Fig. 40 Pd/C catalysed phenoxycarbonylation using N-formylsaccharin as a CO surrogate.258 | ||
In 2019, Franke, Beller and collaborators reported an aqueous-phase protocol that enabled the synthesis of carboxylic acids from a wide range of olefins.259 In addition to the high sustainability of this methodology, the palladium/pyridyl-substituted diphosphine catalytic system could be recycled for 26 runs over 32 days without loss of activity. Under optimized experimental conditions, a mixture of acetic acid and water was used as the solvent to obtain the best results. An excellent TON of >350000 was achieved in the synthesis of industrially relevant propionic acid, demonstrating the potential of the developed system for industrial applications. Also, a 10 g scale synthesis was performed multiple times, resulting in the production of more than 350 g of the hydroxycarbonylation product from 1-dodecene with a TON higher than 11000.
In 2021, Beller and co-workers showcased the efficient carbonylation of 1,3-dienes in a scalable and 100% atom-economical process (Fig. 39).260 Unlike the main industrial route for producing adipate esters – which uses a corrosive mixture and releases stoichiometric amounts of nitrous oxide – a catalytic system based on palladium, bidentate 1,2-bis-di-t-butylphosphin-oxylene (DTBPX) ligand, and p-toluenesulfonic acid (PTSA) acid as the co-catalyst enables diesters to be obtained from 37 different 1,2- and 1,3-dienes in excellent yields. The proposed mechanism reveals two distinct catalytic cycles for the dicarbonylation of dienes, highlighting the pivotal role of the nature of the solvent in the formation of palladium intermediates during the first C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C carbonylation step.
C carbonylation step.
In 2016, Alexanian and collaborators developed a process for the manganese-catalysed cyclization of alkenes with alkyl iodides followed by alkoxycarbonylation or aminocarbonylation (Fig. 41).261 In the particular case of alkoxycarbonylation, EtOH acts as a solvent and nucleophile, while in aminocarbonylation, EtOH acts only as a solvent since the nucleophilicity of amines is higher. The authors described the use of both primary and secondary alkyl iodides as substrates in these reactions with a diverse array of cyclic and acyclic structures. The method was successfully applied to the synthesis of five-, six-, and seven-membered rings including heterocycles (24 examples with 64–89% yield).
 | ||
| Fig. 41 Top: manganese-catalysed carboacylations of alkenes with alkyl iodides.261 Bottom: palladium-catalysed alkoxycarbonylation of unactivated secondary alkyl bromides.262 | ||
In 2016, Alexanian and Sargent reported a palladium-catalysed alkoxycarbonylation of unactivated secondary alkyl bromides that proceeded at low CO pressure under mild conditions (Fig. 41).262 In this transformation, a mixture of n-BuOH and heptane (1:1) was used as the solvent. The reactions efficiently delivered esters from a diverse range of unactivated alkyl bromides representing the first example of the catalytic carbonylation of alkyl bromides with CO. Mechanistic investigations supported a proposed hybrid organometallic–radical pathway instead of more common two-electron transformations.
Even though the examples described present various environmentally friendly alternatives to replace traditional organic solvents, reports on asymmetric carbonylation of olefins using green solvents are scarcer. In this context, in 2021 Guan and collaborators showed a successful example of the palladium-catalysed asymmetric Markovnikov hydroxycarbonylation and alkoxycarbonylation of vinyl arenes (Fig. 42).263 Hydroxycarbonylation was accomplished in a mixture of THF with MTBE, whereas alkoxycarbonylation was achieved in a mixture of THF with an alcohol. The authors synthesized 43 compounds in good to excellent yields (64–96%), excellent b/l ratio (>99:1), and ee (86–96%). Moreover, the gram-scale synthesis of five drug compounds through this methodology was showcased. While the maximum hydroxycarbonylation yield was obtained in pure MTBE, the combination of MTBE/THF gave the highest ee.
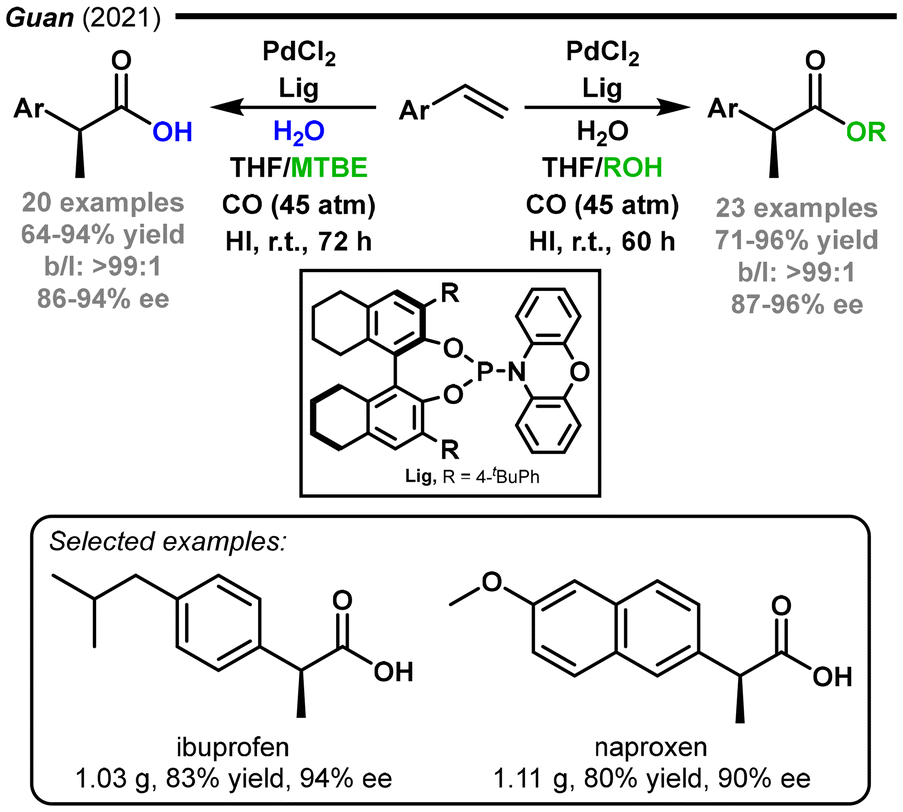 | ||
| Fig. 42 Asymmetric Markovnikov hydroxycarbonylation and hydroalkoxycarbonylation of vinyl arenes.263 | ||
Despite the use of halides and olefins as typical substrates in alkoxycarbonylation, in 2018 Li, Wang, and Wu disclosed a methodology for producing alkyl arylacetates from benzylic alcohols using DMC as both the solvent and in situ activator in the presence of a base (Fig. 43).264 Remarkably, no halogen additive was necessary to accomplish this reaction. The mechanism proposed suggested the formation of a mixed organic carbonate by the reaction of the benzylic alcohol with DMC, followed by its oxidative addition to the palladium catalyst to give an acylpalladium complex, which released the desired product after alcohol insertion. A broad scope of benzylic alcohols were tested as substrates, with 15 esters being obtained in 54–93% yield.
 | ||
| Fig. 43 Direct carbonylation of benzylic alcohols in DMC as the solvent and the activator.264 | ||
In the next year, the same group reported a similar procedure, but instead of using high-pressure CO, this reactant was generated in situ by the base-catalysed decarbonylation of benzyl formates, which were also used as substrates.265 After the formation of the benzyl alcohol and CO from benzyl formate, carbonylation of the benzyl alcohol followed the same pathway as that described above. DMC played the role of both the green solvent and in situ activator. 15 examples were presented in this work demonstrating the synthesis of various alkyl 2-arylacetates (46–86% yield). More information about the use of formates as a CO surrogate can be found in a review article by Fujihara and Tsuji.266
In 2016, Mecking and collaborators reported a single-step process for preparing long-chain α,ω-dicarboxylic acids from unsaturated fatty acids by their isomerization and hydroxycarbonylation (Fig. 44).267 By using water as a nucleophile, the authors successfully performed, for the first time, the isomerizing carbonylation of internal fatty acids – highly immiscible with water compounds – to prepare valuable dicarboxylic acids. To overcome the poor miscibility of fatty acids with water and obtain high yields of the desired products, mixtures of water with several solvents were evaluated. In addition to traditional solvents, green solvents, such as 2-MeTHF, diglyme, tert-butanol, and MEK, were tested. Nevertheless, THF was chosen as the best solvent for the reaction of oleic acid due to it performing best, the possibility of easy removal from the reaction mixture (high volatility), and good miscibility with all components. The method was applied to various fatty acids, such as oleic acid, 10-undecanoic acid, erucic acid, and even high oleic sunflower oil.
 | ||
| Fig. 44 Ruthenium- and palladium-catalysed hydroxycarbonylation of alkenes and alkynes.224,267,268 | ||
In 2021, Beller and co-workers reported the ruthenium-catalysed hydroxycarbonylation of terminal olefins (Fig. 44).224 Using the Ru3(CO)12/PCy3 catalytic system, PTSA as a co-catalyst, and mixture of water/organic solvent as the reaction medium, 12 carboxylic acids were successfully prepared in moderate to good yields. Anisole was also tested as a green solvent, but the reaction showed no conversion. Interestingly, only fluorinated solvents, such as hexafluoroisopropanol (HFIP) and trifluoroacetic acid (TFA), allowed for satisfactory yields of the carboxylic acids. Special experiments revealed that the role of fluorinated solvents was related to their strong H-donor ability.
Recently, Ning, Chen and collaborators disclosed an efficient method for preparing α,β-unsaturated carboxylic acids by the palladium-catalysed hydrocarboxylation of alkynes in a water-based medium in the presence of sodium dodecylbenzenesulfonate (SDBS) as a surfactant (Fig. 44).268 A special picolinamide-derived diphosphine ligand with a hydrophilic sulfonate group was developed to accomplish the process. The success of the ligand was attributed to the ability of the pyridine moiety to shuttle the proton, facilitating the formation of the palladium–hydride species. Several α,β-unsaturated carboxylic acids were synthesized in good to excellent yields, with excellent b/l ratio at room temperature. To demonstrate a feasible application, the gram-scale synthesis of a naproxen intermediate was performed (82% yield, b/l > 99:1).
Although less explored, hydroxycarbonylation of alkynes represents a green pathway to obtain α,β-unsaturated carboxylic acids. One of the main issues to explore with this methodology lies in catalyst recycling. In 2021, to address this challenge, a water-soluble palladium/Sulfoxantphos catalytic system was developed, utilizing water as the sole solvent in the absence of any acid additive (Fig. 45).269 Notably, the authors discovered that the volume of water played a crucial role in the product yield. The increase in the water volume led to the decrease in the concentration of the ligand coordinated to palladium, thus resulting in the formation of palladium black and, therefore, leading to catalyst deactivation. The catalytic system demonstrated tolerance towards a broad scope of symmetric and dissymmetric alkynes, allowing 18 different carboxylic acids to be obtained in 50–99% yield. Isotope labelling experiments provided insights into the reaction mechanism by revealing the incorporation of deuterium from water into the final product. The water-soluble palladium catalyst was easily separated from the product and reused for 5 cycles, with a minimal loss of catalytic activity.
 | ||
| Fig. 45 Water-soluble palladium-catalysed hydroxycarbonylation of alkynes.269 | ||
Less traditional, but no less important, the use of deep eutectic solvents (DESs) in these processes was recently explored by the groups of Liu and Salomone. The eutectic mixtures represent a new class of environmentally benign polar solvents, with promising potential for applications in catalytic processes. In the work mentioned above, the ruthenium-catalysed alkoxycarbonylation of olefins without the use of acid additives was described (Fig. 46).270 The preparation of the DES system was performed by combining MeP(Ph)3Br and glycerol at a molar ratio of 1:3 at 70 °C. The success of this methodology relies on the formation of the [Ru(CO)2Br3]− species through the dissolution of the ruthenium precursor and coordination with the DES. The reaction with 1-hexene and methanol resulted in 99% conversion with 99% selectivity for the ester.
 | ||
| Fig. 46 Alkoxycarbonylation in deep eutectic solvents.270,271 | ||
Salomone and co-workers reported the palladium-catalysed alkoxycarbonylation of aryl iodides in a DES under gas-free conditions using Mo(CO)6 as the CO source (Fig. 46).271 The DES system was prepared from cholinium chloride and urea and the presence of a phosphine ligand was crucial for the success of the process. Competition between the hydroxylated DES component (cholinium chloride) and its alcohol counterpart in carbonylative coupling with aryl iodide, resulting in lower yields of the desired ester, was observed; however, an increase in the alcohol concentration allowed this limitation to be overcome. The method was applied to various aryl iodides and alcohols, with 12 ester products being obtained in 68–99% yield. The recyclability of the system was also evaluated; however, after the second cycle, a progressive decrease in the catalytic activity was observed.
In addition to green solvents, the use of heterogeneous catalysts can also be seen as an advantageous environmental improvement in catalytic processes due to facile catalyst separation and recyclability. In this context, in 2013 Bhanage's group developed an immobilized palladium metal-containing ionic liquid catalyst for alkoxycarbonylation, phenoxycarbonylation, and aminocarbonylation of aryl iodides in a phosphine-free system (Fig. 47).272 In the alkoxycarbonylation reactions, alcohol was used as both the nucleophile and solvent, in the absence of any additional solvents. The authors presented 8 examples for the synthesis of different esters obtained in 70–95% yield. Remarkably, no leaching was observed, as demonstrated by a hot filtration test, with recycling experiments demonstrating the catalyst's stability and a minimal decrease in activity towards alkoxycarbonylation.
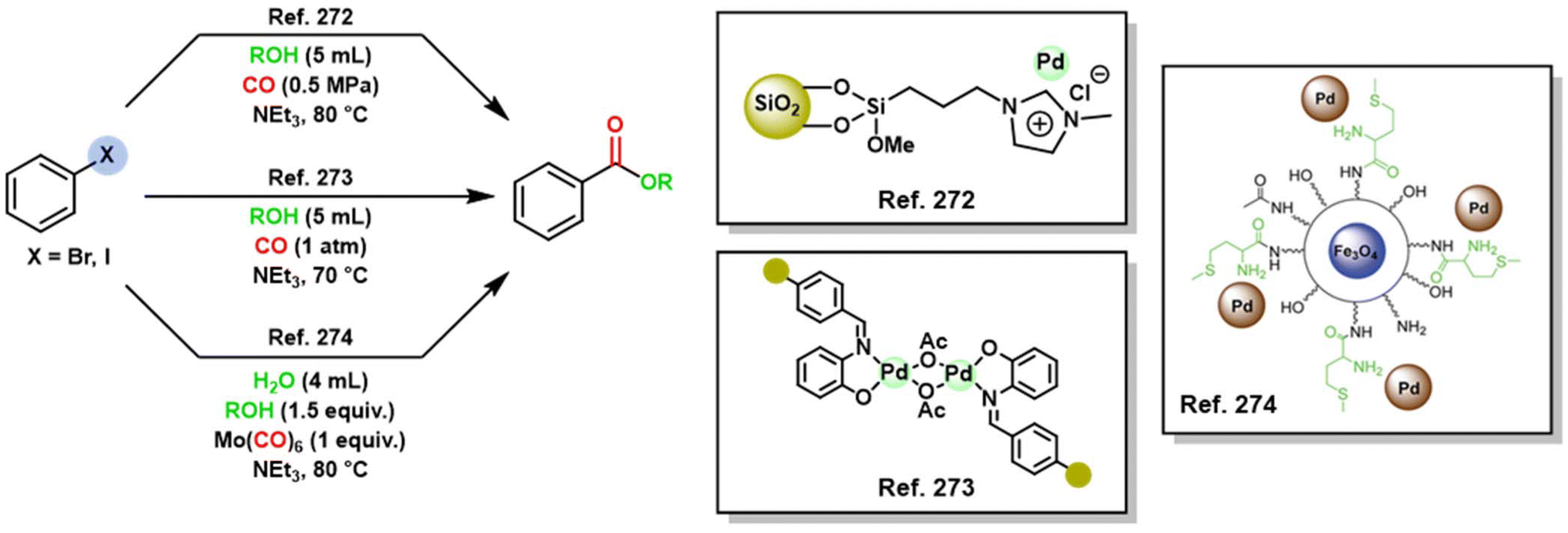 | ||
| Fig. 47 Alkoxycarbonylation of aryl halides over solid catalysts.272–274 This figure has been adapted/reproduced from ref. 274 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Another example of heterogeneous alkoxycarbonylation involves the use of a polymer-supported palladium(II)/Schiff base catalyst for the alkoxycarbonylation of aryl bromides with different alcohols under atmospheric CO pressure (Fig. 47).273 The catalyst showed high catalytic activity and stability with TONs of up to 2475. Moreover, the system was also efficient at aminocarbonylation of bromobenzene and aniline in anisole solutions to give the desired product in a moderate yield of 64%. Later on, immobilized palladium(0)-containing magnetic nanoparticles were prepared and tested for alkoxycarbonylation of aryl iodides with a variety of alcohols using Mo(CO)6 as the CO source under phosphine-free conditions (Fig. 47). For example, the reaction between iodobenzene and methanol in aqueous solutions gave the corresponding methyl ester in 43% yield.274
Recently, through surface engineering Cui and collaborators demonstrated the effect of oxygen vacancies in Fe2O3 heterogeneous catalysts on their performance in the alkoxycarbonylation of aryl iodides (Fig. 48).275 The insertion of oxygen vacancies can modify physical and chemical properties of the material, resulting in enhanced catalytic activity. The formation of three different types of iron sites, which had distinct roles in the reaction mechanism, were shown to participate in different elementary steps: the activation of aryl iodide, CO insertion, and C–O (or C–N in the case of aminocarbonylation) coupling. Using different alcohols as both the reactants and solvents, 27 esters were synthesized in 73–99% yield. A broad scope of alcohols and substituted aryl iodides were successfully tested in this heterogeneously catalysed transformation, demonstrating the positive effect of the oxygen vacancies created on the catalyst's performance.
 | ||
| Fig. 48 Alkoxycarbonylation of aryl iodides catalysed by Fe2O3 with induced oxygen vacancies.275 This figure has been adapted/reproduced from ref. 275 with permission from Springer Nature, copyright 2023. | ||
In 2015, Alper and co-workers tested a phosphaadamantane ligand, CYTOP 292, in a palladium-catalysed alkoxycarbonylation of terminal olefins using Lewis acids, such as SnCl2 or Ti(OiPr)4, to stabilize the formation of Pd–H bonds during the catalytic cycle (Fig. 49).276 The authors tested various solvents (2-butanone, acetonitrile, MEK, acetone, and DME), with the best results being obtained using DME as a co-solvent. An important influence of the nature of the ligand on regioselectivity was observed, with tris(4-methoxyphenyl)-phosphine showing the best results.
 | ||
| Fig. 49 Palladium-catalysed alkoxycarbonylation of olefins and aryl iodides.276–278 | ||
GVL, as a renewable platform molecule, has shown great potential for replacing traditional problematic solvents, as commented on above. In 2020, Mika et al. demonstrated the use of GVL as a reaction medium for the phosphine-free palladium-catalysed aryloxy- and alkoxycarbonylation of aryl iodides (Fig. 49).277 Various other solvents were compared with GVL for phenoxycarbonylation of iodobenzene, but none of them enabled yields and TONs similar to those obtained in GVL to be achieved. The protocol was applied to a broad scope of phenols/alcohols and iodoaromatic compounds allowing for the synthesis of 39 products in 36–99% yield (mostly ≥80%) with high TONs.
Recently, Beller's group reported a highly efficient palladium-catalysed alkoxycarbonylation of olefins using a novel ligand developed by the group (Fig. 49).278 Excellent yields of ester products and catalyst TONs in the range of 106 were obtained. The ligand, called LIKATphos and shown in Fig. 49, was initially tested in methoxycarbonylation of 1-octene with low loadings of Pd(OAc)2 (0.0001–0.005 mol%). The use of Pd2(dba)3 as the metal precursor resulted in an even higher yield and improved thermal stability of the catalytic system at 140 °C. An exceptional yield of 99% of the desired product was achieved using 0.0001 mol% of the catalyst and 0.5 mol% of the ligand. The relatively low regioselectivity (l/b = 3:1) for methoxycarbonylation of 1-octene was explained by the high activity of the catalytic system, which enabled the fast carbonylation of the in situ formed internal olefins. The versatility of the method was evaluated by testing different olefins and alcohols, with 21 ester products being obtained in up to 92% yield and l/b of up to 90/10. Notably, methyl propionate was produced from ethylene with an impressive TON of 237000.278
In the same year, Beller's group reported a cobalt-catalysed alkoxycarbonylation of olefins using secondary phosphine oxides as promoters (Fig. 50).279 Systems with different phosphine oxides demonstrated activity in the transformation of 1-octene, whereas no activity was obtained in the absence of phosphine oxides. Sterically hindered or electron-deficient phosphine oxides displayed inferior results, whereas the promoters containing O-substituents showed a superior performance. Interestingly, the use of Co(OAc)2·4H2O as the catalyst precursor resulted in a higher yield (87%) compared to conventional cobalt compounds, such as Co2(CO)8 (82%), Co4(CO)12 (62%), Co3(PO4)2·H2O (38%), CoCO3 (69%), and Co(OH)2 (83%). It is worth mentioning that the use of cobalt(II) salts is highly advantageous due to operational and cost issues. The authors tested 14 olefins, including terminal, internal, cyclic, and aromatic ones, obtaining the corresponding products in up to 92% yield with an l/b ratio of up to 68:32.
 | ||
| Fig. 50 Cobalt-catalysed methoxycarbonylation of olefins.279 | ||
Shi and colleagues recently developed an efficient palladium-catalysed asymmetric alkoxycarbonylation of vinyl arenes “on water”.280 (R)-DTBM-SEGPHOS (shown in Fig. 51) was found to be an effective ligand for performing the reaction under aqueous conditions. The hydrophobicity of the ligand appeared to be crucial in this transformation, which likely proceeded “on water”. Using this protocol, 17 examples were successfully demonstrated to give the corresponding products in yields of up to 92%. The branched products were obtained preferentially in these reactions with an ee of 96%.
 | ||
| Fig. 51 Top: asymmetric alkoxycarbonylation of vinyl arenes “on water”.280 Bottom: electroreduction of CO2 to generate CO coupled to alkoxycarbonylation reaction.281 | ||
Huang, Dong, and collaborators recently reported a two-compartment system for the palladium-catalysed alkoxycarbonylation of alkynes using CO from the electroreduction of CO2 (Fig. 51).281 The authors used methanol or ethanol as solvents and nucleophiles to afford 26 examples of the α,β-unsaturated esters in moderate to high yields (40–98%) and good regioselectivities to the branched products. The presence of electron-withdrawing groups in the arene substituent significantly reduced the yield of the desired product.
As alcohols (mostly methanol and ethanol) and water, which have good sustainability ranks, participate as nucleophiles in these examples, the focus for these transformations in terms of sustainability is the choice of a co-solvent, if any is used. In this regard, organic carbonates (DMC and PC) are promising candidates, along with γ-valerolactone.
5. Aminocarbonylation
Aminocarbonylation involves the addition of CO and an amine to a substrate, such as an alkene or aryl halide, in the presence of a catalyst, usually a transition metal complex.282 Aminocarbonylation reactions are widely used in the synthesis of amides, which are ubiquitous in pharmaceuticals, agrochemicals, polymers, and natural products. The aminocarbonylation reaction offers several advantages, including high atom economy, the use of simple and readily available starting materials, and the ability to access amides directly in a single step. Reaction conditions are typically mild, making the method suitable for sensitive substrates and functional groups. However, challenges remain in optimizing the reaction conditions to achieve high yields and selectivity. The choice of catalyst, ligand, and reaction conditions can significantly impact the reaction efficiency and the formation of side products. Palladium-based catalysts are commonly used due to their high efficiency and tolerance toward a wide range of functional groups. Other metals, such as nickel,231,283,284 copper,285 and manganese,286 can also be employed in aminocarbonylation reactions. Recent advances have focused on developing more robust and selective catalytic systems, as well as exploring alternative CO sources, as the handling of CO requires careful safety measures due to its toxicity and flammability.287–290 Efforts are also being made to improve the sustainability and green chemistry aspects of these reactions through the use of less toxic solvents and renewable starting materials.In 2016, Mika and collaborators reported the use of GVL, a renewable and nontoxic solvent, as an environmentally benign reaction medium for homogeneous palladium-catalysed aminocarbonylation reactions (Fig. 52).291 GVL was compared with the solvents conventionally employed in industry for these reactions in terms of vapour pressure, an important parameter for the use of solvent on a large scale. The vapour pressure of GVL was found to be two orders of magnitude advantageously lower than those of toluene or DMF. Aminocarbonylation of iodobenzene and its 4-substituted derivatives with tert-butylamine was performed in the presence of Pd(OAc)2 as the catalyst precursor and NEt3 or PPh3 as auxiliary ligands. GVL proved to be a promising solvent for these reactions. Although GVL showed an inferior general performance compared to DMF, some specific advantages were observed, in particular, higher yields of double carbonylated products. A broad scope of carboxamide and ketocarboxamide products were synthesized by the protocol developed in this work.
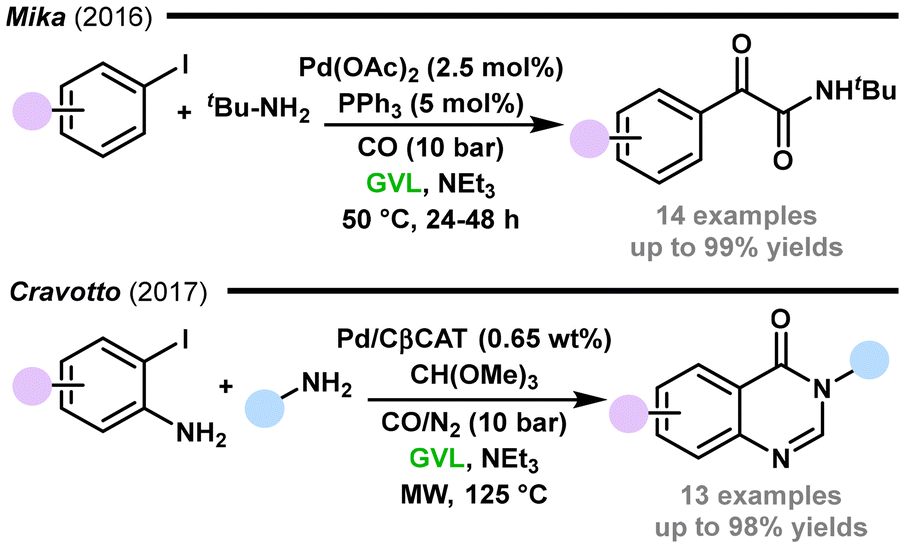 | ||
| Fig. 52 Green synthesis of quinazolinones by carbonylative coupling using GVL as the solvent.291,292 | ||
In 2017, Cravotto and co-workers reported a less conventional application of the aminocarbonylation reaction, which consisted of the microwave-assisted carbonylative coupling of anilines and amines catalysed by a heterogeneous palladium catalyst to give 4(3H)-quinazolinones (Fig. 52).292 The efficacy of GVL as the solvent for these transformations was demonstrated by comparison with traditional organic solvents, such as toluene, acetonitrile, DMF, and 1,4-dioxane. A wide range of quinazolinones were synthesized in good to excellent yields from substituted o-iodoanilines, trimethyl orthoformate, and a variety of amines.
Recently, Bayer et al. published an extensive study on the application of renewable solvents in carbonylation reactions, in particular, in the palladium-catalysed aminocarbonylation of aryl bromides (Fig. 53).49 The model reaction of N-methylaniline with 4-bromobenzonitrile catalysed by the Pd(OAc)2/Xantphos system in the presence of NEt3 was performed in various renewable solvents, most of them showing excellent performances (Fig. 53). The best results were obtained in nonpolar hydrocarbon solvents, such as limonene (99% yield), p-cymene (97% yield), γ-terpinene (94% yield), and α-pinene (97% yield). Nonpolar organic carbonates also gave excellent results: 97% yield in DMC and 94% in DEC. The method was successfully extended to a broad scope of substrates using DMC or limonene as solvents, with many functional groups (such as CHO, CN, CO2Me) being well tolerated under the conditions applied.
 | ||
| Fig. 53 Screening of renewable solvents for the aminocarbonylation of 4-bromobenzonitrile with N-methylaniline.49 This figure has been adapted/reproduced from ref. 49 with permission from the American Chemical Society, copyright 2020. | ||
The replacement of fossil-based solvents by biomass-based solvents in aminocarbonylation was also addressed in the work of Takács and colleagues (Fig. 54).293 The authors studied the potential of alkyl methyl and ethyl levulinates as well as 2-MeTHF and compared the performance of these green solvents with that of traditional DMF in the reaction of iodobenzene with morpholine using the Pd(OAc)2/Xantphos catalytic system. At nearly complete conversion, the reactions in green solvents showed 100% selectivity for the amide product vs. only 88% in DMF. Various amines and iodobenzenes were evaluated as the substrates, with levulinates as the solvents presenting better results in most of the cases compared to 2-MeTHF.
 | ||
| Fig. 54 Aminocarbonylation of iodobenzenes in biomass-based solvents and in DMF.293 | ||
In 2021, Wu and co-workers developed a rhodium-catalysed aminocarbonylation of DMC with nitro compounds (Fig. 55).294 In this novel transformation, DMC was used as a substrate (C1 building block) and as a solvent; while W(CO)6 was used as both the CO source and reductant. Under optimized conditions, the reaction was performed in the presence of water, employing the Rh2(CO)4Cl2/1,3-bis(diphenylphosphino)propane (DPPP) catalytic system, NaI as an additive, and Na3PO4 as a base. A wide range of nitro compounds were successfully tested as co-reagents allowing for the synthesis of various carboxamides in good to excellent yields. The proposed mechanism involves the base-assisted reaction of NaI with DMC to generate CH3I, which further reacts with rhodium species. A subsequently formed acetyl iodide undergoes nucleophilic attack by the amine generated from the nitro compound to give the final acetamide product (Fig. 55).
 | ||
| Fig. 55 Aminocarbonylation of DMC with nitrocompounds.294 | ||
A palladium-catalysed carbonylative coupling of acylhydrazine with aryl bromide resulting in N,N′-diacylhydrazines was reported by the group of Skrydstrup in 2014 (Fig. 56).295 The model reaction of benzhydrazide with 6-methoxy-2-bromonaphthalene in the presence of the Pd(dba)2/Xantphos catalytic system was performed in various solvents. The reaction in the traditional solvent dioxane resulted in 98% yield of the desired coupling product; however, the green solvent anisole also showed excellent performance allowing for 92% yield.
 | ||
| Fig. 56 Palladium-catalysed carbonylative coupling of acylhydrazine with aryl bromide.295 | ||
In 2008, Bhanage's group developed a general and sustainable approach for aminocarbonylation of aryl iodides with amines in water (Fig. 57).296 The reaction was performed in various solvents, including water, using Pd(OAc)2 as the catalyst (without any auxiliary ligand) and NEt3 as a base. It was found that polar solvents demonstrated superior results compared to nonpolar ones. The reactions in water resulted in the highest yields of carboxamides compared to all other solvents. A broad scope of amines, including aliphatic, cyclic, and aromatic amines, were successfully employed as co-reagents in these reactions to give the corresponding products in 62–96% yield.
 | ||
| Fig. 57 Palladium-catalysed carbonylation of aryl halides with amines in water.296,297 | ||
In 2023, the group of Lipshutz reported the palladium-catalysed aminocarbonylation of aryl/heteroaryl halides in an aqueous micellar medium using W(CO)6 as the source of CO (Fig. 57).297 In this interesting work, the authors proposed an efficient green methodology to replace traditional protocols. The system was composed of Pd(OAc)2 as the catalyst precursor, [(t-Bu)3PH]BF4 as the pre-ligand and acid source, TPGS −750-M as a surfactant, and K3PO4 as a base. A series of aryl iodides and aryl bromides were tested in the reactions with different amines, resulting in aminocarbonylation products in good to excellent yields. To demonstrate the applicability of the method, the synthesis of various pharmaceutical precursors and compounds from the Merck Informer Library was performed. To monitor the environmental impact of this approach, the E factor was calculated to be 8.7 (considering the product purification step) and 3.0 (omitting the extraction solvent), which showed that the process generated low amounts of waste.
Using a different approach for CO generation, Schwab and co-workers performed the aminocarbonylation of 5-iodo-1,2,3-triazoles with amines in various sustainable solvents (Fig. 58).298 In their method, a two-chamber reactor was used, in which CO formation was carried out by the reaction of sulphuric and formic acids. Then, the gas was transferred to the other chamber for catalytic aminocarbonylation. Various solvents were evaluated in a model reaction, which gave the aminocarbonylation product in moderate to good yields: DMC (95%), toluene (93%), acetonitrile (trace), anisole (75%), p-cymene (62%), and 2-MeTHF (63%). A wide range of triazoles and amines were successfully employed as substrates in this transformation to give the corresponding products in up to 98% yield. Additional information on the use of formic acid as a CO surrogate can be found in specific reports in the literature.299,300
 | ||
| Fig. 58 Aminocarbonylation of triazoles in sustainable solvents.298 | ||
Although reports on heterogeneous aminocarbonylation processes are scarce in the literature, a few examples can be found. The heterogeneous solvent-free aminocarbonylation of iodobenzene was also performed in the presence of palladium catalysts prepared by the immobilisation of palladium on supported ionic liquid phases (SILPs) (Fig. 59).301 In the first tests, a mixture of amide and α-ketoamide was obtained; however, after catalyst screening and system optimization, the yields of the desired amides were significantly improved. Moreover, under specific conditions (higher pressure and DMF as the solvent), the selective formation of double carbonylation products was achieved.
 | ||
| Fig. 59 Heterogeneous palladium catalysed solvent-free aminocarbonylation of iodobenzene.302 | ||
A silica-supported palladium(0) catalyst was used for aminocarbonylation of iodoarenes with anilines and amines under ligand-free and solvent-free conditions at atmospheric pressure of CO and moderate temperature (Fig. 59).302 Interestingly, the addition of conventional organic solvents (DMSO, THF, DMF, acetonitrile, benzene) decreased the yields of the amide product. Several amides were synthesized in up to 99% yield. The solid catalyst showed good stability during five recycling experiments.
In 2019, Alexanian and Sargent reported a cobalt-catalysed stereospecific aminocarbonylation of unactivated alkyl tosylates using a remarkably broad range of amines for the synthesis of enantioenriched amides (Fig. 60).303 It was found that the use of t-amyl-alcohol as the solvent was critical, whereas polar aprotic solvents (e.g., THF) gave significantly reduced yields of the amide. The reaction occurred with excellent stereospecificity and chemoselectivity to produce amides as well as carboxylic acids and ketones starting from readily available enantioenriched alcohols. The broad scope of the reaction was highlighted using a diverse range of nitrogen nucleophiles, including challenging primary amines and ammonia.
 | ||
| Fig. 60 Top: cobalt-catalysed aminocarbonylation of alkyl tosylates.303 (Middle): cobalt-catalysed aminocarbonylation of (hetero)aryl halides promoted by visible light.304 Bottom: cobalt-catalysed deaminative amino- and alkoxycarbonylation of aryl trialkylammonium salts promoted by visible light.305 | ||
In 2020, Alexanian and Veatch reported the cobalt-catalysed aminocarbonylation of (hetero)aryl halides at low CO pressure and ambient temperature in t-amyl-alcohol as a solvent under visible light irradiation (Fig. 60).304 The authors performed the reactions of various (hetero)aryl and vinyl bromides, chlorides, and triflates with a series of amine nucleophiles, including an ammonia surrogate. In total, 46 examples were reported with 33–99% yield of the desired products. The results of mechanistic investigations were consistent with the hypothesis that the reaction proceeded via intermolecular charge transfer involving a donor–acceptor complex between the substrate and the cobalt catalyst.
In 2022, Alexanian and collaborators reported a cobalt-catalysed aminocarbonylation of aniline-derived trialkylammonium salts, which involved a challenging cleavage of the C(sp2)–N bond (Fig. 60).305 The reaction was promoted by visible light. The authors tested several solvents and solvent combinations in a model reaction under mild conditions and low pressures of CO. In general, the best results were observed with the solvents considered to be green, such as t-amyl-alcohol (61%), 2-butanol (52%), anisole (44%) and EtOAc (40%), while hazardous and problematic solvents, such as benzene (39%), DCM (4%), 1,4-dioxane (18%), DMF (2%), and MeCN (5%), gave inferior results. A range of alkylamines were successfully used as co-reagents in these reactions; also alkoxycarbonylation was demonstrated. Mechanistic studies and DFT calculations supported a mechanism involving a key visible light-induced carbonyl photodissociation and SNAr-type oxidative addition.
In 2024, Duan, Guo, and colleagues reported a copper-catalysed regioselective C–C bond cleavage/aminocarbonylation cascade process for the synthesis of γ-lactam or succinimide skeletons containing an amide group (Fig. 61).285 After extensive solvent evaluation, ethanol was found to be the best solvent, which outperformed traditional solvents, such as CH3CN, THF, and dioxane, providing the desired product in 89% yield. The protocol was applied to a wide range of substrates yielding the corresponding products in 35–98% yield. The advantages of the method are mild conditions, a broad scope of amines, and excellent compatibility of functional groups.
 | ||
| Fig. 61 Copper-catalysed regioselective C–C bond cleavage/aminocarbonylation.285 | ||
In 2024, Alexanian and colleagues reported light-promoted cobalt-catalysed aminocarbonylation and hydroaminomethylation under mild conditions and low CO pressure (Fig. 62).199 After the step that produced amides, PhSiH3 was added to the reaction solution allowing for the sequential cobalt-catalysed reduction of the primarily formed amide resulting in formal hydroaminomethylation of the original alkene. In terms of solvents, MTBE presented the best results, surpassing traditional solvents, such as toluene and MeCN. In a scale-up experiment, the process was successfully performed under neat conditions. Photochemical catalysis demonstrated remarkable versatility, allowing for the use of a wide scope of alkene and amine substrates, achieving high chemo- and regioselectivity. The proposed mechanism involved the formation of hydridocobalt species through photodissociation of the carbonyl ligand, enabling catalytic activity under mild conditions.
 | ||
| Fig. 62 Cobalt-catalysed synthesis of amides from alkenes and amines promoted by light.199 | ||
A wide variety of green solvents were tested in aminocarbonylation reactions. Not surprisingly, the solvent that performed best depended greatly on the specific system. Nonetheless, some are worth mentioning for have given good results in different systems: DMC, γ-valerolactone and anisole.
6. Other processes
6.1. Thiocarbonylation
The thiocarbonylation reaction involves the addition of CO and a sulphur source to a substrate, such as an alkene or alkyne, resulting in the formation of sulphur-containing products. These compounds have significant applications as pharmaceutical and agrochemical products as well as in materials science. For example, thiocarbonyl groups can serve as precursors for various sulphur-containing functional groups, which are important in bioactive molecules and polymers. Palladium306,307 and nickel308–310 complexes are commonly used as catalysts in thiocarbonylation reactions. The presence of ligands, such as phosphines or amines, can significantly influence the reaction selectivity and product yield. The sulphur source is crucial for the nature of the final product, which can be a thioester, thioketone, or thiocarbamate.In 2018, Fleischer and colleagues reviewed the literature on the synthesis of thioesters by transition metal-catalysed reactions, including thiocarbonylation reactions.311,312 Recent advances in thiocarbonylation were also the subject of a paper published by Khayitov and colleagues in 2024.313 Recent research on thiocarbonylation has focused on developing more efficient and selective catalysts as well as greener and safer reaction conditions. For example, the use of milder sulphur sources and CO surrogates309,314–317 can significantly improve process safety and sustainability. Although thiocarbonylation is a less explored reaction in comparison with the processes discussed above, some important examples of using green solvents to accomplish this transformation have been published.
The first report on the use of green solvents in thiocarbonylation was described by the group of Skrydstrup in 2013 (Fig. 63).318 In their work, a two-chamber reactor was employed to generate the required CO ex situ. Then, thiocarbonylation of aryl iodides with thiophenols was performed in an anisole solution containing sodium acetate and a catalytic system composed of Pd(OAc)2 and DPEphos (bis[(2-diphenylphosphino)phenyl] ether). The reaction with 11 electron-deficient aryl iodides in anisole solutions gave the corresponding thioesters in 40–99% yield. Interestingly, the initial screening of the substrate scope started with electron-rich aryl iodides, which reacted with aromatic thiols in DME solutions, providing the corresponding thioesters in good yields. However, the application of these conditions to electron-deficient aryl iodides proved to be problematic due to poor chemoselectivity. After a solvent screening, which included toluene, propionitrile, dioxane, cyclopentyl methyl ether, and trifluorotoluene, anisole was found to be an optimal green medium for this reaction.
 | ||
| Fig. 63 Palladium-catalysed thiocarbonylation of halides in anisole solutions.318,319 | ||
Later on, the same group reported a similar procedure using the same type of two-chamber reactor, but this time with the Pd(PhCN)2Cl2/Xantphos catalytic system, as the most efficient one (Fig. 63).319 The reactions of aryl, vinyl, and benzyl bromides with thiophenols were performed in anisole solutions containing sodium acetate as a base with ex situ generated CO. Other solvents were also tested, such as DME, diglyme, toluene, dioxane, and benzotrifluoride; however, only poor results were obtained. Several examples of reactions with electron-rich and electron-deficient aryl bromides were presented, with the addition of sodium iodide being required in the case of electron-deficient aryl bromides to improve the reaction chemoselectivity. The authors also reported the synthesis of 13C labelled benzothiophenes using 1-mercaptobenzophenones as nucleophiles in the carbonylation step followed by McMurry coupling for ring closure.
In 2019, Liao and colleagues reported the first highly enantioselective palladium-catalysed thiocarbonylation of styrenes using chiral sulfoxide-(P-dialkyl)-phosphine ligands (SOP) under mild reaction conditions (Fig. 64).307 The use of special chiral ligands was a key to the successful execution of this reaction. Thiocarbonylation reactions with a broad scope of substrates proceeded smoothly under mild conditions (1 atm CO and 0 °C). It has also been demonstrated that this transformation can be performed using CO surrogates, greatly improving the safety aspects. The generality and utility of the method was confirmed by its application to the synthetic transformations of thioester products and the direct acylation of cysteine-containing dipeptides. The reaction mechanism was also studied in this work and a plausible proposal for the catalytic cycle was presented.
 | ||
| Fig. 64 Palladium-catalysed enantioselective thiocarbonylation of styrenes.307 | ||
In 2008, Alper and colleagues reported the palladium-catalysed thiocarbonylation of aryl iodides with thiols in phosphonium ionic liquids (PSILs) (Fig. 65).320 The most effective PSILs identified were THP-Br, THP-NTf2, and THP-PF6, which allowed for 84, 82, and 91% isolated yields of the desired product, respectively. In contrast, the same reaction performed in THF produced the desired product in only 29% yield. Among the PSILs evaluated, trihexyl(tetradecyl)phosphonium hexafluorophosphate (THP-PF6) was found to be a particularly effective medium for this reaction. The study demonstrated not only the efficiency of PSILs as solvents but also the recyclability of both the ionic liquid and the active palladium catalyst.
 | ||
| Fig. 65 Palladium-catalysed thiocarbonylation of iodoarenes with thiols in phosphonium salt ionic liquids.320 | ||
6.2. Carbonylative cross-coupling
In 2021, Kégl and colleagues made a nice contribution reviewing the history of studies in the field of catalytic carbonylative coupling reactions in Hungary.321 In 2022, Wu and collaborators reviewed transition metal catalysed carbonylative cross-coupling reactions with the use of alkyl carbon nucleophiles.20 The carbonylative Suzuki–Miyaura reaction is a variant of the classic Suzuki–Miyaura cross-coupling reaction, which involves CO in the coupling process. This reaction enables the formation of carbon–carbon bonds with the introduction of a carbonyl group and results in the synthesis of ketones, esters, or amides, depending on the nucleophile used.6,25,322–324Palladium complexes, often containing phosphine ligands, are the catalysts of choice for this reaction due to their efficiency. However, other transition metals are also known to perform this transformation.325,326 The choice of ligands, bases, and solvents can significantly influence the product yield and reaction selectivity. The carbonylative Suzuki–Miyaura reaction is a powerful tool for the synthesis of carbonyl-containing compounds, particularly, ketones. This reaction is important for the preparation of various fine chemicals, such as pharmaceutical and agrochemical products, where the introduction of carbonyl functionalities is often crucial.
In 2017, Alexanian and Sargent reported a stereospecific cobalt-catalysed carbonylative cross-coupling of alkyl tosylates and dienes to produce enantioenriched dienones (Fig. 66).327 The reaction proceeded under low CO pressure and mild conditions in t-amyl-alcohol solutions. This transformation represents a unique convergent approach for the asymmetric synthesis of valuable carbonyl compounds from easily accessed starting materials.
 | ||
| Fig. 66 Cobalt-catalysed carbonylative cross-coupling of alkyl tosylates and dienes.327 | ||
In 2021, Das, Hajra, and collaborators reviewed recent advances in transition-metal catalysed carbonylative Suzuki–Miyaura coupling reactions.326 In 2024, Hu and Kazemi published a review on the applications of palladium nanocatalysts in this transformation.322 The literature on the use of CO surrogates for palladium-catalysed Suzuki–Miyaura and Sonagashira carbonylative cross-coupling reactions was reviewed by Aronica in 2023.328 The solvent effects in palladium-catalysed cross-coupling reactions and the use of unconventional conditions, including unconventional reaction media, were reviewed in 201929 and 2020,329 respectively. The authors of these reviews described several such types of transformations performed in green solvents; however, none of them involved the participation of CO, except for one example: the carbonylative Sonogashira cross-coupling of aryl iodides performed in an environmentally benign solvent, poly(ethylene glycol) (PEG).330
In one of the early reports in 1998, Suzuki and Miyaura described an efficient method for the palladium-catalysed carbonylative cross-coupling of arylboronic acids with aryl halides under atmospheric pressure of CO (Fig. 67).331 Various solvents, such as anisole, toluene, dioxane, and DMF, were tested as the reaction medium. The use of less polar solvents was more efficient at synthesizing cross-coupling products, with the best results being obtained in anisole solutions. A broad scope of substrates containing different functional groups were tested to give the corresponding unsymmetrical biaryl ketones in moderate to good yields.
 | ||
| Fig. 67 Palladium-catalysed carbonylative Suzuki–Miyaura coupling.331,332 | ||
In 2013, the group of Skrydstrup reported the palladium-catalysed carbonylative Suzuki–Miyaura coupling of iodobenzenes with boronic acids in anisole (Fig. 67).332 To avoid the use of high-pressure CO, the process was performed in a two-chamber reactor to generate CO ex situ. The air tolerance of the system, which operates without the exclusion of oxygen and with the use of non-degassed and non-dried solvents, is an important advantage of the approach developed. The scope of the method was demonstrated through the synthesis of 19 benzophenone products obtained in 50–93% yield. The protocol was also applied for the synthesis and isotope labelling of two pharmaceutical drugs, which were obtained in both regular and isotope-labelled forms: nordazepam in 55% (CO) and 63% (13CO) yield and Tricor in 84% (CO) and 85% (13CO) yield.
In 2014, Han and Zhong developed the first iron-catalysed carbonylative Suzuki reaction (Fig. 68).333 The reactions, with 38 examples of aryl iodides containing electron-donating or electron-withdrawing functionality, proceeded smoothly in solutions of eco-friendly PEG-400, affording the desired products in 71–97% yield. The reaction in other solvents tested, such as DMF (16%), ethanol (0%), dioxane (6%), and DCE (0%), resulted in much lower yields.
 | ||
| Fig. 68 Iron-catalysed carbonylative Suzuki reactions using PEG-400 as a solvent.333 | ||
In 2017, Bhanage and collaborators reported the Pd/C-catalysed carbonylative Suzuki–Miyaura cross-coupling of aryl iodides with N-formylsaccharin as the CO surrogate using PC as the environmentally benign and sustainable polar aprotic solvent (Fig. 69).334 The authors tested various solvents, including dioxane and toluene, as well as DMF/dioxane mixtures, which gave low to moderate yields. In non-polar toluene, for example, the reaction mainly occurred through non-carbonylative Suzuki–Miyaura cross-coupling, while the carbonylation product, biaryl ketone, was obtained in only 9% yield. It is known that the carbonylative Suzuki–Miyaura cross-coupling of aryl halides in polar aprotic solvents typically produces the corresponding aryl carboxylic acids.335 However, the reaction over the Pd/C catalyst in solutions of PC gave the desired biaryl ketone in 74% yield. A variety of biaryl ketones were obtained in this work, including (4-methoxyphenyl)(3,4,5-trimethoxyphenyl)methanone, an antineoplastic agent from the phenstatin family. No co-catalysts, special additives, or ligands were used in this protocol. The Pd/C catalyst was reused five times with only a slight reduction in catalytic activity. Moreover, the reaction was scaled up to a gram-scale production.
 | ||
| Fig. 69 Carbonylative Suzuki–Miyaura cross-coupling using N-formylsaccharin as a CO surrogate.334 | ||
Recently, Panda provided an overview of the literature on the effects of solvent polarity in Sonogashira coupling reactions.336 According to the author, the choice of solvent plays a crucial role in the Sonogashira cross-coupling reaction. The solvents can influence reaction rates, selectivity, and the stability of the catalyst, thereby impacting the overall efficiency and success of the reaction. Polar solvents generally promote higher reaction rates and yields by aiding in the solvation and stabilization of intermediates, whereas nonpolar solvents can enhance selectivity by reducing steric hindrance. Additionally, the effects of solvents on regioselectivity were noted, with polar solvents tending to favor certain regioisomers. In addition, the author suggested that future research could concentrate on creating more sustainable solvent options and refining solvent screening strategies, taking into account both reaction performance and environmental considerations.
In 2019, Bhanage and colleagues reported palladium-catalysed carbonylative Sonogashira and carbonylative Suzuki–Miyaura cross-coupling using PC as the solvent (Fig. 70).337 Although the reactions in traditional solvents, such as toluene, DMF, and dioxane, presented excellent yields of the desired products, it was shown that PC could be used as an alternative green reaction medium, which efficiently combined excellent catalytic performance and high product yields. Under optimized conditions, the aminophosphine pincer complex {[C6H3-2,6-(NHP{piperidinyl}2)2]Pd(Cl)} could be applied as the catalyst at low palladium loadings (10−4–10−6 mol%) resulting in high catalytic turnovers (105–107).
 | ||
| Fig. 70 Aminophosphine palladium pincer-catalysed carbonylative Sonogashira and Suzuki–Miyaura cross-coupling.337 | ||
In addition to these studies, in 2020 Bayer et al. reported the application of a wide range of renewable solvents in the carbonylative coupling of boronic acids with bromoarenes (Fig. 71).49 The model reaction of m-tolylboronic acid with 3-bromoanisole in the presence of Pd(acac)2 as a catalyst precursor and di(1-adamantyl)-n-butylphosphine hydroiodide (CataCXium AHI) as a ligand was performed in various renewable solvents to give the following yields of the corresponding benzophenones: α-pinene (50%), γ-terpinene (50%), limonene (80%), p-cymene (75%), methylal (50%), rose oxide (33%), DMC (16%), and 2-MeTHF (30%). It was concluded that nonpolar hydrocarbons as solvents led to higher product yields compared to nonpolar ethers and carbonates. Interestingly, the best solvent, limonene, which possessed two C–C double bonds, showed no reactivity in Heck-type arylation under the applied conditions. The method was successfully extended to a broad scope of substrates using limonene as the reaction medium.
 | ||
| Fig. 71 Carbonylative Suzuki–Miyaura coupling of aryl halides with boronic acids.49 | ||
In 2016, the Bhanage group reported a palladium-catalysed carbonylative Sonogashira cross-coupling reaction performed in solutions of PEG with different molecular weights ranging from 200 to 1500 (Fig. 72).330 The authors noted that the performance of PEG-400 and PEG-600 in terms of substrate conversion and reaction selectivity for the desired product was better than those of PEG-200 and PEG-1500. Considering the recyclability of the palladium catalyst and PEG, the authors selected more viscous PEG-600 for optimization studies. The system worked for aryl iodides containing both electron-donating and electron-withdrawing groups; however, the substrates with a strong EWG, such as cyano and nitro groups, in the para-position did not undergo carbonylation under the applied conditions.
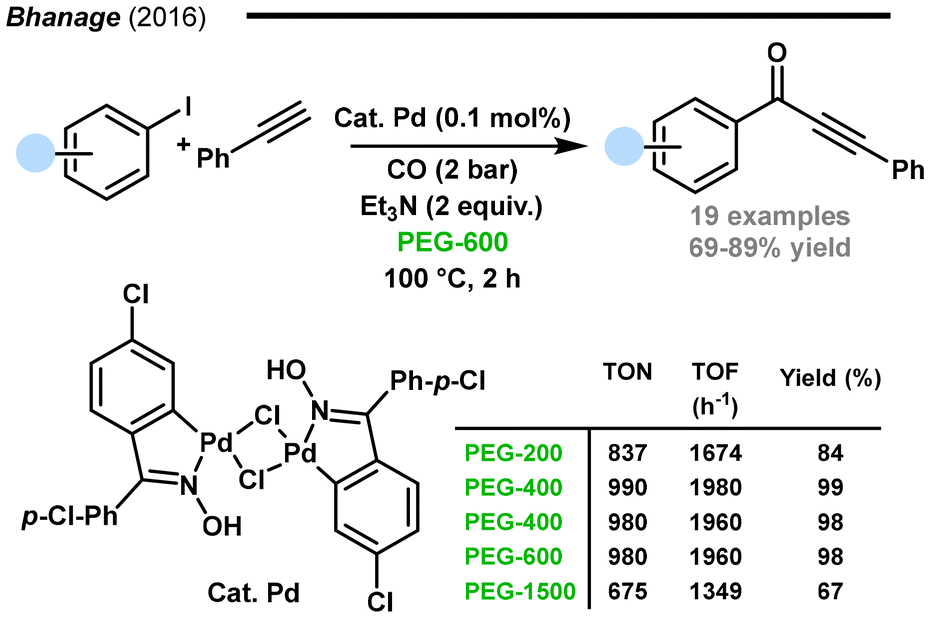 | ||
| Fig. 72 Oxime palladacycle catalysed carbonylative Sonogashira cross-coupling using PEG as a solvent.330 | ||
6.3. Ring expansion carbonylation
Ring expansion carbonylation is a carbonylation reaction in which a cyclic compound, typically containing oxygen, nitrogen, or sulphur, undergoes ring expansion through the incorporation of CO.338–341 This reaction provides a valuable synthetic route to lactones, lactams, and thiolactones by inserting a carbonyl group into smaller cyclic structures, such as epoxides, aziridines, or thiiranes, leading to the formation of larger rings.341,342 The process often requires transition metal catalysts, such as palladium or rhodium complexes, in combination with CO, enabling efficient carbonylation of these cyclic substrates under mild conditions. These reactions feature high atom economy and usually high selectivity; this makes them highly attractive for green chemistry applications.Lactones and thiolactones are valuable intermediates for the synthesis of high-value chemicals, including flavours, fragrances, and biologically active molecules, such as antibiotics and anticancer agents. Moreover, their application in polymer synthesis has significant potential for creating sustainable materials, e.g., polyesters and polythioesters, which are environmentally friendly alternatives to petrochemical-derived plastics. Recent advances in the synthesis of β-lactams involving CO were reviewed by Wang and Liu.343
In 2006, Coates and colleagues studied the mechanism of ring expansion carbonylation of epoxides to give lactones in the presence of the [(salph)Al(THF)2]+[Co(CO)4]− (salph = N,N‘-o-phenylenebis(3,5-di-tert-butylsalicylideneimine)) complex as the catalyst.344,345 The authors showed that the coordinating capacity of the solvent significantly influenced the rate of lactone formation due to the need to stabilize the aluminium cation intermediate formed during lactone production. Coordinating solvents, such as THF or other unhindered ethers, effectively stabilize aluminium in this state, accelerating the carbonylation reaction. In contrast, polar solvents that are weak donors, such as 2-MeTHF, 2,5-dimethyltetrahydrofuran (2,5-diMeTHF), or 1,2-difluorobenzene, provide less efficient stabilization for the aluminium cation, resulting in slower carbonylation. Highly polar solvents, such as CH3CN, tend to bind strongly to aluminium, thus hindering its interaction with epoxides.
In 2022, Gusevskaya, Beller, and collaborators unveiled a process for ring expansion/ring opening of oxetanes using a cobalt/phosphine oxide catalytic system under mild hydroformylation conditions (Fig. 73).346 The authors explored a wide range of traditional and green solvents. The experiments performed in anisole, DMC, and DEC demonstrated efficiency similar to the reaction in toluene. Alternatively, the reaction was completely inhibited in polar solvents, such as DMF, MeCN, and DMSO. The presence of the syngas atmosphere was crucial for the formation of active cobalt species from the Co2(CO)8 pre-catalyst, with CO acting as a ligand for its stabilization, while H2 provided the acidic properties required for oxetane activation. By testing different oxetanes, a significant role of the substituents in the oxetane ring was confirmed, with the reaction leading to either carbonylative ring expansion or reductive ring opening to give γ-lactones or primary alcohols, respectively.
 | ||
| Fig. 73 Carbonylation of heterocycles.346,347 | ||
Using a less conventional but also environmentally friendly approach, an interesting methodology was developed for the synthesis of lactones and keto-amides by a photoinduced copper-catalysed carbonylation reaction (Fig. 73).347 This transformation was accomplished through radical-mediated ring-expansion/aminocarbonylation cascade reactions between a hemiketal hydroperoxide and an amine. Methanol was found to be the optimal solvent, surpassing trifluorotoluene and 1,4-dioxane. A broad scope of amines, including anilines, heterocycles, and alkyl amines with different functional groups were tested in this reaction, giving the corresponding products in moderate to excellent yields.
In 2020, Cai and collaborators developed a heterogeneous palladium-catalysed cyclocarbonylation of 2-iodoanilines with acyl chlorides using 2-MeTHF as the solvent (Fig. 74).348 Among the traditional solvents tested, the best result was achieved in THF: 95% yield of the desired product. Naturally, a green alternative to THF could be 2-MeTHF. In this solvent, the reaction was also efficient providing the desired product in 97% yield, with a great improvement in process sustainability. This strategy was successfully applied to 41 examples, obtaining a wide variety of 2-substituted 4H-3,1-benzoxazin-4-one derivatives in good to excellent yields (62–97%) from commercially easily available starting materials. The [2N-MCM-41-Pd(OAc)2] catalyst could be recovered via a simple filtration process and recycled at least 8 times without any apparent decrease in catalytic efficiency, showing negligible palladium leaching (0.38 mmol g−1 based on the ICP-AES analysis).
 | ||
| Fig. 74 Heterogeneous palladium-catalysed cyclocarbonylation of 2-iodoanilines with acyl chlorides.348 | ||
7. Conclusion and perspectives
Carbonylation reactions, which involve the addition of carbon monoxide to organic substrates in the presence of nucleophiles, represent a powerful and atom-economical way to access carbonyl-containing compounds. Catalytic carbonylation is widely used to produce both bulk and fine chemicals and is especially important for the fine chemicals industry. Hydroformylation (the “oxo process”) is a particular case of carbonylation in which molecular hydrogen is used as a nucleophile to produce aldehydes. This transformation is one of the most important homogeneously catalysed reactions applied in the chemical industry. Most of the carbonylation processes, both in academia and in industry, employ metal complex catalysts in solution; therefore, the solvent used in the process is a critical issue for its sustainability.As sustainability becomes a key concern in the chemical industry, the choice of solvent is increasingly important not only for its effect on the reaction outcome but also for its environmental and toxicological impacts. The replacement of fossil-based and hazardous solvents for environmentally friendly alternatives can lead to the development of greener and more sustainable synthetic practices in catalytic carbonylation. Numerous research works have been published on this challenging topic, most of them in the last ten years, and the present article gives an overview of this information. Hydroformylation and hydroformylation-based tandem processes, e.g., hydroaminomethylation, are a central focus; however, other carbonylation reactions (alkoxycarbonylation, aminocarbonylation, carbonylative cross-coupling, etc.) performed in green media are also addressed.
The literature discussed in this review showed that a wide range of green solvents could be efficiently used as reaction media for carbonylation reactions. The scope of these solvents includes compounds such as organic carbonates, alcohols, anisole, γ-valerolactone, 2-methyltetrahydrofuran, p-cymene, methyl ethyl ketone, and methyl tert-butyl ether. In many cases, the replacement of conventional organic solvents for greener alternatives not only improved the environmental aspects of the process but also had a beneficial chemical impact allowing for better reaction yields, higher selectivities, and milder reaction conditions. However, it should be mentioned that there is no universal green solvent that can be recommended for carbonylative transformation in general: often each specific reaction and even each specific substrate requires thorough research to find an appropriate green solvent. This is especially the case when the aim is a tandem process that combines several chemical reactions, several reagents, and sometimes several catalytic systems, which could be affected by the nature of the solvent.
Beyond direct chemical impacts, solvent effects extend to the practical and environmental aspects of the process. The volatility, toxicity, and sustainability of a solvent can determine how easily a process can be scaled up or adapted to green chemistry standards. Therefore, when selecting an appropriate solvent, it is not enough to consider only its chemical functionality or how it performs in the reaction. A comprehensive evaluation must also include the solvent's environmental impact, potential hazards, and effects on human health, which are critical for developing more sustainable and safer carbonylation processes. Addressing solvent selection more deliberately offers a largely untapped avenue for advancing both the effectiveness and sustainability of catalytic systems. Thus, paying greater attention to solvents in research and industry could unlock significant advancements in chemical innovation.
Additionally, industry practices and economic considerations heavily influence solvent selection. In many cases, the choice of solvent is driven more by cost and availability than by its optimal chemical performance. Solvents that are inexpensive, readily available, and easy to handle are often preferred in industrial processes, even if they are not the best choice for enhancing catalytic efficiency or selectivity. This cost-driven approach can overshadow the potential benefits of selecting more tailored solvents that could improve reaction outcomes or lead to more sustainable processes.
Moreover, environmental regulations and the need for greener chemistry have started to push industry to reconsider solvent choices. Nowadays, the employment of green solvents to reduce environmental damage is among the most important goal for increasing the sustainability of several chemical processes. While some progress has been made toward safer, less toxic solvents, the inertia of economic factors still slows the widespread adoption of innovative alternatives. As a result, solvents—despite their significant impact on reactions—are often chosen based on short-term practicality rather than their full potential for optimizing processes or reducing environmental impact. It should be mentioned that “drop-in” sustainable solvents, i.e., solvents that can just replace the current ones without changing the hardware of the industrial plant, have more chance of being implemented in current industrial processes. Solvents that facilitate catalyst recycling (e.g. biphasic and thermomorphic systems) are highly desirable but normally require a specific plant design.
Ensuring the sustainability of new carbonylation processes through the development of greener and/or more sustainable solvents is crucial for mitigating environmental impact and meeting the demands of a rapidly evolving global economy while maintaining environmental stewardship. By using benign solvents instead of traditional organic solvents, green carbonylation processes can minimize environmental pollution, improve safety, and enhance process efficiency providing the route to essential chemicals and products. This makes solvents indispensable not only for practical purposes but also for driving innovation in eco-friendly processes. We hope that the overview presented in this article will contribute to increasing the attractiveness of the field, inspiring further developments of sustainable catalytic carbonylation processes, and encouraging researchers to develop new reactions employing more recommended solvents from the outset.
Author contributions
F.G.D.: conceptualization, supervision, writing – original draft, writing – review & editing. L.D.A.: writing – original draft, writing – review & editing. G.M.V.: writing – original draft. E.N.S.: supervision, writing – review & editing. E.V.G.: supervision, writing – review & editing.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
CNPq, CAPES, FAPEMIG, and CNPQ-INCT-Catálise (Brazil) are gratefully acknowledged for providing financial support. F. G. D. thanks CNPQ 150340/2022-2, CAPES 88887.980151/2024-00 and 88887.980183/2024-00 for postdoctoral fellowships.References
- X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed.
- Z. Yin, J.-X. Xu and X.-F. Wu, ACS Catal., 2020, 10, 6510–6531 CrossRef CAS.
- D. Das and B. M. Bhanage, Adv. Synth. Catal., 2020, 362, 3022–3058 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS.
- Catalytic Carbonylation Reactions, ed. M. Beller, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, vol. 18 Search PubMed.
- R. M. B. Carrilho, M. J. F. Calvete, G. Mikle, L. Kollár and M. M. Pereira, Chin. J. Chem., 2024, 42, 199–221 CrossRef CAS.
- Y. Zong, R. Zhang, B. Ma, J. Peng, C. Wu, X. Zou, Y. Qian, G.-Q. Chen and X. Zhang, Sci. Adv., 2024, 10, eado9607 CrossRef CAS PubMed.
- L. Gonsalvi, A. Guerriero, E. Monflier, F. Hapiot and M. Peruzzini, in Hydroformylation for Organic Synthesis, Springer Berlin, Heidelberg, Berlin, 1st edn., 2013, pp. 1–47 Search PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- P. Kalck, C. Le Berre and P. Serp, Coord. Chem. Rev., 2020, 402, 213078 CrossRef CAS.
- R. Sang, Y. Hu, R. Razzaq, R. Jackstell, R. Franke and M. Beller, Org. Chem. Front., 2021, 8, 799–811 RSC.
- J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 CrossRef CAS PubMed.
- H. Li, K. Dong, H. Jiao, H. Neumann, R. Jackstell and M. Beller, Nat. Chem., 2016, 8, 1159–1166 CrossRef CAS PubMed.
- P. M. Maitlis, A. Haynes, G. J. Sunley and M. J. Howard, J. Chem. Soc., Dalton Trans., 1996, 2187 RSC.
- A. C. Dimian and A. A. Kiss, Chem. Eng. Technol., 2021, 44, 1792–1802 CrossRef CAS.
- J. De Tommaso and J.-L. Dubois, Polymers, 2021, 13, 2724 CrossRef CAS PubMed.
- B. Zhang, H. Yuan, Y. Liu, Z. Deng, M. Douthwaite, N. F. Dummer, R. J. Lewis, X. Liu, S. Luan, M. Dong, T. Wang, Q. Xu, Z. Zhao, H. Liu, B. Han and G. J. Hutchings, Nat. Commun., 2024, 15, 7837 CrossRef CAS PubMed.
- J.-X. Xu, C.-S. Kuai, B. Chen and X.-F. Wu, Chem. Catal., 2022, 2, 477–498 CrossRef CAS.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- C. Jiménez-González, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2004, 7, 42–50 CrossRef.
- C. S. Slater, M. J. Savelski, W. A. Carole and D. J. C. Constable, in Green Chemistry in the Pharmaceutical Industry, Wiley, 2010, pp. 49–82 Search PubMed.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC.
- W. Fang, H. Zhu, Q. Deng, S. Liu, X. Liu, Y. Shen and T. Tu, Synthesis, 2014, 46, 1689–1708 CrossRef.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS.
- G. Li, B. Wang and D. E. Resasco, Surf. Sci. Rep., 2021, 76, 100541 CrossRef CAS.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- P. M. Piccione, J. Baumeister, T. Salvesen, C. Grosjean, Y. Flores, E. Groelly, V. Murudi, A. Shyadligeri, O. Lobanova and C. Lothschütz, Org. Process Res. Dev., 2019, 23, 998–1016 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760–773 CrossRef CAS.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- M. Tobiszewski, S. Tsakovski, V. Simeonov, J. Namieśnik and F. Pena-Pereira, Green Chem., 2015, 17, 4773–4785 RSC.
- V. Isoni, L. L. Wong, H. H. Khoo, I. Halim and P. Sharratt, Green Chem., 2016, 18, 6564–6572 RSC.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS PubMed.
- E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry, Oxford University Press, Oxford, 2000 Search PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- A. Ismael, A. Gevorgyan, T. Skrydstrup and A. Bayer, Org. Process Res. Dev., 2020, 24, 2665–2675 CrossRef CAS.
- D. de Albuquerque, W. Teixeira, S. Narayanaperumal and R. Schwab, J. Braz. Chem. Soc., 2022, 33, 637–663 CAS.
- C. Árvai and L. T. Mika, Chin. J. Chem., 2024, 42, 406–429 CrossRef.
- B. Zhang, D. Peña Fuentes and A. Börner, ChemTexts, 2022, 8, 2 CrossRef.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed.
- P. Samanta and J. Canivet, ChemCatChem, 2024, 16, e202301435 CrossRef CAS.
- Y. Zhang, M. Sigrist and P. Dydio, Eur. J. Org. Chem., 2021, 2021, 5985–5997 CrossRef CAS.
- C. De, R. Saha, S. K. Ghosh, A. Ghosh, K. Mukherjee, S. S. Bhattacharyya and B. Saha, Res. Chem. Intermed., 2013, 39, 3463–3474 CrossRef CAS.
- T. Vanbésien, E. Monflier and F. Hapiot, Eur. J. Lipid Sci. Technol., 2016, 118, 26–35 CrossRef.
- E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 382–411 CrossRef CAS.
- R. Kumar and S. H. Chikkali, J. Organomet. Chem., 2022, 960, 122231 CrossRef CAS.
- S. Chakrabortty, A. A. Almasalma and J. G. de Vries, Catal. Sci. Technol., 2021, 11, 5388–5411 RSC.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed.
- F. Ungváry, Coord. Chem. Rev., 2007, 251, 2087–2102 CrossRef.
- F. Ungváry, Coord. Chem. Rev., 2007, 251, 2072–2086 CrossRef.
- A. A. Kelkar, in Industrial Catalytic Processes for Fine and Specialty Chemicals, Elsevier, 2016, pp. 663–692 Search PubMed.
- M. M. Pereira, R. M. B. Carrilho, F. M. S. Rodrigues, L. D. Dias and M. J. F. Calvete, in Catalysis for a Sustainable Environment, Wiley, 2024, pp. 301–322 Search PubMed.
- F. G. Delolo, C. Kubis, B. Zhang, H. Neumann, E. N. dos Santos, E. V. Gusevskaya and M. Beller, Catal. Sci. Technol., 2024, 14, 1524–1533 RSC.
- W. A. Beavers, US5043480, 1991.
- W. A. Beavers, US4973741, p. 1990.
- M. A. Murphy, B. L. Smith, A. Aguilo and K. D. Tau, US4873378A, p. 1987.
- J. R. Briggs, J. M. Maher and A. M. Harrison, US5210318, 1993.
- J. R. Briggs, J. M. Maher and A. M. Harrison, US5225387, 1993.
- J. R. Briggs, J. M. Maher and A. M. Harrison, US5030766, 1991.
- K. D. Tau, US5053562, 1991.
- M. A. Murphy, B. L. Smith, A. Aguilo and K. D. Tau, US4935554, 1990.
- M. A. Murphy, US4873379, 1989.
- M. A. Murphy, EP0343944, 1989.
- M. A. Murphy, A. Aguilo and B. L. Smith, EP0257967, 1987.
- W. Alsalahi and A. M. Trzeciak, J. Mol. Catal. A: Chem., 2015, 408, 147–151 CrossRef CAS.
- R. Tudor and A. Shah, Johnson Matthey Technol. Rev., 2017, 61, 246–256 CrossRef CAS.
- D. F. Foster, D. J. Adams, D. Gudmunsen, A. M. Stuart, E. G. Hope and D. J. Hamilton, Chem. Commun., 2002, 722–723 RSC.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72–75 CrossRef PubMed.
- M. Ali, A. Gual, G. Ebeling and J. Dupont, ChemCatChem, 2014, 6, 2224–2228 CrossRef CAS.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- L. Tao, M. Zhong, J. Chen, S. Jayakumar, L. Liu, H. Li and Q. Yang, Green Chem., 2018, 20, 188–196 RSC.
- D. Koch and W. Leitner, J. Am. Chem. Soc., 1998, 120, 13398–13404 CrossRef CAS.
- J. Bianga, K. U. Künnemann, T. Gaide, A. J. Vorholt, T. Seidensticker, J. M. Dreimann and D. Vogt, Chem. – Eur. J., 2019, 25, 11586–11608 CrossRef CAS PubMed.
- A. Behr, D. Obst and B. Turkowski, J. Mol. Catal. A: Chem., 2005, 226, 215–219 CrossRef CAS.
- E. Schäfer, Y. Brunsch, G. Sadowski and A. Behr, Ind. Eng. Chem. Res., 2012, 51, 10296–10306 CrossRef.
- G. Kiedorf, D. M. Hoang, A. Müller, A. Jörke, J. Markert, H. Arellano-Garcia, A. Seidel-Morgenstern and C. Hamel, Chem. Eng. Sci., 2014, 115, 31–48 CrossRef CAS.
- A. Jörke, T. Gaide, A. Behr, A. Vorholt, A. Seidel-Morgenstern and C. Hamel, Chem. Eng. J., 2017, 313, 382–397 CrossRef.
- W. Alsalahi and A. M. Trzeciak, Coord. Chem. Rev., 2021, 430, 213732 CrossRef CAS.
- R. M. Deshpande, S. S. Divekar, B. M. Bhanage and R. V. Chaudhari, J. Mol. Catal., 1992, 77, L13–L17 CrossRef CAS.
- L. Monnereau, D. Sémeril and D. Matt, Eur. J. Org. Chem., 2010, 2010, 3068–3073 CrossRef.
- M. N. Shaikh, M. A. Aziz, A. Helal, M. Bououdina, Z. H. Yamani and T.-J. Kim, RSC Adv., 2016, 6, 41687–41695 RSC.
- Z. Wang and Y. Yang, RSC Adv., 2020, 10, 29263–29267 RSC.
- P. Wang, H. Shi, B. Feng, D. Zhao and D. Yang, Mol. Catal., 2023, 548, 113459 CrossRef CAS.
- Z. Yu, S. Zhang, L. Zhang, X. Liu, Z. Jia, L. Li, N. Ta, A. Wang, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 11955–11967 CrossRef CAS PubMed.
- S. Tian, L. Shi, J. Zhu, L. Li, F. Ye, Z. Xu and L. Xu, Eur. J. Org. Chem., 2025, e202500020 CrossRef.
- P. Neubert, S. Fuchs and A. Behr, Green Chem., 2015, 17, 4045–4052 RSC.
- P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016, 18, 842–847 RSC.
- P. Pongrácz, B. Bartal, L. Kollár and L. T. Mika, J. Organomet. Chem., 2017, 847, 140–145 CrossRef.
- K. C. B. Oliveira, A. de Camargo Faria, A. C. Monteiro, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2016, 523, 139–145 CrossRef CAS.
- A. de Camargo Faria, K. C. B. Oliveira, A. C. Monteiro, E. N. dos Santos and E. V. Gusevskaya, Catal. Today, 2020, 344, 24–31 CrossRef CAS.
- F. G. Delolo, K. C. B. Oliveira, E. N. Santos and E. V. Gusevskaya, Mol. Catal., 2019, 462, 1–9 CrossRef CAS.
- F. G. Delolo, E. N. dos Santos and E. V. Gusevskaya, Green Chem., 2019, 21, 1091–1098 RSC.
- A. de C. Faria, M. P. de Oliveira, A. C. Monteiro, R. L. V. Mota, K. C. B. Oliveira, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2020, 591, 117406 CrossRef.
- M. P. de Oliveira, F. G. Delolo, J. A. A. Villarreal, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2021, 616, 118082 CrossRef CAS.
- F. G. Delolo, G. M. Vieira, J. A. A. Villarreal, E. N. dos Santos and E. V. Gusevskaya, Catal. Today, 2021, 381, 272–279 CrossRef CAS.
- F. G. Delolo, G. M. Vieira, J. A. Avendaño-Villarreal, A. de Oliveira Dias, E. N. dos Santos and E. V. Gusevskaya, J. Catal., 2023, 421, 20–29 CrossRef CAS.
- F. G. Delolo, T. P. Moreira, A. de O. Dias, E. N. dos Santos and E. V. Gusevskaya, J. Catal., 2024, 432, 115437 CrossRef CAS.
- F. G. Delolo, J. Yang, H. Neumann, E. N. dos Santos, E. V. Gusevskaya and M. Beller, ACS Sustainable Chem. Eng., 2021, 9, 5148–5154 CrossRef CAS.
- X. Kang, L. Mao, J. Shi, Y. Liu, B. Zhai, J. Xu, Y. Jiang, E. Lichtfouse, H. Jin and L. Guo, Environ. Chem. Lett., 2024, 22, 815–839 CrossRef CAS.
- J. Peach and J. Eastoe, Beilstein J. Org. Chem., 2014, 10, 1878–1895 CrossRef PubMed.
- H. Jin and B. Subramaniam, Chem. Eng. Sci., 2004, 59, 4887–4893 CrossRef CAS.
- A. Olmos, G. Asensio and P. J. Pérez, ACS Catal., 2016, 6, 4265–4280 CrossRef CAS.
- A. Behr, D. Obst and B. Turkowski, J. Mol. Catal. A: Chem., 2005, 226, 215–219 CrossRef CAS.
- M. S. Shaharun, B. K. Dutta, H. Mukhtar and S. Maitra, Chem. Eng. Sci., 2010, 65, 273–281 CrossRef CAS.
- J. T. Vossen, W. Leitner and A. J. Vorholt, ACS Sustainable Chem. Eng., 2023, 11, 10462–10470 CrossRef CAS.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- J. Hibbel, E. Wiebus and B. Cornils, Chem. Ing. Tech., 2013, 85, 1853–1871 CrossRef CAS.
- P. J. Baricelli, J. C. Pereira and M. Rosales, Catal. Today, 2025, 443, 114969 CrossRef CAS.
- K. E. Naße, F. S. Heinen, N. Pawlowsky, M. Schrimpf, E. Monflier, S. Tilloy, W. Leitner and A. J. Vorholt, Chem. Eng. J., 2024, 497, 154114 CrossRef.
- M. Ferreira, H. Bricout, T. F. H. Roth, T. Seidensticker, S. Tilloy and E. Monflier, Catal. Today, 2024, 442, 114951 CrossRef CAS.
- C. G. Vieira, M. C. de Freitas, K. C. B. de Oliveira, A. de Camargo Faria, E. N. dos Santos and E. V. Gusevskaya, Catal. Sci. Technol., 2015, 5, 960–966 RSC.
- N. Herrmann, J. Bianga, T. Gaide, M. Drewing, D. Vogt and T. Seidensticker, Green Chem., 2019, 21, 6738–6745 RSC.
- J. Bianga, N. Herrmann, L. Schurm, T. Gaide, J. M. Dreimann, D. Vogt and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2019, 122, 1900317 CrossRef.
- N. Herrmann, J. Bianga, M. Palten, T. Riemer, D. Vogt, J. M. Dreimann and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2020, 122, 1900166 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- W. Alsalahi and A. M. Trzeciak, RSC Adv., 2014, 4, 30384–30391 RSC.
- M. Beller and J. G. E. Krauter, J. Mol. Catal. A: Chem., 1999, 143, 31–39 CrossRef CAS.
- A. A. Dabbawala, D. U. Parmar, H. C. Bajaj and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 282, 99–106 CrossRef CAS.
- F. Migliorini, F. Dei, M. Calamante, S. Maramai and E. Petricci, ChemCatChem, 2021, 13, 2794–2806 CrossRef CAS.
- F. G. Delolo, E. N. dos Santos and E. V. Gusevskaya, ChemCatChem, 2024, 16, e202400273 CrossRef CAS.
- H. Liu, L. Liu, W.-D. Guo, Y. Lu, X.-L. Zhao and Y. Liu, J. Catal., 2019, 373, 215–221 CrossRef CAS.
- H. Liu, Y.-X. Yao, H.-Y. Shang, D. Yang and Y.-Y. Tian, React. Chem. Eng., 2023, 8, 778–783 RSC.
- J. Norinder, C. Rodrigues and A. Börner, J. Mol. Catal. A: Chem., 2014, 391, 139–143 CrossRef CAS.
- C. Rodrigues, F. G. Delolo, J. Norinder, A. Börner, A. L. Bogado and A. A. Batista, J. Mol. Catal. A: Chem., 2017, 426, 586–592 CrossRef CAS.
- P. Wang, D.-L. Wang, H. Liu, X.-L. Zhao, Y. Lu and Y. Liu, Organometallics, 2017, 36, 2404–2411 CrossRef CAS.
- Y.-Q. Li, P. Wang, H. Liu, Y. Lu, X.-L. Zhao and Y. Liu, Green Chem., 2016, 18, 1798–1806 RSC.
- C. G. Vieira, J. G. da Silva, C. A. A. Penna, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2010, 380, 125–132 CrossRef CAS.
- M. C. de Freitas, C. G. Vieira, E. N. dos Santos and E. V. Gusevskaya, ChemCatChem, 2013, 5, 1884–1890 CrossRef CAS.
- C. G. Vieira, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2013, 466, 208–215 CrossRef CAS.
- G. A. Carvalho, E. V. Gusevskaya and E. N. dos Santos, J. Braz. Chem. Soc., 2014, 25, 2370–2377 CAS.
- S. R. Khan and B. M. Bhanage, Tetrahedron Lett., 2013, 54, 5998–6001 CrossRef CAS.
- Y.-Q. Li, P. Wang, H. Liu, Y. Lu, X.-L. Zhao and Y. Liu, Green Chem., 2016, 18, 1798–1806 RSC.
- P. Wang, H. Liu, Y.-Q. Li, X.-L. Zhao, Y. Lu and Y. Liu, Catal. Sci. Technol., 2016, 6, 3854–3861 RSC.
- D. Gorbunov, M. Nenasheva, A. Gorbunov, R. Matsukevich, A. Maximov and E. Karakhanov, React. Chem. Eng., 2021, 6, 839–844 RSC.
- Z. Shu, X.-X. Zhao, Z. Zheng, X. Zhang, Y. Zhang, S. Sun, J. Chen, C. Xie, B. Yuan and X. Jia, Chem. Commun., 2023, 59, 5237–5240 RSC.
- Z. Shu, X. Zhang, W. Meng, Z. Yu, Y. Zhou, C. Chen, B. Yuan and X. Jia, ACS Sustainable Chem. Eng., 2024, 12, 2430–2438 CrossRef CAS.
- H. Liu, Y.-X. Yao, H.-Y. Shang, D. Yang and Y.-Y. Tian, React. Chem. Eng., 2023, 8, 778–783 RSC.
- A. J. Sandee, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468–8476 CrossRef CAS PubMed.
- Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, ed. A. Börner and R. Franke, Wiley, 2016 Search PubMed.
- T. Ichihara, K. Nakano, M. Katayama and K. Nozaki, Chem. – Asian J., 2008, 3, 1722–1728 CrossRef CAS PubMed.
- D. Gorbunov, M. Nenasheva, E. Naranov, A. Maximov, E. Rosenberg and E. Karakhanov, Appl. Catal., A, 2021, 623, 118266 CrossRef CAS.
- D. Gorbunov, M. Nenasheva, A. Maximov and E. Karakhanov, Appl. Catal., A, 2024, 670, 119538 CrossRef CAS.
- E. N. dos Santos, C. U. Pittman and H. Toghiani, J. Mol. Catal., 1993, 83, 51–65 CrossRef CAS.
- Y. Ma, S. Qing, Z. Gao, X. Mamat, J. Zhang, H. Li, W. Eli and T. Wang, Catal. Sci. Technol., 2015, 5, 3649–3657 RSC.
- C. O. Oseghale, B. M. Mogudi, C. A. Akinnawo and R. Meijboom, Appl. Catal., A, 2020, 602, 117735 CrossRef CAS.
- C. S. MacNeil, L. N. Mendelsohn, T. P. Pabst, G. Hierlmeier and P. J. Chirik, J. Am. Chem. Soc., 2022, 144, 19219–19224 CrossRef CAS PubMed.
- T. Li, S. Fan, X. Gao, J. Zhang, Q. Ma and T.-S. Zhao, Fuel, 2024, 365, 131192 CrossRef CAS.
- L. Wu, I. Fleischer, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306–14312 CrossRef CAS PubMed.
- I. Fleischer, K. M. Dyballa, R. Jennerjahn, R. Jackstell, R. Franke, A. Spannenberg and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2949–2953 CrossRef CAS PubMed.
- Q. Chen, X. Kang, X. Zhang, Y. Cao and L. He, J. Org. Chem., 2023, 88, 5044–5051 CrossRef CAS PubMed.
- H. Zhou, Y. Sun, L. Deng, W. Yang and Y. Li, J. Organomet. Chem., 2024, 1004, 122927 CrossRef CAS.
- Y. Zhu, Z. Wang, Y. Zhao, X. Zhou, Y. Zhang and Y. Yang, ACS Catal., 2024, 14, 4593–4600 CrossRef CAS.
- D. Konya, K. Q. Almeida Leñero and E. Drent, Organometallics, 2006, 25, 3166–3174 CrossRef CAS.
- W. Huang, X. Tian, H. Jiao, R. Jackstell and M. Beller, Chem. – Eur. J., 2022, 28, e202104012 CrossRef CAS PubMed.
- K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746–18757 CrossRef CAS PubMed.
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393–17400 CrossRef CAS PubMed.
- D. Fuchs, G. Rousseau, L. Diab, U. Gellrich and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 2178–2182 CrossRef CAS PubMed.
- O. Diebolt, C. Müller and D. Vogt, Catal. Sci. Technol., 2012, 2, 773 RSC.
- M. R. L. Furst, V. Korkmaz, T. Gaide, T. Seidensticker, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 4319–4323 CrossRef CAS.
- C. Becquet, F. Berche, H. Bricout, E. Monflier and S. Tilloy, ACS Sustainable Chem. Eng., 2021, 9, 9444–9454 CrossRef CAS.
- H. Shenouda and E. J. Alexanian, Org. Lett., 2019, 21, 9268–9271 CrossRef CAS PubMed.
- A. El Mouat, W. Abdallah, J. Ternel, M. Ferreira, H. Bricout, A. J. Vorholt, H. Stieber, S. Stoertte, E. Monflier, M. Lahcini and S. Tilloy, ChemSusChem, 2025, 18, e202401384 CrossRef CAS PubMed.
- A. Serrano-Maldonado, T. Dang-Bao, I. Favier, I. Guerrero-Ríos, D. Pla and M. Gómez, Chem. – Eur. J., 2020, 26, 12553–12559 CrossRef CAS PubMed.
- A. Weber, L. Porthun and R. Schomäcker, Catalysts, 2022, 12, 773 CrossRef CAS.
- P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3366 CrossRef CAS PubMed.
- I. Tkatchenko, F. G. A. Stone and E. W. Abel, in Comprehensive Organometallic Chemistry: Synthesis, Pergamon, Oxford, UK, 1982, pp. 101–223 Search PubMed.
- S. Fuchs, T. Rösler, B. Grabe, A. Kampwerth, G. Meier, H. Strutz, A. Behr and A. J. Vorholt, Appl. Catal., A, 2018, 550, 198–205 CrossRef CAS.
- A. Cunillera, A. Ruiz and C. Godard, Adv. Synth. Catal., 2019, 361, 4201–4207 CrossRef CAS.
- A. Cunillera, M. D. de los Bernardos, M. Urrutigoïty, C. Claver, A. Ruiz and C. Godard, Catal. Sci. Technol., 2020, 10, 630–634 RSC.
- C. G. Arena, Curr. Org. Chem., 2021, 25, 1831–1852 CrossRef CAS.
- C. Chen, X.-Q. Dong and X. Zhang, Org. Chem. Front., 2016, 3, 1359–1370 RSC.
- A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC.
- S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179 RSC.
- D. Crozet, M. Urrutigoïty and P. Kalck, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- K. Cousin, F. Hapiot and E. Monflier, Eur. J. Lipid Sci. Technol., 2020, 122, 1900131 CrossRef.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989–3996 CrossRef CAS PubMed.
- V. K. Srivastava and P. Eilbracht, Catal. Commun., 2009, 10, 1791–1795 CrossRef CAS.
- S. Gülak, L. Wu, Q. Liu, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 7320–7323 CrossRef PubMed.
- M. Ali, A. Gual, G. Ebeling and J. Dupont, ChemSusChem, 2016, 9, 2129–2134 CrossRef CAS PubMed.
- J. Liu, C. Kubis, R. Franke, R. Jackstell and M. Beller, ACS Catal., 2016, 6, 907–912 CrossRef CAS.
- D. Yang, C.-K. Zhong, L.-Y. Tian, L.-L. Zhang and H. Liu, J. Catal., 2023, 428, 115132 CrossRef CAS.
- C. Qian, Q. Zheng, J. Ji, G. Liang, B. Tu and T. Tu, Chem. Catal., 2023, 3, 100787 CrossRef CAS.
- P. Wang, X. Tian, H. Shi, B. Feng and D. Zhao, J. Organomet. Chem., 2024, 1007, 123027 CrossRef CAS.
- J. Yang, F. G. Delolo, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202112597 CrossRef CAS PubMed.
- M. S. Faculak, A. M. Veatch and E. J. Alexanian, Science, 2024, 383, 77–81 CrossRef CAS PubMed.
- Y. Yuan, Y. Zhang, W. Li, Y. Zhao and X. Wu, Angew. Chem., Int. Ed., 2023, 62, e202309993 CrossRef CAS PubMed.
- M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef CAS PubMed.
- G. Liu, K. Huang, C. Cai, B. Cao, M. Chang, W. Wu and X. Zhang, Chem. – Eur. J., 2011, 17, 14559–14563 CrossRef CAS PubMed.
- S. Li, K. Huang, J. Zhang, W. Wu and X. Zhang, Org. Lett., 2013, 15, 3078–3081 CrossRef CAS PubMed.
- Z. Zheng and L. Wang, Synthesis, 2019, 51, 1585–1594 CrossRef CAS.
- S. Fuchs, M. Steffen, A. Dobrowolski, T. Rösler, L. Johnen, G. Meier, H. Strutz, A. Behr and A. J. Vorholt, Catal. Sci. Technol., 2017, 7, 5120–5127 RSC.
- T. Vanbésien, E. Monflier and F. Hapiot, Green Chem., 2017, 19, 1940–1948 RSC.
- A. de Oliveira Dias, M. G. P. Gutiérrez, J. A. A. Villarreal, R. L. L. Carmo, K. C. B. Oliveira, A. G. Santos, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2019, 574, 97–104 CrossRef CAS.
- H. Liu, D. Yang, Y. Yao, Y. Xu, H. Shang and X. Lin, Mol. Catal., 2020, 485, 110843 CrossRef CAS.
- B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2372–2375 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, M. Zhang, Q. Liu, R. Franke, R. Jackstell and M. Beller, ChemSusChem, 2014, 7, 3260–3263 CrossRef CAS PubMed.
- T. A. Faßbach, F. O. Sommer and A. J. Vorholt, Adv. Synth. Catal., 2018, 360, 1473–1482 CrossRef.
- K. Wittmann, W. Wisniewski, R. Mynott, W. Leitner, C. L. Kranemann, T. Rische, P. Eilbracht, S. Kluwer, J. M. Ernsting and C. J. Elsevier, Chem. – Eur. J., 2001, 7, 4584–4589 CrossRef CAS PubMed.
- B. Hamers, P. S. Bäuerlein, C. Müller and D. Vogt, Adv. Synth. Catal., 2008, 350, 332–342 CrossRef CAS.
- B. Hamers, E. Kosciusko-Morizet, C. Müller and D. Vogt, ChemCatChem, 2009, 1, 103–106 CrossRef CAS.
- A. Behr and R. Roll, J. Mol. Catal. A: Chem., 2005, 239, 180–184 CrossRef CAS.
- A. Behr, A. Kleyensteiber and M. Becker, Chem. Eng. Process. Process Intensif., 2013, 69, 15–23 CrossRef CAS.
- J. Bianga, K. U. Künnemann, L. Goclik, L. Schurm, D. Vogt and T. Seidensticker, ACS Catal., 2020, 10, 6463–6472 CrossRef CAS.
- E. Monciatti, F. Migliorini, G. Romagnoli, M. Vogt, R. Langer, M. L. Parisi and E. Petricci, ACS Sustainable Chem. Eng., 2023, 11, 13387–13397 CrossRef CAS.
- Z. Nairoukh and J. Blum, J. Org. Chem., 2014, 79, 2397–2403 CrossRef CAS PubMed.
- R. Sang, Y. Hu, R. Razzaq, R. Jackstell, R. Franke and M. Beller, Org. Chem. Front., 2021, 8, 799–811 RSC.
- A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef.
- G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed.
- F. Bertoux, E. Monflier, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 1999, 143, 11–22 CrossRef CAS.
- R. Dühren, P. Kucmierczyk, R. Jackstell, R. Franke and M. Beller, Catal. Sci. Technol., 2021, 11, 2026–2030 RSC.
- P. Kalck and M. Urrutigoïty, Inorg. Chim. Acta, 2015, 431, 110–121 CrossRef CAS.
- T.-H. Jing, Y.-Y. Zhuang, X.-X. Zhang, J.-G. Qian, X.-L. Zhao, Y. Lu, H.-J. Wang and Y. Liu, J. Catal., 2024, 432, 115406 CrossRef CAS.
- B. Wang, C. Shen and K. Dong, Org. Lett., 2024, 26, 3628–3633 CrossRef CAS PubMed.
- J. Mehara, M. Anania, P. Kočovský and J. Roithová, ACS Catal., 2024, 14, 5710–5719 CrossRef CAS PubMed.
- N. Liu, X. Wu, C. Wang, J. Qu and Y. Chen, Chem. Commun., 2022, 58, 4643–4646 RSC.
- S. J. Ton, K. T. Neumann, P. Nørby and T. Skrydstrup, J. Am. Chem. Soc., 2021, 143, 17816–17824 CrossRef CAS PubMed.
- X. Chen, G. Chen and Z. Lian, Chin. J. Chem., 2024, 42, 177–189 CrossRef CAS.
- F. Ziyanak, M. Kuş and L. Artok, Adv. Synth. Catal., 2011, 353, 897–902 CrossRef CAS.
- A. A. Seni, L. Kollár, L. T. Mika and P. Pongrácz, Mol. Catal., 2018, 457, 67–73 CrossRef CAS.
- P. Wang, Y. Wang, H. Neumann and M. Beller, Chem. Sci., 2022, 13, 13459–13465 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, ChemCatChem, 2014, 6, 2805–2809 CrossRef CAS.
- H.-J. Ai, Y. Yuan and X.-F. Wu, Chem. Sci., 2022, 13, 2481–2486 RSC.
- X. Zhang, C. Shen, C. Xia, X. Tian and L. He, Green Chem., 2018, 20, 5533–5539 RSC.
- L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5, 3091 CrossRef PubMed.
- M. Luo, Z. Liu, H. Chen, H. Fu, R. Li and X. Zheng, J. Catal., 2024, 433, 115459 CrossRef CAS.
- P. Yang, Y.-H. Zhao and X.-F. Wu, Org. Chem. Front., 2024, 11, 2462–2467 RSC.
- Y. Gao, W. Lu, P. Liu and P. Sun, J. Org. Chem., 2016, 81, 2482–2487 Search PubMed.
- K. Zhao, H. Wang, T. Li, S. Liu, E. Benassi, X. Li, Y. Yao, X. Wang, X. Cui and F. Shi, Nat. Commun., 2024, 15, 2016 Search PubMed.
- K. Dong, R. Sang, Z. Wei, J. Liu, R. Dühren, A. Spannenberg, H. Jiao, H. Neumann, R. Jackstell, R. Franke and M. Beller, Chem. Sci., 2018, 9, 2510–2516 RSC.
- K. Dong, X. Fang, S. Gülak, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Nat. Commun., 2017, 8, 14117 CrossRef PubMed.
- K. Dong, R. Sang, X. Fang, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 5267–5271 CrossRef CAS PubMed.
- R. I. Pugh, P. G. Pringle and E. Drent, Chem. Commun., 2001, 1476–1477 RSC.
- R. I. Pugh and E. Drent, Adv. Synth. Catal., 2002, 344, 837–840 Search PubMed.
- J. G. Knight, S. Doherty, A. Harriman, E. G. Robins, M. Betham, G. R. Eastham, R. P. Tooze, M. R. J. Elsegood, P. Champkin and W. Clegg, Organometallics, 2000, 19, 4957–4967 CrossRef CAS.
- J. Vondran, M. R. L. Furst, G. R. Eastham, T. Seidensticker and D. J. Cole-Hamilton, Chem. Rev., 2021, 121, 6610–6653 Search PubMed.
- C. J. Rodriguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720 RSC.
- S. El Houssame, L. El Firdoussi, S. Allaoud, A. Karim, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 2001, 168, 15–23 CrossRef.
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102–7107 CrossRef CAS PubMed.
- Y. Yamamoto, Adv. Synth. Catal., 2010, 352, 478–492 Search PubMed.
- T. M. Konrad, J. A. Fuentes, A. M. Z. Slawin and M. L. Clarke, Angew. Chem., Int. Ed., 2010, 49, 9197–9200 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- P. H. Gehrtz, V. Hirschbeck and I. Fleischer, Chem. Commun., 2015, 51, 12574–12577 RSC.
- Y. Tan, J. Lang, M. Tang, J. Li, P. Mi and X. Zheng, ChemistrySelect, 2021, 6, 2343–2349 Search PubMed.
- P. Gautam, P. Kathe and B. M. Bhanage, Green Chem., 2017, 19, 823–830 Search PubMed.
- R. Sang, P. Kucmierczyk, R. Dühren, R. Razzaq, K. Dong, J. Liu, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 14365–14373 Search PubMed.
- J. Yang, J. Liu, Y. Ge, W. Huang, F. Ferretti, H. Neumann, H. Jiao, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 9527–9533 CrossRef CAS PubMed.
- C. M. McMahon, M. S. Renn and E. J. Alexanian, Org. Lett., 2016, 18, 4148–4150 CrossRef CAS PubMed.
- B. T. Sargent and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 7520–7523 Search PubMed.
- Y. Yao, X. Zou, Y. Wang, H. Yang, Z. Ren and Z. Guan, Angew. Chem., Int. Ed., 2021, 60, 23117–23122 CrossRef CAS PubMed.
- Y. Li, Z. Wang and X.-F. Wu, Green Chem., 2018, 20, 969–972 RSC.
- X. Qi, M. Lai and X.-F. Wu, Org. Chem. Front., 2019, 6, 3397–3400 RSC.
- T. Fujihara and Y. Tsuji, J. Jpn. Pet. Inst., 2018, 61, 1–9 CrossRef CAS.
- V. Goldbach, L. Falivene, L. Caporaso, L. Cavallo and S. Mecking, ACS Catal., 2016, 6, 8229–8238 CrossRef CAS.
- D. Liu, T. Ru, Z. Deng, L. Zhang, Y. Ning and F.-E. Chen, ACS Catal., 2023, 13, 12868–12876 CrossRef CAS.
- J. Lv, L. Zong, J. Zhang, J. Song, J. Zhao, K. Zhang, Z. Zhou, M. Gao, C. Xie, X. Jia and X. Ren, Catal. Sci. Technol., 2021, 11, 4708–4713 RSC.
- Y. Liu, D. Yang, G. Zeng, K. Li, S. Wei, H. Li, M. Ji and R. Liu, ChemistrySelect, 2023, 8, e202303191 CrossRef CAS.
- F. Messa, A. N. Paparella, D. Veselý, J. Krajčovič, P. Papadia, S. Perrone and A. Salomone, Eur. J. Org. Chem., 2023, 26, e202300309 CrossRef CAS.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287–293 CrossRef CAS.
- S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986–38999 RSC.
- A. R. Hajipour, Z. Tavangar-Rizi and N. Iranpoor, RSC Adv., 2016, 6, 78468–78476 RSC.
- S. Liu, T. Li, F. Shi, H. Ma, B. Wang, X. Dai and X. Cui, Nat. Commun., 2023, 14, 4973 CrossRef CAS PubMed.
- M. Amézquita-Valencia, G. Achonduh and H. Alper, J. Org. Chem., 2015, 80, 6419–6424 CrossRef PubMed.
- J. M. Tukacs, B. Marton, E. Albert, I. Tóth and L. T. Mika, J. Organomet. Chem., 2020, 923, 121407 CrossRef CAS.
- W. Huang, R. Jackstell, R. Franke and M. Beller, Chem. Commun., 2023, 59, 9505–9508 RSC.
- W. Huang, R. Jackstell, A. Spannenberg and M. Beller, Catal. Sci. Technol., 2023, 13, 2475–2479 RSC.
- J. Li, Y. Min and Y. Shi, New J. Chem., 2024, 48, 17381–17384 RSC.
- H. Shi, S. Jia, M. Dong, H. Wu, Z. Huang and K. Dong, Org. Lett., 2024, 26, 8982–8987 CrossRef CAS PubMed.
- C. M. Abdulla Afsina, R. M. Philip, P. V. Saranya and G. Anilkumar, Curr. Org. Synth., 2023, 20, 308–331 CrossRef CAS PubMed.
- G. Mei, Y. Lu, X. Yang, S. Chen, X. Yang, L. Yang, C. Tang, Y. Sun, B. Y. Xia and B. You, Angew. Chem., Int. Ed., 2024, 63, e202314708 CrossRef CAS PubMed.
- V. P. Mahajan, Y. A. Kolekar and B. M. Bhanage, Mol. Catal., 2024, 561, 114167 CrossRef CAS.
- Q.-C. Shan, Y. Zhao, Y. Wu, H.-F. Liu, X.-H. Duan and L.-N. Guo, Sci. China: Chem., 2024, 67, 3798–3806 CrossRef CAS.
- Y.-H. Zhao, X.-W. Gu and X.-F. Wu, Org. Chem. Front., 2024, 11, 442–447 RSC.
- T. Seidensticker, M. R. L. Furst, R. Frauenlob, J. Vondran, E. Paetzold, U. Kragl and A. J. Vorholt, ChemCatChem, 2015, 7, 4085–4090 CrossRef CAS.
- Shaifali, P. Sharma, P. Mehara and P. Das, Chem. – Asian J., 2023, 18, e202201288 CrossRef CAS PubMed.
- P. Halder, V. Talukdar, A. Iqubal and P. Das, J. Org. Chem., 2022, 87, 13965–13979 CrossRef CAS PubMed.
- Shaifali, Sheetal, R. Bains, A. Kumar, S. Ram and P. Das, Org. Biomol. Chem., 2020, 18, 7193–7200 RSC.
- D. Marosvölgyi-Haskó, B. Lengyel, J. M. Tukacs, L. Kollár and L. T. Mika, ChemPlusChem, 2016, 81, 1224–1229 CrossRef PubMed.
- E. Calcio Gaudino, S. Tagliapietra, G. Palmisano, K. Martina, D. Carnaroglio and G. Cravotto, ACS Sustainable Chem. Eng., 2017, 5, 9233–9243 CrossRef CAS.
- N. Uzunlu, P. Pongrácz, L. Kollár and A. Takács, Molecules, 2023, 28, 442 CrossRef CAS PubMed.
- Z.-P. Bao, R.-G. Miao, X. Qi and X.-F. Wu, Chem. Commun., 2021, 57, 1955–1958 RSC.
- T. L. Andersen, W. Caneschi, A. Ayoub, A. T. Lindhardt, M. R. C. Couri and T. Skrydstrup, Adv. Synth. Catal., 2014, 356, 3074–3082 CrossRef CAS.
- P. Tambade, Y. Patil, M. Bhanushali and B. Bhanage, Synthesis, 2008, 2008, 2347–2352 CrossRef.
- J. C. Caravez, M. J. Wong, R. D. Kavthe, B. S. Takale and B. H. Lipshutz, ACS Catal., 2023, 13, 12383–12390 CrossRef CAS.
- D. Y. de Albuquerque, J. R. de Moraes and R. S. Schwab, Eur. J. Org. Chem., 2019, 2019, 6673–6681 CrossRef CAS.
- N. Hussain, A. K. Chhalodia, A. Ahmed and D. Mukherjee, ChemistrySelect, 2020, 5, 11272–11290 CrossRef CAS.
- J. Cao, Z.-J. Zheng, Z. Xu and L.-W. Xu, Coord. Chem. Rev., 2017, 336, 43–53 CrossRef CAS.
- M. Papp, P. Szabó, D. Srankó and R. Skoda-Földes, RSC Adv., 2016, 6, 45349–45356 Search PubMed.
- Q. Hu, L. Wang, C. Wang, Y. Wu, Z. Ding and R. Yuan, RSC Adv., 2017, 7, 37200–37207 RSC.
- B. T. Sargent and E. J. Alexanian, Angew. Chem., Int. Ed., 2019, 58, 9533–9536 CrossRef CAS PubMed.
- A. M. Veatch and E. J. Alexanian, Chem. Sci., 2020, 11, 7210–7213 RSC.
- A. M. Veatch, S. Liu and E. J. Alexanian, Angew. Chem., Int. Ed., 2022, 61, e202210772 CrossRef CAS PubMed.
- H.-J. Ai, F. Zhao, H.-Q. Geng and X.-F. Wu, ACS Catal., 2021, 11, 3614–3619 CrossRef CAS.
- X. Wang, B. Wang, X. Yin, W. Yu, Y. Liao, J. Ye, M. Wang, L. Hu and J. Liao, Angew. Chem., Int. Ed., 2019, 58, 12264–12270 CrossRef CAS PubMed.
- W. Mai, Y. Zhao, M. Lv, W. Zhang, Y. Xiao, J. Yuan and L. Yang, Eur. J. Org. Chem., 2024, 27, e202400026 CrossRef CAS.
- Y. Xiao, W. Lv, J. Yuan, L. Yang, P. Mao and W. Mai, ChemistrySelect, 2022, 7, e202200914 CrossRef CAS.
- S.-Z. Cai, R. Yu, C. Li, H. Zhong, X. Dong, B. Morandi, J. Ye and X. Fang, Org. Lett., 2023, 25, 8683–8687 CrossRef CAS PubMed.
- H. Ai, W. Lu and X. Wu, Angew. Chem., Int. Ed., 2021, 60, 17178–17184 CrossRef CAS PubMed.
- V. Hirschbeck, P. H. Gehrtz and I. Fleischer, Chem. – Eur. J., 2018, 24, 7092–7107 CrossRef CAS PubMed.
- J. Khayitov, Y. T. Rakhmonovich, B. F. Eshburiyevich, I. B. Mahmudjanovich, R. Khusniddin, U. B. Ziyadullayevich, A. N. Maxmudovna and K. E. Yunusov, Chem. Rev. Lett., 2024, 7, 333–345 CAS.
- X. Qi, Z.-P. Bao, X.-T. Yao and X.-F. Wu, Org. Lett., 2020, 22, 6671–6676 CrossRef CAS PubMed.
- M. Li, G. Zhao, S. Xu, D. Miao, S. Li, Y. Qiu, D. Chen, Z. Quan, X. Wang and Y. Liang, Adv. Synth. Catal., 2024, 366, 3367–3371 CrossRef CAS.
- X.-F. Pan, P. Ji, D. Wen, X. Qi and X.-F. Wu, J. Catal., 2024, 437, 115681 CrossRef CAS.
- H. Wang, C. Li, Y. Li, J. Chen, S. Liu and Y. Li, Org. Chem. Front., 2024, 11, 1322–1331 RSC.
- M. N. Burhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2013, 15, 948–951 CrossRef CAS PubMed.
- M. N. Burhardt, A. Ahlburg and T. Skrydstrup, J. Org. Chem., 2014, 79, 11830–11840 CrossRef CAS PubMed.
- H. Cao, L. McNamee and H. Alper, J. Org. Chem., 2008, 73, 3530–3534 CrossRef CAS PubMed.
- T. R. Kégl, L. T. Mika and T. Kégl, Molecules, 2022, 27, 460 CrossRef PubMed.
- P. Hu and M. Kazemi, J. Coord. Chem., 2024, 77, 1324–1348 Search PubMed.
- M. Beller and X.-F. Wu, in Transition Metal Catalyzed Carbonylation Reactions, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 95–114 Search PubMed.
- J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed.
- B. S. Kadu, Catal. Sci. Technol., 2021, 11, 1186–1221 RSC.
- D. Bhattacherjee, M. Rahman, S. Ghosh, A. K. Bagdi, G. V. Zyryanov, O. N. Chupakhin, P. Das and A. Hajra, Adv. Synth. Catal., 2021, 363, 1597–1624 CrossRef CAS.
- B. T. Sargent and E. J. Alexanian, J. Am. Chem. Soc., 2017, 139, 12438–12440 CrossRef CAS PubMed.
- L. A. Aronica, Inorg. Chim. Acta, 2023, 558, 121763 CrossRef CAS.
- M. A. Andrade and L. M. D. R. S. Martins, Molecules, 2020, 25, 5506 CrossRef CAS PubMed.
- P. Gautam and B. M. Bhanage, ChemistrySelect, 2016, 1, 5463–5470 Search PubMed.
- T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS.
- A. Ahlburg, A. T. Lindhardt, R. H. Taaning, A. E. Modvig and T. Skrydstrup, J. Org. Chem., 2013, 78, 10310–10318 CrossRef CAS PubMed.
- Y. Zhong and W. Han, Chem. Commun., 2014, 50, 3874–3877 RSC.
- P. Gautam, R. Gupta and B. M. Bhanage, Eur. J. Org. Chem., 2017, 2017, 3431–3437 CrossRef CAS.
- T. Ishiyama, H. Kizaki, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1993, 34, 7595–7598 CrossRef CAS.
- B. Panda, in Sustainable Chemical Insight in Biological Exploration, ed. H. S. Biswas, A. Ghosh, S. Poddar, S. A. I. S. M. Ghazali and A. Bhaumik, Lincoln University College, Malaysia, 2024, pp. 72–86 Search PubMed.
- P. Gautam, N. J. Tiwari and B. M. Bhanage, ACS Omega, 2019, 4, 1560–1574 CrossRef CAS PubMed.
- K. Khumtaveeporn and H. Alper, Acc. Chem. Res., 1995, 28, 414–422 CrossRef CAS.
- T. L. Church, Y. D. Y. L. Getzler, C. M. Byrne and G. W. Coates, Chem. Commun., 2007, 657–674 Search PubMed.
- I. Omae, Coord. Chem. Rev., 2011, 255, 139–160 CrossRef CAS.
- N. Piens and M. D′hooghe, Eur. J. Org. Chem., 2017, 2017, 5943–5960 CrossRef CAS.
- T. N. Allah, L. Ponsard, E. Nicolas and T. Cantat, Green Chem., 2021, 23, 723–739 RSC.
- P. Wang, D. Yang and H. Liu, Chin. J. Org. Chem., 2021, 41, 3448 CrossRef CAS.
- T. L. Church, C. M. Byrne, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 8156–8162 Search PubMed.
- T. L. Church, Y. D. Y. L. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125–10133 Search PubMed.
- F. G. Delolo, J. Fessler, H. Neumann, K. Junge, E. N. dos Santos, E. V. Gusevskaya and M. Beller, Mol. Catal., 2022, 530, 112621 CrossRef CAS.
- J.-Q. Tao, S. Liu, T.-Y. Zhang, H. Xin, X. Yang, X.-H. Duan and L.-N. Guo, Chin. Chem. Lett., 2023, 109263 Search PubMed.
- W. Hao, Z. Xu, Z. Zhou and M. Cai, J. Org. Chem., 2020, 85, 8522–8532 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |