
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Closed-loop chemically recyclable aromatic polyesters based on asymmetric dicarboxylates obtainable from lignocellulose†
Nitin G.
Valsange
a,
Niklas
Warlin
 a,
Smita V.
Mankar
a,
Nicola
Rehnberg
b,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
a,
Smita V.
Mankar
a,
Nicola
Rehnberg
b,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se; baozhong.zhang@chem.lu.se
bBona Sweden AB, Box 210 74, 200 21 Malmö, Sweden
First published on 18th March 2025
Abstract
Utilizing building blocks derived from lignocellulose is an attractive strategy towards biobased aromatic polyesters. Here, we present the straightforward synthesis of three new asymmetric di-aromatic diester monomers by one-step reactions of lignin-derived hydroxybenzoates (methyl paraben, methyl vanillate and methyl syringate, respectively) with potentially sugar-based methyl 5-chloromethyl-2-furoate. The two diester monomers based on methyl paraben and methyl vanillate were polymerized with 1,6-hexanediol, 1,4-butanediol, and neopentyl glycol, respectively, to produce two series of polyesters with reasonably high molecular weights (12–37 kg mol−1). The fully amorphous polyesters showed high thermal stability (Td,5 > 275 °C), a tunable glass transition temperature (Tg ∼ 36–75 °C), and moderately high mechanical stiffness. In addition, we have successfully developed and demonstrated a pathway for closed-loop chemical recycling of the polyesters through depolymerization using methanolysis, which allowed for the recovery of the native diester and diol monomers. The recovered monomers were subsequently repolymerized to produce a recycled polyester with properties comparable to the original one.
Green foundation1. We have designed and prepared a family of asymmetrical diester monomers from lignocellulosic starting materials intending to develop robust and chemically recyclable biobased polyesters for use as, e.g., packaging materials.2. The polyesters contain paraben, vanillate, syringate and furoate units, respectively, and showed a wide range of advantageous properties. Furthermore, we successfully developed and demonstrated a method for closed-loop chemical recycling of the polyesters via depolymerization by methanolysis. This generated the native diester and diol monomers, which were subsequently efficiently repolymerized to produce a recycled biobased polyester with properties comparable to the original ones. 3. After these achievements, there remains a need to mitigate monomer instability to facilitate the polycondensation process and to scale up and optimize the recycling process to realize the full potential and circularity of these materials. |
1. Introduction
Biobased polymers hold significant promise as a sustainable alternative to traditional fossil-based plastics, offering important benefits such as low greenhouse gas emissions and the use of renewable raw materials.1–3 However, their widespread market introduction faces some critical challenges, including high production costs and limited performance in comparison to fossil-based plastics. Further challenges arise from the limited accessibility of building blocks, the polymerization of heat-sensitive biobased monomers, and the processing of inherently unstable biobased polymers. To outperform low-cost fossil-based plastics and circumvent the high costs of biobased polymers, recent efforts have focused on developing biobased polymers with new and improved properties.4 In addition, increasing attention has been directed toward the recycling of biobased polymers to address end-of-life concerns, promoting both sustainability and recyclability.5,6 Chemical recycling is an attractive approach to achieve closed-loop recycling of polymers by enabling the recovery and reuse of monomers without significantly compromising the material quality. Therefore, the development of chemically recyclable biobased polymers with improved and new properties is crucial to realize the full potential of these materials in a circular economy.7Thermoplastic aromatic polyesters are an important class of high-performance polymers due to their excellent thermo-mechanical properties and good chemical resistance.8–10 Poly(ethylene terephthalate) (PET) and poly(butylene terephthalate) (PBT) are the market-leading aromatic polyesters, widely used in packaging and textile applications.11–14 To a great extent, the superior quality of these polyesters is attributed to the presence of rigid terephthalate units in their backbone, which provides substantial stiffness to the polymer chain and a high melting point, which considerably improves the material properties. However, they are also considered rather problematic materials because of the non-renewable origin of the terephthalic acid (TPA) used in their production.2,15,16 Although the semicrystalline nature of these polymers provides significant mechanical strength to the materials, it limits their applications in cases where high optical clarity is essential. Consequently, developing new biobased aromatic monomers is an attractive strategy to replace terephthalic acid in aromatic polyesters and to produce fully amorphous polyesters.
Among the various naturally occurring renewable resources, lignocellulose is an abundant and readily available source of chemical building blocks for polyesters.1,17,18 Lignin is the richest source of phenolic and aromatic compounds, accounting for ∼15–30% of the lignocellulosic biomass in plants. It is also a by-product of the paper and pulp industry and ∼70 million tons of lignin are produced annually as waste, most of which is incinerated to recover energy.19 Therefore, the exploitation of lignin-based aromatics to develop value-added products has drawn attention in academia and industry over the last few decades.20–22 Various hydroxyaldehydes such as p-hydroxybenzaldehyde, vanillin, and syringaldehyde are obtained after the depolymerization of lignin, and can be directly transformed into their respective hydoxyacids, namely p-hydroxybenzoic acid, vanillic acid, and syringic acid, respectively.23–25 Both hydroxyaldehydes and their corresponding acid derivatives have been used to prepare aromatic monomers for polyester synthesis. For instance, Mialon et al. used vanillin and acetic anhydride to prepare an acetylferulic acid monomer, which was subsequently used to make biobased poly(dihydroferulic acid) (PHFA) with a glass transition temperature (Tg) of 73 °C and a melting temperature (Tm) of 234 °C, hence demonstrating comparable thermal properties to PET.26 In an extension of this work, the same group has reported on hydroxyacid monomers obtained via alkylation of 4-hydroxybenzoic acid, vanillic acid, and syringic acid, respectively, using α,ω-chloroalcohols of varying lengths.27 Next, a series of poly(alkylene 4-hydroxybenzoate)s, poly(alkylene vanillate)s, and poly(alkylene syringate)s with a thermal stability up to 407–478 °C was prepared through polycondensations. The Tg values of the polyesters ranged from 50 to 80 °C and increased when the number of methoxy groups on the aromatic ring increased due to the increasing rotational barrier. In a further study, vanillic acid was reacted with 1,4-dibromoobutane and chloroacetic acid, respectively, to synthesize symmetric and asymmetric diacid monomers. The polycondensation of the resulting monomers with different diols produced amorphous polyesters with Tg values in the range 5–67 °C.28 Cramail et al. have developed divanillin and dimethylvanillate compounds through dimerization of vanillin and methyl vanillate, respectively, using laccase from Trametes versicol.29 They further modified these compounds to obtain polymerizable monomers, i.e., methylated divanillyl diol and dimethylvanillate dimer, and used these to develop aromatic polyesters. For example, the diol monomer was polycondensed with a variety of diacids. The resulting polyesters with terephthalic acid and furandicarboxylic acid showed Tg values of 102 and 139 °C, respectively. Such high Tgs are highly desired for hot-filling packaging applications and for the production of containers for beverages. Consequently, lignin-derived monomers demonstrate significant potential for the preparation of high-performance aromatic polyesters.
Cellulose and hemicellulose account for the major fraction (70–85 wt%) of lignocellulosic biomass and are recognized as crucial renewable resources for the development of value-added chemicals and materials.30 Sugars derived from cellulose and hemicellulose are essential raw materials to produce furanic building blocks, mainly 5-hydroxymethylfurfural (HMF) and furfural.3,31 These furanic compounds are important building blocks for the development of aromatic monomers. For example, HMF is used to produce 2,5-furan dicarboxylic acid (FDCA), which has gained attention as a possible replacement for fossil-based terephthalic acid in PET production, yielding a 100% biobased poly(ethylene 2,5-furandicarboxylate) or PEF.32 Recently, a furfural-based bifuran diacid (BFDCA) has been prepared as a biobased replacement of fossil-based biphenyldicarboxylic acid (BDA) used to improve PET properties.33,34 In another piece of work, a catalyzed two-step synthesis of a difuranic–diacid obtained after the oxidation of a di-HMF intermediate derived from D-fructose was reported for polyester synthesis.35 Furthermore, several bisfuranic diesters and diols with different linkages between the furan rings have been reported for polyester preparation.36,37 Most of the aforementioned lignin- and sugar-based polyesters are semicrystalline, and the corresponding amorphous polyesters remain relatively rare. This motivates the development of new biobased aromatic monomers for the production of amorphous aromatic polyesters.
Herein, we present the synthesis of new asymmetric di-aromatic diester monomers via a facile reaction of lignin-based chemical building blocks, i.e., methyl paraben, methyl vanillate, and methyl syringate, respectively, with biobased methyl 5-chloromethyl-2-furoate (MCMF) obtainable from cellulose (sugar). The monomers based on methyl paraben and methyl vanillate were then employed in polycondensations with different linear and branched aliphatic diols to prepare two series of amorphous polyesters, which were characterized concerning the structural, thermal, and mechanical properties. Furthermore, a chemical recycling process was developed and studied to investigate the possibilities for closed-loop recyclability of the polyesters. We aimed to demonstrate the use of lignin- and sugar-based aromatic building blocks to develop high-performance recyclable amorphous polyesters.
2. Experimental
2.1. Materials
Methylparaben (99%), methylvanillate (99%), methyl syringate (≥98%) K2CO3 (99%), dibutyltinoxide (DBTO, 98%), neopentyl glycol (NPG, 99%), mesitylene (98%), CDCl3 (99.8% atom D, 1% TMS), 1,6-hexanediol (1,6-HD, 97%), and 1,4-butanediol (1,4-BD, 99%) were purchased from Sigma Aldrich. Methyl 5-chloromethyl-2-furoate was purchased from Chemtronica AB Sweden. Xylene was purchased from Scharlau, chloroform (99.8%), DMSO (≥98%), and ethanol (99.1%) were obtained from Fischer, and methanol was supplied by VWR chemicals. All solvents were of analytical grade or higher, and all reagents and chemicals were used without further purification.2.2. Monomer synthesis
1H NMR (400.23 MHz, CDCl3, δ, ppm): 3.88 (s, 3H), 3.90 (s, 3H), 5.11 (s, 2H), 6.55 (d, J = 3.5, 1H), 7.01–6.93 (m, 2H), 7.17 (d, J = 3.5 Hz, 1H), 8.04–7.96 (m, 2H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 52.03, 52.15, 62.42, 111.76, 114.43, 118.84, 123.56, 131.76, 144.84, 153.80, 159.03, 161.72, 166.76.
Mass: HRMS (ESI+, m/z): exact mass calcd for C15H14O6+: 290.0790, found 290.0756.
1H NMR (400.23 MHz, CDCl3, δ, ppm): 3.89 (s, 6H), 3.91 (s, 3H), 5.18 (s, 2H), 6.55 (dd, J = 3.5, 0.7 Hz, 1H), 6.96 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 3.5 Hz, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.64 (dd, J = 8.4, 2.0 Hz, 1H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 52.10, 52.15, 56.14, 63.44, 111.90, 112.80, 113.07, 118.89, 123.37, 124.04, 144.72, 149.40, 151.37, 153.98, 159.05, 166.79.
Mass: HRMS (ESI+, m/z): exact mass calcd for C16H16O7+: 320.0896, found 320.0985.
The synthesis of diester monomer M3 was carried out using a similar procedure as described for diester M1 and M2. 1H NMR (400.23 MHz, CDCl3, δ, ppm): 3.85 (s, 6H), 3.87 (s, 3H), 3.89 (s, 3H), 5.20 (s, 2H), 6.46 (d, J = 3.4, 1H), 7.11 (d, J = 3.5 Hz, 1H), 7.27 (s, 2H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 52.01, 52.37, 56.35, 66.50, 106.77, 111.71, 118.85, 125.99, 140.11, 144.46, 153.26, 155.50, 159.19, 166.74.
Mass: HRMS (ESI+, m/z): exact mass calcd for C17H18O8+: 350.1002, found 350.1106.
2.3. Polyester synthesis
Diesters M1 and M2 were polymerized with the diols 1,6-HD (a), 1,4-BD (b), and NPG (c) to obtain two series of polyesters, P1a–c and P2a–c, respectively (Scheme 1). The polymerization protocol of P1b is described here as a typical example. To a 50 mL three-neck round bottom flask equipped with a mechanical stirrer, a nitrogen inlet, and a vacuum distillation outlet were added monomer M1 (2 g, 6.89 mmol), DBTO (9 mg, 0.5 mol%), and 1,4-butanediol (0.683 g, 7.58 mmol). Subsequently, the reaction mixture was degassed by three successive vacuum–nitrogen cycles at room temperature. Then, 3 mL of xylene was added, and the reaction mixture was heated to 160 °C and stirred under a nitrogen atmosphere for 5 h. Next, the temperature was increased to 180 °C, and mesitylene (5 mL) was added. A high nitrogen flow was applied for 15 h to facilitate the removal of the excess diol. Afterward, the product was dissolved in chloroform and precipitated into methanol to give the polyester P1b as a white solid (1.95 g, 89%). | ||
| Scheme 1 Synthesis of the diester monomers and their polymerizations with a variety of different diols using a modified melt polycondensation method. | ||
1H NMR (400.23 MHz, CDCl3, δ, ppm): 1.99–1.82 (m, 4H), 4.45–4.29 (m, 4H), 5.11 (s, 2H), 6.54 (d, J = 3.5 Hz, 1H), 7.00–6.93 (m, 2H), 7.16 (dd, J = 1.1, 3.5 Hz, 1H), 8.03–7.96 (m, 2H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 25.50, 25.60, 25.62, 25.72, 62.45, 64.30, 64.41, 64.63, 64.74, 111.76, 114.46, 118.81, 123.61, 123.65, 131.76, 144.87, 144.91, 153.87, 153.90, 158.61, 158.64, 161.77, 166.22, 166.24.
The other polyesters were synthesized using a similar procedure as described for P1a. However, in the synthesis of P1c and P2c, a molar ratio of diester to diol of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was used, and both the transesterification and polycondensation steps were carried out for 15 h each.
:2 was used, and both the transesterification and polycondensation steps were carried out for 15 h each.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 25.67, 25.74, 25.85, 25.93, 28.66, 28.71, 28.77, 28.81, 62.46, 64.78, 64.83, 65.10, 65.14, 111.72, 114.42, 118.65, 123.80, 131.72, 145.04, 153.79, 158.74, 161.72, 166.33.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 21.58, 21.88, 22.00, 22.12, 35.31, 35.35, 35.38, 62.39, 69.46, 69.52, 69.69, 69.77, 111.74, 111.77, 114.48, 118.83, 118.86, 123.37, 123.41, 131.75, 144.61, 144.64, 153.97, 153.99, 154.01, 158.46, 158.51, 161.78, 161.81, 166.02, 166.06.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 25.68, 25.73, 25.82, 25.87, 28.67, 28.71, 28.78, 28.80, 56.17, 63.47, 64.95, 65.06, 111.87, 112.83, 113.03, 118.71, 123.30, 124.28, 144.93, 149.40, 151.39, 153.96, 158.75, 166.37.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 25.49, 25.58, 25.61, 25.70, 56.17, 63.44, 64.46, 64.56, 64.65, 111.90, 112.82, 113.02, 118.85, 123.33, 124.09, 144.75, 144.78, 149.41, 151.44, 154.03, 158.62, 158.64, 166.27.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 21.86, 22.00, 22.14, 35.30, 35.39, 35.45, 56.13, 56.14, 63.36, 69.50, 69.52, 69.66, 69.69, 111.91, 112.80, 112.94, 118.91, 118.93, 123.28, 123.84, 123.87, 144.50, 144.53, 149.39, 151.47, 154.14, 158.46, 158.48, 166.06, 166.08.
2.4. Characterization
1H and 13C NMR measurements were performed on a Bruker DRX400 spectrometer at 400.13 MHz and 100.61 MHz, respectively, with the samples dissolved in chloroform-d. Chemical shifts were reported as δ values (ppm). Size exclusion chromatography (SEC) of the polyesters were carried out using a Malvern OMNISEC instrument equipped with one TGuard, Org Guard, col 10 × 4.6 mm as a guard column, 2 × T6000M, general mixed org. 300 × 8.0 mm as an analytical column, and a refractive index (RI) detector using THF as the eluent at 40 °C at an elution rate of 1 mL min−1. Calibration was performed with narrow dispersity polystyrene standards (Mn = 96, 52.4, 30, 17.5, 3.5 and 3.0 kg mol−1). Thermogravimetric analysis (TGA) was carried out on a TA instrument mode TGA Q500. The measurement was performed under a nitrogen atmosphere by heating from 50 to 600 °C with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was performed on a DSC Q2000 analyzer from TA instruments. Data were recorded between −50 and 200 °C, and the glass transition temperature (Tg) was determined from the second heating cycle. The polyesters were hot pressed into films (35 mm × 5 mm × 1 mm) for dynamic mechanical analysis (DMA). Hot pressing was performed at 50–60 °C above the Tg of the polymer, after which the samples were rapidly cooled to room temperature. Dynamic mechanical analysis (DMA) was performed in the stretching mode using a TA Instruments Q800 analyzer. The hot-pressed samples were heated from −20 °C to 120 °C at a heating rate of 3 °C min−1 and a frequency of 1 Hz with a constant strain of 0.1%.2.5. Chemical recycling
Chemical recycling of the polyesters was investigated using polyester P2a as a respresentative example.:20), yielding crude 1,6-HD after solvent removal and a solid residue exclusively containing the diester monomer. The crude diol was recrystallized from t-butyl methyl ether to obtain the recycled diol (r-1,6-HD) as a pale white solid (0.91 g, 72%). Subsequently, the solid residue and the solid product from the evaporation of the mother liquor (from the initial diester recrystallization) were combined and purified further by recrystallization to yield additional r-M2 (0.79 g, 23%), increasing the overall yield of the recycled diester to 2.53 g (74%).
3. Results and discussion
3.1. Monomer and polyester synthesis
Lignin-derived hydroxyacids and hydroxybenzoates are generally considered having low toxicity due to their natural occurrence and metabolic compatibility. As a result, these compounds are widely used in the food, cosmetic, and pharmaceutical industries. Therefore, these building blocks represent a safer and sustainable alternative for polymer development compared to the corresponding petroleum-based compounds.38–40 Three diester monomers M1, M2, and M3 were synthesized by simple and straightforward SN2 reactions of methyl 5-chloromethyl-2-furoate (MCMF, 4) with three different hydroxybenzoates, namely methyl paraben (1), methyl vanillate (2) and methyl syringate (3), respectively (Scheme 1). The reactions were performed at room temperature for 24 h using K2CO3 as a base in dimethyl sulfoxide (DMSO). The choice of DMSO was made because of its ability to dissolve the base and, thus, to efficiently facilitate the deprotonation of the phenol group. The slurries obtained after the reactions were poured into ice-cold water to isolate the crude monomer products, which were subsequently purified via straightforward recrystallization in ethanol to give the diester monomers M1, M2, and M3 in good yields, i.e., 75, 76 and 77%, respectively.The diester monomers were investigated in polycondensations with various biobased diols, i.e., 1,6-hexanediol (1,6-HD), 1,4-butanediol (1,4-BD) and neopentyl glycol (NPG). The polymerizations were conducted using a two-step modified melt polycondensation method including transesterification and polycondensation steps, employing DBTO as a catalyst. The transesterification step was performed under a low nitrogen flow using xylene as a solvent, and the subsequent polycondensation step was conducted under a high nitrogen flow in the presence of mesitylene as a solvent. Xylene was used in the transesterification to reduce the evaporation of the diol and achieve complete transesterification, and mesitylene was used in the polycondensation step for the azeotropic removal of the excess diol, which efficiently increased the molecular weights of the polymers.
The presence of electron-donating substituents on the aromatic monomers often promotes their degradation and favours unwanted side reactions during polycondensations at high temperatures. This usually imparts significant colouration and branching/crosslinking of the resulting polymeric material. The diesters M1–M3 all possess an electron-donating methylene group on the furan ring and varying methoxy substitutions on the phenolic ring, which may influence the thermal stability during polycondensations. Therefore, before examining the polymerizability of each diester, temperature screening was performed to identify suitable polymerization conditions. The screening experiments were investigated using diester M2 after hypothesizing that the monomer thermal stability would decrease from diester M1 to M3, i.e., with increasing methoxy substitutions. Therefore, the optimized polymerization conditions of the ‘intermediate’ diester (M2) might also be favourable for both diesters M1 and M3. Diester M2 was polymerized with 1,6-HD at different temperatures to obtain the corresponding polyester, P2a. A 10% molar excess of 1,6-HD in relation to the diester was used in all cases. Three polymerizations involving different combinations of temperatures for transesterification and polycondensation steps were investigated (Table 1, entries 1–3). The transesterifications were performed for 5 h at a temperature interval of 20 °C between 160–200 °C, and the subsequent polycondensations were conducted for 15 h at 20 °C above the transesterification temperature. As can be seen, transesterification at 160 °C and polycondensation at 180 °C showed the best results (entry 2) and gave a reasonably high molecular weight (Mn = 25.0 kg mol−1) and a high isolated yield (84%). A comparatively lower molecular weight (Mn = 11.7 kg mol−1) and yield (77%) were obtained when the corresponding steps were carried out at 140 and 160 °C, respectively (entry 1). Moreover, transesterification at 180 °C and polycondensation at 200 °C resulted in significant monomer degradation and, consequently, a partially crosslinked product was formed (entry 3). Additionally, the molecular weight of the soluble portion (uncrosslinked) was lower (Mn = 20.0 kg mol−1) than what was obtained for entry 2. The higher monomer degradation was also reflected in the physical appearance of the product, which had a dark yellow colour. Even though no degradation/crosslinking was detected for entries 1 and 2, a pale-yellow tint was observed in these products. This might imply a very minor monomer degradation during the polymerization. Still, a fine balance between sufficient monomer stability and the formation of a high molecular weight polymer was achieved for entry 2. Therefore, the transesterification at 160 °C and polycondensation at 180 °C were identified as the best polymerization conditions and were subsequently used in the polycondensations involving diesters M1–M3.
| Entry | Temperaturea (°C) | Yieldb (%) | M n (kg mol−1) | M w (kg mol−1) | Đ | |
|---|---|---|---|---|---|---|
| Transesterification (step 1) | Polycondensation (step 2) | |||||
| a Polymerization conditions: temperature for transesterification and polycondensation steps (step 1 and 2, respectively). Transesterification was carried out for 5 h under a low nitrogen flow using xylene as a solvent. Next, polycondensation took place under a high nitrogen flow for 15 h with mesitylene as a solvent. b Calculated from the weight of the polyester obtained after purification. c Measured by SEC in THF against polystyrene standards. d Molecular weight of the soluble polymer fraction isolated from the partially crosslinked product. | ||||||
| 1 | 140 | 160 | 77 | 11.7 | 33.2 | 2.84 |
| 2 | 160 | 180 | 84 | 25.0 | 58.0 | 2.32 |
| 3 | 180 | 200 | 52 | 18.0d | 53.1d | 2.95d |
In the next step, diesters M1 and M3 were employed in polymerizations with 1,6-HD using the optimized conditions. Diester M1 successfully gave polyester P1a in a reasonably good yield (80%) and high molecular weight (Mn = 36.7 kg mol−1). The product was almost white, indicating no monomer degradation during the polymerization. In contrast, diester M3 suffered from severe degradation during the polymerization and consequently formed a completely crosslinked product. No crosslinking/gelation was observed when the polymerization was performed at lower temperatures and shorter times. However, these products showed significantly lower molecular weight and a prominent brown colouration, indicating a very low thermal stability of diester M3. Therefore, the initial assessment of the polymerization of the diesters revealed that an increasing methoxy substitution on the phenolic group decreased the thermal stability of the diester monomers and favoured side reactions that yielded coloured or crosslinked products. Still, diesters M1 and M2 showed excellent stability under the optimized conditions to produce reasonably high molecular weight polymers without any significant colouration or gelation. Therefore, only diesters M1 and M2 were investigated in the further polymerizations with 1,4-BD and NPG. The polymerizations of M1 and M2 with 1,4-BD were conducted using similar conditions to those with 1,6-HD, with a slight excess of diol (10 mol%) to obtain polyesters P1b and P2b. However, in the polymerizations with NPG, only a significant excess of diol (100 mol%) and a longer transesterification time (15 h) yielded polyesters P1c and P2c with reasonably high molecular weights. The use of a small NPG excess resulted in incomplete transesterification and the formation of low molecular weight polymers. In all cases, the viscous mixtures obtained after the polymerizations were dissolved in chloroform and the products were then precipitated in methanol to give two series of polyesters, i.e., P1a–c and P2a–c in 73–89% and 54–84% isolated yields, respectively. The molecular weights of the polyesters were measured by SEC and the results showed the formation of reasonably high molecular weight polymers with Mn ranging between 15.0 and 36.7 for P1a–c and 11.9 and 25.0 kg mol−1 for P2a–c. Furthermore, the polydispersity (Đ) of the polyesters in both series was in an acceptable range, i.e., 1.86–2.37, indicating successful polymerizations without any significant side reactions.
The chemical structure of the monomers and polyesters was confirmed by 1H NMR spectroscopy and further supported by 13C NMR investigations (Fig. S1 and S2 and S8–S13†). Moreover, the monomer formation was validated by HRMS analysis (Fig. S5–S7†). The stacked 1H NMR spectra of the M1 and M2, along with the respective polyesters, are depicted in Fig. 1A–H. The 1H and 13C NMR spectra of M3 are shown in the ESI (Fig. S3 and S4†). In the 1H NMR spectrum of M1 (Fig. 1A), the multiplets between 7.96–8.04 and 6.93–7.01 ppm were assigned to the phenolic protons (b and c, respectively). In addition, the signals corresponding to the protons of the furanoate units were observed as two doublets at 7.17 (f) and 6.55 ppm (e), respectively, and a singlet at 5.11 (d) ppm. The signals of the methyl ester group attached to the phenolic ring appeared as a singlet at 3.90 (a), while the methyl ester group of the furanoate units was observed at 3.88 (g) ppm. In the 1H NMR spectrum of M2 (Fig. 1E), the doublet of doublet at 7.64 (b) ppm and two doublets at 7.57 (h) and 6.96 (c) ppm were attributed to the protons of the phenyl ring. In addition, the singlet of the methoxy group was observed at 3.89 (i) ppm, which was merged with the signal of the methyl ester group (g) of the furanoate units. On the other hand, the signal of the methyl ester group linked to the phenolic ring appeared as an isolated singlet at 3.91 (a) ppm. The protons of the furanoate units (e and f) were observed at almost identical chemical shifts as in the case of M1, except for proton d, which was slightly downshifted. After the polymerizations (Fig. 1B–D and F–H), the methyl ester signals (a and g) almost disappeared, indicating complete transesterification. In addition, the signals corresponding to the aliphatic diol residues (1–3), the phenolic protons (b, c, and h), the furanoate unit protons (e, f, and d) and the methoxy group (i) were also observed in the 1H NMR spectra of the polyesters, confirming their structures. Notably, isolated signals were observed for the methylene groups (1) of the diol unit attached to the ester groups in polyesters P1c and P2c, attributed to the asymmetric structure of the diester units. In particular, P1c exhibited four distinct signals for the methylene protons (1), indicating the presence of possible dyad structures along the polymer chains. Similarly, the methyl group protons (2) displayed dyad sensitivity and split into multiple signals both in P1c and P2c. For polyesters P1a–b and P2a–b, the signals of the methylene groups (1) of the diol unit attached to the ester groups of the phenyl and furan rings merged and were observed as multiplets. A similar pattern was observed for methylene protons 2 and 3 of the diol units in the respective polymers. Furthermore, the 13C NMR spectra (Fig. S8–S13†) of all the polyesters revealed multiple resonances for each diol carbon resulting from the different dyad structures in the polymer chain due to the asymmetric nature of the diester units. Notably, no signals indicating the presence of cyclic products were detected. Still, the possibility of their formation at a negligible level cannot be excluded.
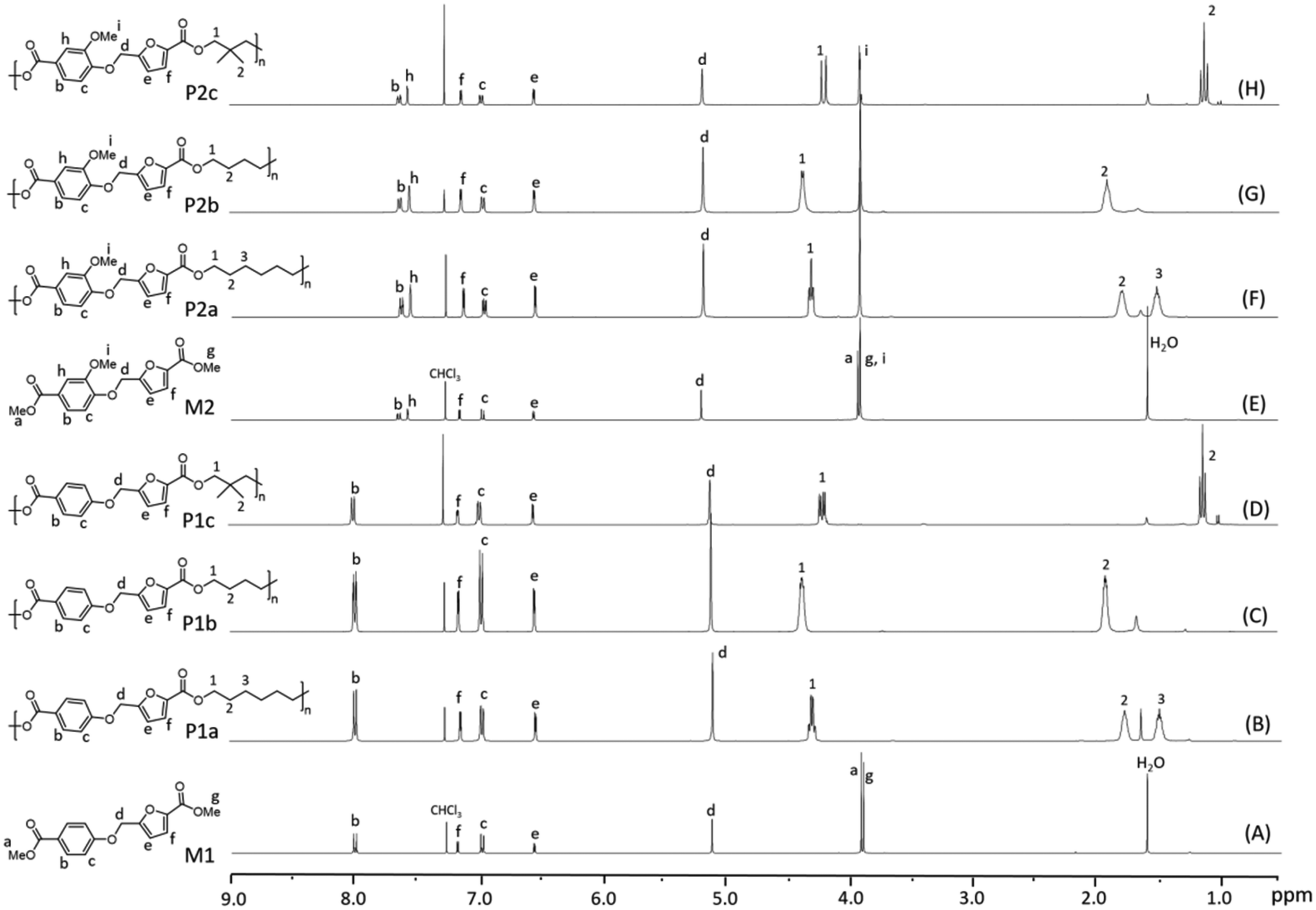 | ||
| Fig. 1 1H NMR spectra of monomers M1 and M2 and polyesters P1a–c and P2a–c recorded in CDCl3. | ||
3.2. Thermal properties
The thermal stability of the polyesters was investigated by thermogravimetric analysis (TGA) under nitrogen (Fig. 2a and b, Table 2). All the polyesters exhibited relatively high initial thermal decomposition temperatures (Td,5 > 275 °C), indicating the possibility for melt processing. Oddly, polyesters P1a–c showed three decomposition rate maxima in contrast to the polyesters in the P2a–c series, which only showed two. In both series, the second decomposition rate maximum at Td ∼ 380 °C indicated a major decomposition step and may be ascribed to the degradation of the ester bonds.41 The first decomposition rate maxima at Td ∼ 325 and 250 °C for P1a–c and P2a–c, respectively, may be attributed to the degradation of the ether bonds in the backbone. The presence of a third decomposition rate maximum in P1a–c was difficult to explain. Compared to the P1a–c polyesters, the P2a–c polyesters exhibited slightly lower thermal stability due to the electron-donating methoxy groups, which was consistent with the observations made with other methoxy-containing polymers.42,43 When compared to other aromatic polyesters, the thermal stability of the present polyesters was lower than that of PET (Td,5 = 382 °C), but close to PEF (Td,5 = 329 °C), especially for the P1a–c series polyesters (Td,5 = 309–312 °C).44,45 | ||
| Fig. 2 TGA weight loss (a) and derivative weight loss (b) curves of the polyesters recorded under a N2 atmosphere. | ||
| Polyester | Yielda (%) | M n (kg mol−1) | M w (kg mol−1) | Đ | T d,5 (°C) | T g (°C) |
|---|---|---|---|---|---|---|
| a Calculated from the weight of the polyester obtained after purification. b M n, Mw, and Đ were measured by SEC in THF. c T d,5 was measured by TGA. d T g was measured from the second DSC heating curve. | ||||||
| P1a | 80 | 37 | 70 | 1.9 | 312 | 36 |
| P1b | 89 | 27 | 53 | 2.0 | 314 | 50 |
| P1c | 73 | 15 | 32 | 2.1 | 309 | 75 |
| P2a | 84 | 25 | 58 | 2.3 | 290 | 48 |
| P2b | 84 | 17 | 40 | 2.3 | 281 | 61 |
| P2c | 54 | 12 | 27 | 2.3 | 274 | 75 |
The thermal transitions of the polymers were investigated by differential scanning calorimetry (DSC), and the data obtained are shown in Fig. 3 and Table 2. As can be seen, no melting endotherm was observed for any of the polymers, indicating their amorphous nature. The Tg values of the two series of polyesters increased with the decreasing length of the diol, which was consistent with the increased backbone rigidity. In general, the Tg values for the polyesters in the P2a–c series were higher than those recorded for the corresponding samples in the P1a–c series, suggesting that the increased bulkiness due to the presence of the methoxy substituent in P2a–c reduced the segmental mobility of the polymer backbones.46,47 However, polyester P2c showed a similar Tg to P1c (75 °C) despite possessing the bulky methoxy substituent on the backbone. This may be due to the lower molecular weight of P2c (12 kg mol−1) than that of P1c (15 kg mol−1). The obtained Tg value of 75 °C for P1c and P2c was comparable to that of commercial PET (76 °C), indicating an excellent heat resistance of the polymers.9 The wide Tg range of the current polyesters suggests a potential use in both flexible and rigid packaging applications. Moreover, their fully amorphous nature implies a high degree of biodegradability, potentially making them a biodegradable alternative to semicrystalline materials such as PET and PEF.
 | ||
| Fig. 3 DSC traces of the polyesters obtained during the second heating cycle with the Tg values indicated. | ||
3.3. Dynamic mechanical properties
The mechanical properties of the polyesters were studied by DMA analysis of hot-pressed samples. An oscillatory deformation was applied to the samples while heating at a rate of 3 °C min−1 from −20 to 120 °C. The storage moduli (E′) of the P1a–c and P2a–c series at the glassy plateau (20 °C) were in the range of 1.97–2.01 and 2.05–2.69 GPa, respectively (Fig. 4a; Table 3), demonstrating a moderately high stiffness. As can be seen, the E′ values for both series of polyesters (except for P1c and P2c) increased with decreasing chain length of the diol unit, which can be correlated with the increased stiffness of the polymer backbone. Furthermore, polyesters P2a–c displayed comparatively higher E′ than the corresponding polymers in the P1a–c series due to the additional rigidity from the bulky methoxy substituents. The lower E′ of samples P1c and P2c, compared to their longer-chain analogues (P1a–b and P2a–b, respectively), may be explained by their lower molecular weight that prevented efficient chain entanglements and consequently made these samples softer. Notably, the E′ values of the polyesters were comparable to those of PET (E′ = 2.0 GPa) and PEF (E′ = 2.5 GPa), indicating their high stiffness.48 | ||
| Fig. 4 DMA data showing the storage (a) and loss modulus (b) of the polyesters (1 Hz, 0.1% strain). | ||
The Tg values were taken at the local maximum of the loss moduli (E′′) in the glass transition region, and the results showed a decreasing trend as the length of the diol units increased (Fig. 4b, Table 3). This observation was consistent with the DSC results. Moreover, the Tg values obtained by DMA were in good agreement with those measured by DSC, indicating a good correlation between the two methods.
3.4. Chemical recycling
The ester links of the aromatic polyesters can be broken by methanolysis to recover the starting methyl diester and diol monomers, which can then be repolymerized to generate the original polymer structure, enabling closed-loop chemical recycling. In the present case, the chemical recyclability was investigated using polyester P2a as a representative case. The depolymerization was conducted in a pressure tube at 120 °C for 8 h, using Zn(OAc)2 as a catalyst in an excess of methanol (Fig. 5a). These conditions were identified after initial small-scale optimization reactions carried out in an exploratory manner, which suggested that more than 97% of the polymer was degraded into its constituent monomers under these conditions. The high degree of depolymerization observed under relatively mild conditions may to a high degree be attributed to the amorphous nature of the polyesters. In contrast, semicrystalline polyesters like PET typically require much higher temperatures and pressures for complete depolymerization.49 As can be seen, the P2a sample completely disappeared after the methanolysis, resulting in a homogeneous solution to indicate complete depolymerization (Fig. 5b). Notably, the diester monomer partly precipitated from the solution during overnight storage at room temperature, facilitating straightforward isolation through filtration. In contrast, the diol monomer remained soluble in methanol, along with a fraction of unprecipitated diester monomer, as confirmed by the 1H NMR spectrum of the filtrate. Although the obtained diester was crystalline, suggesting high purity, it was further recrystallized in ethanol to ensure sufficient monomer purity for a successful repolymerization. This process produced a recycled diester (r-M2) with a yield of 51%. The diol monomer was isolated from the filtrate by evaporating the solvent (methanol) and washing the resulting semi-solid with a water–ethanol mixture (80:20) to extract 1,6-hexanediol. After solvent removal, the crude product was purified via simple recrystallization in t-butyl methyl ether to obtain the recycled diol (r-1,6-HD) in 72% isolated yield. The solid residue remaining after the extraction of the diol monomer exclusively contained the diester monomer. This residue, together with the product obtained from the mother liquor in the first recrystallization process, was recrystallized to obtain additional r-M2 monomer. This step improved the yield of r-M2 to 74%. Both recycled monomers were analyzed by 1H NMR spectroscopy, and the resulting spectra were identical to those of the original monomers, confirming the recovery of high-purity monomers after the depolymerization process (Fig. 5c and d).
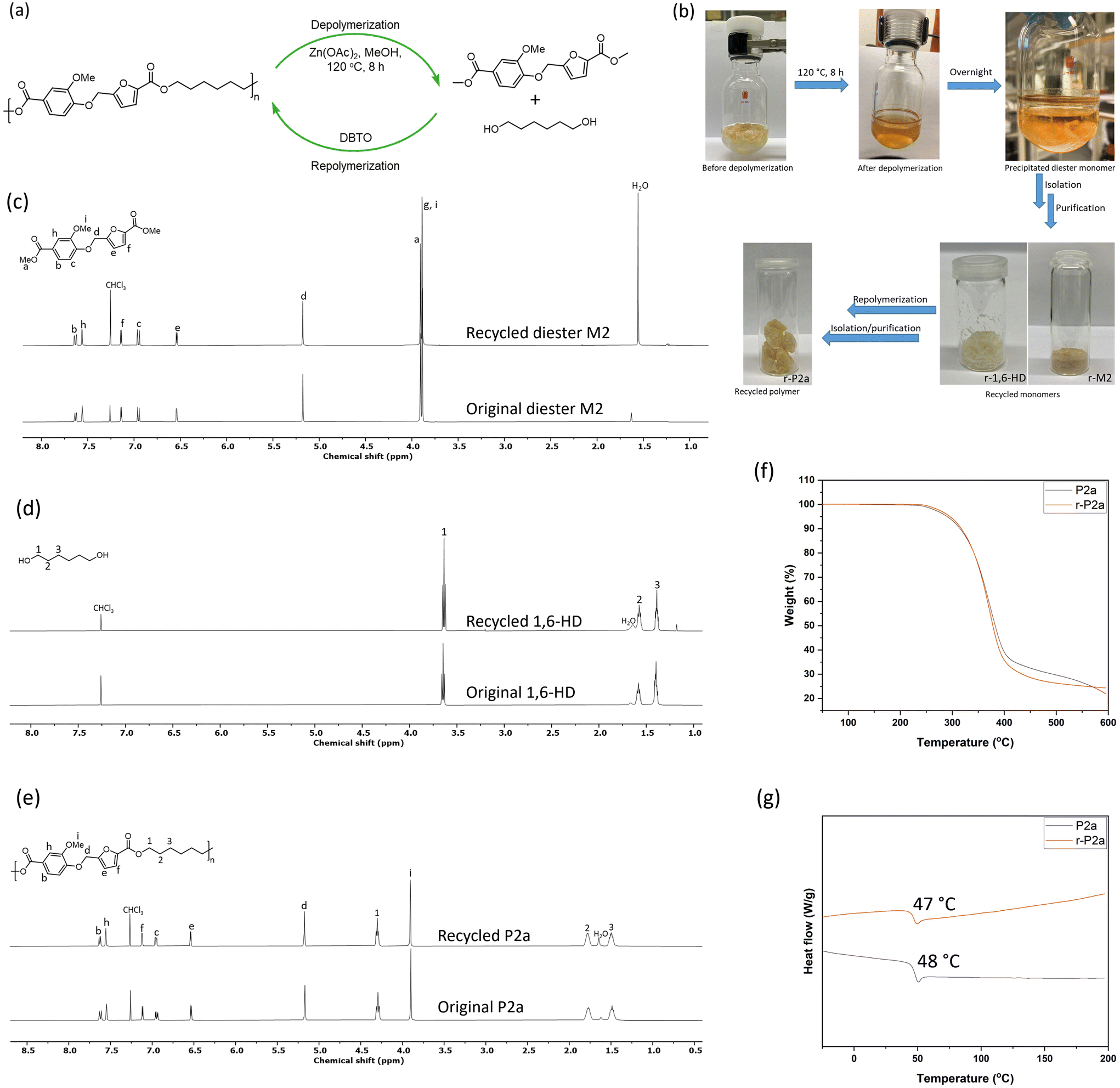 | ||
| Fig. 5 Illustration of the chemical recycling process of polyester P2a, including both the de- and repolymerization steps (a), photographic images showing the depolymerization (before and after methanolysis), the recycled monomers, and the recycled polymers (b), stacked 1H NMR spectra to compare the data of the recycled monomers (r-M2 and r-1,6-HD) and the recycled polymer (r-P2a) with their respective original counterparts (c–e), TGA thermograms (f) and DSC traces from the second heating cycle (g) of the initial P2a and recycled r-P2a samples, respectively. | ||
To close the recycling loop, the two recycled monomers (r-M2 and r-1,6-HD) were repolymerized by following a similar modified melt polycondensation protocol as used for the initial polymer P2a, involving transesterification at 160 °C and polycondensation at 180 °C for 5 h and 15 h, respectively. This produced a recycled polymer (r-P2a) with an isolated yield of 80%, obtained after precipitating the product in methanol. Measurement by SEC gave a molecular weight of Mn = 23.4 kg mol−1, which was close to that of the initial P2a (Mn = 25 kg mol−1), indicating a successful repolymerization. This was further supported by the 1H NMR spectrum of the recycled polymer (Fig. 5e). The spectrum of the recycled polymer was in complete agreement with that of the original polymer, showing the disappearance of the methyl ester signals (a and g) and the presence of backbone signals corresponding to the aliphatic diol residues (1–3), the phenolic protons (b, c, and h), the furanoate unit protons (e, f, and d) and the methoxy group (i). Furthermore, the thermal properties of the recycled polymer were investigated by TGA and DSC analysis (Fig. 5f and g). The measured Td,5 and Tg values of the recycled polymer were 295 and 47 °C, respectively, nearly identical to those of the original polymer (Td,5 = 290 °C and Tg = 48 °C). These findings strongly suggest the efficient chemical recyclability of these new polymers without compromising their molecular structure and material properties, thus paving the way for closed-loop recycling. Still, the limited thermal stability of the monomers, which decreases with the number of electron-donating methoxy substituents on the phenyl ring, is a crucial issue when considering the feasibility to recycling these polymers. In addition, the cost of the biobased building blocks and the chemical recycling process will affect the overall sustainability of these polymers. Consequently, a thorough investigation focusing on mitigating monomer instability and optimizing the recycling process is needed to realize the robustness and sustainability of the polymer recycling.
4. Conclusions
This work demonstrated the successful utilization of lignin- and sugar-derived building blocks for the preparation of amorphous aromatic polyesters. Three asymmetric di-aromatic dicarboxylates with varying methoxy-substitutions were synthesized by reacting sugar-based methyl 5-chloromethyl-2-furoate with lignin-based hydroxybenzoates, i.e. methyl paraben, methyl vanillate, and methyl syringate. The two former dicarboxylate monomers were successfully polymerized with different diols under optimized conditions to yield two series of amorphous polyesters with reasonably high molecular weights. These polyesters showed excellent thermal stability and Tgs in the range 36–75 °C. The highest Tg was achieved by the two polyesters based on neopentyl glycol and is comparable to the Tg of commercial PET. The possibility to depolymerize the polyesters through methanolysis into the original monomers was successfully demonstrated. Furthermore, it was possible to repolymerize the recovered monomers to produce recycled polyesters with comparable molecular weights and properties to the original polymers, indicating viable closed-loop recyclability of the polymers. This circular plastic approach not only offers a solution for effectively managing plastic wastes, but also addresses the end-of-life issues connected with these polymers, promoting more sustainable practices in the use of plastics, thus contributing to a circular economy.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swedish Foundation for Strategic Environmental Research (Mistra) through the “STEPS” project (2016-1489), the Swedish Research Council for Sustainable Development, Formas (2023-00893) and the Royal Physiographic Society in Lund.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed
- N. Hernández, R. C. Williams and E. W. Cochran, Org. Biomol. Chem., 2014, 12, 2834–2849 RSC
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS
- H. Ariffin, H. Nishida, M. A. Hassan and Y. Shirai, Biotechnol. J., 2010, 5, 484–492 CrossRef CAS PubMed
- S. Kakadellis and G. Rosetto, Science, 2021, 373, 49–50 CrossRef CAS PubMed
- S. Muñoz-Guerra, C. Lavilla, C. Japu and A. Martínez De Ilarduya, Green Chem., 2014, 16, 1716–1739 RSC
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS PubMed
- F. Pacheco-Torgal, Y. Ding and S. Jalali, Constr. Build. Mater., 2012, 30, 714–724 CrossRef
- H. K. Webb, J. Arnott, R. J. Crawford and E. P. Ivanova, Polymers, 2013, 5, 1–18 CrossRef
- C. Lavilla, A. M. De Ilarduya, A. Alla, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Macromolecules, 2012, 45, 8257–8266 CrossRef CAS
- C. Lavilla and S. Muñoz-Guerra, Green Chem., 2013, 15, 144–151 RSC
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC
- A. Pellis, E. Herrero Acero, L. Gardossi, V. Ferrario and G. M. Guebitz, Polym. Int., 2016, 65, 861–871 CrossRef CAS
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C. A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed
- P. J. De Wild, W. J. J. Huijgen and R. J. A. Gosselink, Biofuels, Bioprod. Biorefin., 2012, 6, 246–256 CrossRef
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS
- C. H. R. M. Wilsens, J. M. G. A. Verhoeven, B. A. J. Noordover, M. R. Hansen, D. Auhl and S. Rastogi, Macromolecules, 2014, 47, 3306–3316 CrossRef CAS
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable
Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed
- E. Xanthopoulou, Z. Terzopoulou, A. Zamboulis, L. Papadopoulos, K. Tsongas, D. Tzetzis, G. Z. Papageorgiou and D. N. Bikiaris, ACS Sustainable Chem. Eng., 2021, 9, 1383–1397 CrossRef CAS
- J. D. P. Araújo, C. A. Grande and A. E. Rodrigues, Chem. Eng. Res. Des., 2010, 88, 1024–1032 CrossRef
- L. Mialon, A. G. Pemba and S. A. Miller, Green Chem., 2010, 12, 1704–1706 RSC
- L. Mialon, R. Vanderhenst, A. G. Pemba and S. A. Miller, Macromol. Rapid Commun., 2011, 32, 1386–1392 CrossRef CAS PubMed
- C. Pang, J. Zhang, Q. Zhang, G. Wu, Y. Wang and J. Ma, Polym. Chem., 2015, 6, 797–804 RSC
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 6058–6066 RSC
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC
- C. Rosenfeld, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef PubMed
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC
- T. P. Kainulainen, T. I. Hukka, H. D. Özeren, J. A. Sirviö, M. S. Hedenqvist and J. P. Heiskanen, Biomacromolecules, 2020, 21, 743–752 CrossRef CAS PubMed
- Y. Lei, S. Zhang, G. Shen, J. Zhu, J. W. Xue, Z. Chen and G. Yin, Ind. Eng. Chem. Res., 2020, 59, 19876–19883 CrossRef CAS
- A. S. Amarasekara, L. H. Nguyen, N. C. Okorie and S. M. Jamal, Green Chem., 2017, 19, 1570–1575 RSC
- A. M. Ahmed, T. P. Kainulainen, J. A. Sirviö and J. P. Heiskanen, Biomacromolecules, 2022, 23, 1803–1811 CrossRef CAS PubMed
- S. Hbaieb, W. Kammoun, C. Delaite, M. Abid, S. Abid and R. El Gharbi, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 365–373 CrossRef CAS
- M. G. Soni, S. L. Taylor, N. A. Greenberg and G. A. Burdock, Food Chem. Toxicol., 2002, 40, 1335–1373 CrossRef CAS PubMed
- F. A. Khan, R. Irshad, N. Tanveer, S. Yaqoob, Razaullah, R. Ali, N. Ali, J. Saifullah, K. Ali Hasan, S. Naz, A. Qadir, A. Jabeen and Y. Wang, Bioorg. Chem., 2024, 145, 107254 CrossRef CAS PubMed
- C. Srinivasulu, M. Ramgopal, G. Ramanjaneyulu, C. M. Anuradha and C. Suresh Kumar, Biomed. Pharmacother., 2018, 108, 547–557 CrossRef CAS PubMed
- F. Samperi, C. Puglisi, R. Alicata and G. Montaudo, Polym. Degrad. Stab., 2004, 83, 11–17 CrossRef CAS
- M. Hasegawa, Y. Tsujimura, K. Koseki and T. Miyazaki, Polym. J., 2008, 40, 56–67 CrossRef CAS
- B. G. Harvey, A. J. Guenthner, W. W. Lai, H. A. Meylemans, M. C. Davis, L. R. Cambrea, J. T. Reams and K. R. Lamison, Macromolecules, 2015, 48, 3173–3179 CrossRef CAS
- B. A. Alshammari, F. S. Al-Mubaddel, M. R. Karim, M. Hossain, A. S. Al-Mutairi and A. N. Wilkinson, Polymers, 2019, 11, 1–20 CrossRef PubMed
- J. G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Storti and M. Morbidelli, Nat. Commun., 2018, 9, 2701 CrossRef PubMed
- O. Bonjour, H. Nederstedt, M. V. Arcos-Hernandez, S. Laanesoo, L. Vares and P. Jannasch, ACS Sustainable Chem. Eng., 2021, 9, 16874–16880 CrossRef CAS PubMed
- J. A. Emerson, N. T. Garabedian, D. L. Burris, E. M. Furst and T. H. Epps, ACS Sustainable Chem. Eng., 2018, 6, 6856–6866 CrossRef CAS
- R. J. I. Knoop, W. Vogelzang, J. van Haveren and D. S. van Es, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4191–4199 CrossRef CAS
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511–525 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05572a |
| This journal is © The Royal Society of Chemistry 2025 |