
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced mineral carbonation on surface functionalized MgO as a proxy for mine tailings†
Rajeev Kumar
Rai‡
 a,
Rui
Serra-Maia‡
a,
Yingjie
Shi
b,
Peter
Psarras
b,
Aleksandra
Vojvodic
*b and
Eric A.
Stach
*a
a,
Rui
Serra-Maia‡
a,
Yingjie
Shi
b,
Peter
Psarras
b,
Aleksandra
Vojvodic
*b and
Eric A.
Stach
*a
aDepartment of Materials Science and Engineering, University of Pennsylvania, PA, USA-19104. E-mail: stach@seas.upenn.edu
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, PA, USA-19104
First published on 24th March 2025
Abstract
The escalating demands of industrialization and development underscore the necessity for an efficient and scalable carbon capture and storage (CCS) methodology. Mineral carbonation of MgO presents itself as a promising solution due to its considerable theoretical capacity for CO2 adsorption. However, the sluggish kinetics of the carbonation process pose a significant challenge. Consequently, a comprehensive understanding of the structural and chemical alterations occurring during carbonation is imperative for material design. In this study, we conduct a thorough structural and chemical investigation of the MgO (sourced from different mine tailings) carbonation process using electron microscopic techniques. Our findings demonstrate that treating MgO with polar solvents enhances its degree of carbonation significantly, offering a promising avenue for improvement. Moreover, we observe a particle size dependency in MgO carbonation and note that the inclusion of additional materials, such as Si-based compounds, further accelerates the carbonation. Density functional theory (DFT) calculations provide insight into surface functionalization as a result of solvent treatment and its mechanistic effect on the origin of the enhanced carbonation of polar solvent-treated MgO, revealing a stronger interaction between CO2 and the treated MgO (100) surface as compared to the non-polar solvent treated surfaces. These discoveries showcase an alternative approach for enhancing MgO carbonation, thereby offering a potential method for sequestering atmospheric CO2 more effectively using mine waste rich in MgO.
Environmental significanceThe study presented in the manuscript describe the structural and chemical changes occurring during direct carbonation of MgO under normal atmosphere which helps in understanding the reaction mechanism and can further pave path for the future development of enhanced Carbon capture and storage (CCS) technologies and contributing to the goal of limiting global warming. The study leverages waste materials (mine tailings) rich in MgO for carbonation, which otherwise would remain unused. This reduces the need for mining new materials, lowering the environmental footprint associated with mining activities. By improving the kinetics of carbonation and utilizing low-cost materials, this approach could be scaled to capture significant amounts of CO2, thus contributing to broader carbon capture and storage (CCS) efforts. The product of the carbonation process results in the formation of a stable and non-toxic magnesium carbonate which has potential for industrial application. Further lowers down the need for landfilling of disposal of the mine tailings. |
1 Introduction
With the rapid development and ever-increasing energy demand in the past decade, the anthropogenic release of CO2 into the atmosphere has increased its concentration by 20 parts per million (ppm) to reach 421 ppm.1 This alarming rate of CO2 release has drawn global attention to the need for effective and scalable carbon capture and storage (CCS) methods.2–4 Current strategies involve capturing CO2 emitted from industrial flues (point source capture) before it is released into the atmosphere.4–9 The most commonly used adsorbents for CO2 capture from natural and flue gases are liquid amines, such as monoethanolamide, aqueous ammonia, or alkali hydroxide.10–14 However, liquid sorbents are limited and require high operational energy and cost.15 In addition to controlling CO2 emissions, there is also a need to develop methods for directly capturing CO2 from the atmosphere and storing it.In this regard, carbon mineralization has shown significant promise as a chemically and economically effective method of carbon sequestration.16–18 Carbon mineralization is a natural geological process in which minerals and industrial waste containing alkaline metals (Mg, Ca) interact with CO2 to form stable carbonates: MO + CO2 → MCO3, where M represents the divalent ions Mg2+ and Ca2+.19,20 Mineral carbonation offers potential advantages over other CCS methods, as the end products of mineralization are solids, environmentally friendly and have industrial applications. Typically, minerals rich in Ca and Mg in the form of oxides or silicates are used for carbon mineralization.21,22
Among reactive minerals for the carbonation process, MgO has gained significant attention due to its relatively high theoretical CO2 capture capacity, abundance, low cost, non-toxicity, and wide operating range.23,24 Another advantage of carbonation using MgO is its relatively low regeneration temperature compared to other mineral oxides such as CaO, which implicates lower energy requirement during regeneration. The negative ΔG for the carbonation of MgO indicates the thermodynamic feasibility of carbonation under ambient conditions. However, the carbonation of MgO is kinetically limited.25,26 In addition, the low atmospheric CO2 concentration and the formation of a passivating MgCO3 layer on the MgO surface result in a slow reaction rate for natural carbonation. The MgCO3 layer has been proposed to acts as a barrier, hindering CO2 diffusion to the MgO surface for further reaction to occur,27,28 once surface being carbonated, the lack of an effective primary active site further decrease the carbonation kinetics of MgO. The main challenge associated with carbon mineralization using these minerals is accelerating the kinetics, namely, achieving accelerated carbonation.29,30 The conversion of MgO to MgCO3 occurs through a series of chemical reactions, and it has been hypothesized that the thermodynamics and kinetics can be influenced by factors such as hydration (relative humidity (RH)), carbonation duration, and the presence of other chemical components.24,31 Further several efforts have been made to modify and improve the kinetics of MgO carbonation including varying the particle size, increasing the water content,16 or surface functionalization by incorporating metal salts such as K2CO3, KNO3, and NaNO3 or amine.32–35 Recent work by Bracco et al. has demonstrated that increasing relative humidity (RH) promotes the formation of a disordered hydrated reaction front on MgO during carbonation reactions.36 The morphology of hydration products over MgO have been shown to depend on the rate of dissolution and diffusion. The slow rate of diffusion and dissolution leads to the epitaxial growth of Mg(OH)2 and faster rates lead to the bulk growth.37–39 Surface functionalization has also been shown to enhance CO2 uptake. For instance, Alkadhem et al. reported that amine functionalization of MgO surfaces enables stronger interactions between amine groups and CO2, resulting in improved carbonation adsorption.35 Similar functionalization strategies have been explored for other materials, such as carbon molecular sieves, with positive outcomes for CO2 adsorption.40 Furthermore, combined theoretical and experimental investigations have revealed that organic polar groups can adsorb onto MgO surfaces and undergo partial dissociation,41–43 contributing to surface functionalization. This phenomenon offers potential for further optimization of carbonation processes. Despite these advancements, a comprehensive nano- and atomic-scale understanding of the structural and chemical transformations occurring during mineral carbonation remains elusive and warrants further investigation.
Industrial mine wastes rich in alkali ions are another inexpensive yet less exploited alternative material for mineral carbonation.44,45 Despite the existence of a cheap and cost-effective feedstock supply, extensive-scaled application of mine waste for carbonation suffers from low reactivity.46,47 Therefore, even a slight increase in reactivity of the mine waste could open a new paradigm for industrial-scale mineral carbonation. Thus, it is crucial to understand the mineral carbonation mechanism structurally and chemically under ambient conditions. Fundamental insight into the mechanism may help to accelerate mineral carbonation efforts.
The purpose of this work is to provide a mechanistic understanding of the structural changes that occur during the carbonation of surface functionalized MgO minerals for proxy mine waste using a combination of experimental and computational tools and have been compared with non-functionalized MgO. The use of organic polar molecules as a solvent provides an added advantage to just functionalize the surface without much alteration to the initial structure of MgO due to extensive hydration and provides evidence of surface functionalization effects for the carbonation process. The minerals were obtained directly from the mines and contained various impurities, primarily Si-based, which we found have variable impact on carbonation. While this complicates the analyses presented herein, we focus on generally observed trends.
2 Materials and method
2.1 Material synthesis
Two MgO samples, hereafter referred to as sample A and sample B, were prepared by calcinating MgCO3 rich minerals as a proxy for mine tailings obtained from the Calix mine in Australia and the Premier mines of Nevada, USA, respectively. The minerals obtained were calcined at 600 °C for 2 h to regenerate the MgO. TEM studies of carbonation kinetics and mechanism in MgO derived from mine tails were studied by preparing the TEM grid under two different conditions. One set of the TEM grid was prepared by sprinkling the dry powder on a carbon-coated copper grid, hereafter referred to as the “dry sample”, and the other was prepared by dispersing the MgO samples in methanol or isopropyl alcohol (IPA) using sonication, followed by drop-casting a drop of solution on TEM grids and drying, hereafter referred to as the “treated sample”. TEM grids were kept in an open-air container to undergo the natural carbonation process under ambient humidity and air, and were examined at different intervals under the electron microscope.2.2 Characterization
The phase and crystallinity of the initial samples were characterized using X-ray diffraction (XRD) Rigaku MiniFlex 6G theta-2theta vertical goniometer benchtop powder diffraction system. The morphology, microstructure, and structural evolution during the carbonation process were investigated using transmission electron microscopy (TEM). TEM imaging was performed using a JEOL F200, operating at 200 kV. Scanning transmission electron microscopy (STEM), energy dispersive spectroscopy (EDS), and electron energy loss spectroscopy (EELS) were performed using JEOL NEOARM, operating at 200 kV with a convergence angle of 27 mrad. STEM images were acquired with a 4 cm camera length and a probe current of 160 pA. EDS and EELS were acquired in dual spectrum imaging mode (SI) with a probe current of 160 pA to acquire sufficient signal without damaging the samples. A camera length of 2 cm and 0.1 eV per channel or 0.25 eV per channel dispersion was used to obtain EELS spectra with an enhanced signal-to-noise ratio. EELS spectra were acquired with a dual GIF camera and a K2-IS camera operating in the summit mode, provided by GATAN Inc., using a 5 mm aperture fitted at the Gatan imaging filter (GIF) entrance.2.3 Computational methods
All modeled systems were setup and managed using the atomic simulation environment (ASE).48 First-principles non-spin polarized density functional theory (DFT) calculations were performed with the Quantum espresso software package,49,50 using the Bayesian error estimation exchange correlation functional with van der Waals correlation (BEEF-vdW).51A plane-wave basis set was used to expand the Kohn–Sham wave functions, with 500 and 5000 eV kinetic energy and density cutoff, respectively. Ultrasoft pseudopotentials52 were used to approximate core-electrons. The bulk lattice constant of the cubic MgO crystal53 was found to be 4.282 Å, which is close to the experimentally reported values of 4.21–4.24 Å.54,55 A 4 × 4 × 1 (4 × 4 × 4) Monkhorst–Pack k-point grid54 was used for all surface (bulk) calculations. The (100) and (110) surfaces of MgO were modeled using a 2 × 2 repeated cell in the x- and y-directions, a 4-layer slab, and a 10 Å vacuum spacing between periodic images in the z-direction. The atomic positions of the two lower slab layers were constrained to their bulk lattice coordinates, allowing only the atoms in the top two slab layers to relax. The individual methanol, isopropanol (IPA), hexane, water, CO2, and H2 molecules were modeled in a supercell with 10 Å vacuum in all directions since these energies are needed as reference energies to obtain the thermodynamics (see details below).Herein, we will computationally model the reactivity and carbonation of the (100) and (110) MgO surface facets which are the two predominant surface facets observed experimentally using XRD (see Fig. S4 in ESI†). Hence, we assume that the other less pronounced surface facets, e.g. the (111) and (311) surfaces, will have minimal impact on the overall carbonation performance. The treatment of the material with the polar and non-polar solvents was hypothesized to modify the termination of the (100) and (110) surface facets through adsorption of the intact or partially dissociated solvent molecule, which could result in different smaller species adsorbed on the surface. The different species are modeled to adsorb on the bulk-cleaved surfaces and the corresponding Gibbs free energy of adsorption is defined in eqn (2). This assumes that the surfaces are in fast equilibrium with both carbon and oxygen elements, hence the change in solvent chemical potential only affects the chemical potential of hydrogen. The considered dissociated species include individual sub-components of the solvent molecules and combinations of multiple sub-components. Specifically, this analysis included the following single species H, O, OH, CHx, CHxO, and H2O as well as mixed combinations including H + OH, H + CHxO, OH + CHx at coverage 0.125 ML and 1 ML (see legend of Fig. 8). Here, x is either 2 or 3, depending on the nature of the solvent reservoir. The monolayer coverage is defined by an (or a pair of) adsorbate per MgO unit. We further assume that the entropic contributions of the crystal structures and the volume changes are negligible. The Gibbs free energy of adsorption is given by
| ΔGads = ΔFads + PΔV − nsolvμsolv ≈ (Esys − Eslab − Gsolv) − nsolvμsolv | (1) |
 | (2) |
 | (3) |
| Gsolv(T, P) = Hsolv(T) − TSsolv(T, P), | (4) |
After the treatment of the material with the solvents, which could result in a functionalized material, the remaining solvent is retrieved from the system and the solid–liquid equilibrium transforms into a solid–air equilibrium. For the new solid–air equilibrium, the dissociatively adsorbed species no longer obey boundary condition with the treatment solvent reservoir, leading to a possibility for these adsorbed species to desorb from the surface. To assess the stability of the adsorbed dissociates species at the new equilibrium, we perform a desorption energy analysis. The desorption energy of a single/pair of the adsorbate species for a given coverage is defined by:
| ΔEdes = En−1 − En + ∑ Ei, | (5) |
Finally, to investigate the carbonation thermodynamics of the solvent-modified (100) and (110) MgO surfaces, we performed a comprehensive computational screening of CO2 adsorption on multiple possible adsorption sites and initial configurations of the molecule.57 These results are compared with the bare untreated surfaces, that is, clean bulk-cleaved surfaces. One specific adsorption map for the bare MgO (100) surface is presented in Fig. 1. The adsorption maps for other differently faceted and terminated surfaces can be found in Fig. S12–S18 in the ESI.† The CO2 adsorption energy is defined by:
| ΔEads = ECO2+slab − Eslab − ECO2, | (6) |
 | ||
| Fig. 1 Adsorption site and orientation map for considered starting configurations for CO2 adsorption on the clean bulk-cleaved MgO (100) surface (top-down view). Mg and oxygen atoms are represented in green and red, respectively. | ||
3 Results
3.1 Carbonation of MgO nanoparticless in dry as-produced conditions
Morphological analyses of the dry samples in their original condition (day-0) were imaged (Fig. 2 and S1†), revealing clusters of crystalline MgO nanoparticles. The high-resolution image of MgO shows the predominant (100) surface of MgO. Periodic microscopic observations were conducted to track the carbonation process of the dry samples. After 120 days, no structural changes associated with carbonation were observed in the dry sample A and aggregates similar to those observed on day-0 were still present (Fig. 2(c and d)). Electron energy loss spectroscopy of the dry sample A confirmed the initial findings from the STEM images. Initially, the freshly dispersed samples exhibited strong O K edge peaks at 538.8 eV, 547 eV, and 557.5 eV, which are characteristic of MgO, with a minor impurity peak of Ca, and have very small quantity of adventitious carbon, indicating the samples were clean. After 120 days, sample A displayed no structural changes and the strong crystalline peaks of the O K edge for MgO remained unchanged. Adventitious carbon was found on the sample surface on day-120, as evidenced by the presence of a broad amorphous C K edge, contrary to what was observed for the as-prepared condition. A similar carbonation behavior was also observed for dry sample B (Fig. S1†). However, in contrast to the sample A, the dry sample B exhibited evidence of water adsorption after 6 months, as identified through EELS acquisition, showing an additional pre-peak around ∼531.6 eV near the O K edge. The pre-peak before the main O K edge can be corroborated to the presence of oxygen molecules from the decomposition of H2O or (–OH) groups adsorbed on the MgO surface.58,59 These observations indicate that untreated MgO minerals exhibit slow carbonation kinetics. | ||
| Fig. 2 High-angle annular dark-field scanning transmission electron micrographs of dry-sprinkled MgO sample A (a–c) day-0, (d and e) day 120, and (f) corresponding EELS spectra (inset: magnified O K edge). | ||
3.2 Carbonation of MgO nanoparticles dispersed and sonicated in polar solvents: methanol and IPA
It has been suggested that carbonation kinetics can be enhanced by adding surface functionalization, moisture/humidity, seed nuclei or the presence of secondary minerals.60 To understand the impact of surface functionalization on carbonation, we prepared MgO nanoparticles functionalized with different functional groups by treating them with different solvents. The crystalline MgO sample A and sample B were dispersed in a polar solvent (methanol or IPA), drop-casted onto a TEM grid, dried and stored in an open container similar to the dry-sprinkled MgO. The carbonation of MgO dispersed in polar solvent and sonicated proceeds sporadically for well-dispersed MgO in contact with ambient air as it converts into amorphous MgCO3 (Fig. 3). The amorphous carbonate that is formed has a very smooth and round surface, consistent with previously observed amorphous spherules formed from CaO carbonation.61 The area occupied by the newly formed MgCO3 is approximately the same as the area initially occupied by MgO before undergoing carbonation, which indicates slight or no area expansion during carbonation (Fig. S2†). In addition, the general shape of the original MgO aggregates is maintained throughout the aggregate's carbonation and after an aggregate has fully carbonated with the collapse of the individual MgO nanoparticles to one large grouping of carbonates. The amorphous carbonate features initiate and grow from the surface of the crystalline MgO aggregates. As the carbonate grows, the MgO gets depleted, eventually leading to the complete conversion of the original MgO aggregates. The EELS signature of the O K edge of the aggregates can also be used to characterize the formation of carbonate phase. The EELS signature for the O K feature of carbonate is different from the one for MgO, as shown in the insets of Fig. 3. | ||
| Fig. 3 Representative images of MgO carbonation. (a) MgO was prepared by calcinating mined MgCO3 and freshly dispersed on a TEM grid (day-0), inset: O K EELS spectra. (b) MgO aggregate dispersed on a TEM grid undergoing carbonation, inset: selected area electron diffraction (SAED) pattern from the carbonated and uncarbonated region. (c) Fully carbonated MgO aggregate (MgCO3) dispersed on a TEM grid, inset: O K EELS spectra of fully carbonated aggregates. | ||
The formed carbonate phase are electron beam sensitive. If amorphous carbonate phase is exposed to the electron beam in vacuum conditions, it rapidly bubbles off the CO2 and de-carbonates, regaining a nanocrystalline MgO structure (Fig. S3†). Although the beam induced MgO has similar crystal structure to the pre-carbonated MgO, its morphology differs, and the newly formed MgO has small crystallite sizes (<10 nm). This process is fast and complete conversion is observed in <10 s unless the electron beam dose and dose rate are very low.
A qualitative difference in carbonation was observed for the polar solvent treated sample A and sample B. The carbonation process is faster for the treated sample A (Fig. 4). The vast majority of the treated sample A was fully converted to the amorphous carbonate even after only one month, while in the case of treated sample B, most aggregates were not fully converted during the same carbonation duration. To dive deeper into difference in the degree of carbonation of treated sample A and sample B, XRD analysis was performed (Fig. S4†). XRD analysis showed that both samples are structurally periclase and exhibit similar levels of preferential orientation, favouring the (100) facets, consistent with high-resolution TEM imaging (Fig. S5†). This is one of the primary facets that was used in our computational models. The broader XRD peaks for sample A as compared to sample B depict the particle size difference between sample A and sample B. Using the Scherrer equation τ = Kλ/(cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θβ) on the intense 200 reflection of the XRD pattern, an average particle size was calculated, where τ is the crystallite size, K is the shape factor β is the full width at half maxima of the peak and θ is the Bragg angle. An average particle size of 12 nm was found for sample A, compared to 34 nm for sample B. The estimated particle size is consistent with the observations from TEM analysis of freshly dispersed sample A and sample B (Fig. S6†). In summary, sample A, with smaller particles of the two samples, shows more carbonation when treated with the polar solvent. While both MgO samples carbonated sporadically and quickly after dispersed in methanol or IPA (Fig. 3 and 4) and drop-casted onto a TEM grid, neither showed carbonation when left as untreated powder form in contact with air for an extended period (Fig. 2 and S1†).
θβ) on the intense 200 reflection of the XRD pattern, an average particle size was calculated, where τ is the crystallite size, K is the shape factor β is the full width at half maxima of the peak and θ is the Bragg angle. An average particle size of 12 nm was found for sample A, compared to 34 nm for sample B. The estimated particle size is consistent with the observations from TEM analysis of freshly dispersed sample A and sample B (Fig. S6†). In summary, sample A, with smaller particles of the two samples, shows more carbonation when treated with the polar solvent. While both MgO samples carbonated sporadically and quickly after dispersed in methanol or IPA (Fig. 3 and 4) and drop-casted onto a TEM grid, neither showed carbonation when left as untreated powder form in contact with air for an extended period (Fig. 2 and S1†).
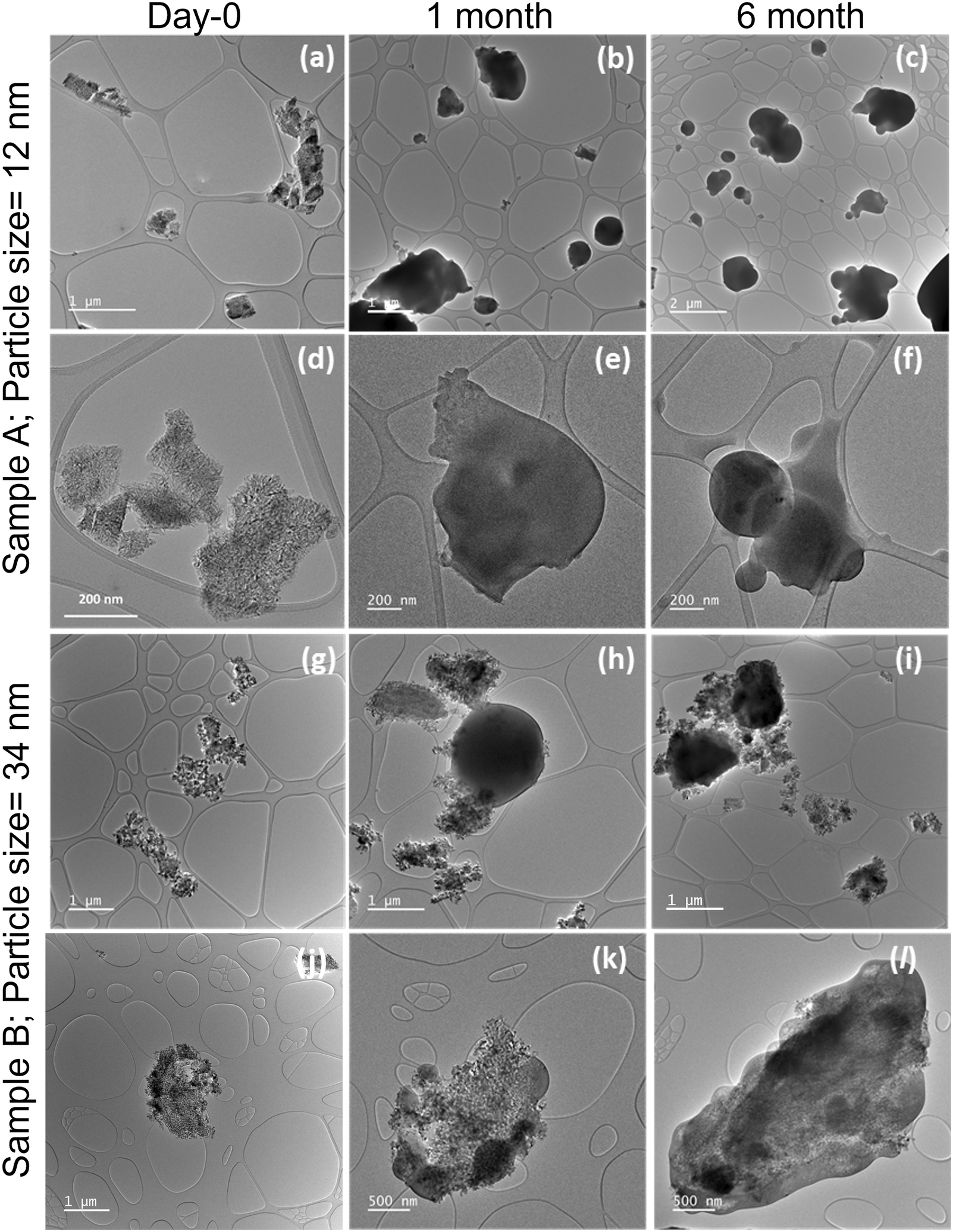 | ||
| Fig. 4 Time-evolution of two MgO samples with different average particle size. (a and d) Freshly dispersed sample A; (b and e) sample A after 1 month of dispersion on the TEM grid; (c and f) sample A after 6 months of dispersion on TEM grid; (g and j) freshly dispersed sample B; (h and k) sample B after 1 month of dispersion on TEM grid; (i and l) sample B after 6 months of dispersion on the TEM grid. | ||
The carbonation of MgO aggregates does not happen at the same rate for all MgO aggregates. Likewise, it does not occur homogeneously over each aggregate. While certain aggregates show complete conversion after two months some remain completely uncarbonated (Fig. 5(a and b)). The aggregates that remain as MgO and do not carbonate are small. For example, the average aggregate size of uncarbonated MgO for freshly dispersed is <1 μm (n = 30). After two months, virtually all large aggregates have carbonated to MgCO3 (Fig. 4 and 5(a–f)). The average aggregate size of the remaining uncarbonated material is only 100 nm (n = 30), showing that only very small aggregates are left uncarbonated (Fig. 4 and 5(a)). In contrast, the large aggregates exhibit extensive carbonation in all cases. Furthermore, the uncarbonated aggregates typically exhibit significant evidence of adventitious carbon at their surface, which is in contrast to the large MgO aggregates that have undergone carbonation and seldomly have surface adventitious carbon (Fig. 5(c–f)). These findings indicate that larger MgO aggregate sizes are more prone to convert to MgCO3 at the tested conditions.
 | ||
| Fig. 5 (a) TEM image of amorphous carbonate and smaller uncarbonated aggregates marked in circles as region 1 and 2, respectively; (b) SAED pattern from the marked region 1 and 2 of (a); (c and d) STEM image of uncarbonated aggregates and corresponding C K EELS spectra showing the adventitious C features (e and f) STEM image of carbonated aggregates and corresponding C K EELS spectra from the marked region as 1 and 2; (g–i) STEM image, C K and O K EELS spectra from carbonated and uncarbonated regions for an MgO aggregates. | ||
The carbonation of MgO can also be influenced by the presence of certain impurities which could act as hydrating agents by adsorbing the moisture and maintaining the hydration of the MgO. The mine-tailed samples have SiO2-based impurities, which when dispersed in solvent, surround the MgO in various regions and can facilitate the carbonation of MgO in those aggregates. Fig. S7† shows the STEM-EDS maps of the solution dispersed sample A showing the presence of SiO2-based impurities layering over the MgO aggregates which were aggregated in dry powder. The aggregates that carbonated more quickly have a significant fraction of SiO2-based impurities in the nearby area and have attracted moisture (Fig. 5(g–i)). The presence of moisture in the carbonating MgO aggregates can be identified from the EELS spectra of carbonated and uncarbonated regions (Fig. 5(g–i)). The carbonated area shows a pre-edge peak in O K edge, corresponding to the gaseous release of O2 due to presence of either H2O or (–OH).58,59 At the same time, the uncarbonated MgO regions have less Si in those aggregates (Fig. S7†). The observed SiO2-modified carbonation indicates that the hygroscopic nature of SiO2 can also further affect the carbonation of the MgO aggregates by maintaining enough hydration around the aggregates. MgO is shown to form metal-silicate hydrate upon reaction with SiO2-based mineral in presence of water further reinforcing our observation.62,63 However, the exact quantification ratio of MgO to SiO2 needed to maximize the carbonation was not achieved as the amount of SiO2 keept changing due to the surface diffusion of SiO2 over the aggregates. The hygroscopic nature of SiO2 can be further supported by observing formation of amorphous regions between the aggregates after an extended period (Fig. S8†). A layering of SiO2 was observed between the MgO aggregates, which was absent in the freshly prepared samples.
The impact of sonication is indirectly related to the interaction of solvent with the MgO nanoparticles. A drop of IPA was added to the dry-sprinkled MgO nanoparticles and examined over an extended period. The carbonation kinetics were found to be slower as compared to the sonication-induced dispersion. Very few regions of MgO show carbonation, presumably due to insufficient solvent interaction with MgO and hence small quantities of functionalization which could influence the reaction. Most areas showed uncarbonated MgO with adventitious carbon (Fig. S9†).
The influence of solvent polarity was investigated by dispersing MgO nanoparticles in the non-polar solvent hexane. The hexane-dispersed MgO nanoparticle aggregates showed no structural changes and retained their high crystallinity following dispersion, as confirmed by electron energy loss spectroscopy (EELS) analysis of the O K-edge (Fig. S10(a, b and e)†). No adventitious carbon contamination was detected initially. However, upon exposure to air for approximately one month, the MgO aggregates exhibited significant adsorption of adventitious carbon, as evidenced by the C K-edge EELS spectra (Fig. S10(e)†). Prolonged air exposure of hexane-treated samples led to partial carbonation, although some uncarbonated MgO remained even after two years of atmospheric contact (Fig. S10(f–h)†). The non-polar nature of hexane, lacking functional groups conducive to surface interactions, does not provide any additional benefit for carbonation. On the contrary, it may hinder the carbonation process by facilitating the adsorption of adventitious carbon, leading to surface passivation. Interestingly, unlike samples treated with polar solvents, hexane-treated MgO surfaces were devoid of silica (SiO2) deposition. In contrast, polar solvent-treated samples typically exhibited diffuse SiO2 based layers surrounding the MgO aggregates, as shown in Fig. S11.† This difference underscores the role of solvent polarity in surface chemistry and structural interactions.
3.3 Understanding MgO carbonation at the nanoscale level
To gain a deeper understanding of the MgO carbonation process, we employed electron energy loss spectroscopy (EELS) to analyze the evolution of MgO aggregates into MgCO3 (Fig. 6). The EELS images revealed a clean surface of MgO with well-defined crystal edges (Fig. 6(a)). To facilitate carbonation, we dispersed the MgO on a TEM grid and introduced air by deliberately exhaling humidified CO2 onto the grid. High concentration of CO2 with humidity have been shown to enhanced the carbonation process64,65 and, in less than 1 hour, an amorphous carbonated layer was observed at the surface of freshly dispersed sample A aggregates (Fig. 6(b)). The freshly dispersed MgO aggregates undergoing carbonation initially did not contain any carbon on their surface. However, during the carbonation process, a clear EELS carbonate signature emerged in the amorphous carbonated materials on the surface of the MgO aggregates, indicating the uptake of CO2 during the carbonation process (Fig. 6(c)). The carbon K edge spectra further revealed an additional peak at approximately ∼287 eV, in addition to the conventional carbonate carbon K edge. This additional peak can be attributed to C–OH groups, implying the formation of bicarbonate species along with the carbonation process. | ||
| Fig. 6 Carbonation of MgO upon being exposed to exhaled air. (a) Freshly dispersed MgO Calix with clean surface. (b) Freshly dispersed sample A (MgO Calix) upon being exposed to human exhaled air. (c) EELS C-edge for region t 1 (fully carbonated) and region 2 (front of carbonation) in panel (b). | ||
In Fig. 7, we observe the aggregates undergoing the natural carbonation process, and the point EELS spectra of the carbon K edge display a systematic variation between carbonate and bicarbonate, indicating the possible formation of carbonates via a transition zone of bicarbonates. The fully carbonated region of the aggregates shows characteristic peak features of carbonates. However, as we move towards the uncarbonated MgO there is an increase in the intensity of the additional peak corresponding to bicarbonates, suggesting a concentration gradient of these different species within the aggregates. Continuing inward, the carbon peak entirely vanishes in the MgO region of the aggregates, as MgO does not inherently contain carbon before the carbonation process starts. This finding supports the notion that the carbonation process progresses via a carbonation front, which starts at the surface of the MgO aggregate and propagates inward into the bulk of the aggregate. Overall, the EELS analysis provides critical insights into the carbonation mechanism of MgO, demonstrating the simultaneous formation of bicarbonate species and the subsequent conversion to carbonates. The identification of a carbonation front, that starts at the surface of the MgO nanoparticle aggregate and propagates inward, further contributes to our understanding of the spatial dynamics of the process.
 | ||
| Fig. 7 Carbonation front of an MgO sample. (a) Low-magnification carbonation front of a Calix MgO sample. (b) High-magnification carbonation front of a Calix MgO sample. (c) EELS spectra from the MgO through the carbonation front to the MgCO3 (1–4). | ||
3.4 Solvent-modified MgO surface functionalization
To understand the possibility of surface functionalization depending on the solvent and its effect on the experimentally enhanced carbonation process, we performed DFT calculations. We modeled the stability of functionalized MgO (100) and (110) surfaces as a result of MgO being treated with and in contact with a polar (methanol, IPA) and non-polar (hexane) solvent. Fig. 8(a–c) shows the thermodynamically favored surface termination and coverage of different solvent derived species, which taken together define the functionalization, when MgO (100) is exposed to the solvent as a function of the chemical potential for each solvent. The changes in Gibbs free energies for a full monolayer coverage (solid lines in Fig. 8) were normalized by the number of adsorbate pairs so that the average energy of adsorbing each pair was obtained. The chemical potentials corresponding to the saturated vapor are shown by the dashed black line. Near the saturation conditions, the termination components vary in response to different treatment solvents. For a methanol-treated MgO (100) surface, a low coverage of –H and –OH pairings is the most stable dominating functionalization. For an IPA-treated MgO (100) surface, the termination components are diverse, consisting of a mixture of –H, –OH, and –CHxO species. On the other hand, the hexane-treated MgO (100) surface prefers to be un-functionalized as none of the solvent derived species stabilize it. | ||
| Fig. 8 Stability phase diagrams for an MgO (100) surface exposed to different solvents (a) methanol, (b) isopropanol, and (c) hexane, resulting in adsorption of different solvent-derived species as indicated by the legend. (d) Desorption energetics of the methanol/IPA-treated MgO (100) surface in equilibrium with vacuum. | ||
Fig. 8(d) shows the desorption thermodynamics of the adsorbed solvent-derived species from the MgO (100) surface once the remainder of the solvent is removed post treatment. The MgO (100) surface readily loses the adsorbed species when they are not in contact with the solvent reservoir (only exception is in the scenario of sparsely distributed (0.125 ML) H–OH pairings (ΔEdes of +0.231 eV)). It is worth noting that 0.125 ML –OH species (without H) alone are unstable (−1.983 eV), highlighting the pivotal role of H species as stabilizers. From Fig. 8(a and b), we conclude that the methanol-treated MgO (100) surface maintains its predominant termination when exposed to the air, while the IPA-treated MgO (100) surface transforms from a multi-component system to a surface covered predominantly with H-OH paired species due to their weaker propensity for desorption, suggesting the nature of irreversible adsorption of H–OH pairings on the MgO (100) surfaces.
Similarly, decorating the (110) surfaces with H–OH pairings originating from a methanol or IPA treatment is also readily thermodynamically favored (see Fig. S19(c and d)†). However, the H–CHxO pairings are competitively adsorbing, and this trend is more significant for the IPA-treated case. The desorption analysis shows both these adsorbate pairings are stable on the MgO (110) surfaces (ΔEdes of +1.951 eV for H–OH and +0.541 eV for H–CHxO). The irreversible binding of the H–CHxO species indicates the MgO (110) surfaces are likely susceptible to adventitious carbon. It is also worth noting that the hexane-treated MgO (110) surface accumulates CHx groups, implying the potential of MgO (110) surfaces for attracting adventitious carbon that could hinder carbonation.
3.5 Mechanistic carbonation analysis on functionalized MgO surfaces
CO2 adsorption was studied on bare untreated MgO (100) surface and functionalized methanol/IPA-treated MgO (100) surfaces (terminated with 0.125 ML of H–OH pairings). In addition, these conditions were compared to the scenario where explicit water molecules are present, as to simulate the moisture component in the air. The complete list of considered adsorption configurations and energies are given in Tables S1–S8.†Fig. 9 summarizes the lowest adsorption energies specific to each terminated surface. On the bare MgO (100) surface, we only found CO2 interacting weakly with the surface in a physisorbed state (ΔEads of −0.292 eV). On the methanol/IPA-treated MgO (100) surface, there were three possible stable CO2 adsorption states: CO2 adsorbs with a linear geometry (−0.609 eV) (Fig. S20(a and b)†); it could be adsorbed in a bent geometry forming a C–O bond with a lattice oxygen and transforming into a carbonate-like species (−0.852 eV) (Fig. 10(c and d)); or it could form a C–OH bond with a surface OH group and become a bicarbonate-like species (−0.714 eV) (Fig. 10(e and f)). To verify if CO2 predominantly interacts with the surface and the adsorbed species and not with other CO2 molecules as present through the mirrored images due to the periodic boundary conditions, we performed the same calculation on an expanded (4 × 4) supercell. We find similar adsorption results with a value of −0.894 eV compared to −0.852 eV for carbonate-like species and −0.744 eV compared to −0.714 eV for bicarbonate-like species formation. This confirms that the dominating interactions are primarily a consequence of adsorbate–surface interaction and not adsorbate-adsorbate interactions. We next investigated if the improved CO2 adsorption thermodynamics can be attributed to the proximity of the adsorbing CO2 molecule to the H–OH functional groups. To do this, we adsorbed the CO2 as far from OH as possible in the 4 × 4 supercell. The resulting ΔEads is −0.290 eV, which is very close to the adsorption value on a bare untreated surface (−0.292 eV). The redistribution (gain and loss) of electrons of each atom caused by the carbonation processes is shown in Fig. 10 (isosurface level was set to 0.003 e Å−3 for all plots). The electron charge density analysis confirms the capability of CO2 chemisorbing on the methanol/IPA-treated (100) surface but not on the bare surface. Two viable pathways for carbonation on a methanol/IPA-treated MgO (100) surface (Fig. 10): (1) H first recombines with OH to form a water molecule, and then interacts with O in CO2 and stabilizes its structure in a bent configuration, and (2) OH stays dissociated from H, and directly forms a C–OH bond. In both pathways, OH groups are indispensable, but the importance of isolated H is unclear. Therefore, we performed additional calculations to isolate OH from its H counterpart and performed an adsorption analysis on the surface covered with OH only. The calculated ΔEads values are −0.469 eV for linear CO2, −0.447 eV for carbonate-like species, and −0.810 eV for bicarbonate-like species (see Fig. S23†). Notably, forming carbonate-like species becomes less favored when the surface lacks recombined water. Instead, the isolated OH group allows a surface Mg to rumple and then facilitates stabilizing CO2, however, this effect has a smaller influence on the adsorption than water. On the other hand, OH likely has more freedom to interact with C without electron transfer with H, hence ΔEads is more negative. We have also modeled CO2 adsorption on the surface with full (1ML) H–OH coverage for comparative purposes, even though this starting surface is not thermodynamically favored. The ΔEads is −10.394 eV (or −1.299 eV per H–OH pair), but no evidence for chemisorption is found (see Fig. S20(c and d)†). Instead, the very negative energy is due to surface relaxation and not adsorption.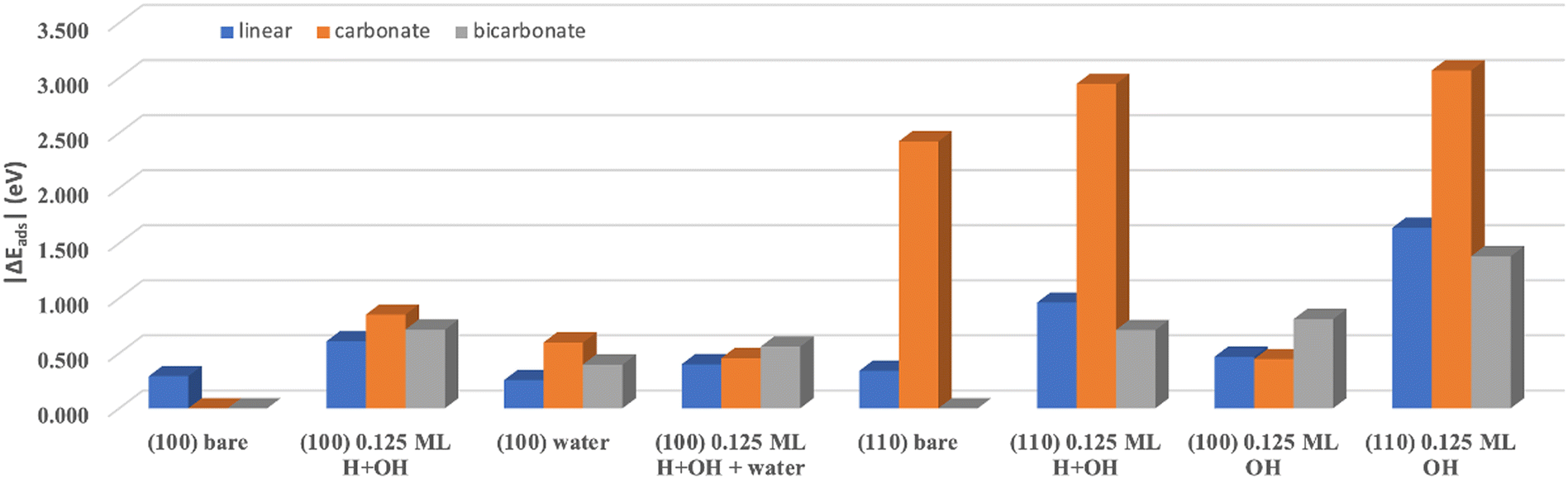 | ||
| Fig. 9 The lowest CO2 adsorption energy on bare and functionalized (100) and (010) surfaces with the CO2 in different configurations: linear, carbonate and bicarbonate. | ||
 | ||
| Fig. 10 Visualized charge difference upon CO2 interacting with an MgO (100) bare surface (a) top-down view, (b) side view, methanol/IPA-treated surface with carbonate-like species formation (c) top-down view, (d) side view, methanol/IPA-treated surface with bicarbonate-like species formation (e) top-down view, (f) side view. | ||
When a system is exposed to air, water can participate as a reactant and is particularly critical for lowering the carbonation energy barriers.66 To examine the role of water, we assume a 1:1 H2O:CO2 ratio. The bare MgO (100) surface was found to have weak interaction with a water molecule (ΔEads of −0.486 eV), while the water interaction with the methanol/IPA-treated (100) surface was much stronger (−1.024 eV). This could be due to the hydrogen bonding between water and surface OH groups. With these adsorbed water configurations on the bare MgO (100) surfaces, adsorption of CO2 was modeled. The resulting observed CO2 configurations were both carbonate-like (ΔEads of −0.598 eV) and bicarbonate-like species (−0.398 eV), in addition to linearly adsorbed CO2 (−0.257 eV) (see Fig. S21†). For carbonation on the methanol/IPA-treated MgO (100) surfaces with explicit water, we find that the adsorbed linear CO2 is more favorable (−0.400 eV), but the thermodynamics also shift toward favoring the formation of bicarbonate-like species (−0.560 eV) over carbonate-like species (−0.455 eV) (see Fig. S22†). Prior to the carbonate-like species formation, H and OH recombination takes place first and subsequently triggers the carbonation. Although the presence of H–OH pairings seemingly has no significant effect in lowering ΔEads as long as molecular water exists, the relatively unfavorable thermodynamics (−0.486 eV) for adsorbing water directly on a bare MgO (100) surface implies diminished feasibility in initiating the carbonation process. More importantly, bicarbonate-like species ΔEads is much less negative when a water molecule is pre-adsorbed in contrast to the surfaces without adsorbed water molecules but with H–OH pairings. This is because there is an energy cost for breaking the H–OH bond in water to subsequently form a C–OH bond with CO2. This highlights the role of an OH surface species that outperforms a molecular water for carbonation. Note that bulk water does not favor a spontaneous dissociation over the bare MgO (100) surfaces (see Fig. S19(a)†). In other words, treating the MgO (100) surface with water wouldn't result in the same favorable conditions (H–OH pairs) with enhanced carbonation as polar organic solvents can achieve.
The adsorption energy calculations were repeated on the MgO (110) surfaces. On the bare MgO (110) surface, the ΔEads was found to be much more negative (−2.424 eV) than in any other cases. The superior reactivity can be attributed to the presence of surface four-coordinated oxygen (O4c) site, which readily forms a C–O bond with CO2 and turns into a carbonate-like species. The superior thermodynamics likely results in an excessively reactive surface, making it susceptible to passivation by unreactive carbon deposition.28,36,67 The O4c sites are likely occupied, hindering further carbonation on the bare (110) surface. Finally, we modeled the interaction between organic groups and the O4c sites by studying the adsorption behaviors of H–CHxO groups. When the (110) surface is covered with 0.125 ML H–CHxO, the ΔEads becomes less negative with respect to the distance between the CO2 adsorption site and the position of CHxO. When CO2 adsorbs on a lattice O that is 3.195 Å away from CHxO, ΔEads is −1.673 eV. When the distance is 4.854 Å, the ΔEads value becomes −2.134 eV, and at the distance of 5.121 Å we find ΔEads is −2.443 eV. Hence, we conclude that the presence of CHxO screens the surface toward CO2 adsorption, but the effect drops significantly as the proximity increases. When the surface is fully covered with H–CHxO, there is only physisorption (−0.223 eV). This suggests a monotonic decrease in the CO2 adsorption thermodynamics with respect to the coverage of H–CHxO pairings. Conversely, the presence of H–OH pairings could enhance the interaction between CO2 and surface species, similar to (100) surfaces. The ΔEads of adsorbing linear CO2 and forming carbonate-like species improves from −0.341 eV to −0.960 eV and from −2.424 eV to −2.945 eV, respectively. Regardless, we expect a smaller tendency towards carbonation as the ΔEads values indicate due to the aforementioned passivation effect. Because of the passivated (110) surfaces, no carbonation was experimentally reported. With methanol/IPA treatment, we have insufficient experimental evidence to show if carbonation has occurred on (110) surfaces. If so, it is likely through the formation of bicarbonate-like species (−0.711 eV), which is less affected by the passivation effect.
4 Discussions
The findings of our study show that surface functionalization of MgO nanoparticles plays a crucial role in facilitating the carbonation process of proxy mine tailings rich in MgO. We observed that the as-produced dry MgO powder samples showed no signs of carbonation when exposed to air, whereas the carbonation process accelerated significantly once the MgO nanoparticles were dispersed in a polar solvent, sonicated, dried, and then exposed to air. Despite the thermodynamic feasibility of carbonation, the slow carbonation rate observed in the dry sprinkled MgO samples can be attributed to the low CO2 concentration in the atmosphere and inadequate water content.24,68 When MgO contacts with methanol/IPA it spontaneously forms surface reactive H–OH groups, which can greatly alter the reaction with CO2 to form MgCO3.69–71 On the other hand, dispersing the minerals in a non-polar solvent like hexane led to the accumulation of adventitious carbon, hindering the access of CO2 and H2O to the surface of MgO. Furthermore, our Rietveld analysis revealed that both samples exhibited a similar extent of preferential orientation or texture, favoring (100) facets in both cases, which is consistent with our TEM analysis. The particle size difference between sample A and sample B can corroborate the difference in the kinetics of the MgO samples. The carbonation process of MgO is surface-driven, with smaller crystal sizes showing more pronounced effects. However, we couldn't experimentally identify a specific initiation site, such as a crystal corner or edge, that could propagate the reaction. Morphological observations also supported the surface-driven process, as early stages of carbonation consistently formed discrete surface features on the MgO aggregates. Unfortunately, imaging the carbonation reaction front was very challenging due to rapid beam-induced changes in the MgCO3 area. Nonetheless, our results suggest that the reaction front is not homogeneous, pointing towards mass-diffusion limitations in the conversion process.72 This is supported by the fact that the shape of an aggregate is preserved during and after the conversion to MgCO3, indicating that the reaction occurs entirely in the solid state without dissolution and re-precipitation. Throughout our investigations, we detected the presence of elements like Si, Ca, Fe, and F. Ca was well mixed within the MgO aggregates, while Si-based impurity aggregates spread out when dispersed in a polar solvent. The hydrophilic nature of SiOx may further promote the carbonation of MgO aggregates by maintaining a hydrating environment around the MgO particles.73 The initial solvation step is crucial in enhancing carbonation kinetics, as the surface polarity increases with the adsorption of the –OH group, attracting moisture from the ambient atmosphere and promoting carbonation.24 This corroborates the enhanced carbonation achieved for MgO treated with methanol/IPA, which, as discussed above, induces the formation of surface Mg–OH groups.71 EELS analysis of the carbonating aggregates revealed that the transformation from MgO to MgCO3 involved the formation of mixed carbon-containing phases rather than a direct conversion. Additionally, during the transition, a peak corresponding to bicarbonate ion was detected, indicating that bicarbonates are an intermediate species in the conversion of MgO to MgCO3. Further our DFT calculations show that functionalization is possible but dependent on the solvent being polar vs. non-polar. Importantly, the DFT findings also confirm our experimental observation of enhanced carbonation over solvent-treated i.e. functionalized MgO. Specifically, partially the H–OH covered MgO (100) surface shows an increased propensity towards water and CO2 moiety and subsequently undergoes carbonation process. The presence of H and OH on the surface together with explicit H2O molecules stemming from the atmosphere results in the formation of bicarbonate along with the carbonate, which is in agreement with the experimental observation. Overall, our study demonstrates the significance of surface functionalization, particle size, and other impurities in influencing the carbonation process of MgO. These findings have implications for understanding and optimizing the carbonation potential of mine tailings rich in MgO for environmental applications.5 Conclusion
We conducted a comprehensive study on the natural carbonation process of MgO derived from Mg-ion-rich mine tailings. While the carbonation of dry MgO at ambient atmospheric conditions was found to kinetically slow although thermodynamically feasible, it can be significantly improved by dispersing in a polar, OH-containing solvent. Notably, we observed that the carbonation process of MgO is size-dependent, with smaller particle size aggregates of MgO carbonate more rapidly than larger ones. Moreover, we discovered that the presence of hygroscopic minerals, such as SiOx, further enhances the carbonation process by maintaining a hydrated atmosphere surrounding the MgO aggregates. In our investigation, we utilized electron energy loss spectroscopy (EELS) to study the carbonation mechanism and the intermediates involved. We found that the carbonation of MgO does not occur directly; instead, it forms mixed phases of carbon-containing substances. Particularly, the naturally carbonating aggregates revealed that the carbonation takes place through the formation of an intermediate bicarbonate phase. DFT simulations show existence of functionalized surfaces stemming from polar solvent treatment and further strengthen our hypothesis of enhanced carbonation of these modified MgO surfaces. A favorable decomposition of polar solvent over MgO (100) to partially cover the surface with H and OH. Adsorbed isolated H and OH are shown to be crucial for further adsorption of both H2O and CO2 to undergo the carbonation process. These findings shed new light on the carbonation behavior of MgO and provide valuable insights into the role of particle size and hygroscopic minerals in enhancing this process. Our study contributes to a better understanding of the natural carbonation potential of MgO, which may have significant implications for various applications in carbon sequestration and environmental remediation.Data availability
Data supporting this study have been included as a part of ESI† file.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Vagelos Institute for Energy Science and Technology (University of Pennsylvania) for support through a seed grant. This work was carried out in part at the Singh Center for Nanotechnology, which is supported by NSF National Nanotechnology Coordinated Infrastructure Program under grant NNCI-2025608. Additional support from the NSF through the University of Pennsylvania Materials Research Science and Engineering Center (MRSEC) (DMR-1720530; DMR-2309043) is acknowledged. A. V. acknowledges the Canadian Institute for Advanced Research (CIFAR) for support through the The Accelerated Decarbonization Program.Notes and references
- T. P. X. Lan and K. W. Thoning, Trends in globally-averaged CO2 determined from NOAA Global Monitoring Laboratory measurements, version 2024-01, DOI:10.15138/9N0H-ZH07.
- J. Pires, F. Martins, M. Alvim-Ferraz and M. Simões, Recent developments on carbon capture and storage: An overview, Chem. Eng. Res. Des., 2011, 89(9), 1446–1460 CrossRef CAS.
- R. S. Haszeldine, Carbon capture and storage: how green can black be?, Science, 2009, 325(5948), 1647–1652 CrossRef CAS PubMed.
- N. Bahman, M. Al-Khalifa, S. Al Baharna, Z. Abdulmohsen and E. Khan, Review of carbon capture and storage technologies in selected industries: potentials and challenges, Rev. Environ. Sci. Biotechnol., 2023, 1–20 Search PubMed.
- Y. Zhu and H. Frey, Integrated gasification combined cycle (IGCC) power plant design and technology, in Advanced power plant materials, design and technology, Elsevier, 2010, pp. 54–88 Search PubMed.
- D. Cebrucean and I. Ionel, Biomass co-firing with carbon capture, 2022 Search PubMed.
- C. Song, Q. Liu, S. Deng, H. Li and Y. Kitamura, Cryogenic-based CO2 capture technologies: State-of-the-art developments and current challenges, Renewable Sustainable Energy Rev., 2019, 101, 265–278 CrossRef CAS.
- U. W. Siagian, A. Raksajati, N. F. Himma, K. Khoiruddin and I. Wenten, Membrane-based carbon capture technologies: Membrane gas separation vs. membrane contactor, J. Nat. Gas Sci. Eng., 2019, 67, 172–195 CrossRef CAS.
- D. Fu and J. Xie, Absorption capacity and viscosity for CO2 capture process using [N1111][Gly] promoted K2CO3 aqueous solution, J. Chem. Thermodyn., 2016, 102, 310–315 CrossRef CAS.
- A. Gautam and M. K. Mondal, Review of recent trends and various techniques for CO2 capture: Special emphasis on biphasic amine solvents, Fuel, 2023, 334, 126616 CrossRef CAS.
- M. Wilson, P. Tontiwachwuthikul, A. Chakma, R. Idem, A. Veawab and A. Aroonwilas, et al., Test results from a CO2 extraction pilot plant at boundary dam coal-fired power station, in Greenhouse Gas Control Technologies-6th International Conference, Elsevier, 2003, pp. 31–36 Search PubMed.
- D. P. Hagewiesche, S. S. Ashour, H. A. Al-Ghawas and O. C. Sandall, Absorption of carbon dioxide into aqueous blends of monoethanolamine and N-methyldiethanolamine, Chem. Eng. Sci., 1995, 50(7), 1071–1079 CrossRef CAS.
- G. T. Rochelle, Amine scrubbing for CO2 capture, Science, 2009, 325(5948), 1652–1654 CrossRef CAS PubMed.
- Y. Mao, X. Yang and T. V. Gerven, Amine-assisted simultaneous CO2 absorption and mineral carbonation: effect of different categories of amines, Environ. Sci. Technol., 2023, 57(29), 10816–10827 CrossRef CAS PubMed.
- M. Ramezan, T. J. Skone, N. Nsakala, G. Liljedahl, L. Gearhart and R. Hestermann, et al., Carbon dioxide capture from existing coal-fired power plants, National Energy Technology Laboratory, DOE/NETL Report, 2007, vol. 401, p. 110907 Search PubMed.
- G. Gadikota, Carbon mineralization pathways for carbon capture, storage and utilization, Commun. Chem., 2021, 4(1), 23 CrossRef CAS PubMed.
- I. M. Power, A. L. Harrison, G. M. Dipple, S. Wilson, P. B. Kelemen and M. Hitch, et al., Carbon mineralization: from natural analogues to engineered systems, Rev. Mineral. Geochem., 2013, 77(1), 305–360 CrossRef CAS.
- J. M. Matter, M. Stute, S. Ó. Snæbjörnsdottir, E. H. Oelkers, S. R. Gislason and E. S. Aradottir, et al., Rapid carbon mineralization for permanent disposal of anthropogenic carbon dioxide emissions, Science, 2016, 352(6291), 1312–1314 CrossRef CAS PubMed.
- W. J. J. Huigen and R. N. J. Comans, Carbon dioxide sequestration by mineral carbonation: Literature Review, Energy Research Centre of the Netherlands, 2003, https://publications.ecn.nl/E/2003/ECN-C--03-016 Search PubMed.
- K. S. Lackner, Carbonate chemistry for sequestering fossil carbon, Annu. Rev. Energy, 2002, 27(1), 193–232 CrossRef.
- A. M. Kierzkowska, R. Pacciani and C. R. Müller, CaO-based CO2 sorbents: from fundamentals to the development of new, highly effective materials, ChemSusChem, 2013, 6(7), 1130–1148 CrossRef CAS PubMed.
- A. Ababou, M. Ajbary, M. Taleb and A. Kherbeche, Direct mineral carbonation of new materials for CO2 sequestration, J. Mater. Environ. Sci., 2017, 8(9), 3106–3111 CAS.
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, Progress in MgO sorbents for cyclic CO2 capture: A comprehensive review, J. Mater. Chem. A, 2019, 7(35), 20103–20120 RSC.
- N. McQueen, P. Kelemen, G. Dipple, P. Renforth and J. Wilcox, Ambient weathering of magnesium oxide for CO2 removal from air, Nat. Commun., 2020, 11(1), 3299 CrossRef CAS PubMed.
- S. Gregg and J. Ramsay, Adsorption of carbon dioxide by magnesia studied by use of infrared and isotherm measurements, J. Chem. Soc. A, 1970, 2784–2787 RSC.
- R. Philipp and K. Fujimoto, FTIR spectroscopic study of carbon dioxide adsorption/desorption on magnesia/calcium oxide catalysts, J. Phys. Chem., 1992, 96(22), 9035–9038 CrossRef CAS.
- T. Papalas, I. Polychronidis, A. N. Antzaras and A. A. Lemonidou, Enhancing the intermediate-temperature CO2 capture efficiency of mineral MgO via molten alkali nitrates and CaCO3: Characterization and sorption mechanism, J. CO2 Util., 2021, 50, 101605 CrossRef CAS.
- J. Weber, V. Starchenko, K. Yuan, L. M. Anovitz, A. V. Ievlev and R. R. Unocic, et al., Armoring of MgO by a Passivation Layer Impedes Direct Air Capture of CO2, Environ. Sci. Technol., 2023, 57(40), 14929–14937 CrossRef CAS PubMed.
- W. Seifritz, CO2 disposal by means of silicates, Nature, 1990, 345(6275), 486 CrossRef.
- P. C. Chiang, S. Y. Pan, P. C. Chiang and S. Y. Pan, Principles of accelerated carbonation reaction, Carbon dioxide mineralization and utilization, 2017, pp. 71–96 Search PubMed.
- S. Ó. Snæbjörnsdóttir, B. Sigfússon, C. Marieni, D. Goldberg, S. R. Gislason and E. H. Oelkers, Carbon dioxide storage through mineral carbonation, Nat. Rev. Earth Environ., 2020, 1(2), 90–102 CrossRef.
- M. Rekhtina, M. Krödel, Y. H. Wu, A. Kierzkowska, F. Donat and P. M. Abdala, et al., Deciphering the structural dynamics in molten salt–promoted MgO-based CO2 sorbents and their role in the CO2 uptake, Sci. Adv., 2023, 9(26), eadg5690 CrossRef CAS PubMed.
- Y. Xu, F. Donat, C. Luo, J. Chen, A. Kierzkowska and M. A. Naeem, et al., Investigation of K2CO3-modified CaO sorbents for CO2 capture using in-situ X-ray diffraction, Chem. Eng. J., 2023, 453, 139913 CrossRef CAS.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Alkali metal nitrate-promoted high-capacity MgO adsorbents for regenerable CO2 capture at moderate temperatures, Chem. Mater., 2015, 27(6), 1943–1949 CrossRef CAS.
- A. M. Alkadhem, M. A. Elgzoly and S. A. Onaizi, Novel amine-functionalized magnesium oxide adsorbents for CO2 capture at ambient conditions, J. Environ. Chem. Eng., 2020, 8(4), 103968 CrossRef CAS.
- J. N. Bracco, G. Camacho Meneses, O. Colón, K. Yuan, J. E. Stubbs and P. J. Eng, et al. Reaction Layer Formation on MgO in the Presence of Humidity, ACS Appl. Mater. Interfaces, 2023, 16(1), 712–712 CrossRef PubMed.
- S. O. Baumann, J. Schneider, A. Sternig, D. Thomele, S. Stankic and T. Berger, et al., Size effects in MgO cube dissolution, Langmuir, 2015, 31(9), 2770–3776 CrossRef CAS.
- J. M. Hanlon, L. B. Diaz, G. Balducci, B. A. Stobbs, M. Bielewski and P. Chung, et al., Rapid surfactant-free synthesis of Mg (OH)2 nanoplates and pseudomorphic dehydration to MgO, CrystEngComm, 2015, 17(30), 5672–5679 RSC.
- P. Liu, P. M. Abdala, G. Goubert, M. G. Willinger and C. Copéret, Ultrathin single crystalline MgO (111) nanosheets, Angew. Chem., Int. Ed., 2021, 60(6), 3254–3260 CrossRef CAS PubMed.
- S. Cho, H. R. Yu, T. H. Choi, M. J. Jung and Y. S. Lee, Surface functionalization and CO2 uptake on carbon molecular sieves: experimental observation and theoretical study, Appl. Surf. Sci., 2018, 447, 8–14 CrossRef CAS.
- S. A. Fuente, C. A. Ferretti, N. F. Domancich, V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, et al., Adsorption of 2-propanol on MgO surface: A combined experimental and theoretical study, Appl. Surf. Sci., 2015, 327, 268–276 CrossRef CAS.
- R. Kakkar, P. N. Kapoor and K. J. Klabunde, First principles density functional study of the adsorption and dissociation of carbonyl compounds on magnesium oxide nanosurfaces, J. Phys. Chem. B, 2006, 110(51), 25941–25949 CrossRef CAS PubMed.
- D. Smith and A. Tench, Reactions of alcohols adsorbed on MgO. Part I. Heterogeneous reactions with hydrogen atoms and trapped electrons at room temperature: a new technique in surface chemistry, Can. J. Chem., 1969, 47(8), 1381–13819 CrossRef CAS.
- V. Molahid, F. Kusin and M. Soomro, Mineral carbonation for carbon dioxide capture and storage using mining waste as feedstock material, in IOP Conference Series: Earth and Environmental Science, IOP Publishing, 2023, vol. 1205, p. 012011 Search PubMed.
- A. L. Harrison, I. M. Power and G. M. Dipple, Accelerated carbonation of brucite in mine tailings for carbon sequestration, Environ. Sci. Technol., 2013, 47(1), 126–134 CrossRef CAS PubMed.
- J. Sipilä, S. Teir and R. Zevenhoven, Carbon dioxide sequestration by mineral carbonation Literature review update 2005–2007, Report Vt., 2008, vol. 1, p. 2008 Search PubMed.
- D. P. Butt, K. S. Lackner, C. H. Wendt, S. D. Conzone, H. Kung and Y. C. Lu, et al., Kinetics of thermal dehydroxylation and carbonation of magnesium hydroxide, J. Am. Ceram. Soc., 1996, 79(7), 1892–1898 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen and M. Dułak, et al. The atomic simulation environment–a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29(27), 273002 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car and C. Cavazzoni, et al., et al., ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli and M. Calandra, et al., Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29(46), 465901 CrossRef CAS PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis and J. K. Nørskov, et al., Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(23), 235149 CrossRef.
- D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41(11), 7892 CrossRef PubMed.
- R. M. Hazen, Effects of temperature and pressure on the cell dimension and X-ray temperature factors of periclase, Am. Mineral., 1976, 61(3–4), 266–271 CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188 CrossRef.
- Z. Liu, X. He, Z. Mei, H. Liang, L. Gu and X. Duan, et. al., Polarity poly-manipulation based on nanoscale structural transformation on strained 2D MgO, J. Phys. D: Appl. Phys., 2014, 47(10), 105303 CrossRef.
- R. Michalsky, Y. J. Zhang, A. J. Medford and A. A. Peterson, Departures from the adsorption energy scaling relations for metal carbide catalysts, J. Phys. Chem. C, 2014, 118(24), 13026–13034 CrossRef CAS.
- P. Hu, S. Wang and Y. Zhuo, Research on CO2 adsorption performances of metal-doped (Ca, Fe and Al) MgO, Sep. Purif. Technol., 2021, 276, 119323 CrossRef CAS.
- R. Wirth, Water in minerals detectable by electron energy-loss spectroscopy EELS, Phys. Chem. Miner., 1997, 24(8), 561–568 CrossRef CAS.
- B. Winkler, M. Avalos-Borja, V. Milman, A. Perlov, C. Pickard and J. Yates, Oxygen K-edge electron energy loss spectra of hydrous and anhydrous compounds, J. Phys.: Condens. Matter, 2013, 25(48), 485401 CrossRef CAS PubMed.
- A. F. A. Fuhaid and A. Niaz, Carbonation and corrosion problems in reinforced concrete structures, Buildings, 2022, 12(5), 586 CrossRef.
- T. Yang, B. Keller, E. Magyari, K. Hametner and D. Günther, Direct observation of the carbonation process on the surface of calcium hydroxide crystals in hardened cement paste using an atomic force microscope, J. Mater. Sci., 2003, 38, 1909–1916 CrossRef CAS.
- Y. Jia, J. Zhang, Y. Zou, Q. Guo, M. Li and T. Zhang, et al., Development and Applications of MgO-activated SiO2 system—achieving a low carbon footprint: A review, Green Energy and Resources, 2024, 100072 CrossRef.
- G. B. Singh, C. Sonat, E. Yang and C. Unluer, Performance of MgO and MgO–SiO2 systems containing seeds under different curing conditions, Cem. Concr. Compos., 2020, 108, 103543 CrossRef.
- D. P. Prentice, O. AlShareedah, M. Sarkar, J. Arabit, I. Mehdipour and S. Afzal, et al., Process modeling guides operational variables that affect CO2 utilization during the accelerated carbonation of concrete, AIChE J., 2024, 70(5), e18387 CrossRef CAS.
- S. Kwon, M. Fan, H. F. Dacosta, A. G. Russell and C. Tsouris, Reaction kinetics of CO2 carbonation with Mg-rich minerals, J. Phys. Chem. A, 2011, 115(26), 7638–7644 CrossRef CAS PubMed.
- M. G. Gardeh, A. A. Kistanov, H. Nguyen, H. Manzano, W. Cao and P. Kinnunen, Exploring mechanisms of hydration and carbonation of MgO and Mg (OH)2 in reactive magnesium oxide-based cements, J. Phys. Chem. C, 2022, 126(14), 6196–6206 CrossRef CAS PubMed.
- K. Rausis, A. R. Stubbs, I. M. Power and C. Paulo, Rates of atmospheric CO2 capture using magnesium oxide powder, Int. J. Greenhouse Gas Control, 2022, 119, 103701 CrossRef CAS.
- P. B. Kelemen, N. McQueen, J. Wilcox, P. Renforth, G. Dipple and A. P. Vankeuren, Engineered carbon mineralization in ultramafic rocks for CO2 removal from air: Review and new insights, Chem. Geol., 2020, 550, 119628 CrossRef CAS.
- A. Scott, C. Oze, V. Shah, N. Yang, B. Shanks and C. Cheeseman, et al., Transformation of abundant magnesium silicate minerals for enhanced CO2 sequestration, Commun. Earth Environ., 2021, 2(1), 25 CrossRef.
- H. K. Bharadwaj, J. Y. Lee, X. Li, Z. Liu and T. C. Keener, Dissolution kinetics of magnesium hydroxide for CO2 separation from coal-fired power plants, J. Hazard. Mater., 2013, 250, 292–297 CrossRef PubMed.
- Z. Song and H. Xu, Splitting methanol on ultra-thin MgO (100) films deposited on a Mo substrate, Phys. Chem. Chem. Phys., 2017, 19(10), 7245–7251 RSC.
- I. Bena, M. Droz, K. Martens and Z. Racz, Reaction–diffusion fronts with inhomogeneous initial conditions, J. Phys.: Condens. Matter, 2007, 19(6), 065103 CrossRef.
- C. Gunathilake and M. Jaroniec, Mesoporous calcium oxide–silica and magnesium oxide–silica composites for CO2 capture at ambient and elevated temperatures, J. Mater. Chem. A, 2016, 4(28), 10914–10924 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional structural characterization and theoretical calculation data are present in ESI† file. See DOI: https://doi.org/10.1039/d4en00940a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |