
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerization-like mechanism for fixation of CO2 with epoxides by multifunctional organocatalysts†
Mingan
Chen
 ,
Hui
Yang
and
Ming Wah
Wong
*
,
Hui
Yang
and
Ming Wah
Wong
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmwmw@nus.edu.sg
First published on 28th June 2022
Abstract
The commonly accepted mechanism of CO2 fixation of epoxides to cyclic carbonates catalyzed by multifunctional non-halide organocatalysts is challenged by our computational DFT-D3 study, which revealed a new polymerization-like mechanism comprising alternate epoxide and CO2 activation steps and a nested CO2 activation pathway. We investigated a recently reported CO2 coupling with epoxide reaction catalyzed by a bis-phenolic multifunctional catalyst. The predicted cis/trans product ratio is in excellent agreement with experimental results. The general applicability of the new mechanism is supported by another diamine-diacid catalyzed CO2 fixation reaction.
CO2 fixation with epoxides to synthesize cyclic carbonates represents one of the successful strategies to utilize the greenhouse gas. In the wake of the harm to the environment, there has been a growing interest in developing efficient organocatalysts for the chemical fixation processes of industrial feedstock, particularly the green halide-free catalysts.1–3 One of the major challenges in catalyst design is the lack of thorough understanding of the mechanism of activation, which holds the key to further catalyst activity improvement. Although many multifunctional halide-free organocatalysts have recently been reported,1–11 not much progress has been made in the in-depth understanding of the catalytic mechanism.
Three mechanisms are commonly proposed for CO2 fixation with epoxides, namely epoxide activation, CO2 activation and dual activation (Scheme 1). The first is the most commonly proposed one, with experimental and computational support.2 In this pathway, the catalysts act as Lewis or Brønsted acids to activate the oxygen atoms of epoxides, which makes them more susceptible to nucleophilic ring opening. The nucleophiles are usually halides but are increasingly replaced by other nucleophilic groups, such as onium salts, hydrogen bond donors and tertiary amines, which are either a part of the acid catalysts or added separately. The second CO2 activation mechanism involves nucleophilic activation of CO2 in the first step, generating reactive carboxylate intermediates that open the epoxides. It requires the catalyst to be nucleophilic towards CO2, but not towards epoxides. For the dual activation mechanism, with simultaneous activation of CO2 and epoxides, it involves an initial nucleophilic addition of the catalysts to CO2 instead of to the epoxides (Scheme 1).
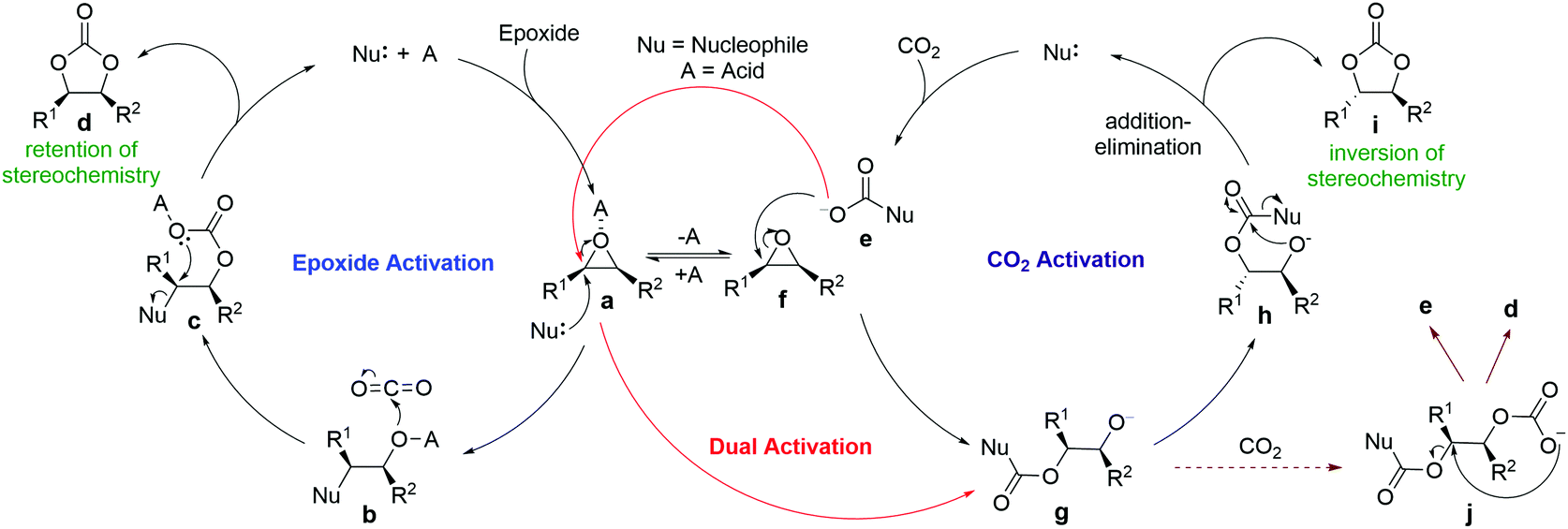 | ||
| Scheme 1 Generally proposed reaction mechanisms of organocatalyzed CO2 fixation. | ||
To date, the majority of computational studies in the literature support the epoxide activation mechanism.12–15 However, proposals of CO2 activation and dual activation mechanisms have increased substantially in recent reports on halide-free multifunctional organocatalyzed CO2 fixation.4,7,10,16,17 One major supporting evidence for such proposals is the experimental detection and sometimes isolation of catalyst-CO2 adducts, although scarcely any computational studies have confirmed the CO2 activation pathway in such cases. For example, a DFT study on NHC-catalyzed CO2 fixation found that the earlier-proposed CO2 activation mechanism is less favorable and the catalysis is likely to follow the epoxide activation mechanism.12 In fact, it is likely that the catalyst-CO2 adducts may just be a resting state of the catalyst, and the cycloaddition reaction follows a different mechanism.6
Another important piece of supporting evidence for the CO2 activation pathway comes from studies of the stereochemistry of deuterated products of terminal epoxides. Young et al. proposed that the epoxide activation mechanism involves two SN2 reactions at the same carbon (bound to R1 in species a and c, Scheme 1), resulting in a retention of stereochemistry in the products.18 In contrast, the CO2 activation pathway involves only one SN2 reaction (between e and f), which leads to an inversion of the product stereochemistry. One excellent example is the recent work by North and co-workers, who reported salophen N,N′-phenylenebis(5-tert-butylsalicylideneimine) 1-catalyzed cycloaddition of CO2 with epoxides.8 Their stereochemical study revealed that trans-epoxide 2a yields almost equal amounts of trans- and cis-products (Scheme 2), which suggests that both the epoxide and CO2 activation mechanisms are in operation. A novel bi-CO2 activation mechanism was proposed to explain the uncommon stereochemistry result, which involves another CO2 activation after intermediate g to form j in Scheme 1, yielding product d with retention of stereochemistry.8 Interestingly, a recent computational study of this reaction by Jiao and co-workers concluded that the epoxide activation mechanism is still favored over the CO2 activation mechanism.19 However, the authors failed to rationalize the distribution of product stereochemistry based on their proposed epoxide activation mechanism. The inconsistency in activation pathway and product stereochemistry distribution between theory and experiment undermines the confidence in the currently proposed mechanisms and a thorough computational mechanistic study is warranted.
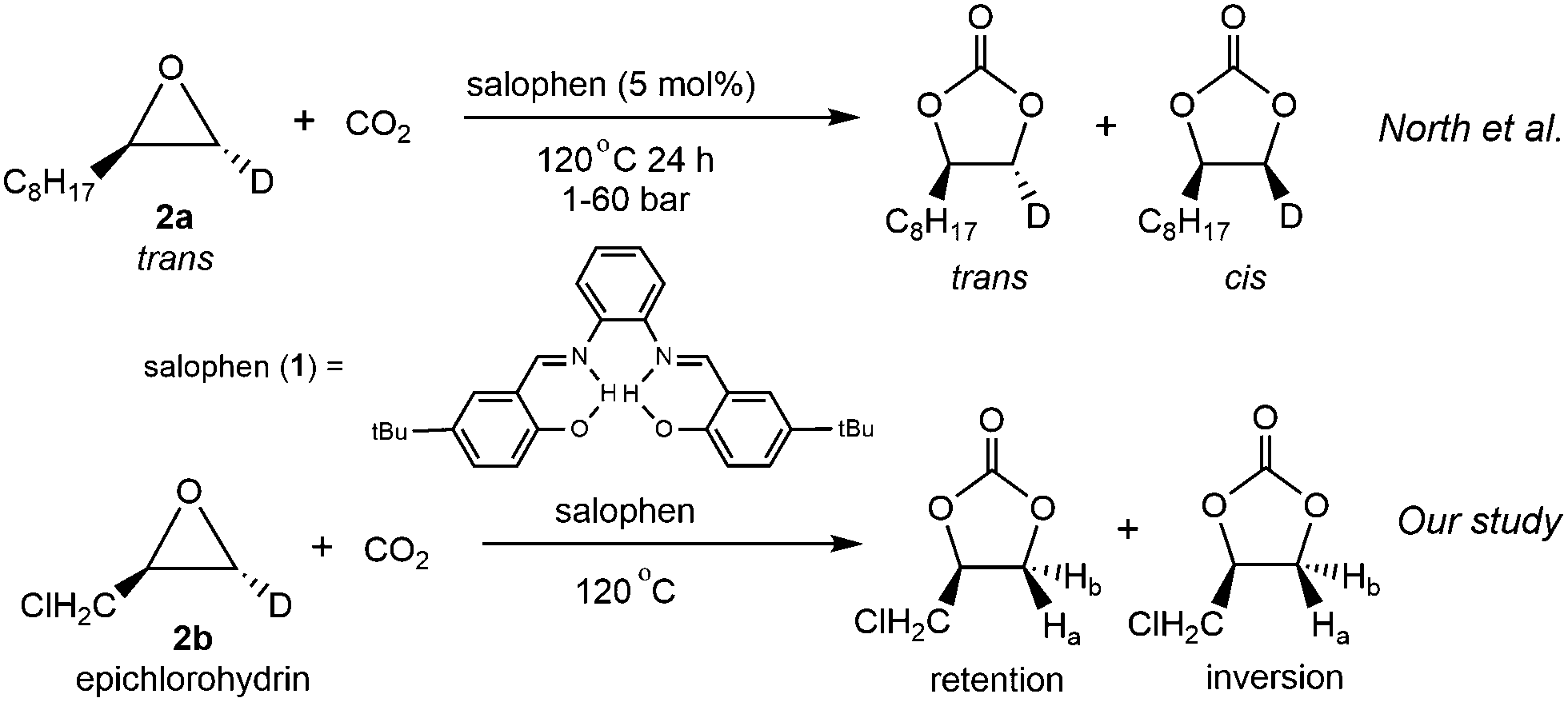 | ||
| Scheme 2 Stereochemistry studies with salophen catalyst 1. | ||
Previous mechanistic proposals hypothesized that the catalytic cycle initiates either through activating the epoxide substrate or CO2 by the catalyst, followed by nucleophilic reaction with the other substrate. To better understand the intriguing stereochemistry results, we first re-examined both mechanisms of 1-catalyzed CO2 fixation with epichlorohydrin (2b) using M06-2X20 theory with D321 dispersion correction in the solution phase with the SMD22 solvation model (see ESI† for rationales for the DFT method and solvent choices). Transition state (TS) conformation sampling was carried out using our recently developed docking program QMTSDock23 at the Hartree–Fock level. The low-energy conformers obtained from docking were subjected to further optimization at the DFT level. Several important improvements on the key TS structures were obtained, with significantly lower activation barriers for the CO2 activation pathway compared to those reported by Jiao et al (see ESI† for detailed discussion). The results are summarized in Scheme 3. In addition to comparing various activation modes, we also compared the two possible nucleophilic sites of catalyst 1, namely the hydroxyl and imine groups, and calculated their reactivities towards the epoxide electrophile. Our calculations showed that the hydroxyl group is a better nucleophile when the imine serves as a Brønsted base to deprotonate it (see ESI† for the pathway utilizing the imine group as the nucleophile). The second hydroxyl group is essential for a low activation barrier, via transferring a proton to the developing oxyanion in the transition state TS-A-1 (Fig. 1). In agreement with the computational study by Jiao et al., the epoxide activation pathway still has a significantly lower activation barrier than that of the CO2 activation pathway (46.6 vs. 57.6 kcal mol−1). In other words, the observed product stereochemistry distribution cannot be explained by the conventional CO2 activation pathway catalyzed by 1.
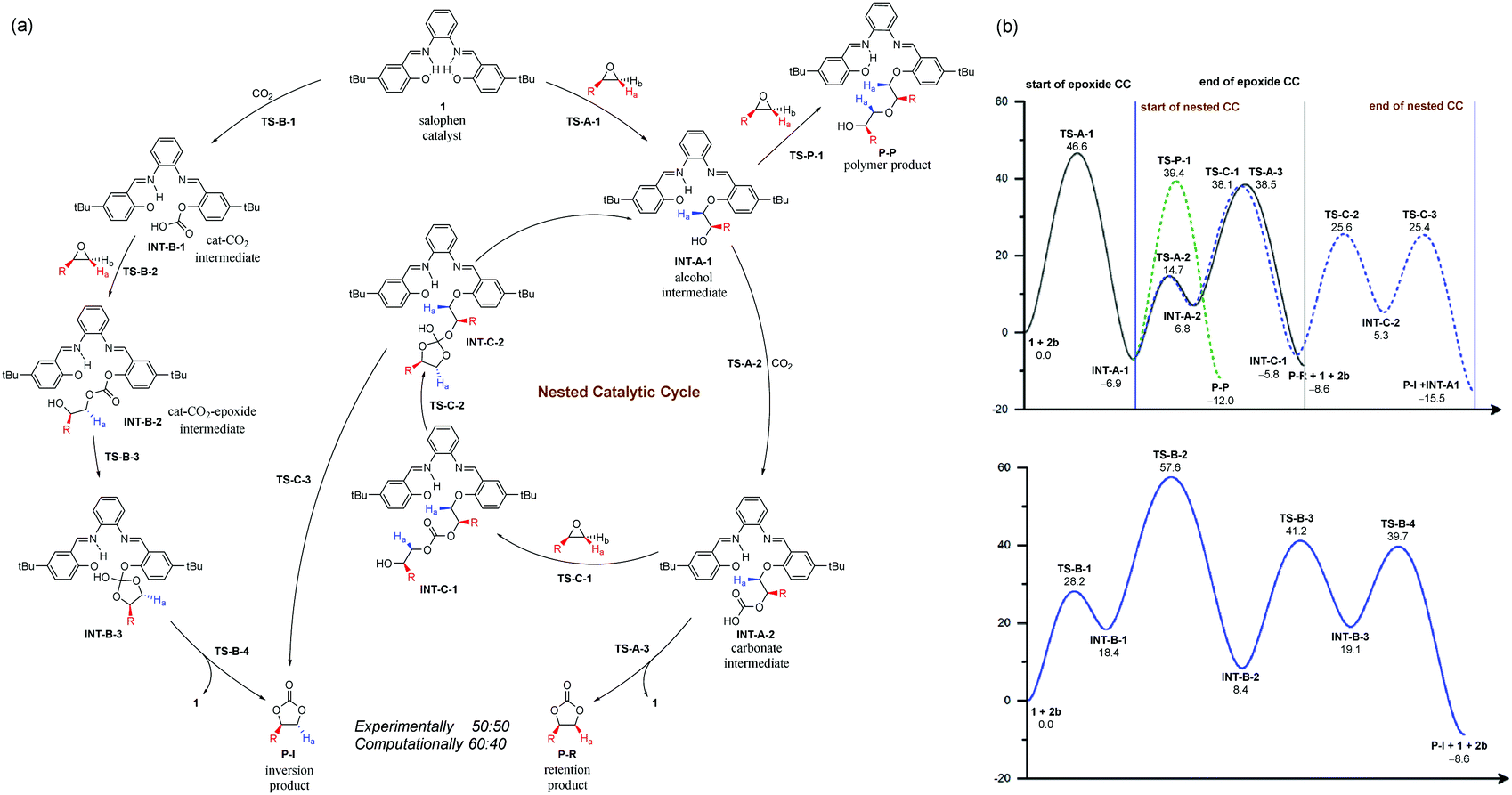 | ||
| Scheme 3 (a) Explored reaction pathways with a nested catalytic cycle (CC), (b) (top) calculated reaction profiles with initial activation of the epoxide substrate and (bottom) calculated reaction profile for the conventional CO2 activation mechanism. R = CH2Cl and relative free energies are in kcal mol−1. Proton transfer reactions are omitted for clarity. | ||
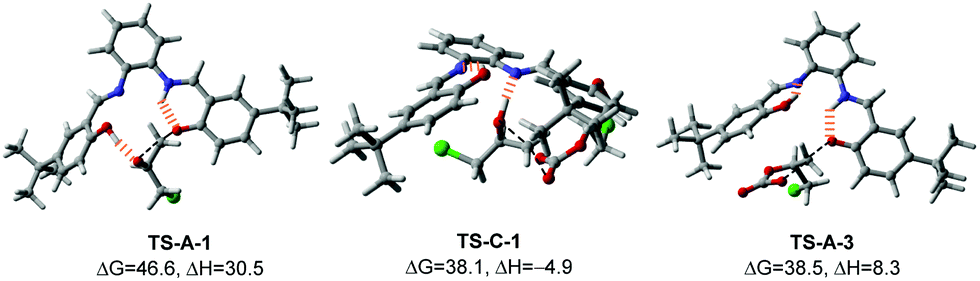 | ||
| Fig. 1 Optimized geometries of key transition states (activation barriers in kcal mol−1). Hydrogen bonds are shown as hashed lines. | ||
To find an alternative mechanism that explains the inversion product, it is instructive to gain a deeper understanding of the conventional CO2 mechanism first. One major hypothesis of CO2 activation is that the generated CO2 adduct that has a formal charge separation is a more potent nucleophile to epoxide addition than the original catalyst. Indeed, our calculated result is consistent with this hypothesis. The barrier for adding CO2-catalyst adduct INT-B-1 to epoxide through TS-B-2 is 39.2 kcal mol−1, 7.4 kcal mol−1 lower than that of TS-A-1 in the epoxide activation mechanism. However, addition of catalyst 1 to CO2 is highly unfavorable, with product INT-B-1 being 18.4 kcal mol−1 higher in energy. In other words, although adduct INT-B-1 is a better nucleophile than catalyst 1, it is thermodynamically unfavorable to form in the first place.
We noticed that in the epoxide activation mechanism, intermediate INT-A-1 also adds to CO2 but yields adduct INT-A-2 that is more favorable energetically, only 13.7 kcal mol−1 higher. Based on these findings, we envisage that imino alcohol INT-A-1 could be a better catalyst than 1 to promote CO2 activation, yielding the inversion product P-I. In this case, the CO2 activation cycle catalyzed by INT-A-1 would be nested inside the conventional epoxide activation cycle, sharing the CO2 addition step from INT-A-1 to INT-A-2 (Scheme 3) with it. The two cycles diverge from INT-A-2 and thus the product stereochemistry distribution would depend on the relative energies of TS-C-1versusTS-A-3. Recognizing the similarity between the two TSs, both belonging to an SN2 reaction at a carbon center with an oxygen atom leaving group and the ring strain of the epoxide molecule, we hypothesize that the product stereochemistry could be rationalized by comparing the newly proposed nested CO2 activation pathway and the conventional epoxide activation pathway. A similar mechanism leading to polymeric carbonates is well known in metal-catalyzed CO2-fixation reactions with epoxides.24,25 However, it has not been proposed or studied computationally in organocatalyzed reactions, probably due to the preconception that a metal center is needed to sustain polymeric chain growth.
The calculated reaction profile of the nested CO2 activation pathway is shown in Scheme 3 and the optimized key TSs are shown in Fig. 1. The two TSs responsible for product stereochemistry distribution, namely TS-C-1 and TS-A-3, are indeed close in energy, with a ΔΔG‡ difference of just 0.4 kcal mol−1 in favor of the former. This is in good agreement with the observed almost equal yields of the inversion (P-I) and retention (P-R) products. The predicted small energy difference between the two stereoselective TSs is confirmed by higher level computational theories, namely double-hybrid DFT and DLPNO-CCSD(T) methods (see ESI† for benchmarking details). The calculated overall barriers are somewhat high compared to the experimental conditions. These slightly higher values are likely due to the systematic error of the DFT method (see ESI† for benchmark detail). Although TS-C-1 is entropically less favorable than TS-A-3 due to addition to another molecule from INT-A-2, it is 13.2 kcal mol−1 lower in enthalpy, which offsets the entropic disadvantage. Unlike TS-A-1 that prefers the simultaneous assistance of two hydrogen bonds, TS-C-1 and TS-A-3 prefer only one hydrogen bond assistance to the substrate (Fig. 1). It is important to note that the hydrogen bonding of the imine groups plays a crucial role of proton transfer in the catalytic mechanism (see ESI† for detailed examples).
Although the nested catalytic cycle in Scheme 3 gives an excellent explanation of the product stereochemistry, it is natural to ask if a polymeric carbonate pathway as in metal-catalyzed reactions24 would lead to polymer formation here. To this end, we investigated the pathways of adding one and two more repeating units, CO2 and then epichlorohydrin in that order, to INT-C-1 (see ESI† for more detail). The computational result showed that further polymer growth is increasingly difficult, possibly due to the small size of catalyst 1, which makes it increasingly entropically unfavorable to stabilize the growing polymer chain through a hydrogen bond from the imine group. Besides, the epoxides are being added away from the catalytic system, in contrast to the insertion from a typical metal center (initiator). Another pathway leading to polyether formation was also investigated and the associated transition state TS-P-1 was found to be 1.3 kcal mol−1 higher than TS-C-1 (Scheme 3). The closeness of the reaction barriers between the polymer and cyclic carbonate pathways makes it hard to exclude the former confidently. It is likely that minor polymer products are formed together with the major cyclic carbonate product. In other words, the proposed mechanism is only polymerization-like but without significant polymer formation. This may explain why the observed yield of cyclic carbonate is about 8% lower than the total conversion.8 It is worth noting that the cyclic carbonate can be formed by a backbiting mechanism from a growing polymer chain.26 However, this alternative pathway is unlikely due to increasing difficulty in the polymer growth (see ESI†).
One key feature of the new mechanism is the organocatalytic cascade leading to the formation of the stereochemically inverted trans product through the CO2 activation pathway catalyzed by a reactive intermediate, namely INT-A-1. The second key feature is that the proposed CO2 and epoxide activation pathways are both an integral part of a polymerization-like pathway, differing in their exit points. It seems clear that the two activation pathways are not locked in an either-or relationship, but rather a stereochemically complementary and mechanistically integrated one. With the ongoing development of more efficient multifunctional organocatalysts, we expect to see more such examples. Due attention should be paid to leverage this complementary and integrated relationship, whether to understand the mechanism or design new catalysts. To demonstrate the general applicability of this polymerization-like mechanism, we chose to study another recently reported reaction catalyzed by a multifunctional diamine-diacid catalyst BCEDA (short for N,N′-bis(4-carboxyphenyl)ethylenediamine).11 Our preliminary theoretical investigation, based on the same M06-2X-D3 theory (Scheme 4), suggests that the difference between the nested CO2 activation pathway and the epoxide activation pathway is only 2.4 kcal mol−1 (difference between TS-D-3 and TS-E-1). In contrast, the conventional CO2 activation mechanism proposed by the authors is ∼15 kcal mol−1 higher in energy than the epoxide pathway.
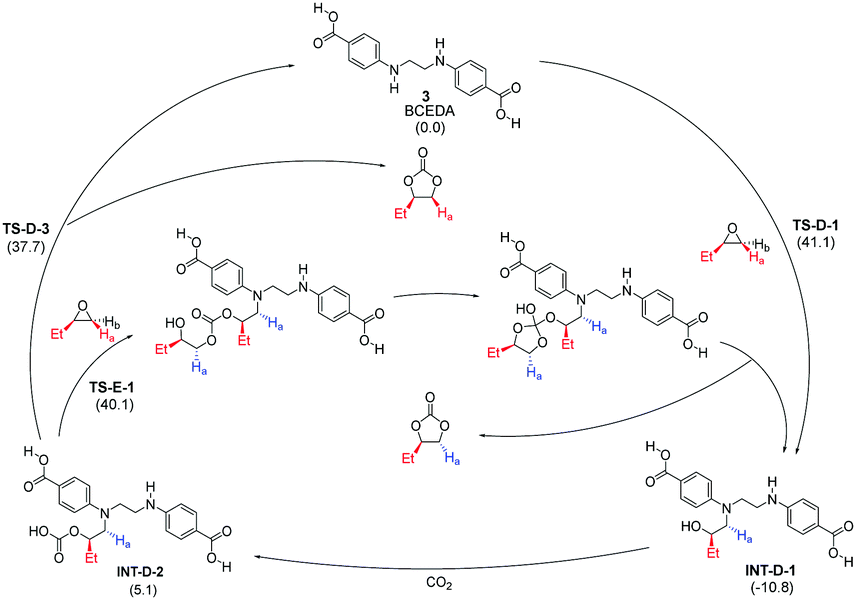 | ||
| Scheme 4 Calculated mechanism of multifunctional BCEDA-catalyzed CO2 fixation with 1,2-epoxybutane. Relative free energies in kcal mol−1. | ||
In summary, we have shown through comprehensive computational investigation that along with the conventional epoxide activation pathway, a simultaneous polymerization-like mechanism can be in operation during the salophen-catalyzed CO2 coupling with epoxides. The proposed new mechanism comprises a nested CO2 activation pathway that is catalyzed by an alcohol intermediate. The predicted cis/trans product ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 (see ESI† for detailed calculations) is in excellent agreement with the experimental finding. This is the first time that the CO2 activation pathway has been demonstrated computationally to be competitive to the epoxide pathway in organocatalyzed CO2 fixation with epoxide. The applicability of the new polymerization-like mechanism was confirmed by studying another organocatalyzed CO2 fixation reaction. We believe that the new computational insights of the relationship between the two mechanisms will lead to better understanding of the experimental results and designing of more efficient multifunctional organocatalysts for CO2 fixation.27
:60 (see ESI† for detailed calculations) is in excellent agreement with the experimental finding. This is the first time that the CO2 activation pathway has been demonstrated computationally to be competitive to the epoxide pathway in organocatalyzed CO2 fixation with epoxide. The applicability of the new polymerization-like mechanism was confirmed by studying another organocatalyzed CO2 fixation reaction. We believe that the new computational insights of the relationship between the two mechanisms will lead to better understanding of the experimental results and designing of more efficient multifunctional organocatalysts for CO2 fixation.27
This research was supported by Singapore Ministry of Education (Grant no: A-008483-00-00).
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Tech., 2017, 7, 2651–2684 RSC.
- L. P. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- F. Zhang, Y. Wang, X. Zhang, X. Zhang, H. Liu and B. Han, Green Chem. Eng., 2020, 1, 82–93 CrossRef.
- P. Goodrich, H. Q. N. Gunaratne, J. Jacquemin, L. Jin, Y. Lei and K. R. Seddon, ACS Sustainable Chem. Eng., 2017, 5, 5635–5641 CrossRef CAS.
- L. Guglielmero, A. Mezzetta, C. S. Pomelli, C. Chiappe and L. Guazzelli, J. CO2 Util., 2019, 34, 437–445 CrossRef CAS.
- A.-H. Liu, Y.-L. Dang, H. Zhou, J.-J. Zhang and X.-B. Lu, ChemCatChem, 2018, 10, 2686–2692 CrossRef CAS.
- A. A. Pawar and H. Kim, J. CO2 Util., 2019, 33, 500–512 CrossRef CAS.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- S. Yue, X.-J. Hao, P.-P. Wang and J. Li, Mol. Catal., 2017, 433, 420–429 CrossRef CAS.
- Z. Yue, M. Pudukudy, S. Chen, Y. Liu, W. Zhao, J. Wang, S. Shan and Q. Jia, Appl. Catal., A, 2020, 601, 117646 CrossRef CAS.
- F. Zhang, S. Bulut, X. J. Shen, M. H. Dong, Y. Y. Wang, X. M. Cheng, H. Z. Liu and B. X. Han, Green Chem., 2021, 23, 1147–1153 RSC.
- M. J. Ajitha and C. H. Suresh, Tetrahedron Lett., 2011, 52, 5403–5406 CrossRef CAS.
- H.-G. Kim, C.-S. Lim, D.-W. Kim, D.-H. Cho, D.-K. Lee and J. S. Chung, Mol. Catal., 2017, 438, 121–129 CrossRef CAS.
- W. Natongchai, J. A. Luque-Urrutia, C. Phungpanya, M. Solà, V. D'Elia, A. Poater and H. Zipse, Org. Chem. Front., 2021, 8, 613–627 RSC.
- S. Ryu, Bull. Korean Chem. Soc., 2019, 40, 1033–1038 CrossRef CAS.
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497–18501 CrossRef CAS PubMed.
- G. Yuan, Y. Zhao, Y. Wu, R. Li, Y. Chen, D. Xu and Z. Liu, Sci. China Chem., 2017, 60, 958–963 CrossRef CAS.
- C. Beattie, M. North, P. Villuendas and C. Young, J. Org. Chem., 2013, 78, 419–426 CrossRef CAS PubMed.
- C.-H. Guo, M. Liang and H. Jiao, Catal. Sci. Tech., 2021, 11, 2529–2539 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Phys. Chem. A, 2019, 123, 10303–10314 CrossRef CAS PubMed.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- A. Brege, B. Grignard, R. Méreau, C. Detrembleur, C. Jerome and T. Tassaing, Catalysts, 2022, 12, 124 CrossRef CAS.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed computational results, total energies and Cartesian coordinates of all calculated structures; choice of DFT method and solvent; comparison with computational work of Jiao et al.; other mechanistic pathways. See DOI: https://doi.org/10.1039/d2cc03409c |
| This journal is © The Royal Society of Chemistry 2022 |