
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecycling of two molecular catalysts in the hydroformylation/aldol condensation tandem reaction using one multiphase system†
Marc
Strohmann
a,
Jeroen T.
Vossen
a,
Andreas J.
Vorholt
 *a and
Walter
Leitner
ab
*a and
Walter
Leitner
ab
aMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: andreas-j.vorholt@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringer Weg 1, 52074 Aachen, Germany
First published on 6th November 2020
Abstract
Tandem reactions are of great importance to efficiently execute multiple conversions in one synthesis step. Herein we present a multiphase system for the hydroformylation/aldol condensation, which is able to recycle both optimized catalysts multiple times. The system consists of an organometallic rhodium/sulfoXantphos hydroformylation catalyst and basic NaOH as aldol condensation initiator, which are both immobilized in a polyethylene glycol phase. Under reaction conditions, NaOH is converted to sodium formate, which is still able to catalyse the aldol condensation. The reaction and recycling are demonstrated by the conversion of 1-pentene to the corresponding aldol product in a recycling experiment. During nine consecutive runs, no significant loss of activity is found with an overall TON of 8700 in regard to the rhodium catalyst and an average rhodium leaching of only of 0.07% per run is observed.
Introduction
Tandem catalysis provides a powerful tool for improving molecular transformations, as it allows a reduction in energy and material and saves overall process time. Therefore, it is not surprising to see tandem catalysis as a growing field in current research.1–6 In comparison to one-pot reactions, tandem systems are characterized by having all components present at the beginning of the reaction. Furthermore, tandem reactions differ from domino or cascade reactions as such that more than one catalyst cycle is involved. Tandem catalysis can be further subdivided into auto-tandem and orthogonal tandem catalysis, depending on whether only one or multiple catalyst species are used.7,8Hydroformylation is the reaction of olefins with hydrogen and carbon monoxide (synthesis gas) to linear and branched aldehydes. It has been well investigated and is nowadays one of the largest homogeneously catalysed processes in industry.9 The produced aldehydes are usually not the final products, but are further converted in subsequent reaction steps. Additionally, aldehydes are very versatile and can be transformed in many ways. Therefore, the hydroformylation reaction is particularly suitable as a part of a tandem catalysis network.10,11 One possible tandem sequence is a double bond isomerization followed by hydroformylation, since terminal olefins are hydroformylated much faster than internal ones.12,13 However, more common are tandem reactions in which hydroformylation is the first reaction step, followed by, hydrogenation (hydrohydroxymethylation), reductive amination (hydroaminomethylation), acetalization, acyloin reaction or aldol condensation (Scheme 1).10,11,14–17
 | ||
| Scheme 1 Possible tandem reactions involving hydroformylation. | ||
The hydroformylation/aldol condensation tandem reaction allows the formation of α,β-unsaturated aldehydes, which are an important molecule class for cosmetics, agrochemicals and pharmaceuticals.18 Nevertheless, it has received less attention compared to hydrohydroxymethylation or hydroaminomethylation. One reason might be the increased complexity of this reaction sequence because of the additional catalyst required for the aldol condensation step. In 2014 Beller and co-workers presented a homogeneous catalyst system for the hydroformylation/aldol condensation tandem reaction.19 The group took advantage of a rhodium/phosphine hydroformylation catalyst in combination with a pyrrolidine-based organocatalyst for the aldol step. With this system, they were able to convert a variety of olefins to the corresponding aldol products with high conversions and selectivity. Additionally, they showed cross aldol condensation with aromatic aldehydes. Ethyl acetate or N-methyl-2-pyrrolidon (NMP) were used as solvent in the tandem reactions. For this homogeneous reaction system, no recycling of the catalysts was discussed.
A different strategy to approach the hydroformylation/aldol condensation reaction was presented by Jasra and co-workers.20–22 They prepared multifunctional catalysts by immobilizing the organometallic species RhH(CO)(PPh3)3 onto a basic carbonate, a hydrotalcite of the form [Mg1−xAlx(OH2)x+(CO32−)x/n·mH2O]. This catalyst was able to convert linear olefins from ethylene to 1-decene to the corresponding aldol derivatives in one-pot. However, with increasing length of the olefin substrates the selectivity to the aldol product decreases as side reactions such as isomerization and hydrogenation of the starting material took place. Additionally, hydrogenation of the aldol product to the saturated aldehyde was observed. As a heterogeneous catalyst, it could be separated from the product mixture by simple filtration. Unfortunately, the basic support loses its activity for the aldol condensation during catalysis because of structural changes, thus not allowing a successful reuse of the multifunctional catalyst over multiple runs.
The difficulty of catalyst removal and especially catalyst recycling is one of the major hurdles to bring homogeneous catalysed reactions into application. The presence of two different catalysts increases the difficulty level of recycling in tandem reactions in general and thus also that of the hydroformylation/aldol condensation tandem reaction.
Multiphase systems present an elegant way of recycling molecular catalysts by immobilizing the catalyst in one of the liquid phases, while the product forms the other phase. This recycling strategy has found application in industry for example in the oligomerization of ethylene (Shell Higher Olefine Process)23 or the hydroformylation of propene (Ruhrchemie/Rhône-Poulenc process).24 Water is the most common choice for the polar catalyst phase in multiphase catalysis, but other polar solvents such as 1,4-butanediol, methanol,25 polyethylene glycol (PEG),26 ionic liquids27,28 or deep eutectic solvents29 have been applied in industry and academia, as well. Among these, especially water and polyethylene glycol are widely considered as green solvents.30,31 Furthermore, aqueous multiphase catalysis has been successfully used for catalyst recycling in the hydroaminomethylation tandem reaction.32
While hydroformylation and aldol condensation alone have often been studied in liquid/liquid systems, to our knowledge, the tandem reaction has not been demonstrated in such a manner. In this work, we therefore set out to apply multiphase catalysis to perform the hydroformylation/aldol condensation tandem reaction with special focus on recycling of both catalysts simultaneously. 1-Pentene was chosen as model substrate. Water and polyethylene glycol are investigated as solvents for the catalyst phase and the recyclability of the orthogonal catalyst system is demonstrated in recycling experiments.
Results and discussion
Catalysis in water
The conversion of 1-pentene served as model reaction for our investigation of the multiphase hydroformylation/aldol condensation tandem reaction. 1-Pentene is first hydroformylated to n-hexanal and then undergoes self-aldol condensation to form the desired aldol product 4-butyloctenal (Scheme 2). Also displayed are typical side reactions including the hydrogenation of pentene to pentane, of hexanal to hexanol and of the aldol product to the saturated aldehyde 4-butyloctanal (saturated aldol). Another side product is the iso-hexanal (2-methylpentanal) which forms during hydroformylation but does not undergo self-condensation because of its branching. In order to achieve a high conversion towards the aldol product, a good n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :iso ratio as well as a low hydrogenation activity are necessary. For our biphasic reaction system, we initially chose water as catalyst phase and [Rh(acac)(CO)2] with sulfoXantphos (4,5-bis(diphenylphosphi-no)-9,9-dimethyl-2,7-disulfoxanthene disodium salt) as in situ hydroformylation catalyst system. Xantphos and its sulfonated, water-soluble variation sulfoXantphos are well known hydroformylation ligands that give high linear to branched ratios due to their wide bite angle (111°) and were successfully applied for hydroformylation reactions in the past.32–34 We then focussed on finding a suitable catalyst for the aldol condensation step. Inorganic bases, such as sodium or potassium hydroxide are the most common catalysts in aldol reactions. However, the reaction also proceeds via enamine catalysis. In terms of enamine catalysis, amines such as pyrrolidine or amino acids can be used. A couple of different bases and organocatalysts have been selected and tested in the biphasic aldol condensation of hexanal (Fig. 1).
:iso ratio as well as a low hydrogenation activity are necessary. For our biphasic reaction system, we initially chose water as catalyst phase and [Rh(acac)(CO)2] with sulfoXantphos (4,5-bis(diphenylphosphi-no)-9,9-dimethyl-2,7-disulfoxanthene disodium salt) as in situ hydroformylation catalyst system. Xantphos and its sulfonated, water-soluble variation sulfoXantphos are well known hydroformylation ligands that give high linear to branched ratios due to their wide bite angle (111°) and were successfully applied for hydroformylation reactions in the past.32–34 We then focussed on finding a suitable catalyst for the aldol condensation step. Inorganic bases, such as sodium or potassium hydroxide are the most common catalysts in aldol reactions. However, the reaction also proceeds via enamine catalysis. In terms of enamine catalysis, amines such as pyrrolidine or amino acids can be used. A couple of different bases and organocatalysts have been selected and tested in the biphasic aldol condensation of hexanal (Fig. 1).
 | ||
| Scheme 2 Desired conversion of 1-pentene to the C12-aldol product by hydroformylation/aldol condensation tandem reaction and typical side products that occur. | ||
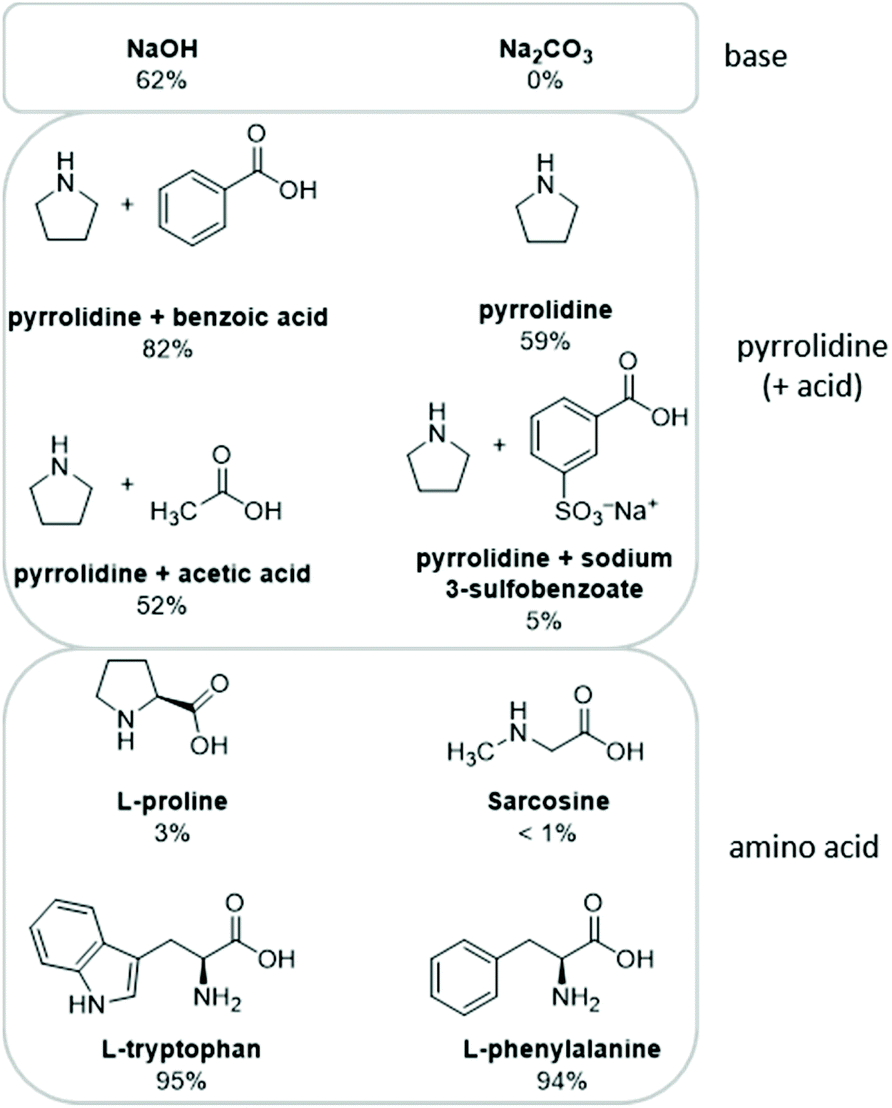 | ||
| Fig. 1 Hexanal conversion in the aqueous biphasic aldol condensation using different catalysts. Conditions: Hexanal (8.2 mmol, 1 mL), H2O (0.4 mL), catalyst (5 mol%), 65 °C, 1.5 h. Conversions were determined by GC-FID. | ||
The basic catalyst NaOH gave a moderate conversion of 62% after the reaction time of 1.5 h, whereas no conversion was observed for the less basic Na2CO3. Although NaOH seems to be a suitable catalyst for the aldol condensation, it is known that the rate of hydroformylation is reduced with increasingly basic pH-values.43 Additionally, in the work by Fang et al. NaOH turned out to be an ineffective aldol catalyst, when used under hydroformylation conditions.19 In the same work, the authors found a combination of pyrrolidine and benzoic acid to be the most successful aldol catalyst for their reaction system. The pyrrolidine enables enamine catalysis, while the benzoic acid accelerates the enamine formation. Thus, pyrrolidine in combination with different organic acids was tried for our system, as well. A good conversion of 82% was reached in the case of benzoic acid, confirming the suitability of this organocatalytic system. In comparison, pyrrolidine alone only gave 59% conversion. With catalyst recycling in mind, we attempted the reaction with acetic acid and the sulfonated benzoic acid (sodium 3-sulfobenzoate) as acidic additive in combination with pyrrolidine. Due to the higher solubility of the named acids in water, they are more likely to remain immobilized in the water phase after the reaction. Unfortunately, both acidic additives lead to lower conversions compared to pyrrolidine without additives, 52% in the case of acetic acid and only 5% in the case of sulfonated benzoic acid. It is assumed, that the reaction proceeds slower with these catalysts, because the aldol reaction takes place in the organic phase. If an acidic additive is added to the reaction, it will form an adduct with pyrrolidine and depending on the water solubility of the acid, the adduct will be solvated mostly in the water phase with barely any transition of pyrrolidine to the organic phase.
The amino acid L-proline combines the functionalities of the pyrrolidine ring and a carboxylic acid and is known for asymmetric aldol condensations.35 However, only 3% of hexanal was converted by proline under the chosen reaction conditions. Sarcosine, also bearing a secondary amine and a carboxylic acid group, showed even less activity with less than 1% conversion. The low activity of these two amino acids might be a result of their high solubility in water and therefore low tendency to shift to the organic phase for the catalysis. In a work by Ostrowski et al. all 20 proteinogenic amino acids were compared for their catalytic activity in aldol condensations of aldehydes in ethanol.36L-Tryptophan and L-phenylalanine performed best and tryptophan was successfully applied in a water system, too. Similarly, in our biphasic system both amino acids performed excellently with hexanal conversions of 95% and 94%, respectively. NaOH, pyrrolidine/benzoic acid and tryptophan were selected for further investigation, since they were the most active of each of the three catalyst types.
We continued by searching for suitable reaction conditions for the hydroformylation step, before adding the aldol catalysts. Table 1 summarizes the results of the hydroformylation of 1-pentene with and without the selected aldol catalysts. A low temperature of 65 °C was not enough for the hydroformylation step, as no conversion of pentene was observed. Increasing the temperature to 100 °C and 120 °C then lead to conversions of 18% and 58%, respectively (Table 1, entries 1–3). Next, we added the different aldol catalyst to investigate the whole tandem reaction at 120 °C. In presence of NaOH less pentene (48%) was converted compared to the reaction without NaOH (entry 4). In addition, no aldol product was observed after the reaction. It turned out that NaOH is deactivated under reaction conditions by a reaction with CO to sodium formate (Scheme 3a), which was confirmed by the presence of the formate peak at around 8.5 ppm in the 1H-NMR spectrum (see Fig. S1†). With a pKa-value of only 3.8 (acidity constant of formic acid), the basicity of sodium formate does not seem to be enough to catalyse the aldol condensation itself. The formation of formate was also observed at a lower temperature of 100 °C and a lower pressure of 10 bar and proceeds faster than the hydroformylation. Since CO is essential for the tandem reaction and cannot be replaced, this deactivation excluded NaOH as possible aldol catalyst in the water system. When pyrrolidine/benzoic acid or tryptophan were used as catalyst, higher pentene conversions (92% and 93%) were reached compared to the 58% conversion in the reaction without additional aldol catalyst (entries 5 and 6). In addition, the formation of aldol product was observed in these cases, however, less than what was expected from the previous experiments. The higher pentene conversions are probably due to the amphiphilic nature of these catalysts. With similar structures, they may act in the same way as surfactants and accelerate the hydroformylation by forming micelles, which increase the surface area between the two phases.37 While in the case of pyrrolidine/benzoic acid a pentene conversion of 92% was reached, only 10% of aldol product were formed. The low yield could again be traced back to catalyst deactivation. During catalysis pyrrolidine forms an enamine with hexanal which can be irreversibly hydrogenated to the corresponding amine by the rhodium catalyst (Scheme 3b). The presence of the amine was observed by GC with a yield of around 5%, which equals the amount used as catalyst, hence indicating that pyrrolidine was fully deactivated during the reaction. In the reaction with tryptophan, 29% aldol product was observed, which is the highest yield of the three selected aldol catalyst, but still most hexanal formed during the reaction did not react further. It is not certain, weather tryptophan undergoes a similar deactivation as pyrrolidine, as the corresponding amine could not be observed during the reaction. However, the low yields of aldol product compared to the preliminary experiments indicate that tryptophan is at least partially deactivated.
 | ||
| Scheme 3 Deactivation pathways of NaOH (a) and pyrrolidine (b) under hydroformylation conditions. | ||
| Entry | Aldol catalyst | T [°C] | X [%] | Hexanal [%] (n:iso) |
Aldol [%] |
|---|---|---|---|---|---|
| a Conditions: Pentene (8.2 mmol, 0.9 mL), H2O (0.4 mL), [Rh(acac)(CO)2] (0.1 mol%), sulfoXantphos (0.2 mol%), aldol catalyst (5 mol%), H2/CO (25 bar each), 16 h. b PyH = pyrrolidine. | |||||
| 1 | — | 65 | 0 | 0 | 0 |
| 2 | — | 100 | 18 | 18 (60:40) |
0 |
| 3 | — | 120 | 58 | 57 (97:3) |
0 |
| 4 | NaOH | 120 | 48 | 48 (83:17) |
0 |
| 5 | PyH/PhCOOHb | 120 | 92 | 71 (97:3) |
10 |
| 6 | L-Tryptophan | 120 | 93 | 59 (97:3) |
29 |
Catalysis in polyethylene glycol
Since none of the three aldol catalysts showed promising results in water, we decided to alter the reaction system by switching the catalyst phase to polyethylene glycol (PEG) which is known as a green alternative polar phase for multiphase catalysis.38,39 The work by Zhao et al. demonstrated that multiphase hydroformylation can be performed with PEG as catalyst phase and that a successful catalyst recycling is also possible.26 A comparison of different chain lengths showed that the shortest PEG with an average molecular weight of 200 g mol−1 (PEG-200) gave the best results. Furthermore, all catalysts are soluble in PEG-200, which makes it a possible alternative to water.Therefore, the tandem reaction was attempted again with the same aldol catalysts as before in PEG-200 (Table 2). The reaction was indeed accelerated by the solvent change, as pentene conversions of ≥97% were achieved within the same reaction time of 16 h in all cases. For pyrrolidine/benzoic acid as aldol catalyst, a similar aldol product yield of 12% as for the water system was observed, despite the higher pentene conversion (Table 2, entry 1). This is because pyrrolidine is fully deactivated in a similar manner as in the water system (Scheme 3b). Lowering the temperature to 100 °C lead to a slightly higher yield of 31% but could not prevent the deactivation (entry 2). In the case of tryptophan 73% of the formed hexanal undergoes aldol condensation, however, 41% of the aldol product is further hydrogenated to the saturated aldol (entry 3). In order to reduce the formation of the saturated aldol, the experiment was repeated at a lower temperature. Indeed, at 100 °C, only 6% of the saturated aldol were formed and the yield of the aldol product reached 76% (entry 4). Unfortunately, the use of tryptophan caused an increased miscibility of the two phases, which was evident from the fact that the catalyst phase was smaller after the reaction and the product phase turned yellow. The associated loss of PEG and catalyst inevitably prevents successful recycling of the catalyst phase (see Fig. S4†).
| Entry | Aldol catalyst | T [°C] | X [%] | Hexanal [%] (n:iso) |
Aldol [%] | Sat. aldol [%] |
|---|---|---|---|---|---|---|
| a Conditions: Pentene (8.2 mmol, 0.9 mL), PEG-200 (0.4 mL), [Rh(acac)(CO)2] (0.1 mol%), sulfoXantphos (0.2 mol%), aldol catalyst (5 mol%), H2/CO (25 bar each), 16 h. b PyH = pyrrolidine. | ||||||
| 1 | PyH/PhCO2Hb | 120 | 99 | 60 (97:3) |
12 | 4 |
| 2 | PyH/PhCO2Hb | 100 | 98 | 51 (97:3) |
31 | 2 |
| 3 | L-Tryptophan | 120 | 98 | 15 (96:4) |
32 | 41 |
| 4 | L-Tryptophan | 100 | 97 | 12 (98:2) |
76 | 6 |
| 5 | NaOH | 120 | 97 | 7 (98:2) |
69 | 18 |
| 6 | NaOH | 100 | 97 | 12 (98:2) |
80 | 2 |
In contrast to the water-based system, NaOH showed a good activity for the aldol condensation in PEG. At 120 °C most hexanal underwent aldol condensation, giving 69% of aldol product and 18% of saturated aldol (entry 5). At 100 °C the yield of aldol product could be further increased to 80%, however, more hexanal remained unreacted in the product mixture, as well (entry 6). These results raised the question whether there is less or no deactivation of NaOH in the PEG system. The formation of the formate anion was found by 1H-NMR analysis of the catalyst phase after the reaction repeatedly, indicating the deactivation of NaOH at both temperatures (see Fig. S2†). In order to find a way to avoid deactivation, we took a more detailed look at the temperature dependence of the tandem reaction by varying the reaction temperature between 75 °C and 135 °C (Fig. 2). At 75 °C, a pentene conversion of 80% was reached after the reaction time of 16 h. It was confirmed by 1H-NMR that even at 75 °C deactivation of NaOH takes place in PEG-200, which is why no improvement could be achieved by lowering the reaction temperature. With increased temperature, the conversion also increases up to 98% at 130 °C. While at low temperatures hexanal is the main product, with increasing temperatures more aldol product is formed. The maximum of aldol product yield is obtained at 100 °C with 80%. Above 100 °C the aldol yield drops again, as the further hydrogenation to the saturated aldehyde becomes more and more significant. The yield of additional side products, mostly pentane and hexanol, stays low, at around 5% at all temperatures up to 120 °C. Above 120 °C side product formation increases strongly reaching up to 31% at 135 °C. The data indicates that 100 °C is the best temperature to maximize the aldol product yield, since the consecutive reaction to the saturated aldehyde is still mostly suppressed. The sum of aldol product and saturated aldol is high with 88% at 125 °C.
 | ||
| Fig. 2 Temperature variation in the hydroformylation/aldol condensation reaction of 1-pentene in PEG-200 with Rh/SulfoXantphos and NaOH. Side products include pentane and alcohols. Conditions: Pentene (8.2 mmol, 0.9 mL), PEG-200 (0.4 mL), [Rh(acac)(CO)2] (0.1 mol%), sulfoXantphos (0.2 mol%), NaOH (5 mol%), H2/CO (25 bar each), 16 h. | ||
To foster the aldol reaction a closer look was given to the deactivation of the base catalyst. Different bases were employed and examined whether they react to formates during the reaction. Therefore, we looked at a series of catalysts with similar or slightly lower basic strength as NaOH (Table 3). KOH showed a similar reactivity as NaOH and was also deactivated (Table 3, entry 2). KOSiMe3 is less basic than NaOH with a pKa of 11. However, a comparable result was achieved with this catalyst, even reaching a slightly higher aldol yield of 82% (entry 3). Unfortunately, the formation of the formate anion was also observed when using KOSiMe3. We assumed that formate is formed from water in this case, CO and the basic catalysts as illustrated in Scheme 4. This reaction sequence can be generalized for all strong bases, as the presence of water cannot be avoided since it is a by-product in the aldol condensation. Surprisingly, a reaction with sodium methoxide resulted in much less aldol product in comparison to the previously tested bases, with only 35%, although it is also deactivated by the reaction to the corresponding formate in the same way (entry 4). Trimethylamine also performed worse than NaOH with 54% yield of aldol product, but it was the only base investigated that did not show the formation of formates during the reaction (entry 5). Both results are probably connected to the fact, that triethylamine is a neutral and not an ionic base and intensifies the mixing of the two phases instead of supporting their separation. In fact, the reaction mixture was monophasic after the reaction when using triethylamine as a base. We also wanted to verify that sodium formate is not active as a catalyst in the aldol condensation, which was concluded for the water system. Nevertheless, sodium formate lead exactly to the same product distribution as NaOH (entry 6). This indicated that sodium formate is the actual catalyst in the reaction and not remaining NaOH. Consequently, recycling and reuse of the aldol catalyst should be possible. We suspect that the aldol condensation is catalysed by formate in PEG but not in water, because formate is more basic in PEG. The pKa value for formate in the comparable solvent 1,2-ethanediol is much higher with 7.3 than in water.40,41 Given that 1,2-ethanediol and PEG have similar functionalities, it is likely that a higher basicity is found in PEG, too. Finally, we also tested lithium and caesium formate to see the influence of the counterion. While lithium formate performed worse with only 56% aldol product, caesium formate resulted in the same aldol product yield as sodium formate with 80% (entries 7 and 8).
 | ||
| Scheme 4 Deactivation pathway of strong bases in presence of CO and H2O. | ||
| Entry | Base catalyst | pKab | X [%] | Hexanalc [%] | Aldol [%] | Deactivation? |
|---|---|---|---|---|---|---|
|
a Conditions: Pentene (8.2 mmol, 0.9 mL), PEG-200 (0.4 mL), [Rh(acac)(CO)2] (0.1 mol%), sulfoXantphos (0.2 mol%), base (5 mol%), H2/CO (25 bar each), 100 °C, 16 h.
b Acidity constant of the corresponding acid in water.
c Both isomers, n:iso = 98:2.
d Metal formate. Lithium formate was used as monohydrate.
|
||||||
| 1 | NaOH | 14 | 97 | 12 | 80 | Yes |
| 2 | KOH | 14 | 98 | 13 | 78 | Yes |
| 3 | KOSiMe3 | 11 | 97 | 11 | 82 | Yes |
| 4 | NaOMe | 15 | 98 | 56 | 35 | Yes |
| 5 | NEt3 | 10 | 99 | 43 | 54 | No |
| 6 | NaHCO2d | 3.8 | 97 | 12 | 80 | — |
| 7 | LiHCO2d | 3.8 | 97 | 37 | 56 | — |
| 8 | CsHCO2d | 3.8 | 97 | 13 | 80 | — |
After understanding the current catalysis system better, it was tested for catalyst recycling. In order to verify the recyclability of both catalysts, we therefore performed recycling experiments over multiple runs with NaOH as initiator for the aldol reaction. After each run the product phase separated from the catalyst phase which was then extracted with pentane and treated with high vacuum to remove any remaining product traces. Afterwards new substrate was added for the next run. After each run the product phase was analysed via GC-FID for conversion and yields. The results for a nine-run experiment with NaOH at 100 °C are displayed in Fig. 3. As it can be seen, the pentene conversion stays constant between 96% and 97% throughout all nine runs. Only in run 8, it drops slightly to 94%, but increases to 97% in the final run again. A similar behaviour is found for the yields of the products. The amount of aldol product drops slightly from 81% in the first to 79% in the second run but then only decreases one additional percentage point to 78% in the final run. In contrast, the hexanal yield increases from 9% to 12% in the first two runs and varies between 11% and 13% afterwards. The amount of additional side products, including pentane and hexanol, fluctuates between 2% and 4%. The data clearly indicates the recyclability of this catalyst system, as catalytic activity is maintained over nine experiments for both reactions. All nine runs considered a total turnover number of almost 8700 was reached regarding rhodium compared to the 970 for a single run. The total turnover number regarding sodium formate also increased from 16 to 150 due to the recycling. In addition, we analysed the rhodium leaching by conducting ICP-MS measurements of the product phase after each run. On average, a low rhodium leaching of only 800 ppb was observed, which equals a rhodium loss of 0.07% of the initial rhodium amount per run. As far as we are aware, our system features the first successful recycling concept of the hydroformylation/aldol condensation tandem reaction.
 | ||
| Fig. 3 Recycling experiment of the hydroformylation/aldol condensation of 1-pentene in PEG-200 with NaOH. For conditions see Table 3, entry 1. Side products include pentane and alcohols. | ||
Conclusions
An orthogonal, multiphase catalyst system was developed for the hydroformylation/aldol condensation tandem reaction with the possibility to recycle both catalysts present in the reaction mixture. Rhodium/sulfoXantphos initiates hydroformylation and NaOH aldol condensation, using polyethylene glycol as polar phase. The applicability of this system was demonstrated in the conversion of 1-pentene to 2-butyloctenal with multiple reuse of the catalysts. Besides NaOH, pyrrolidine and amino acids were also tested as aldol catalysts, but they all suffered from irreversible deactivation under hydroformylation conditions. NaOH, too, is transformed under the conditions by a reaction with CO to the less basic sodium formate. Nevertheless, and despite being not active in water, formate is able to catalyse the aldol condensation when polyethylene glycol is used as catalyst phase. Therefore, it was possible to reuse the catalyst system. In nine consecutive runs, the recyclability was shown and a total turnover number regarding rhodium of 8700 was achieved, with an average rhodium leaching of 0.07% of the initial rhodium amount per run.For future investigations, the substrate scope of the tandem reaction will be expanded. Furthermore, in order to enable cross aldol reactions, the aldol condensation must be accelerated to be faster in comparison to the hydroformylation step. Additionally, the reaction could be transferred to a continuous flow set-up in which the long-term stability of the catalyst can be investigated. For this, a solution must be established to continuously remove the water, which is formed as side-product, for example by membrane techniques.
Experimental section
General
All reactions were carried out under argon inert gas atmosphere by using standard Schlenk-techniques or a glovebox. Chemicals were degassed before use and air-sensitive substances were stored under argon. Hydrogen (5.0) and carbon monoxide were supplied by Westfalen. All experiments including the use of gases were conducted in 10 mL stainless steel high-pressure autoclaves with glass inserts.Aldol condensation reactions in water
In general, the aldol catalyst (5 mol%) was dissolved in water (0.4 mL). The catalyst solution was heated to 65 °C and the reaction was started by the addition of hexanal (1.0 mL, 8.2 mmol). The mixture was then stirred for 1.5 h, cooled down and the organic phase was separated and analysed by GC-FID.Hydroformylation/aldol condensation tandem reaction
In general, the aldol catalyst (5 mol%) was weighed into a glass insert, which was transferred to a 10 mL autoclave. The autoclave was filled with argon before adding the catalyst solution consisting of [Rh(acac)(CO)2] (2.1 mg, 0.1 mol%) and sulfoXantphos (12.8 mg, 0.2 mol%) in 0.4 mL water or PEG-200. After the addition of 1-pentene (0.9 mL, 8.2 mmol) the autoclave was pressurized with CO (25 bar) and H2 (25 bar). The mixture was stirred at 100 °C or 120 °C for a reaction time of 16 h, before the autoclave was cooled down to room temperature and vented carefully after the reaction. Then the product phase was separated and analysed via GC-FID. Products were not isolated, instead GC-Yields are presented in the text.Recycling experiments
The tandem reaction was conducted as described before. After separation of the product phase, the catalyst phase was washed with pentane to remove remaining product. Afterwards, the catalyst solution was treated with high vacuum to remove the rest of pentane. Then new 1-pentene was added, and the mixture was pressurized with CO and H2 for the next run. After each run, the product phase was analysed via GC-FID and ICP-MS to determine the product distribution and the Rh-leaching, respectively.Analytics
Gas chromatography measurements were performed on a Shimadzu Chromatograph Nexis GC-2030 equipped with a CP Wax 52 CB column and an FID detector. Samples were prepared by diluting 0.1 mL of product solution with 1 mL heptane. The response factors of all compounds either were determined by calibration or estimated using Sternberg's effective carbon method.421H-NMR spectra were recorded on a Bruker AV400 (400.2 MHz) spectrometer using CDCl3 or D2O as solvent.
ICP-MS measurements were performed on a Shimadzu ICPMS-2030. The measurement provided the Rh-content in mg per L of product solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge basic support by the Max Planck Society. We also would like to thank the SusChemSys initiative for financial support. Open Access funding provided by the Max Planck Society.Notes and references
- H. Pellissier, Adv. Synth. Catal., 2016, 358, 2194–2259 CrossRef CAS.
- J. E. Camp, Eur. J. Org. Chem., 2017, 425–433 CrossRef CAS.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC.
- R. Kourist and J. González-Sabín, ChemCatChem, 2020, 12, 1903–1912 CrossRef CAS.
- H. Pellissier, Org. Prep. Proced. Int., 2019, 51, 311–344 CrossRef CAS.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS.
- B. P. Bondžić, J. Mol. Catal. A: Chem., 2015, 408, 310–334 CrossRef.
- P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3366 CrossRef CAS.
- T. Gaide, J. Bianga, K. Schlipköter, A. Behr and A. J. Vorholt, ACS Catal., 2017, 7, 4163–4171 CrossRef CAS.
- M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706–1724 CrossRef CAS.
- P. Kalck and M. Urrutigoity, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS.
- P. Eilbracht and A. M. Schmidt, in Catalytic Carbonylation Reactions, ed. M. Beller, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 65–95, DOI:10.1007/3418_017.
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS.
- K. A. Ostrowski, T. A. Faßbach, D. Vogelsang and A. J. Vorholt, ChemCatChem, 2015, 7, 2607–2613 CrossRef CAS.
- T.-S. Jiang and J.-H. Li, Chem. Commun., 2009, 7236–7238, 10.1039/B917782E.
- X. Fang, R. Jackstell, R. Franke and M. Beller, Chem. – Eur. J., 2014, 20, 13210–13216 CrossRef CAS.
- S. K. Sharma, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2009, 301, 31–38 CrossRef CAS.
- S. K. Sharma, V. K. Srivastava, R. S. Shukla, P. A. Parikh and R. V. Jasra, New J. Chem., 2007, 31, 277–286 RSC.
- V. K. Srivastava, S. K. Sharma, R. S. Shukla and R. V. Jasra, Catal. Commun., 2006, 7, 879–884 CrossRef CAS.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef CAS.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- Y.-l. Liu, J.-g. Zhao, Y.-j. Zhao, H.-M. Liu, H.-y. Fu, X.-l. Zheng, M.-l. Yuan, R.-x. Li and H. Chen, RSC Adv., 2019, 9, 7382–7387 RSC.
- Y. Zhao, Y. Liu, J. Wei, H. Fu, X. Zheng, M. Yuan, R. Li and H. Chen, Catal. Lett., 2018, 148, 438–442 CrossRef CAS.
- X. Jin, J. Feng, Q. Ma, H. Song, Q. Liu, B. Xu, M. Zhang, S. Li and S. Yu, Green Chem., 2019, 21, 3267–3275 RSC.
- S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS.
- J. Esteban, H. Warmeling and A. J. Vorholt, Catal. Commun., 2019, 129, 105721 CrossRef CAS.
- M. Vafaeezadeh and M. M. Hashemi, J. Mol. Liq., 2015, 207, 73–79 CrossRef CAS.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC.
- T. A. Faßbach, F. O. Sommer and A. J. Vorholt, Adv. Synth. Catal., 2018, 360, 1473–1482 CrossRef.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC.
- B. Hamers, P. S. Bäuerlein, C. Müller and D. Vogt, Adv. Synth. Catal., 2008, 350, 332–342 CrossRef CAS.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS.
- K. A. Ostrowski, D. Lichte, M. Stuck and A. J. Vorholt, Tetrahedron, 2016, 72, 592–598 CrossRef CAS.
- T. Rösler, T. A. Faßbach, M. Schrimpf, A. J. Vorholt and W. Leitner, Ind. Eng. Chem. Res., 2019, 58, 2421–2436 CrossRef.
- V. S. Shende, V. B. Saptal and B. M. Bhanage, Chem. Rec., 2019, 19, 2022–2043 CrossRef CAS.
- Á. Molnár and A. Papp, Coord. Chem. Rev., 2017, 349, 1–65 CrossRef.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- J.-D. Yang, http://ibond.nankai.edu.cn.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
- H. Warmeling, D. Hafki, T. von Söhnen and A. J. Vorholt, Chem. Eng. J., 2017, 326, 298–307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03392h |
| This journal is © The Royal Society of Chemistry 2020 |