
The catalytic activity and mechanism of oxygen reduction reaction on P-doped MoS2†
Xiaoming
Zhang
ab,
Shaodong
Shi
a,
Tianwei
Gu
a,
Leyi
Li
a and
Shansheng
Yu
 *a
*a
aState Key Laboratory of Automotive Simulation and Control, Department of Materials Science, Jilin University, Changchun 130012, China. E-mail: yuss@jlu.edu.cn
bDivision of Fuel Cell & Battery, Dalian National Laboratory for Clean Energy, Dalian Institution of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 22nd May 2018
Abstract
With the approaching commercialization of proton exchange membrane fuel cell technology, developing active, non-precious metal oxygen reduction reaction (ORR) catalyst materials to replace currently used Pt-based catalysts is a necessary and essential requirement in order to reduce the overall system cost. Here, we report a single-atom doped molybdenum disulfide sheet (short as X-MoS2) catalyst for the ORR using a dispersion-corrected density functional theory method. Of all the eleven X-MoS2 (X = B, C, N, O; Al, Si, P; Ga, Ge, As, and Se) systems, only the phosphorus atom doped molybdenum disulfide (P-MoS2) has an O2 adsorption energy close to that of a Pt(111) surface. We have further explored the detailed ORR mechanism of P-MoS2. Along the four-electron reaction pathway, the reduction of OH to H2O is the rate-limiting step with the largest diffusion barrier of 0.79 eV.
1. Introduction
In a proton exchange membrane fuel cell (PEMFC), the rate of the oxygen reduction reaction (ORR) at the cathode is much lower than that of the anode reaction,1,2 and many studies have focused on improving the ORR activities using various catalysts. At present, platinum based materials are well known as the most efficient catalysts for the ORR.3–6 However, as a noble metal, the high price and rarity of the platinum element limit the commercialization of PEMFCs.7 Recently, 2D materials with a large surface area, including graphene8–11 and MoS2 based materials,12–23 have received much attention as alternative catalysts. MoS2 is a transition metal chalcogenide that displays a layered structure similar to graphene, with an extended network of alternating Mo and S atoms. The MoS2 nanosheets can be fabricated with controlled sizes, doped with heteroatoms, and surface-modified with other materials for unique crystal structures and high anisotropy. Moreover, the electronic structure of MoS2 is highly dependent on the coordination of the Mo metal and its d-electrons, making it an important candidate with many potential uses in the field of catalysis. Furthermore, the edges of MoS2 nanosheets have unsaturated coordination and dangling bonds that play an important role in surface-active applications. Consequently, these unexpected properties of MoS2 nanosheets offer opportunities for new fundamental and technological research in a large variety of fields.24–30Wang et al.12 demonstrated that the MoS2 particles with the smallest size (∼2 nm) showed outstanding activity for the oxygen evolution reaction (OER) and ORR, in contrast to the inert bulk MoS2. They concluded that the Mo edges on the extremely small MoS2 nanoparticles should contribute to the four-electron reaction process of the ORR on the modified electrode. The incorporation of a heterogenous atom into MoS2 may be a useful method to enhance the ORR performance. Controllable engineering of high electronegativity oxygen heteroatoms into MoS2 ultrathin nanosheets was realized via a facile post-modification process by Huang et al.16 The incorporated oxygen atoms impart a dramatically enhanced ORR activity to the pristine nanosheets, with a 7.8-fold current increase as well as 180 mV and 160 mV positive shifts in both onset and half-wave potentials that are almost comparable to the commercial Pt/C catalyst. The same research group also achieved ultrathin phosphorus-doped MoS2 nanosheets,15 which exhibit dramatically enhanced catalytic activity for the ORR. Both the onset and half-wave potentials are 160 mV more positive than the pristine form and the current density increased by 7-fold. Particularly, a change in the ORR selectivity from the two-electron to a four-electron route was observed. In addition, the proposed material demonstrates an improved stability and extra methanol tolerance compared to commercial Pt/C. The lower electronegativity of phosphorus atoms in the plane of MoS2 nanosheets leads to an abundance of active sites, which may prefer to absorb oxygen molecules in the electrolyte and further accelerate the subsequent reduction processes. Xiao and coworkers17 predicted that Co or Ni doped MoS2 can be used as suitable electrocatalysts for the ORR using a density functional theory (DFT) method because the doped MoS2 can effectively adsorb O2. Zhao et al.31 investigated the ORR activity of N- and P-doped MoS2 monolayers in acidic medium by means of systematical DFT computations. Their results revealed that N doping provides the MoS2 monolayer with better catalytic performance than P doping due to its moderate binding strength with the ORR species. Especially, the energy barrier and overpotential of the ORR on the N-doped MoS2 monolayer are even lower than those on the Pt-based catalysts. The improved catalytic activity of the MoS2 monolayer mainly originates from the induced suitable spin density due to N and P doping. Although attention has been devoted to understanding the ORR mechanism on pristine or doped 2D MoS2 at the molecular level, the reason for enhancing or limiting the ORR activity by dopants is still inconclusive. More effort is needed to further increase the ORR activity of these catalysts.
First-principles methods have been proven to be extremely successful in the rational design of solid catalysts.32 In this work, we have done calculations with a dispersion-corrected DFT (DFT-D) method to understand the ORR activity on X-MoS2, and further to identify the mechanism and the kinetics of the involved ORR elementary reaction. The aim of this article is to explore the origins of the enhancement and limitation of dopants and provide a reference for designing better catalysts in the future.33
2. Computational method
The spin-polarized DFT calculations were performed using the DMol3 code embedded in Materials Studio (Accelrys, San Diego, CA)34 with a long-range dispersion correction via Grimme's scheme.35 The generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) functional is employed to describe exchange and correlation effects.36 The DFT semi-core pseudopods core treatment method is implemented for relativistic effects, which replace core electrons by a single effective potential and introduce some degree of relativistic correction into the core.37 The double numerical atomic orbital augmented by a polarization function was chosen as the basis set.38 During geometrical optimization, the basis set cut-off was chosen to be 4.9 Å. The convergence tolerances for the geometry optimization were set to 10−5 Ha for the energy, 0.002 Ha Å−1 for the force, and 0.005 Å for the displacement. The electronic SCF tolerance was set to 10−6 Ha. A Fermi smearing parameter of 0.005 Ha was used in the calculations. The reciprocal space was sampled with a 4 × 4 × 1 k-point grid generated automatically using the Monkhorst–Pack method39 for the relaxation calculations and a (12 × 12 × 1) k-point grid was used for electronic structure computations. Complete linear synchronous transit/quadratic synchronous transit (LST/QST) calculations were performed to locate transition states (TSs). Moreover, transition states were identified by the number of imaginary frequencies (NIMG) with NIMG = 1, the vibrational modes, and the ‘‘TS conformation’’ implemented in DMol3. Our DFT calculated lattice parameter for MoS2 is 3.20 Å, which is consistent with the experimental value40 and other computational results.17,41,42 A more than 15 Å thick vacuum was added to avoid the artificial interactions between MoS2 and its images. The metastable 1T phase persists after exfoliation and coexists with the 2H phase. However, since the metastable 1T phase can be diminished by heating or aging,43 the monolayer MoS2 with the stable 2H phase was adopted and modeled as a periodically repeated (4 × 4) supercell. In all of the structure optimization calculations, all the atoms are fully relaxed.The adsorption energies (ΔEads) of the adsorbates are calculated through
| ΔEads = Eads/X-MoS2 − EX-MoS2 − Eads |
3. Results
3.1 The adsorption structures of O2 on X-MoS2
We have firstly investigated the adsorption of O2 on the X-MoS2 (X = B, C, N, O, Al, Si, P, Ga, Ge, As, and Se) nanosheets. There are four kinds of possible adsorption geometry for the pristine MoS2 and doped MoS2, which are the end-on site, side-on site, top site and hollow site on X atoms, as shown in Fig. S1 (ESI†). The most stable adsorption geometries, adsorption energies, Hirshfeld charges and p band centers are listed in Table 1. For the dopants in the same group, we find that the O2 adsorption strength has a decreasing trend when the doping elements move from left to right: B > C > N–O (Al > Si > P > S; Ga > Ge > As > Se). The p band center of X atoms decreases in the same order: B > C > N > O (Al > Si > P > S; Ga > Ge > As > Se). This means that the more active the X atom (p band center close to the Fermi level), the stronger the binding energies of O2. However, the Hirshfeld charges of X atoms do not have obvious trends. Furthermore, it is well known that the O2 molecule possesses triplet states in which two single electrons occupy 2px* and 2py*. The empty bands of X atoms offer an orbital for their anti-orbital electrons and this induces a stronger binding between O2 and X-MoS2.| Geometry structure | Adsorption mode | ΔEads | d O–O | Δq (Xbare) | ε p |
|---|---|---|---|---|---|
| B-MoS2 | Side-on | −2.52 | 1.60 | −0.17 | −0.70 |
| C-MoS2 | End-on | −1.21 | 1.35 | −0.26 | −1.69 |
| N-MoS2 | Top | −0.09 | 1.23 | −0.32 | −2.83 |
| O-MoS2 | Top | −0.11 | 1.23 | −0.29 | −3.27 |
| Al-MoS2 | Side-on | −2.69 | 1.48 | 0.16 | −0.33 |
| Si-MoS2 | Side-on | −2.57 | 1.59 | −0.004 | −0.80 |
| P-MoS2 | Side-on | −0.87 | 1.58 | −0.14 | −1.17 |
| MoS2 | Hollow | −0.02 | 1.23 | −0.11 | −2.01 |
| Ga-MoS2 | Side-on | −1.53 | 1.44 | 0.21 | −0.20 |
| Ge-MoS2 | Side-on | −1.13 | 1.55 | 0.07 | −0.47 |
| As-MoS2 | Side-on | 0.27 | 1.55 | −0.06 | −0.90 |
| Se-MoS2 | Side-on | 2.45 | 1.59 | −0.04 | −1.65 |
Oxygen molecule adsorption is the first step in the ORR and the adsorption strength should be within a suitable range. If the adsorption is too strong, it is not conducive to product desorption. On the other hand, if the adsorption strength is too weak, the coverage of O2 may be too low to have a high reaction rate and the O–O bond would be too strong to be broken. Previous works have shown that platinum demonstrates a high ORR activity. According to previous theoretical studies of O2 adsorbed on a Pt surface, the adsorption energy is suggested to vary from −0.49 to −1.02 eV44–46 based on different models and methods. Therefore, the adsorption energy on the Pt surface can be used as an important reference to evaluate ORR activities of other catalysts. In all doping systems, only the adsorption energy of O2 on the P-MoS2 sheet falls in the range [−0.49, −1.02] (eV). Therefore, in the further study, we only consider the ORR mechanism on the P-MoS2 sheet. Our results are different from those of Zhao et al.31 due to the differences in calculation methods. In the experiment in ref. 31, the influence of the water environment was considered. The oxygen adsorption energy of the P-MoS2 catalyst was −0.93 eV in ref. 31, slightly higher than −0.87 eV in our experiment. However, the difference in oxygen adsorption energy of the N-MoS2 catalyst is more significant (−0.37 eV vs. −0.09 eV). The response to the water environment is different for different adsorption structures and this will be our next focus. In this experiment, we did not consider the ORR activity of the N-MoS2 catalyst, given its very low oxygen adsorption energy. Our results give different activity explanations for the P-MoS2 catalyst, which is the meaning of this article.
3.2 The adsorption of various ORR involved species on P-MoS2
We have firstly investigated the adsorption of various ORR involved species on P-MoS2, including single adsorption of O2, O, OH, OOH, and H2O, and co-adsorption of O&O, O2&H, O&OH, OH&OH, H2O&O, H2O&OH, and OH&H. Different adsorption sites have been tested for various adsorption configurations. The optimized geometries and the calculated ΔEads values for the most stable configurations are summarized in Fig. 1 and Table 2. | ||
| Fig. 1 Atomic structures of the relaxed geometries for various ORR chemical species adsorbed on P-MoS2. (a) O2, (b) co-adsorption of two O atoms, (c) O2 and H co-adsorption, (d) OOH species, (e) co-adsorption of an O atom and a OH species, (f) two OH species co-adsorption, (g) O and H2O co-adsorption, (h) OH species and H2O co-adsorption, (i) atomic O species, (j) OH species, (k) H atom and OH species co-adsorption, and (l) H2O. The ΔEads values (in eV) for the related species are marked in the corresponding figures. | ||
| Structures | Species | ΔEads | Δq (ads) | Δq (P) | Δq (P-Mo3) | Δq (P-Mo3S6) |
|---|---|---|---|---|---|---|
| Bare MoS2 | — | — | — | −0.14 | 0.49 | −0.16 |
| Fig. 1(a) | O2 | −0.87 | −0.25 | 0.06 | 0.66 | 0.07 |
| Fig. 1(b) | 2O | −3.40 | −0.54 | 0.20 | 0.88 | 0.35 |
| Fig. 1(c) | O2&H | −0.41 | −0.30, 0.10 | 0.03 | 0.61 | 0.10 |
| Fig. 1(d) | OOH | −2.31 | −0.05 | −0.001 | 0.62 | −0.05 |
| Fig. 1(e) | O&OH | −4.30 | −0.26, −0.08 | 0.16 | 0.82 | 0.22 |
| Fig. 1(f) | 2OH | −5.70 | −0.06, −0.14 | 0.16 | 0.75 | 0.09 |
| Fig. 1(g) | O | −5.53 | −0.29 | 0.04 | 0.65 | 0.08 |
| Fig. 1(h) | OH | −4.01 | −0.03 | 0.01 | 0.63 | −0.07 |
| Fig. 1(i) | OH&H | 0.18 | −0.11, −0.02 | 0.08 | 0.68 | 0.01 |
| Fig. 1(j) | H2O | −0.23 | −0.10 | −0.03 | 0.53 | −0.12 |
| Fig. 1(k) | H2O&O | −5.83 | −0.05, −0.28 | 0.04 | 0.65 | 0.09 |
| Fig. 1(l) | H2O&OH | −4.84 | 0.09, −0.11 | 0.02 | 0.64 | −0.05 |
After the hydrogenation of oxygen molecules, the formed OOH species in Fig. 1(d) is negatively charged by 0.05 e and the adsorption energy is −2.31 eV. Upon the OOH adsorption, the O atom without H binds with P, the O–O bond points to the Mo top site and the charge transfer number of the fragment P-Mo3S6 in this system is about 0.11 e. Fig. 1(g) and (h) present the most stable configurations resulting from the O and OH adsorption, respectively. It is found that O and OH both prefer to be bound strongly on the P top sites, with the ΔEads values of −5.53 and −4.01 eV, respectively. The adsorbed atomic O and OH are negatively charged by 0.29 and 0.03 e, respectively, observed from the Hirshfeld charge analysis. For the H2O adsorption in Fig. 1(j), the ΔEads value is −0.23 eV. The weak interactions indicate that the water can be easily released as the final product of the ORR.
3.3 The ORR element reactions on the P-MoS2 nanosheet
As mentioned above, an O2 molecule can be adsorbed on the P atom, and the chemisorption of the O2 molecule is believed to be the first necessary step to initiate the ORR on the catalyst.47–51 Upon the chemisorption of the O2 molecule, there are two possible reaction pathways for the adsorbed O2 molecule: dissociation into two atomic O and hydrogenation into OOH species.48,50,52 Then, the dissociation product (2O) and the hydrogenation product (OOH) can be further hydrogenated and dissociated respectively to form the adsorbed O and OH intermediates. Similar to the OOH species, O and OH intermediates can undergo further hydrogenation to form 2(OH) species or O and H2O species, respectively. Thus, the four-electron pathways in the ORR on the P-MoS2 nanosheet can proceed through several possible reaction pathways, as shown in Fig. 2.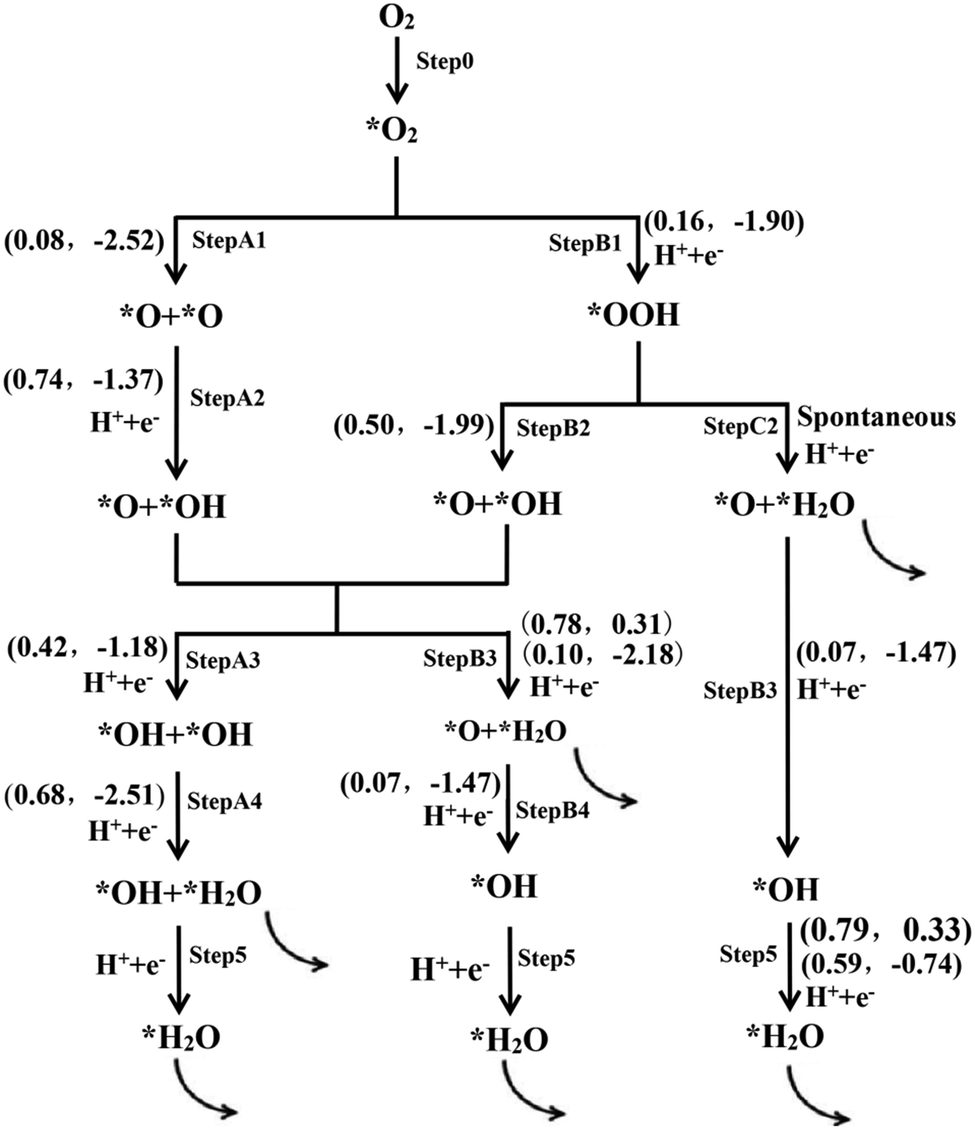 | ||
| Fig. 2 The possible reaction pathways for the ORR on the P-MoS2 catalyst. In the parentheses, the numbers on the left are the energy barrier, Eb, while those on the right are the reaction energy (in eV), Er. The symbol * denotes that the ORR species is adsorbed on the catalyst surface. | ||
From the calculation of the transition states (TSs), the reaction energy and reaction barrier are obtained. The reaction energy, Er, which is defined as the change in total energies between initial states (ISs) and final states (FSs), can be used to evaluate the feasibility of the reaction. Furthermore, the reaction barrier, Eb, is defined as the change in total energies between transition states and final states and it can be used to assess the ease of the reaction.
 | ||
| Fig. 3 The diffusion of the H atom from the P–Mo bridge site to the S atom near to P and the formation of H2O from the hydrogenation of OH, and the release of H2O. | ||
According to the reaction stages presented above, five possible paths for the ORR on the P-MoS2 support are proposed in the following and summarized in Fig. 2, which give an overall picture of the entire ORR on the P-MoS2 support.
Path I: Step0 + StepA1 + StepA2 + StepA3 + StepA4 + Step5;
Path II: Step0 + StepA1 + StepA2 + StepB3 + StepB4 + Step5;
Path III: Step0 + StepB1 + StepB2 + StepB3+ StepB4 + Step5;
Path IV: Step0 + StepB1 + StepB2 + StepA3 + StepA4 + Step5;
Path V: Step0 + StepB1 + StepC2 + StepB4 + Step5.
In Path I, the progression of the ORR on P-MoS2 starts from O2 dissociation. Our TS calculation shows that the dissociation of O2 is kinetically favorable, with a small reaction barrier. With two continuous hydrogenation reactions, the two atomic O will convert into two OH fragments, and finally form a H2O molecule. The rate-limiting step in Path I is Step5 with a reaction barrier of 0.79 eV, corresponding to the diffusion of the H atom in the OH hydrogenation reaction. In Path II, following the O2 dissociation, the two atomic O will be sequentially hydrogenated to form H2O. The rate-limiting steps in Path I and Path II are the same. Path III, Path IV and Path V all start from the hydrogenation of the adsorbed O2 molecules to form OOH. Upon dissociation of the OOH species, the formed O and OH would be hydrogenated into two OH (for Path III) and O&H2O (for Path IV), respectively. In addition to the direct hydrogenation reaction of OOH, H&OOH will spontaneously form O&H2O (for Path V). Upon the release of H2O, P-MoS2 is recovered. The rate-limiting step of Path III, Path IV and Path V is also Step5 with the reaction barrier of 0.79 eV, corresponding to the diffusion of the H atom in the OH hydrogenation reaction.
For all the elementary reactions, the energy barrier for the hydrogenation reaction is particularly high, especially if the H-atom is localized at the P–Mo bridge site. This is the main factor limiting the ORR activity of P-MoS2 nanosheets. In order to explore the origin of this phenomenon, we calculated the density of states (DOS) and orbital information, as shown in Fig. 4. It was found that the orbitals of the P atom and the Mo atom are hybridized to form strong bonds, especially near the highest occupied orbital. The calculations show that the density of electrons is mainly localized at the P–Mo bridge site, and these rich electrons have an anchor effect on the H atom. In addition, we also calculated the deformation charge density map, and found that the P–Mo bridge site is a negative charge accumulation zone, which has a strong interaction with the H atom.
 | ||
| Fig. 4 The electronic structures of the P-MoS2 nanosheet: (a) DOS of P, S and Mo atoms and orbitals at a specific energy level; (b) deformation charge density and Hirshfeld charge. | ||
The activation energy of the rate-determining step of the ORR is 0.79 eV on Pt(111),51 0.80 eV on Pt(100),53 and 0.56 eV on FeN4-gra.54 The current DFT calculations show that P-MoS2 could promote the ORR with an energy barrier of 0.79 eV for the rate-determining step of the most preferred pathway, which suggests that the catalytic activity of P-MoS2 could be comparable to those of the Pt and FeN4-gra catalysts. All of the elementary steps of the five pathways are exothermic, suggesting that P-MoS2 could promote the ORR via five kinds of pathways with catalytic activity comparable to that of the Pt catalyst.
4. Conclusions
The detailed kinetic behaviors of the ORR on P-MoS2 are studied via a DFT-D method. It is found that the dopant P plays a key role in initiating the ORR and the P-Mo3S6 moiety is the active center for all possible elementary steps. Summarizing our calculation results of activation energies, it is concluded that V is the most feasible reaction pathway. The largest reaction barrier is 0.79 eV and comparable to the Pt surface. However, the P–Mo bridge site restricts the diffusion of H atoms and this limits the ORR activity of the P-MoS2 nanosheet. To break this limitation, reducing the P–H–TM binding energy or enhancing the binding strength between H and the neighboring site S (or change to X) via a co-doping method is a potential solution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Yu S. S. is grateful for the financial support from Innovation Foundation of the 46th Research Institute of China Electronics Technology Group Corporation Program (Grant No. CJ20160902) and the National Natural Science Foundation of China (Grant No. 51602305). The DFT calculations utilized resources of High Performance Computing Center, Jilin University.References
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- W. C. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, N. M. Markovic and P. N. Ross, Electrochim. Acta, 2002, 47, 3787–3798 CrossRef.
- H. R. Colón-Mercado and B. N. Popov, J. Power Sources, 2006, 155, 253–263 CrossRef.
- J. Greeley, J. Rossmeisl, I. E. L. Stephens, I. Chorkendorff, A. S. Bondarenko, J. K. Nørskov, T. P. Johansson, H. A. Hansen and T. F. Jaramillo, Nat. Chem., 2009, 1, 552–556 CrossRef PubMed.
- J. Christoph, T. G. Noh, J. Lee, P. Strasser and M. Eiswirth, Surf. Sci., 2009, 603, 1652–1661 CrossRef.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef PubMed.
- D. S. Geng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 Search PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X.a. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef PubMed.
- M. Park, T. Lee and B. S. Kim, Nanoscale, 2013, 5, 12255–12260 RSC.
- T. Wang, J. Zhuo, Y. Chen, K. Du, P. Papakonstantinou, Z. Zhu, Y. Shao and M. Li, ChemCatChem, 2014, 6, 1877–1881 CrossRef.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef PubMed.
- C. Du, H. Huang, X. Feng, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 7616–7622 Search PubMed.
- H. Huang, X. Feng, C. Du and W. Song, Chem. Commun., 2015, 51, 7903–7906 RSC.
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 16050–16056 Search PubMed.
- B. B. Xiao, P. Zhang, L. P. Han and Z. Wen, Appl. Surf. Sci., 2015, 354, 221–228 CrossRef.
- R. Illathvalappil, S. M. Unni and S. Kurungot, Nanoscale, 2015, 7, 16729–16736 RSC.
- C. Suresh, S. Mutyala and J. Mathiyarasu, Mater. Lett., 2016, 164, 417–420 CrossRef.
- E. Lee and Y.-U. Kwon, RSC Adv., 2016, 6, 47468–47473 RSC.
- Y. Z. Huang, L. M. Wu, X. T. Wu, L. H. Li, L. Chen and Y. F. Zhang, J. Am. Chem. Soc., 2010, 132, 12788–12789 CrossRef PubMed.
- P. Bothra, M. Pandey and S. K. Pati, Catal. Sci. Technol., 2016, 6, 6389–6395 Search PubMed.
- X. J. Chua, J. Luxa, A. Y. S. Eng, S. M. Tan, Z. Sofer and M. Pumera, ACS Catal., 2016, 6, 5724–5734 CrossRef.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef.
- X. Huang, Z. Y. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- Q. Fu and X. H. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 RSC.
- G. Zhang, H. J. Liu, J. H. Qu and J. H. Li, Energy Environ. Sci., 2016, 9, 1190–1209 Search PubMed.
- H. Y. Zhang, Y. Tian, J. X. Zhao, Q. H. Cai and Z. F. Chen, Electrochim. Acta, 2017, 225, 543–550 CrossRef.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- T. Jacob and W. A. Goddard, ChemPhysChem, 2006, 7, 992–1005 CrossRef PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- T. Boker, R. Severin, A. Muller, C. Janowitz, R. Manzke, D. Voss, P. Kruger, A. Mazur and J. Pollmann, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 235305 CrossRef.
- P. Johari and V. B. Shenoy, ACS Nano, 2012, 6, 5449–5456 CrossRef PubMed.
- D. Le, T. B. Rawal and T. S. Rahman, J. Phys. Chem. C, 2014, 118, 5346–5351 Search PubMed.
- G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. W. Chen and M. Chhowalla, ACS Nano, 2012, 6, 7311–7317 CrossRef PubMed.
- M. P. Hyman and J. W. Medlin, J. Phys. Chem. C, 2007, 111, 17052–17060 Search PubMed.
- L. Qi, X. Qian and J. Li, Phys. Rev. Lett., 2008, 101, 146101 CrossRef PubMed.
- J. A. Keith, G. Jerkiewicz and T. Jacob, ChemPhysChem, 2010, 11, 2779–2794 CrossRef PubMed.
- W. Orellana, J. Phys. Chem. C, 2013, 117, 9812–9818 Search PubMed.
- J. Zhang, Z. Wang and Z. Zhu, J. Power Sources, 2014, 255, 65–69 CrossRef.
- H. Zhang, Z. Fu, R. Zhang, Q. Zhang, H. Tian, D. Legut, T. C. Germann, Y. Guo, S. Du and J. S. Francisco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11082–E11091 CrossRef PubMed.
- S. Kattel, Z. Y. Duan and G. F. Wang, J. Phys. Chem. C, 2013, 117, 7107–7113 Search PubMed.
- Z. Duan and G. Wang, Phys. Chem. Chem. Phys., 2011, 13, 20178 RSC.
- S. Kattel and G. Wang, J. Phys. Chem. Lett., 2014, 5, 452–456 CrossRef PubMed.
- Z. Y. Duan and G. F. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 Search PubMed.
- S. Kattel and G. Wang, J. Phys. Chem. Lett., 2014, 5, 452–456 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp01294f |
| This journal is © the Owner Societies 2018 |