DOI:
10.1039/C5RA23805F
(Paper)
RSC Adv., 2016,
6, 4070-4076
Synthesis of donor–acceptor copolymer using benzoselenadiazole as acceptor for OTFT†
Received
11th November 2015
, Accepted 16th December 2015
First published on 21st December 2015
Abstract
Donor–acceptor-based poly(E)-4-(3,4′-didodecyl-5′-(2-(3-dodecylthiophen-2-yl)vinyl)-2,2′-bithiophen-5-yl)-7-(4-dodecylthiophen-2-yl)benzo[c][1,2,5]selenadiazole (11) has been synthesized by a Stille coupling reaction. This polymer has a low energy band gap between the HOMO and LUMO energy levels of 1.75 eV. The polymer exhibited good thermal stability. An OTFT prepared using this polymer displayed high hole mobility of 0.097 cm2 V−1 s−1 at 200 °C, a high on/off ratio of 7.8 × 104, and a low threshold voltage of 11.2 V. When compared with as-cast films, annealed films exhibited higher mobility, which was attributed to an increase in crystallinity with an increase in the annealing temperature.
Introduction
During recent years, organic semiconductors have emerged as an alternative to inorganic semiconductors due to their superior advantages over inorganic semiconductors.1–5 Among organic semiconductors, organic field-effect transistors (OFETs) have received significant attention because of their high potential for low-cost, large area, and flexible electronic devices via solution processes. They have several applications in fields such as flexible displays,6 radio frequency identification (RFID) tags,7 and sensors.8 In order to increase the charge carrier mobility, several strategies have been applied. The mobility can be increased by the introduction of donor–acceptor units in the molecules.9
The energy band gap between the HOMO and LUMO levels can be lowered by the introduction of donor–acceptor-based units. Several donor units have been designed and synthesized. Until now, in comparison with donor moieties, acceptor moieties are much less available. The common acceptor moieties are diketopyrrolopyrrole (DPP),10,11 isoindigo (iI),12,13 benzothiadiazole (BTD),14,15 and benzobis(thiadiazole),16 etc. There is therefore a need to develop new types of acceptor units for D–A-type low-band-gap molecules. In comparison with small molecules, polymers have superior solution processability properties and mechanical strength, so most of the research in OTFTs has been focused on polymer materials.
A vinylene linkage is not only helpful for reducing the band gap but also increases the degree of coplanarity in a molecule.17–21 Alkyl chains on a vinylene linkage promote the solubility of a polymer. One research group has reported the synthesis of a copolymer containing a vinylene group that has displayed a hole mobility of 1.05 cm2 V−1 s−1.22 A donor–donor-type copolymer synthesized by using a vinylene group has also exhibited a hole mobility of 3.91 cm2 V−1 s−1.23 Benzoselenadiazole is an electron acceptor formed by the replacement of sulphur in benzothiadiazole. Benzoselenadiazole compounds have a lower HOMO energy level than compounds containing benzothiadiazole due to the larger size and strong electron affinity of selenium.24 Moreover, selenium-containing compounds exhibit a red shift in the UV-visible spectrum compared with sulphur-containing compounds.25–27 Strong Se–Se interactions dramatically enhance inter-chain interactions.28,29 In the literature, OTFTs prepared from benzoselenadiazole are not much studied when compared with solar cell applications.30 By considering the abovementioned issues, the acceptor unit benzoselenadiazole has been synthesized. This acceptor moiety was copolymerized with a donor unit containing thiophenes connected by a vinylene linkage. The introduction of alkyl chains on the donor group helped solubilise the polymer in common organic solvents such as chloroform.
The synthesis of the compounds is illustrated in Scheme 1. Compounds 2, 4 and 9 were synthesized by following literature procedures.31–33 Compound 5 was obtained by the Suzuki coupling reaction of compounds 4 and 3. Compound 5, on bromination, produced compound 6. Compound 10 was obtained by the stannylation of compound 9. The polymer 11 was obtained by the Stille coupling polymerisation reaction of compounds 6 and 10. The molecular weight of the polymer as measured in THF was Mn = 6663 g mol−1, Mw = 8387 g mol−1. The optical properties were investigated by UV-visible absorption spectroscopy; the thermal properties were analyzed by TGA and DSC analysis. The polymer exhibited good hole mobility and a high on/off ratio.
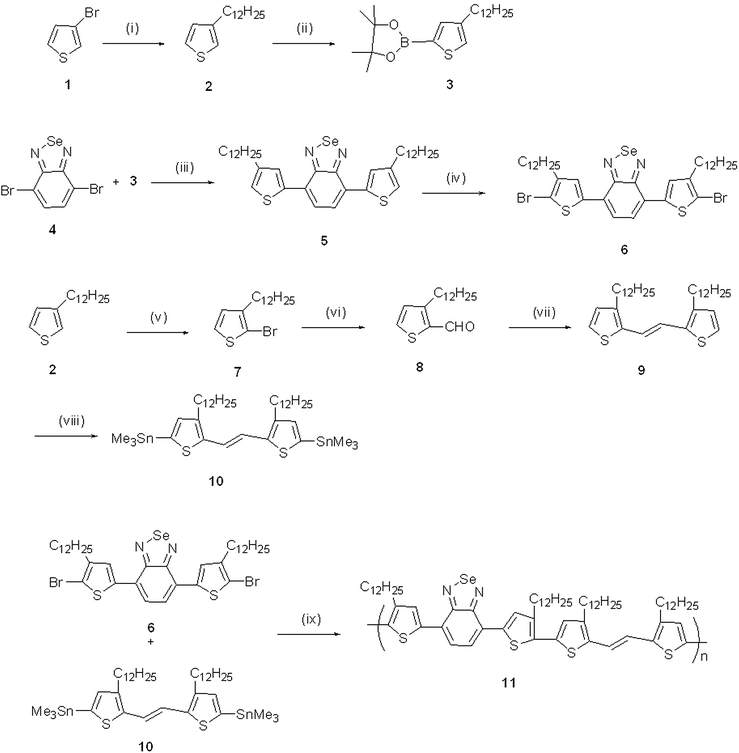 |
| | Scheme 1 Synthesis of polymer (11). (i) C12H25Br, Mg, (dppp)NiCl2, THF; (ii) n-BuLi, IPTMD, THF; (iii) K2CO3, Pd(PPh3)4, toluene; (iv) NBS, THF; (v) NBS, DMF; (vi) Mg, MeMgBr, DMF, THF; (vii) TiCl4, Zn, THF; (viii) n-BuLi, Me3SnCl, THF; (ix) Pd2(dba)3, P(o-tolyl)3, chlorobenzene. | |
Experimental
General and materials
All reagents and chemicals were purchased from Sigma Aldrich Co., TCI, or Alfa Aesar Co. Solvents such as tetrahydrofuran (THF), diethyl ether, toluene, and methylene chloride were used after distillation in the presence of sodium/benzophenone or calcium hydride under nitrogen gas.
Measurements
1H and 13C NMR spectra were recorded on 300 MHz and 75 MHz spectrometers, respectively. The values of the chemical shift were reported in δ units (ppm). A Shimadzu FT-IR spectrophotometer was used for IR analysis. Mass spectrometry analysis was carried out on a JEOL JMS-700. UV-visible absorption was recorded using a Shimadzu UV-1065PC UV-vis spectrophotometer. Cyclic voltammogram measurements were carried out on an Epsilon E3 at room temperature in a tetrabutylammonium perchlorate (0.1 M) solution in chloroform under a nitrogen atmosphere at a scan rate of 50 mV s−1. Pt wire and Ag/AgCl electrodes served as the counter and reference electrodes, respectively. Melting points were measured with an Electrothermal Mode 1307 digital analyzer and were uncorrected. Differential scanning calorimetry (DSC) measurements were carried out under nitrogen using a TA Instruments 2100 differential scanning calorimeter by heating from 0 °C to 300 °C at a rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed under nitrogen using a TA Instruments 2050 thermogravimetric analyzer by heating at a rate of 10 °C min−1 from 30 °C to 800 °C.
Device fabrication
The electrical properties of compound 11 were verified using a top-contact bottom-gated OFET. A 3000 Å thick SiO2 dielectric was thermally oxidized on a heavily N-doped silicon substrate as the gate electrode. The substrate was modified by octadecyltrichlorosilane (ODTS) to decrease the number of hydroxyl groups and increase the hydrophobicity of the SiO2 surface after piranha cleaning (98% concentrated H2SO4 (60 mL)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 (40 mL)). The active layer of compound 11 for the OFET was spin-coated at 3000 rpm from a 0.2 wt% chloroform solution. Gold source/drain electrodes were deposited (100 nm) with width-to-length ratios of 1000 μm/100 μm. The OFET device was tested with a probe station and a Keithley 2400/236 source/measuring unit. The transfer curves were obtained in the saturation region (drain voltage = −80 V).
:H2O2 (40 mL)). The active layer of compound 11 for the OFET was spin-coated at 3000 rpm from a 0.2 wt% chloroform solution. Gold source/drain electrodes were deposited (100 nm) with width-to-length ratios of 1000 μm/100 μm. The OFET device was tested with a probe station and a Keithley 2400/236 source/measuring unit. The transfer curves were obtained in the saturation region (drain voltage = −80 V).
2-(4-Dodecylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3)
n-Butyllithium (8.41 mL of a 2.5 M solution in hexane, 38.03 mmol) was added dropwise to a solution of compound 2 (8.0 g, 31.69 mmol) in THF (160 mL) at −78 °C and stirred for 2 h. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.41 mL, 41.20 mmol) was added in one portion and the mixture was stirred for 1 h at −78 °C and allowed to stand overnight at room temperature under nitrogen. The mixture was poured into water, extracted with dichloromethane, dried over anhydrous MgSO4, concentrated and purified by column chromatography using hexane/ethyl acetate (10:1, v/v) to obtain compound 3 as a colourless liquid. Yield: 6.10 g (50.87%); IR (KBr, cm−1): 3090 (sp2 C–H), 1450 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1370 (C–O); 1H NMR (300 MHz, CDCl3, ppm): δ 7.50 (s, 1H), 7.23 (s, 1H), 2.64 (t, J = 6 Hz, 2H), 1.59–1.69 (m, 2H), 1.36 (s, 12H), 1.27–1.28 (m, 18H), 0.91 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3, ppm): δ 144.73, 138.51, 127.59, 83.99, 31.96, 30.70, 30.00, 29.70, 29.61, 29.50, 29.40, 29.34, 24.83, 24.78, 22.73, 14.17; EI, MS m/z (%): 278 (26, M+).
C), 1370 (C–O); 1H NMR (300 MHz, CDCl3, ppm): δ 7.50 (s, 1H), 7.23 (s, 1H), 2.64 (t, J = 6 Hz, 2H), 1.59–1.69 (m, 2H), 1.36 (s, 12H), 1.27–1.28 (m, 18H), 0.91 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3, ppm): δ 144.73, 138.51, 127.59, 83.99, 31.96, 30.70, 30.00, 29.70, 29.61, 29.50, 29.40, 29.34, 24.83, 24.78, 22.73, 14.17; EI, MS m/z (%): 278 (26, M+).
4,7-Bis(4-dodecylthiophen-2-yl)benzo[c][1,2,5]selenadiazole (5)
Compound 4 (1.0 g, 2.93 mmol) and compound 3 (2.66 g, 7.03 mmol) were dissolved in toluene (35 mL) and 2 M K2CO3 (30 mL) solution. N2 was bubbled for 10 min. Then, Pd(PPh3)4 (0.2 g, 0.176 mmol) was added and the flask was degassed, and the resulting mixture was heated at 100 °C for 24 h. After cooling, the reaction mixture was poured into water and extracted with methylene chloride. The organic layer was washed with brine and dried over MgSO4. The solvent was evaporated and the residue was purified by column chromatography on silica gel with hexane/methylene chloride (4:1, v/v) to give compound 5 as a red solid. Yield: 1.68 g (83.72%); IR (KBr, cm−1): 2950 (sp2 C–H), 1470 (CC); 1H NMR (300 MHz, CDCl3, ppm): δ 7.89 (d, J = 1.5 Hz, 2H), 7.76 (s, 2H), 7.06 (d, J = 1.2 Hz, 2H), 2.70 (t, 4H), 1.69–1.74 (m, 4H), 1.28 (m, 36H), 0.90 (t, J = 6.3 Hz, 6H); 13C NMR (75 MHz, CDCl3, ppm): δ 158.22, 144.04, 139.33, 128.91, 127.45, 125.80, 121.88, 31.97, 30.68, 30.56, 29.74, 29.71, 29.68, 29.56, 29.42, 22.75, 14.19; HRMS (FAB), calculated for C38H56N2S2Se: 684.3050, found: 684.3051.
4,7-Bis(5-bromo-4-dodecylthiophen-2-yl)benzo[c][1,2,5]selenadiazole (6)
Compound (5) (0.10 g, 0.146 mmol) was dissolved in THF (10 mL). N-Bromosuccinimide (0.055 g, 0.307 mmol) was added in one portion to the reaction mixture, which was stirred at room temperature for 5 h. The reaction mixture was poured into water and extracted with methylene chloride. The organic layer was washed with brine and dried over MgSO4. The solvent was evaporated and the residue was purified by column chromatography on silica gel with hexane/methylene chloride (8:1, v/v) to give compound 6 as a dark red solid. Yield: 0.098 g (79.61%); IR (KBr, cm−1): 2950 (sp2 C–H), 1470 (CC); 1H NMR (300 MHz, CDCl3, ppm): δ 7.67 (s, 2H), 7.66 (s, 2H), 2.64 (t, J = 7.5 Hz, 4H), 1.63–1.70 (m, 4H), 1.28–1.74 (m, 36H), 0.89 (t, J = 6.6 Hz, 6H); 13C NMR (75 MHz, CDCl3, ppm): δ 157.63, 142.61, 138.71, 127.58, 126.60, 124.81, 112.28, 31.97, 29.84, 29.73, 29.70, 29.66, 29.50, 29.42, 29.35, 22.74, 14.19; HRMS (FAB), calculated for C38H54Br2N2S2Se 840.1260, found: 840.1256.
(E)-1,2-Bis(3-dodecyl-5-(trimethylstannyl)thiophen-2-yl)ethene (10)
Compound 9 (1 g, 1.89 mmol) was dissolved in THF (26 mL) and cooled to −78 °C. n-BuLi (1.6 M) (2.6 mL, 4.16 mmol) was added dropwise and the mixture was stirred for 30 min. Then, the reaction mixture was warmed to room temperature and stirred for 1 h, and again cooled to −78 °C. Then, trimethyltin chloride (1 M) in THF (4.73 mL, 4.73 mmol) was added to the resulting reaction mixture, which was stirred for 10 min at the same temperature, then warmed to room temperature and stirred overnight. The reaction mixture was poured into saturated ammonium chloride solution. The aqueous layer was extracted with ether. The combined organic extracts were washed with water and dried over MgSO4. After removing the solvent, the crude product was extracted with isopropyl alcohol to obtain compound 10 as a pale yellow solid. Yield: 0.98 g (60.66%); 1H NMR (300 MHz, CDCl3, ppm): δ 7.02 (s, 2H), 6.94 (s, 2H), 2.68 (t, J = 7.8 Hz, 4H), 1.61 (m, 4H), 1.27 (br, 36H), 0.90 (t, J = 6.0 Hz, 6H), 0.39 (s, 18H); 13C NMR (75 MHz, CDCl3, ppm): δ 142.10, 141.73, 138.19, 135.29, 119.41, 31.96, 31.16, 29.73, 29.69, 29.65, 29.57, 29.54, 29.40, 28.38, 22.73, 14.18, −8.23.
Poly(E)-4-(3,4′-didodecyl-5′-(2-(3-dodecylthiophen-2-yl)vinyl)-2,2′-bithiophen-5-yl)-7-(4-dodecylthiophen-2-yl)benzo[c][1,2,5]selenadiazole (11)
Compound 6 (0.45 g, 0.54 mmol) and compound 10 (0.46 g, 0.54 mmol) were dissolved in chlorobenzene (20 mL) and the flask was degassed, then Pd2(dba)3 (0.02 g, 0.02 mmol) and P(o-tolyl)3 (0.026 g, 0.086 mmol) were added and again the flask was degassed. The reaction mixture was heated at 100 °C for 48 h. The reaction mixture was cooled and added to a solution of methanol/HCl (200/40) and stirred for 4 h. After filtering, the crude product was purified by Soxhlet extraction using methanol, hexane and chloroform sequentially. The chloroform fraction was concentrated and added to methanol. After filtration, the polymer 11 was obtained as a dark green solid. Yield: 0.412 g (63.65%). The molecular weight of the polymer as measured in THF was Mn = 6663 g mol−1, Mw = 8387 g mol−1.
Results and discussion
Synthesis of compounds
The synthesis of the compounds is illustrated in Scheme 1. Compounds 2, 4 and 9 were synthesized by following literature procedures.31–33 The polymer 11 was obtained by the Stille coupling polymerisation reaction of compounds 6 and 10.
Optical properties
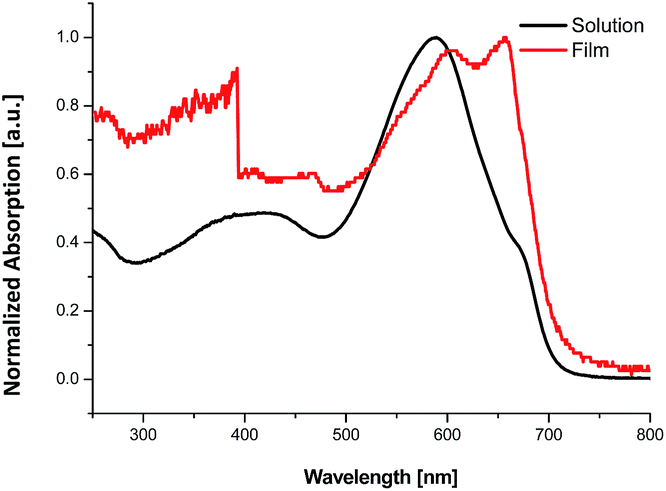
The optical properties of the synthesized 11 were analyzed using UV-visible absorption spectra in a CHCl3 solution as well as in thin films. Fig. 1 presents the UV-visible spectrum of 11. The absorption values are listed in Table 1. Polymer 11 exhibited two absorption bands, which is characteristic of donor–acceptor-type compounds. The short-wavelength band corresponds to π–π transitions and the long-wavelength band corresponds to intramolecular charge transfer (ICT). In solution, the lower absorption band was broad, extending from 360 nm to 450 nm, whereas the intramolecular charge transfer band had an absorption value of 589 nm. In the film state the absorption values were red-shifted; the intramolecular charge transfer (ICT) band had an absorption maximum at 657 nm and a shoulder peak at 604 nm. The red shift in the film state arose due to intermolecular interactions. The optical band gap of 11 was calculated from the cut-off wavelength of the film state spectrum to be 1.75 eV. When compared with donor–donor-type polymers containing a vinylene group, poly[(E)-1,2-(3,3′-ditetradecyl-2,2′-dithienyl)ethylene-alt-dithieno(3,2-b:2′,3′-d)-thiophene] (P14) and poly[(E)-1,2-(3,3′-dioctadecyl-2,2′-dithienyl)ethylene-alt-dithieno(3,2-b:2′,3′-d)thiophene] (P18) (band gap of 1.78 eV),23 polymer 11 has a smaller band gap. This originated from the donor–acceptor nature of polymer 11.
 |
| | Fig. 1 UV-vis absorption spectra of compound 11 in chlorobenzene solution and in the film state. | |
Table 1 Optical, thermal and electrochemical properties of 11
| Compound |
λabs, sol (nm) |
λabs, film (nm) |
HOMOa (eV) |
LUMOb (eV) |
Egc (eV) |
Td (5%)d (°C) |
Tme (°C) |
| Estimated from the onset of the oxidation potential. Calculated from (HOMO level − band gap). Calculated from the wavelength of the absorption edge. Decomposition temperature at 5% weight loss. Tm represents the endothermic melting point. |
| 11 |
360–450, 589 |
604, 657 |
−5.80 |
−4.05 |
1.75 |
444 |
275, 220 |
Electrochemical properties
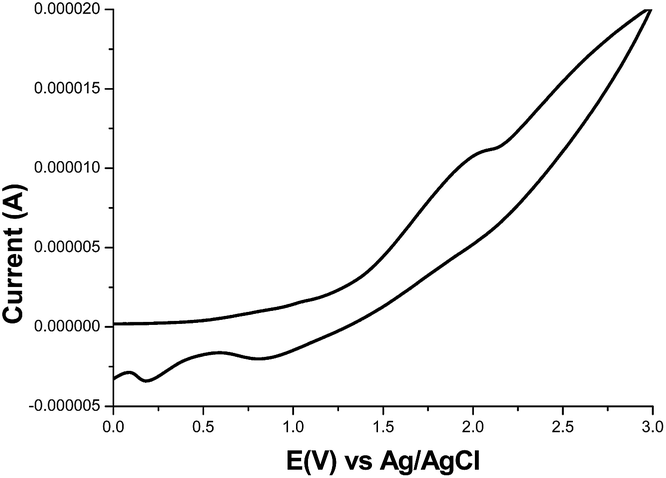
The HOMO and LUMO energy levels were investigated by using cyclic voltammetry (CV) measurements. A cyclic voltammogram of 11 is shown in Fig. 2. CV was performed in a 1.0 × 10−3 M solution of the compound in CHCl3 containing 0.1 M Bu4NClO4. The HOMO energy level of 11 was calculated from the onset of its oxidation potential to be −5.80 eV. The LUMO energy level of 11 was calculated from its HOMO obtained from CV and its optical band gap obtained from UV-vis to be −4.05 eV. A decrease in the LUMO energy level leads to a narrowing of the energy band gap between the HOMO and LUMO energy levels, which is suitable for charge conduction. The HOMO and LUMO values are listed in Table 1. From the lower energy band gap and lower LUMO energy level, we can expect that OTFTs prepared from these compounds can exhibit good mobility. When compared with donor–donor-type polymers containing a vinylene group (P14 and P18; HOMO and LUMO energy levels are 4.80 and 3.02 eV, respectively),23 polymer 11 has decreased HOMO and LUMO energy levels. The donor and acceptor groups in polymer 11 contributed to the decrease in the HOMO and LUMO energy levels, respectively.
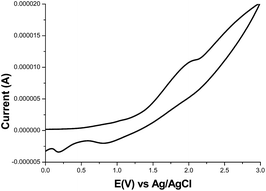 |
| | Fig. 2 Cyclic voltammogram of compound 11. | |
Thermal properties
The thermal properties of 11 were analyzed by thermogravimetric (TGA) and differential scanning calorimetry (DSC) analysis. Fig. 3 presents the TGA and DSC curves. The decomposition temperature at 5% weight loss was observed at 444 °C, which indicates that polymer 11 was thermally highly stable. In the DSC analysis, the endothermic melting temperature was observed at 275 °C in the first heating and at 220 °C in the second heating. The TGA and DSC values are listed in Table 1.
 |
| | Fig. 3 TGA and DSC curves of compound 11. | |
OTFT characterization
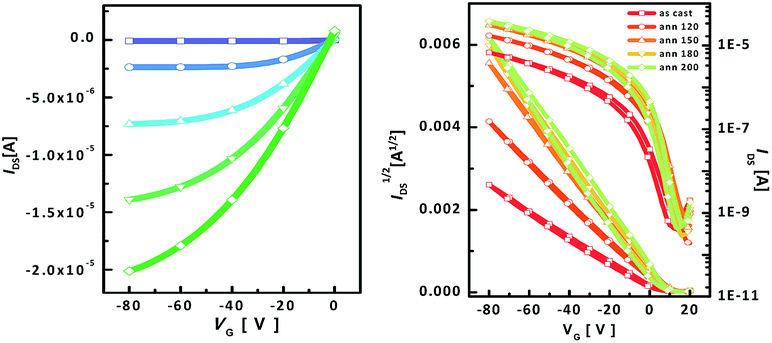
An OTFT based on 11 was fabricated using bottom gate-top contact device geometry. The OTFT film was prepared by spin-coating a solution of the polymer in chloroform. The mobility was calculated from the saturation regime by using μsat = (2IDSL)/(WC(Vg − Vth)2) where IDS is the saturation drain current, C is the capacitance of the dielectric, Vg is the gate voltage and Vth is the threshold voltage. Fig. 4 presents both the output curves and transfer curves. The OTFT prepared using compound 11 exhibited high hole mobility. The as-cast film displayed a maximum mobility of 0.022 cm2 V−1 s−1 and an average mobility of 0.019 cm2 V−1 s−1. After thermal annealing, the carrier mobility was increased, which was due to the increase in crystallinity. After annealing at 120 °C, the maximum mobility reached 0.059 cm2 V−1 s−1. On further annealing at 180 °C, the mobility reached 0.094 cm2 V−1 s−1. After heating at 200 °C, the mobility slightly increased to 0.097 cm2 V−1 s−1. The increase in mobility with an increase in the annealing temperature is in good agreement with AFM analysis and 2D-GIXD analysis. The device exhibited a high on/off ratio. The device made of as-cast film displayed a low threshold voltage of 3.5 V, whereas by increasing the temperature to 180 °C the value of the threshold voltage increased to 11.9 V. The OTFT characteristics are listed in Table 2.
 |
| | Fig. 4 OTFT characteristics of compound 11: (a) output curves and (b) transfer curves. | |
Table 2 OTFT characteristics
| |
μAVG (cm2 V−1 s−1) |
μM (cm2 V−1 s−1) |
On/off ratio |
VTH_AVG (V) |
VON |
SS |
| As-cast |
0.019 |
0.022 |
1.1 × 104 |
3.5 |
12 |
4.3 |
| Ann 120 °C |
0.050 |
0.059 |
2.1 × 104 |
8.9 |
18 |
5.5 |
| Ann 150 °C |
0.075 |
0.085 |
3.7 × 104 |
8.6 |
18 |
5.1 |
| Ann 180 °C |
0.078 |
0.094 |
7.1 × 104 |
11.9 |
17 |
4.5 |
| Ann 200 °C |
0.076 |
0.097 |
7.8 × 104 |
11.2 |
16 |
4.2 |
Morphological characteristics
The morphology of the films was analyzed using atomic force microscopy (AFM). The as-cast film displayed a relatively small root-mean-square (RMS) roughness of 1.12 nm. On the other hand, with an increase in the temperature the RMS roughness increased. The maximum RMS roughness of 1.69 nm was observed for the film annealed at 180 °C; with a further increase in the temperature to 200 °C, the RMS roughness slightly decreased. The annealed films exhibited high RMS roughness caused by the increased grain size, which led to an increase in crystallinity; as a result, the annealed films displayed higher mobility compared with devices made of pristine films. The AFM images are shown in Fig. 5.
 |
| | Fig. 5 AFM images recorded for 11 at different annealing temperatures. | |
The crystalline properties and molecular orientations were investigated using 2D-GIXD analysis. Fig. 6 presents the 2D-GIXD analysis of films annealed at different temperatures. In the as-cast film, the lamellar peak (100) was observed at 0.261 Å−1 in the qz direction, which corresponds to a d-spacing of 24.1 Å. The diffraction peak in the qxy direction was observed at 1.72 Å−1, which corresponds to a π–π stacking distance of 3.65 Å. With an increase in the temperature to 150 °C, the d-spacing decreased to 22.8 Å; on further increasing the temperature to 180 °C, the d-spacing further decreased to 22.4 Å. At 150 °C and 180 °C, π–π stacking distances were observed of 3.65 Å and 3.67 Å, respectively. All the films exhibited lamellar peaks of (200) and (300) in the qz direction; with an increase in the temperature, the intensity of these peaks increased, which indicated an increase in crystallinity. These 2D-GIXD data are in good agreement with the OTFT mobilities. The presence of diffraction peaks due to π–π stacking in only the qxy plane indicates the edge-on orientation of the molecules. The 2D-GIXD values are listed in Table S1.†
 |
| | Fig. 6 2D-GIXD patterns of compound 11 at different annealing temperatures: (a) as-cast film, (b) 120 °C, (c) 150 °C, (d) 180 °C, and their graphical representation in the qz and qxy planes. | |
Conclusions
A donor–acceptor-based copolymer containing benzoselenadiazole as the acceptor and thiophenes having a vinylene linkage as the donor was designed and synthesized by a Stille coupling reaction. The polymer exhibited a low energy band gap between the HOMO and LUMO levels and good thermal stability. An OTFT prepared using this polymer displayed high hole mobility of 0.097 cm2 V−1 s−1 at 200 °C, a high on/off ratio of 7.8 × 104, and a low threshold voltage of 11.2 V. When compared with the as-cast films, the annealed films exhibited higher mobility, which was contributed by an increase in crystallinity.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant Number: 2015032802).
References
- J. Lee, H. J. Cho, N. S. Cho, D.-H. Hwang, J.-M. Kang, E. Lim, J.-I. Lee and H.-K. Shim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2943 CrossRef CAS.
- R. Yang, R. Tian, Q. Hou, Y. Zhang, Y. Li, W. Yang, C. Zhang and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 823 CrossRef CAS.
- J. Lee, B. J. Jung, S. K. Lee, J. I. Lee, H. J. Cho and H. K. Shim, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1845–1857 CrossRef CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15 CrossRef CAS.
- H. E. Katz, Z. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359 CrossRef CAS PubMed.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- P. F. Baude, D. A. Ender, M. A. Haase, T. W. Kelley, D. V. Muyres and S. D. Theiss, Appl. Phys. Lett., 2003, 82, 3964 CrossRef CAS.
- S. Lai, M. Demelas, G. Casula, P. Cosseddu, M. Barbaro and A. Bonfiglio, Adv. Mater., 2013, 25, 103 CrossRef CAS PubMed.
- D. Bharath, S. Chithiravel, M. Sasikumar, N. R. Chereddy, B. Shanigaram, K. Bhanuprakash, K. Krishnamoorthy and V. J. Rao, RSC Adv., 2015, 5, 94859 RSC.
- S. Broll, F. N. Bling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481–7488 CrossRef CAS.
- H. Yu, K. H. Park, I. Song, M.-J. Kim, Y.-H. Kim and J. H. Oh, J. Mater. Chem. C, 2015, 3, 11697 RSC.
- S. Nishinaga, H. Mori and Y. Nishihara, Chem. Lett., 2015, 44, 998 CrossRef CAS.
- T. Hasegawa, M. Ashizawa and H. Matsumoto, RSC Adv., 2015, 5, 61035 RSC.
- F. Wu, S. Chen, L. Chen and Y. Chen, Polymer, 2015, 78, 154 CrossRef CAS.
- M. Zhou, M. Wang, L. Zhu, Z. Yang, C. Jiang, D. Cao and Q. Li, Macromol. Rapid Commun., 2015, 36, 2156 CrossRef CAS PubMed.
- M. Mamada, H. Shima, Y. Yoneda, T. Shimano, N. Yamada, K. Kakita, T. Machida, Y. Tanaka, S. Aotsuka, D. Kumaki and S. Tokito, Chem. Mater., 2015, 27, 141 CrossRef CAS.
- F. Goldoni, R. A. J. Janssen and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4629 CrossRef CAS.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS.
- B. Lim, K. J. Baeg, H. G. Jeong, J. Jo, H. Kim, J. W. Park, Y. Y. Noh, D. Vak, J. H. Park, J. W. Park and D. Y. Kim, Adv. Mater., 2009, 21, 2808 CrossRef CAS.
- S. Y. Jang, B. Lim, B. K. Yu, J. Kim, K. J. Baeg, D. Khim and D. Y. J. Kim, Mater. Chem., 2011, 21, 11822 RSC.
- L. Zhang, L. Tan, Z. Wang, W. Hu and D. Zhu, Chem. Mater., 2009, 21, 1993 CrossRef CAS.
- J. Kim, B. Lim, K.-J. Baeg, Y.-Y. Noh, D. Khim, H.-G. Jeong, J.-M. Yun and D.-Y. Kim, Chem. Mater., 2011, 23, 4663 CrossRef CAS.
- S.-Y. Jang, I.-B. Kim, J. Kim, D. Khim, E. Jung, B. Kang, B. Lim, Y.-A. Kim, Y. H. Jang, K. Cho and D.-Y. Kim, Chem. Mater., 2014, 26, 6907 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012 CrossRef CAS.
- R. Qin, W. Li, C. Li, C. Du, C. Veit, H.-F. Schleiermacher, M. Andersson, Z. Bo, Z. Liu, O. Inganas, U. Wuerfel and F. Zhang, J. Am. Chem. Soc., 2009, 131, 14612 CrossRef CAS PubMed.
- L. Huo, J. Hou, S. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500 CrossRef CAS PubMed.
- A. Patra, Y. H. Wijsboom, S. S. Zade, M. Li, Y. Sheynin, G. Leitus and M. Bendikov, J. Am. Chem. Soc., 2008, 130, 6734 CrossRef CAS PubMed.
- S. Toksabay, S. O. Hacioglu, N. A. Unlu, A. Cirpan and L. Toppare, Polymer, 2014, 55, 3093 CrossRef CAS.
- B. Zhang, J. Xu, L. Hu, G. Chen and W. Yang, Mater. Lett., 2015, 160, 9 CrossRef CAS.
- J. C. Speros, H. Martinez, B. D. Paulsen, S. P. White, A. D. Bonifas, P. C. Goff, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2013, 46, 5184 CrossRef CAS.
- R. Yang, R. Tian, Q. Hou, W. Yang and Y. Ca, Macromolecules, 2003, 36, 7453 CrossRef CAS.
- J. Kim, B. Lim, K.-J. Baeg, Y.-Y. Noh, D. Khim, H.-G. Jeong, J.-M. Yun and D.-Y. Kim, Chem. Mater., 2011, 23, 4663 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 2D-GIXD characteristic properties. See DOI: 10.1039/c5ra23805f |
| ‡ These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.