DNA compaction: fundamentals and applications
André
Estévez-Torres
a and
Damien
Baigl
*bcd
aLaboratoire de photonique et de nanostructures, LPN-CNRS, route de Nozay, 91460, Marcoussis, France. E-mail: aestevez@lpn.cnrs.fr
bDepartment of Chemistry, Ecole Normale Supérieure, 75005, Paris, France. E-mail: damien.baigl@ens.fr; Fax: +33 1 4432 2402; Tel: +33 1 4432 2405
cUniversité Pierre et Marie Curie Paris 6, 75005, Paris, France
dUMR 8640, CNRS, France
First published on 14th May 2011
Abstract
Compaction is the process in which a large DNA molecule undergoes a transition between an elongated conformation and a very compact form. In nature, DNA compaction occurs to package genomic material inside tiny spaces such as viral capsids and cell nuclei. In vitro, several strategies exist to compact DNA. In this review, we first provide a physico-chemical description of this phenomenon, focusing on the modes of compaction, the types of compaction agents and the chemical and physical parameters that control compaction and its reverse process, decompaction. We then describe three main kinds of applications. First, we show how regulated compaction/decompaction can be used to control gene activity in vitro, with a particular emphasis on the use of light to reversibly control gene expression. Second, we describe several approaches where compaction is used as a way to reversibly protect DNA against chemical, biochemical, or mechanical stresses. Third, we show that compact DNA can be used as a nanostructure template to generate nanomaterials with a well-defined size and shape. We conclude by proposing some perspectives for future biochemical and biotechnological applications and enumerate some remaining challenges that we think worth being undertaken.
 André Estévez-Torres | André Estévez-Torres was born in Vigo, Spain. He obtained a MSc in chemistry and physics in 2003 at Ecole normale supérieure, Paris and a PhD in chemistry in 2007 working with Prof. Ludovic Jullien at Université Pierre et Marie Curie in Paris. He then spent a year at the Department of Physics at Kyoto University and two years at Princeton University, working, respectively, with Profs. Kenichi Yoshikawa and Robert H. Austin. He has recently moved to the Laboratoire de photonique et de nansotructures in Marcoussis, France, where he carries out research on the engineering of chemical dynamic systems. |
 Damien Baigl | Damien Baigl is a professor at UPMC and group leader at the Department of Chemistry of Ecole Normale Supérieure (ENS) in Paris. He did a PhD under the supervision of C. E. Williams in the laboratory of P.-G. de Gennes at College de France (2001–2003) and a post-doc in the group of K. Yoshikawa at Kyoto University (2003–2005). He is now developing bottom-up physico-chemical approaches to understand and control complex and biological systems. His research interests include DNA compaction, photocontrol of gene expression, artificial cell models, microfluidics for cell and systems biology, and photo-actuation of micro- and macrofluidic systems. |
1. Introduction
Less than 60 years after the description of the DNA double-helix structure,1DNA has become a very familiar molecule. In the news, DNA is used to identify criminals or to assess the authenticity of a piece of hair. In cinema pictures, it is used to resurrect dinosaurs or to create avatars. In laboratories, DNA is amplified by biochemists, stretched by physicists, modified by chemists. With the opening era of personal genomics, DNA is also the cornerstone of great economical and health challenges. The aim of this review is therefore not to unveil unknown aspects, if any, of DNA neither to make an exhaustive enumeration of experimental results, theoretical considerations, and numerical simulations on DNA but to present in a concise way one phenomenon, DNA compaction, from a physico-chemical point of view and to stress out some potential applications as diverse as nanomaterials fabrication, DNA manipulation, and gene regulation.In living cells, genomic DNA is a long, highly charged, and rather stiff polymer that has to undergo a strong compaction process to fit within tiny available spaces (e.g., the nucleus in eukaryotic cells). This process, also called DNA condensation, can be reproduced and studied in vitro. Several particularly interesting review articles have been published in this field. ‘DNA-inspired electrostatics’ is a short review describing in accessible words delicate physical concepts (such as like-charged attraction) involved in DNA compaction.2 Both physical and biochemical aspects of DNA compaction have been remarkably summarized by Bloomfield.3,4Ref. 5 is a detailed review on DNA compaction/decompaction strategies. Ref. 6 focuses on the nanostructure organization of compacted DNA. The application of DNA compaction in gene delivery, which is of great importance for the success of gene therapy protocols, is well documented in the literature7 and it is out of the scope of this review. Herein, we shall give a brief physico-chemical description of in vitroDNA compaction (modes of DNA compaction, compaction agents, reversibility) before discussing three selected applications: gene regulation (DNA conformation as a trigger of biochemical switches), DNA manipulation (compaction as a protection strategy), and nanostructure fabrication (compact DNA as a nanostructure template).
2. In vitro DNA compaction and decompaction
2.1. Physico-chemical ID of a familiar molecule
Double-stranded DNA is organized into a double-helical structure with a diameter d = 2 nm. Each base pair (bp) has a size a = 0.34 nm and possesses two negatively charged phosphate groups, corresponding to an average distance between charges b = 0.17 nm. DNA persistence length lp, which is of the order of 50 nm (about 150 bp), provides a local stiffness to the molecule.8 To each phosphate group corresponds a cationic counter-ion, which can be free in solution or electrostatically interacting with the DNA molecule. In the framework of the Manning–Oosawa condensation theory,9 a fraction of counter-ions localize in the vicinity of the DNA chain to decrease the electrostatic potential created along the highly charged chain. In average, one DNA phosphate group remains effectively charged every Bjerrum length, lB, the distance at which the electrostatic energy between two elementary charges equals kBT (lB = e2/(4πεkBT) with e the elementary charge, ε the solvent dielectric constant, kB the Boltzmann constant, T the temperature). In the case of counter-ions having a valency Z, for a same entropic cost, the neutralization is Z-fold more effective and the distance between effectively charged DNA monomers becomes ZlB. The fraction of effectively charged DNA monomers is thus feff = b/(ZlB) and the neutralization rate is θ = 1 − feff = 1 − b/(ZlB). For instance, in pure water at 25 °C, lB = 0.71 nm and θ = 0.76, 0.88, 0.92, and 0.94, for Z = 1, 2, 3, and 4, respectively. These figures indicate that over 75% of DNA phosphate groups are neutralized by DNA monovalent counter-ions and that this neutralization significantly increases with an increase in Z. The Manning–Oosawa condensation theory, although obtained from a quite unrealistic case (an infinitely long charged rod) and still strongly debated, has the advantage to provide essential physical ingredients as well as a good estimate of the effective charge of a DNA molecule.For this review, we shall mainly focus on long genomic DNA molecules (significantly larger than lp) in a dilute solution, that is, DNA molecules are not concentrated in the solution and have few interactions between each other. The compaction will thus be unimolecular (that is, involving one DNA molecule) and will result from intramolecular DNA monomer–monomer attractions, usually induced by the addition of an appropriate compaction agent. In such case, the compaction behaviour will be essentially independent of the DNA chain length.
2.2. Three modes for DNA compaction
In water solution, DNA adopts an elongated coil conformation due to the strong repulsion between negatively charged phosphate groups. Upon addition of appropriate compaction agents (see Section 2.3), DNA undergoes a strong compaction process. The identification of the modes of compaction was possible thanks to the remarkable contribution of the Yoshikawa group who brought the analysis of DNA compaction at the level of individual molecules. Fig. 1 shows three pathways that can be followed by DNA to go from the elongated coil state to the compact state. The first type is an all-or-none compaction process where there is no intermediate state but coexistence between the elongated coil state and the compact state.10,11 This process is usually observed when attraction is induced between DNA monomers all along the chain, either by adding small multi-valent counter-ions or by inducing unfavorable contacts between DNA monomers and the solvent (e.g., addition of a poor solvent such as ethanol or addition of neutral polymers that exclude volume to DNA). This is similar to the first-order phase transition between a disordered gas phase (coil state) and a highly condensed solid phase (compact state).11 The second mode of compaction is a progressive transition from the elongated coil state to the compact state. This usually occurs when a strong attraction between several consecutive DNA monomers can be induced locally, typically upon complexation with polycations longer than 10 monomers.12 The two precedent modes of compaction account for a collapse of DNA resulting from DNA monomer–monomer attractive interactions. The highly packaged structure of DNA inside viruses probably results from a combination of these two modes. The third possible route is an assisted, hierarchical compaction by DNA adsorption and wrapping around nanoscale objects. This is the mode of compaction of DNA into chromatin in eukaryotic cells and it is observed in vitro when DNA is compacted by cationic nanoparticles13,14 or dendrimers.15 Clearly, other pathways are possible and intermediate routes between the three cases shown in Fig. 1 are usually observed. For example, DNA compaction by cationic surfactants proceeds through the coexistence between DNA in the fully compact state and DNA shrunk coils, indicating an intermediate route between an all-or-none transition and a progressive compaction.16,17 Segregated states, where the same DNA molecule is composed of compact and unfolded parts, can also be observed under specific conditions.18,19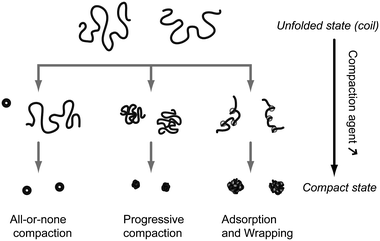 | ||
| Fig. 1 Schematic representation of the 3 principal modes of in vitro unimolecular DNA compaction. | ||
2.3. Compaction agents
Compaction agents are molecules that can induce DNA monomer–monomer attraction and/or provoke unfavorable interaction between DNA and the solvent.Other metal cations such as Al3+ (ref. 27 and 28), lanthanide ions (La3+, Eu3+, Tb3+) (ref. 29), Ga3+ (ref. 28), Cr3+ (ref. 30) and Fe3+ (ref. 31) have also been used. In most cases, these multivalent counter-ions induce an all-or-none compaction.10,11,31 Since these multivalent counter-ions are in competition with monovalent salts present in the medium, an excess of condensing agent is necessary to induce DNA compaction. The critical compaction agent over DNA charge ratio to induce full compaction, ρ*, is thus much larger than 1 (Fig. 2). For similar reasons, ρ* increases with an increase in the concentration of low valence salts (monovalent or valency smaller than Z) present in the medium.20 Conversely, since neutralization becomes more effective for higher Z, ρ* strongly decreases with an increase in Z to reach ρ* ≈ 1 for approximately Z ≈ 10 (Fig. 2).12
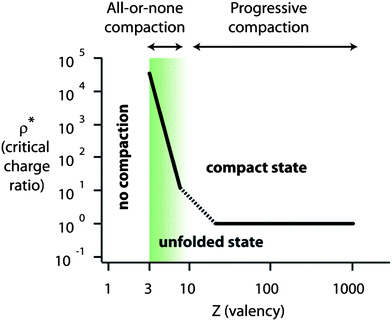 | ||
| Fig. 2 Schematic representation of the mode of compaction (all-or-none or progressive) and critical compaction agent over DNA charge ratio necessary to induce full compaction (ρ*) as a function of the valency of the compaction agent (Z) in the case of purely electrostatic interactions. Inspired from ref. 12. | ||
2.4. Compact state: size, shape and stability
The shape of the compacted DNA results from a balance between surface energy and DNA rigidity. This last parameter can be modified through the addition of monovalent salts yielding larger DNA condensates.47 A toroid with a diameter twice the persistence length is the most common shape,6,23,48 although spherical globules49 are also frequent, and rods,50 flowers51 and racket-shaped52 condensates have also been reported. For DNAs shorter than 40 kbp, single toroids are obtained upon compaction with multivalent cations and their internal diameter decreases with increasing DNA length. Beyond this length, multiple toroids are formed from a single DNA molecule.55The formation of the compact state is counterintuitive for two reasons. First, it is surprising to obtain a stable and dense condensate of a highly charged object (at the onset of compaction DNA still bears 10% of its original charge). Second, it is remarkable that the condensates display a well-defined size. The stability of the compact state is explained in Bloomfield's review.56 Three repulsive contributions to the total free energy need to be considered: (i) bending, coming from the intrinsic rigidity of dsDNA and accounting for ∼+1/300 kBT per bp; (ii) entropic demixing of polymer and solvent, evaluated to +1/150 kBT per bp;57 and (iii) electrostatic repulsion, estimated using Oosawa's framework9 to be +0.24 kBT per bp. Electrostatic attractive interactions are ruled out in the framework of Debye–Hückel and Poisson–Boltzmann descriptions and one needs to consider correlated counterion fluctuations at short distances that are estimated to be −0.3 kBT per bp.58 Adding up repulsive and attractive contributions, the free energy of the compact state is of the order of −0.05 kBT per bp, or −0.1 kJ (mol of bp)−1, compatible with a stable compact state.
Two possible causes have been evoked to explain the limited-size of DNA condensates.59 The first one, thermodynamic, calls for a repulsive free energy coming from topological defects intrinsic to the winding of a linear polymer inside a toroid. The second, kinetic, arises from the energy barrier that two randomly oriented charged rods have to overcome to attain the parallel, attractive, configuration at small separations; a barrier that increases with the size of the condensate. Both contributions become more positive with an increase in toroid size, which could explain the limited size of DNA condensates. Experimentally, toroids have been typically reported to measure around 90–100 nm, which is slightly smaller than 2lp. This value mainly depends on the salt concentration47 and the presence of nucleation loops.47,60 While compaction agent concentration has usually a minor effect on the size of the compact state, it was shown to significantly affect the size of DNA globules and toroids in the case of polyethyleneimine as a compaction agent.61
The first observation of a toroid-like DNA condensate was reported by electron microscopy by Gosule and Schellman in 1976 using spermidine as a condensing agent23 and it was later described in exquisite detail by Hud and coworkers47,48 (Fig. 3A), showing that, in some of the toroids, DNA is hexagonally packed with an interchain distance of 2.6 nm. A related structure has been reported for tightly condensed DNA inside T7 virus capsids; a spool instead of a toroid is observed in this case53 (Fig. 3B). When the compaction process is progressive, condensates are globular with a more disordered, liquid-like structure, although much less data are available.
 | ||
| Fig. 3 Comparison of in vitro and in vivo structures of compacted DNA observed by transmission electron microscopy. (A) Cryoelectron image of a toroidal λ DNA condensate in the presence of Co(NH3)63+. The plane of the toroid is parallel to the image and the fringes represent DNA strands (obtained from ref. 48, copyright 2001 National Academy of Sciences, USA). (B) Average of 77 cryoelectron images of T7 bacteriophage heads from the complete tail-deletion mutant where DNA is compacted in a spool conformation perpendicular to the image plane and 2.5 nm spaced fringes of densely packed DNA are clearly visible (obtained with permission from ref. 53, copyright 1997, Elsevier). (C) T4 DNA compacted in the presence of poly(L-lysine)-covered silica nanoparticles 15 ± 4 nm in diameter at a concentration of 5 × 10−4 wt%. Detail of DNA, dark line, wrapped around a single particle is shown on the left (obtained from ref. 14, copyright 2007, American Chemical Society). (D) Freeze-dried image of a chromatin fiber extracted from rat liver (obtained from ref. 54, © F. Thoma et al., 1979. Originally published in J. CellBiol., 83, 403–427.). Nucleosomes appear as dark circles linked by DNA lines. | ||
The third mode of compaction depicted in Fig. 1 corresponds to the adsorption and wrapping of DNA around nanoscale objects. This mechanism, which is in play in the formation of the nucleosomes, is called complexation by some authors,59 to distinguish it from pure compaction where the volume fraction of monomers in the condensed state is close to 1, while it is 10−2 in the adsorption and wrapping mechanism and 10−5 in a DNA random coil. This process is highly hierarchical and its elemental step is the wrapping of DNA around the nanoscale object. Many theoretical articles have addressed the complexation of DNA with nanoscale objects, as summarized in a comprehensive review by Schiessel.62 The first and systematic experimental study was made by Zinchenko and coworkers13,14 who studied the compaction of DNA in the presence of cationic nanoparticles of sizes ranging between 10 and 100 nm and monovalent salt concentrations spanning 10−2 to 1 M. Three compaction modes were observed depending on the particle size: (i) adsorption of DNA on the particles larger than 40 nm; (ii) wrapping of DNA around particles of size 15 nm (Fig. 3C) in a way similar to chromatin (Fig. 3D) and (iii) adsorption of 10 nm particles onto DNA.13,14 The formation of the compact state for all nanoparticle sizes depended on the salt concentration in a similar way: low compaction at low and high salt concentrations and optimal compaction at intermediate salt concentrations.13,14 This optimum is explained as the interplay between attractive and repulsive electrostatic interactions. At low salt, the increased rigidity of DNA due to the electrostatic contribution to the persistence length hinders compaction. At high salt, κ−1 becomes so small that the attractive electrostatic interaction between the DNA and the nanoparticles is screened. A similar salt effect is observed for the in vitro reconstitution of chromatin, i.e., salt-induced complexation at intermediate salt concentration63 and the salt-induced release of DNA from nucleosome core particles at high salt concentration.64
2.5. Control parameters of DNA compaction/decompaction
As discussed above, a variety of agents are able to induce the compaction of DNA. Here we discuss how physico-chemical parameters, such as salt concentration, solvent dielectric constant, temperature and other external stimuli, affect compaction and decompaction.2.6. Reversible photocontrol of DNA compaction
A particularly interesting experimental parameter to control the compaction state of DNA is light because it is non-invasive and tunable in time and space. Photoreversible DNA condensation was first demonstrated by Le Ny and Lee in 2006.73 They used a cationic surfactant carrying an azobenzene moiety, AzoTAB, standing for azobenzene trimethylammonium bromide, whose conformation changes upon illumination from a more hydrophobic trans isomer to a more hydrophilic cis form (Fig. 4). As a result, the affinity of the surfactant for DNA changes and DNA condensation could be tuned by light at constant AzoTAB concentration. This process is reversible and selective on the illumination wavelength: trans to cisisomerization occurred at 365 while cis to transisomerization happened at 434 nm. We have later demonstrated that the compaction of T4-DNA with AzoTAB is a first order transition and that it is a suitable strategy for controlling single-molecule DNA conformation inside a biomimetic micro-environment using light.17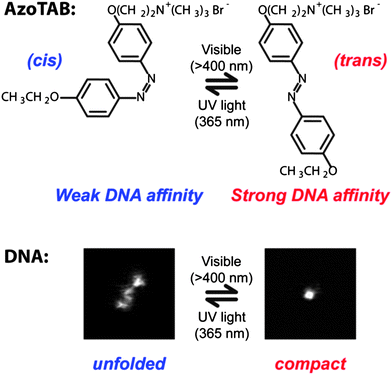 | ||
| Fig. 4 Azobenzene trimethylammonium bromide, AzoTAB, reversibly compacts DNA using light at two different wavelengths. Top: light illumination induces a cis/trans conformational transition that changes the dipolar moment of the surfactant resulting in a differential affinity for DNA. Bottom: DNA compaction can be tuned by light at constant AzoTAB concentration. Pictures are fluorescence microscopy images of an individual T4-DNA molecule stained with YOYO-1 in the presence of AzoTAB (700 μM) in 10 mM TE buffer, after visible (right) and UV (left) illumination. Image sizes are 5 μm × 5 μm. | ||
The concentration of AzoTAB resulting in full DNA compaction was relatively high, typically 700 μM, in these studies.17,73 As a result, subsequent work has attempted to develop similar species with a lower critical compaction concentration. The picture that emerges is that increasing the hydrophobicity of the surfactant tail efficiently reduces this critical concentration, as demonstrated for gemini surfactants74 and derivatives with an increasing number of methyl moieties,40 but it also reduces the reversibility of the photoinduced decompaction. A good balance was achieved when the linker between the trimethylammonium and the azobenzene consisted of 5 (ref. 40) or 4 (ref. 75) methyl groups. These AzoTAB derivatives induced 100% compaction at 100 and 150 μM, respectively, in 10 mM buffer. Moreover, two different approaches combining a cationic AzoTAB derivative and anionic species resulted in a significant decrease of the critical compaction concentration. Catanionic vesicles with a net positive charge formed with an AzoTAB derivative and sodium dodecylbenzenesulfonate at concentrations of 48 and 19 μM, respectively, were capable of the photoreversible condensation of DNA.76 AzoTAB in the presence of 10−3 wt% anionic silica nanoparticles reversibly compacted DNA at a concentration of 200 μM.41 In both cases, the decrease in the critical concentration was attributed to a cooperative effect induced by the anionic species that facilitates the aggregation of the cationic surfactant.
In addition to the reversibility of compaction it is important to consider the kinetics of the process, which of course depends on the photon flux. The photo-isomerization rate constant can be written as k = εI0φ, where ε is the molar absorption coefficient at a given wavelength, I0 the radiative flux of light and φ the quantum yield of the photo-induced reaction. Typical values of ε in the AzoTAB series are in the range 1–3 × 103 m2 mol−1 and φ, measured for a triethyleneglycol derivative,77 is about 1 and 0.7 for the trans to cis and cis to transisomerizations, respectively. Photon fluxes of 10−3 (mol of photons) m−2s−1 (corresponding to a 500 W Hg lamp) resulted in isomerization rates for an AzoTAB derivative of 3 and 2 s−1 for the trans to cis and the cis to transisomerizations, respectively.75 These conditions resulted in the compaction and decompaction of 166 kbp long T4-DNA in 1 s without apparent DNA damage, indicating that the rate-limiting process is the DNA conformational transition.78
2.7. DNA origami
Although it is not usually considered a compaction technique, we would like to include here DNA origami as a sequence-directed strategy to obtain compact DNA structures. DNA origami consists of controlling the shape of a scaffold ssDNA several kbp long using hundreds of short ssDNA sequences as if they were staples that clamp two non-contiguous sequences of the scaffold backbone in a certain geometric configuration. Rothemund first proposed this idea and reported two-dimensional structures such as a smiley face and a map of the western hemisphere with a pixel size of 6 nm, with great reproducibility and relatively short folding times (∼1 h).79 Later 3D nanoscale objects with diverse shapes were reported, for which much longer times, of the order of a week, were required for proper folding.80 It is interesting to compare these very long assembly times to obtain the final nanoscale object to the typical second-scale formation of toroids by unimolecular DNA compaction.2.8. Summary on the fundamental aspects of DNA compaction
We saw that DNA compaction and decompaction can be controlled by a variety of physico-chemical stimuli. Table 1 summarizes for the main types of compaction agents the associated modes of compaction and the parameters that can be used to induce decompaction. Hereafter, we will focus on applications of DNA compaction/decompaction.| Compaction agent | Mode of compaction | Decompaction |
|---|---|---|
| Multivalent cation (3 ≤ Z ≤ 10) | All-or-none11 | [Salt] ↑,20T ↓,67,68ε ↑,21,22,65Z ↓ (e.g., oxido-reduction,31 pH70) |
| Polycation (Z > 10) | Progressive12 | Polyanions |
| Cationic nanoparticle | Adsorption and wrapping13,14 | [Salt] ↑↓13,14 |
| Cationic surfactant | All-or-none + progressive16,17,39 | Light,17,73 adding cyclodextrine,81 anionic82,83 and non-ionic84surfactants |
| Cationic vesicle | Adding detergent72 | |
| Neutral polymer | All-or-none45 | [Salt] ↓,44,45T ↑69 |
3. Reversible compaction for gene regulation
An important part of gene regulation in an organism occurs at the level of transcription and one can distinguish two principal strategies. On the one hand, a ligand (called a trans factor, such as a transcription factor or the bacterial σ factors) binds to a regulatory DNA sequence (cis element) and tunes the transcription activity of one or several genes in a sequence-dependent manner. On the other hand, the higher-order structure of the chromosome may modify the affinity of the trans factor or the RNA polymerase for the DNA sequence, by blocking its access for instance. This second strategy is expected to regulate gene activity over larger sets of genes and in a way that is less sensitive to the sequence. This structural influence on gene regulation has long ago been observed in the silencing properties of heterochromatin in eukaryotes85 and its importance in bacteria has been revealed in the last decade:86,87 supercoiling and DNA condensation play important roles in the regulation of gene expression.The first to study the effect of DNA condensation on transcription were Baeza et al. in 1987.88 They reported an enhancement of transcription in circular plasmids condensed with spermidine. Taking into account the low salt conditions of their experiments and the results described below we can now argue that their interpretation was probably wrong and the enhancement might have been due to spermidine–protein interactions. More recently, Tsumoto et al. demonstrated that the compaction of a 40 kbp long DNA, bearing a T7 promoter at half length, resulted in the sharp inhibition of transcriptional activity, using both spermine and PEG as compacting agents.89 A comparable on/off switching of transcription due to compaction was very recently demonstrated in water-in-oil microdroplets coated with a phospholipid membrane using an elegant FRET assay for detecting single molecule mRNAs.90 Results in a similar direction have been obtained when T4-DNA was complexed with cationic nanoparticles, although here the inhibition of transcription with increasing concentration of nanoparticles was more gradual.91
4. Photocontrol of gene expression based on light-induced nucleic acid conformational changes
We have recently demonstrated that photocontrol of the compaction of nucleic acids (DNA, mRNA) allows to control gene expressionin vitro using light at both transcription and translation levels92 (Fig. 5 and 6). When AzoTAB is added to the gene expression system, DNA (respectively mRNA) folds and transcription (respectively translation) is switched off; after a short UV illumination (1–3 minutes at 365 nm), DNA (respectively mRNA) unfolds back and transcription (respectively translation) is switched on again.92 We have demonstrated that this method is potentially applicable to any DNA template, regardless of its length (from 100 bp to 100 kbp) and its sequence, as well as to bacterial (e.g., E. coli) or viral (e.g., T7) polymerases. This method does not require any covalent modification of the substrates and it is reversible, which is an advantage over photo-uncaging strategies. In all cases, the compaction state of the nucleic acid correlated well with the level of RNA/protein produced. In the case of transcription, RNA production was inhibited by addition of AzoTAB and fully recovered upon UV illumination (Fig. 6A). Translation was also strongly reduced by AzoTAB and enhanced 3- to 6-fold by UV illumination (Fig. 6B). Moreover, this robust approach allows dynamic ON and OFF photoswitches using sequential UV and visible illumination pulses, respectively (Fig. 6C). This is thus, to our knowledge, the only approach allowing both temporal and reversible control, in a sequence-independent way. By coupling this method to gene silencing using specific miRNAs, selective photocontrol was possible and the light-induced production of different combinations of a few target proteins was reported.93 Lee and coworkers went a step forward and applied photoreversible DNA compaction to gene delivery inside mammalian cells.76 In their in vivo studies, protein expression from an internalized plasmid increased 2-fold after UV illumination.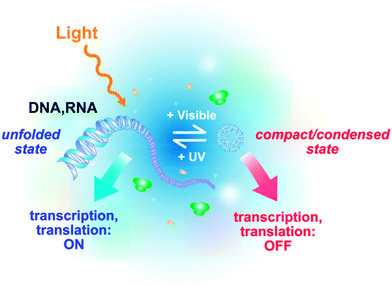 | ||
| Fig. 5 Schematic principle of the reversible photocontrol of gene activity (transcription and translation) based on light-induced DNA/RNA conformational changes. | ||
![AzoTAB allows the reversible photocontrol of transcription and translation activity in vitro. (A) Production of RNA by in vitrotranscription from a linearized plasmid coding for transcripts of two different lengths, 900 b and 5 kb at different AzoTAB concentrations, in the presence and in the absence of UV light (365 nm). Left, a denaturant RNA electrophoresis gel; right, normalized transcriptional activity. For [AzoTAB] ≥ 2 mM (dashed line), DNA is compacted and transcription is inhibited; upon UV illumination, DNA unfolds and transcription is recovered. (B) Normalized EGFP translation activity obtained in a cell-free in vitro expression system containing mRNA for different AzoTAB concentrations in the presence (blue triangles) and in the absence (red squares) of UV. (C) The production of RNA from a 144 bp dsDNA fragment condensed with 2 mM AzoTAB is dynamically controlled using light pulses that switch transcription ON (UV light) and OFF (visible light). Adapted from ref. 92.](/image/article/2011/SM/c1sm05373f/c1sm05373f-f6.gif) | ||
| Fig. 6 AzoTAB allows the reversible photocontrol of transcription and translation activity in vitro. (A) Production of RNA by in vitrotranscription from a linearized plasmid coding for transcripts of two different lengths, 900 b and 5 kb at different AzoTAB concentrations, in the presence and in the absence of UV light (365 nm). Left, a denaturant RNA electrophoresis gel; right, normalized transcriptional activity. For [AzoTAB] ≥ 2 mM (dashed line), DNA is compacted and transcription is inhibited; upon UV illumination, DNA unfolds and transcription is recovered. (B) Normalized EGFP translation activity obtained in a cell-free in vitro expression system containing mRNA for different AzoTAB concentrations in the presence (blue triangles) and in the absence (red squares) of UV. (C) The production of RNA from a 144 bp dsDNA fragment condensed with 2 mM AzoTAB is dynamically controlled using light pulses that switch transcription ON (UV light) and OFF (visible light). Adapted from ref. 92. | ||
5. Compaction for protection
5.1. Protection against chemical or biochemical stress
In the unfolded state, genomic DNA is a very long molecule exposing a huge number of monomers (of the order of 106 in the case of human genomic DNA) to its physico-chemical micro-environment. In contrast, in the compact state DNA monomers are confined in a very dense state making them hardly accessible for other molecules present in the medium. For instance, it has been shown that in the toroidal condensate DNA is organized into a hexagonal array with an interhelix spacing ranging between 2 and 3 nm,48,94–97 leaving a free space between DNA consecutive rows that is smaller than 1 nm (DNA diameter is 2 nm). DNA monomers in such a highly packed structure are thus hardly accessible for surrounding chemical species. As a consequence, reversible DNA compaction can be used as a strategy to temporarily protect DNA from an external chemical or biochemical stress by applying the following procedure. In the absence of stress, DNA can be used in an unfolded and “reactive” state. Should a stress be applied, DNA can be folded into a compact and “silent” state and protected against reaction by stress molecules. When the stress is over, DNA unfolding allows recovering the initial “reactive” state. For instance, it has been shown that DNA compaction by multivalent metal cations (Al3+, Co3+),27 short polyamines (mainly spermine (4+) and spermidine(3+))98,99 and protamine100 offers marked protection against fast neutron98 or gamma ray99 radiation-induced single- and double-strand DNA breakage, which has been explained by the reduced accessibility of DNA bases for radiation-induced reactive species.100Polyamines (mainly spermidine)101,102 and protamine103 have also regularly been used to protect DNA during the delivery into cells by bombardment. DNA compaction by spermidine is also known to inhibit DNA fragmentation by endonucleases, which prevents the onset of apoptosis.104 Finally, DNA compaction by polyamines and analogs has been shown to offer marked protection against oxidative stress.105,1065.2. Protection against mechanical stress
We saw that DNA compaction was a way to reversibly hide DNA monomers from their chemical environment and therefore to ensure protection against chemical and biochemical stresses. The dramatic change of DNA size upon compaction can also be exploited as a way to protect DNA against a mechanical stress. Basic manipulations, such as mixing, pipetting, or pumping/injecting, induce shear forces down to the characteristic Kolmogorov scale η, which is typically of the order of a few μm. According to the polymer-scission theory, η is both the minimal extended polymer length to get significant shear-induced chain scission and the size at which chain fragmentation occurs.107 When unfolded genomic DNA molecules, which are much longer than η, are subjected to the above-mentioned manipulations, they thus experience intense molecular tension along their backbone and strong fragmentation into μm sized fragments. Reversible compaction, which brings each DNA molecule to an overall size much smaller than η, has been demonstrated to be a very efficient way to protect DNA against breakage by shearing stress (Fig. 7).108 The protection against DNA breakage by compaction agent was reported for the first time by Kaiser et al.109 and later confirmed by Cai et al.110 and Kovacic et al.111 The role of reversible folding transition in this protection effect was mentioned by Mizuno and Katsura112 and precisely quantified by Cinque et al. who performed systematic DNA size measurements on tens of thousands of individual genomic molecules.108 We can thus anticipate that the implementation of reversible DNA compaction strategies in biological protocols involving the manipulation of long DNA molecules shall greatly improve the feasibility and accuracy of analyses requiring the preservation of genetic information contiguity, such as DNA mapping and chromosome rearrangement studies.108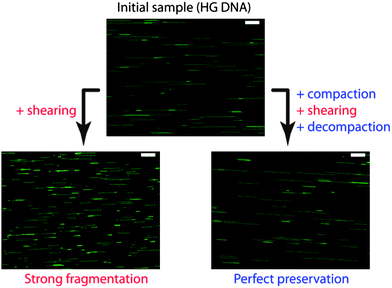 | ||
| Fig. 7 Example of DNA protection by compaction against mechanical stress. Human genomic (HG) DNA sample was submitted to controlled shear stress. Without compaction, molecules are strongly fragmented. If compaction is applied before shear stress, DNA molecules are perfectly preserved after shearing. Pictures are fluorescence images of individual HG DNA molecules combed on a silanized glass substrate. Scale bars are 10 μm. Adapted from ref. 108. | ||
6. Compact DNA as a nanostructure template
Unfolded DNA has been widely used as a template for nanostructure fabrication, enabling a broad variety of applications, as remarkably summarized in the recent review by Becerril and Woolley.113 One of the strategies consists in the localization of transition metal cations through electrostatic interactions with and/or chelation by DNA bases prior to reduction to get a DNA-templated metallic nanostructure. Other involved interactions are π-stacking (DNA–organic molecule interaction) and DNA base pairing. Typical examples of DNA-templated realizations include synthesis of metal nanowires114,115 and nanoparticle assembly on DNA scaffold.116,117 Surprisingly, the use of compact DNA as a nanostructure template has been much less explored. This is all the more surprising that compact DNA offers readily available nanoscale shapes and organizations that can be very difficult to realize through classical strategies. This concept was demonstrated for the first time by Zinchenko et al. who used DNA compacted into toroids by spermine as templates for the one-pot synthesis of silver nanorings with a well-defined shape and size. Later, this strategy was used to produce palladium nanoparticles,118gold nanostructures,119 and photoluminescent nanorings.120 Because DNA-templated nanomaterial deposition can be applied to many atoms including Au,121Ag,114,122Pd,123Pt,124Cu,125,126Ni,127Co,128oxides such as Fe3O4,129 and semi-conductors,130 and due to the broad variety of nanoscale shapes that can be obtained either from unimolecular DNA compaction (toroids, rods, rackets, etc.) or using programmed assembly such as in the origami method, the use of compact DNA as a nanostructure template seems to be a strategy worth being developed and shall open the route to the controlled and programmed preparation of nanostructures with immense possibilities in terms of shape and composition.7. Conclusions
In this review, we first provided a short physico-chemical description of DNA compaction (i) to provide essential fundamental understanding of the process which brings highly charged and semi-flexible DNA chain into a dense and highly organized nanostructure and (ii) to describe and rationalize the possible strategies to control DNA compaction and decompaction. For more details related to one or both of these aspects, other reviews might be consulted.3,5,6,56 The fields of gene delivery and transfection, which are important applications of DNA compaction, were not described here but are well described in dedicated reviews.7We saw that many strategies have been developed to control DNA compaction and decompaction. Among them, the most remarkable one is probably the photocontrol method initiated by Le Ny and Lee73 and further developed by Baigl et al.17,40,74,92 In this approach, without changing the chemical composition of DNA solution, DNA conformation can be controlled using light. In the presence of a photosensitive nucleic acid binder called AzoTAB, DNA is in a compact state under dark conditions. Upon UV illumination, DNA unfolds and stays in the unfolded state if kept in the dark. Upon visible illumination, DNA folds back to the compact state. This method has the great advantage to be reversible and several cycles of compaction/decompaction can be realized by successive visible/UV illuminations. Moreover, light is an ideal external trigger to control DNA conformation as it offers unique advantages: high spatio-temporal resolution of the excitation, tunability of the intensity, low perturbation of the biochemical environment, biocompatibility, and high potentiality for biotechnological applications.
In nature, DNA compaction has two main roles: packaging and regulation of gene expression. Transposed in vitro, we showed that these two properties can be declined in several kinds of applications (Fig. 8). In the process of DNA packaging, DNA folds into highly organized and well-defined structures. On the one hand, because this is a reversible process, it can be used to reversibly protect DNA against mechanical, chemical, or biochemical stresses. Compaction-based protection of DNA has been mainly considered in fundamental studies. Implemented in biochemical protocols, it shall greatly improve the yield and precision of biological procedures such as genomic DNA extraction, manipulation, sequencing, and mapping.108
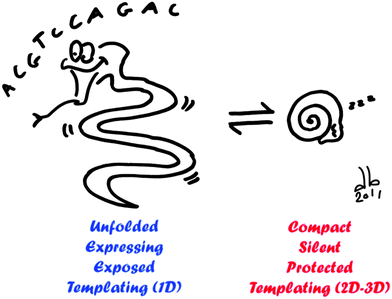 | ||
| Fig. 8 Schematic overview of possible applications of DNA compaction/decompaction. When DNA is unfolded (left), gene expression is activated, DNA is exposed to its environment and it can be used as a template for 1D nanostructure. When DNA is compacted (right), gene expression is silenced, DNA is protected over different biochemical and physical stresses and it can serve as a template for 2D and 3D nanostructures with a well-defined size and shape. | ||
On the other hand, the well-defined morphologies of compact DNA (e.g., toroids, rods) can be used as templates to construct nanostructures with a well-defined size, shape, and composition.122 Beside the naturally occurring DNA compact morphologies, a broad variety of shapes can be obtained by the origami method,79 which considerably increases the variety of realizable templates in terms of shape, size, and spatial organization.
Finally, directly inspired by the natural role of DNA higher-order structure in gene regulation, DNA compaction can be used to control biochemical reactions involved in gene expression. This approach is particularly interesting when it is combined with the photocontrol method.92 Very active research has been devoted in the past few years to the control of DNA transcription activity or gene expression by light.131 Photocaged molecules have been widely and successfully applied but do not allow a reversible control.131 Another strategy has been based on DNA modification with photoactivable groups, which is hardly applicable to in vivo studies and requires specific chemical modification of DNA.132,133 A third approach consists in the construction of a light-switchable gene promoter system, which has the advantage to be compatible in vivo but requires heavy gene construction protocols and is directed to one specific gene.134 All these strategies are based on a sequence-dependent regulation and thus have to be adapted for each particular transcription/translation system. In contrast, by using light to control nucleic acid conformation, gene expression can be photocontrolled at both transcription and translation levels in a reversible and sequence-independent way.92 This strategy shall find many applications for the dynamic photocontrol of gene expression of many kinds of machineries and target gene(s). The main remaining challenge is its implementation for in vivo and reversible photocontrol of gene expression.
Notes and references
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CAS.
- W. M. Gelbart, R. F. Bruinsma, P. A. Pincus and V. A. Parsegian, Phys. Today, 2000, 53, 38–44 CrossRef CAS.
- V. A. Bloomfield, Curr. Opin. Struct. Biol., 1996, 6, 334–341 CrossRef CAS.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269–282 CrossRef CAS.
- A. Gonzalez-Perez and R. S. Dias, Front. Biosci., 2009, 1, 228–241.
- A. Zinchenko, D. Baigl and K. Yoshikawa, in Polymeric Nanostructures and Their Applications, ed. H. S. Nalwa, American Scientific Publishers, Stevenson Ranch (CA), USA, 2007 Search PubMed.
- B. Demeneix, Z. Hassani and J.-P. Behr, Curr. Gene Ther., 2004, 4, 445–455 Search PubMed.
- N. Makita, M. Ullner and K. Yoshikawa, Macromolecules, 2006, 39, 6200–6206 Search PubMed.
- G. S. Manning, J. Chem. Phys., 1969, 51, 924–933 CrossRef CAS; F. Oosawa, in Polyelectrolytes, ed. M. Dekker, New York, 1971, ch. 5 Search PubMed.
- M. Takahashi, K. Yoshikawa, V. V. Vasilevskaya and A. R. Khokhlov, J. Phys. Chem. B, 1997, 101, 9396–9401 CrossRef CAS.
- K. Yoshikawa, M. Takahashi, V. V. Vasilevskaya and A. R. Khokhlov, Phys. Rev. Lett., 1996, 76, 3029–3031 CrossRef.
- T. Akitaya, A. Seno, T. Nakai, N. Hazemoto, S. Murata and K. Yoshikawa, Biomacromolecules, 2007, 8, 273–278 CrossRef CAS.
- A. A. Zinchenko, K. Yoshikawa and D. Baigl, Phys. Rev. Lett., 2005, 95, 228101 CrossRef.
- A. A. Zinchenko, T. Sakaue, S. Araki, K. Yoshikawa and D. Baigl, J. Phys. Chem. B, 2007, 111, 3019–3031 CrossRef CAS.
- W. Chen, N. J. Turro and D. A. Tomalia, Langmuir, 2000, 16, 15–19 CrossRef CAS.
- S. M. Mel'nikov, V. G. Sergeyev and K. Yoshikawa, J. Am. Chem. Soc., 1995, 117, 2401–2408 CrossRef CAS.
- M. Sollogoub, S. Guieu, M. Geoffroy, A. Yamada, A. Estevez-Torres, K. Yoshikawa and D. Baigl, ChemBioChem, 2008, 9, 1201–1206 CrossRef CAS.
- Y. Yoshikawa, Y. S. Velichko, Y. Ichiba and K. Yoshikawa, Eur. J. Biochem., 2001, 268, 2593–2599 CrossRef CAS.
- K. Yoshikawa, Y. Yoshikawa, Y. Koyama and T. Kanbe, J. Am. Chem. Soc., 1997, 119, 6473–6477 CrossRef CAS.
- R. W. Wilson and V. A. Bloomfield, Biochemistry, 1979, 18, 2192–2196 CrossRef CAS.
- P. G. Arscott, C. L. Ma, J. R. Wenner and V. A. Bloomfield, Biopolymers, 1995, 36, 345–364 CrossRef CAS.
- D. Baigl and K. Yoshikawa, Biophys. J., 2005, 88, 3486–3493 CrossRef CAS.
- L. C. Gosule and J. A. Schellman, Nature, 1976, 259, 333–335 CAS.
- D. K. Chattoraj, L. C. Gosule and J. A. Schellman, J. Mol. Biol., 1978, 121, 327–337 CrossRef CAS.
- J. Widom and R. L. Baldwin, J. Mol. Biol., 1980, 144, 431–453 CrossRef CAS.
- J. Widom and R. L. Baldwin, Biopolymers, 1983, 22, 1595–1620 CrossRef CAS.
- M. Spotheimmaurizot, F. Garnier, R. Sabattier and M. Charlier, Int. J. Radiat. Biol., 1992, 62, 659–666 Search PubMed.
- R. Ahmad, M. Naoui, J. F. Neault, S. Diamantoglou and H. A. TajmirRiahi, J. Biomol. Struct. Dyn., 1996, 13, 795–802 Search PubMed.
- H. A. Tajmirriahi, R. Ahmad and M. Naoui, J. Biomol. Struct. Dyn., 1993, 10, 865–877 Search PubMed.
- H. Arakawa, R. Ahmad, M. Naoui and H. A. Tajmir-Riahi, J. Biol. Chem., 2000, 275, 10150–10153 CrossRef.
- Y. Yamasaki and K. Yoshikawa, J. Am. Chem. Soc., 1997, 119, 10573–10578 CrossRef CAS.
- U. K. Laemmli, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4288–4292 CrossRef CAS.
- R. S. Dias, A. Pais, M. G. Miguel and B. Lindman, J. Chem. Phys., 2003, 119, 8150–8157 CrossRef CAS.
- V. A. Kabanov, V. G. Sergeyev, O. A. Pyshkina, A. A. Zinchenko, A. B. Zezin, J. G. H. Joosten, J. Brackman and K. Yoshikawa, Macromolecules, 2000, 33, 9587–9593 CrossRef CAS.
- C.-F. Ke, S. Hou, H.-Y. Zhang, Y. Liu, K. Yang and X.-Z. Feng, Chem. Commun., 2007, 3374–3376 RSC.
- M. W. Hsiang and R. D. Cole, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4852–4856 Search PubMed.
- M. García-Ramírez and J. A. Subirana, Biopolymers, 1994, 34, 285–292 Search PubMed.
- K. Hayakawa, J. P. Santerre and J. C. T. Kwak, Biophys. Chem., 1983, 17, 175–181 CrossRef CAS.
- R. Dias, S. Mel'nikov, B. Lindman and M. G. Miguel, Langmuir, 2000, 16, 9577–9583 CrossRef CAS.
- A. Diguet, N. Mani, M. Geoffroy, M. Sollogoub and D. Baigl, Chem. Eur. J., 2010, 16, 11890–11896 CrossRef CAS.
- S. Rudiuk, K. Yoshikawa and D. Baigl, Soft Matter, 2011 10.1039/c1sm05314k.
- K. B. Roy, T. Antony, A. Saxena and H. B. Bohidar, J. Phys. Chem. B, 1999, 103, 5117–5121 CrossRef CAS.
- S. M. Mel'nikov, M. O. Khan, B. Lindman and B. Jonsson, J. Am. Chem. Soc., 1999, 121, 1130–1136 CrossRef CAS.
- L. S. Lerman, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1886–1890 CAS.
- V. V. Vasilevskaya, A. R. Khokhlov, Y. Matsuzawa and K. Yoshikawa, J. Chem. Phys., 1995, 102, 6595–6602 CrossRef CAS.
- U. K. Laemmli, J. R. Paulson and V. Hitchins, J. Supramol. Struct., 1974, 2, 276–301 Search PubMed.
- C. C. Conwell, I. D. Vilfan and N. V. Hud, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9296–9301 CrossRef CAS.
- N. V. Hud and K. H. Downing, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14925–14930 CrossRef CAS.
- V. V. Vasilevskaya, A. R. Khokhlov, S. Kidoaki and K. Yoshikawa, Biopolymers, 1997, 41, 51–60 CrossRef CAS.
- P. G. Arscott, A. Z. Li and V. A. Bloomfield, Biopolymers, 1990, 30, 619–630 CAS.
- H. G. Hansma, Annu. Rev. Phys. Chem., 2001, 52, 71–92 CrossRef CAS.
- K. Yoshikawa, Y. Yoshikawa and T. Kanbe, Chem. Phys. Lett., 2002, 354, 354–359 Search PubMed.
- M. E. Cerritelli, N. Cheng, A. H. Rosenberg, C. E. McPherson, F. P. Booy and A. C. Steven, Cell, 1997, 91, 271–280 CrossRef CAS.
- F. Thoma, T. Koller and A. Klug, J. Cell Biol., 1979, 83, 403–427 Search PubMed.
- N. Miyazawa, T. Sakaue, K. Yoshikawa and R. Zana, J. Chem. Phys., 2005, 122, 4 Search PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269–282 CrossRef CAS.
- S. C. Riemer and V. A. Bloomfield, Biopolymers, 1978, 17, 785–794 Search PubMed.
- R. Marquet and C. Houssier, J. Biomol. Struct. Dyn., 1991, 9, 159–167 CAS.
- W. M. Gelbart, in Electrostatic Effects in Soft Matter and Biophysics, ed. C. Holm, P. Kekicheff and R. Podgornik, 2001, vol. 46, pp. 53–85 Search PubMed.
- M. R. Shen, K. H. Downing, R. Balhorn and N. V. Hud, J. Am. Chem. Soc., 2000, 122, 4833–4834 CrossRef CAS.
- P. Erbacher, T. Bettinger, P. Belguise-Valladier, S. M. Zou, J. L. Coll, J. P. Behr and J. S. Remy, J. Gene Med., 1999, 1, 210–222 CrossRef CAS.
- H. Schiessel, J. Phys.: Condens. Matter, 2003, 15, R699 CrossRef CAS.
- E. C. Uberbacher, V. Ramakrishnan, D. E. Olins and G. J. Bunick, Biochemistry, 1983, 22, 4916–4923 Search PubMed.
- T. D. Yager, C. T. McMurray and K. E. Van Holde, Biochemistry, 1989, 28, 2271–2281 CrossRef CAS.
- S. Flock, R. Labarbe and C. Houssier, Biophys. J., 1996, 70, 1456–1465 Search PubMed.
- M. Ueda and K. Yoshikawa, Phys. Rev. Lett., 1996, 77, 2133 CrossRef CAS.
- T. Saito, , et al., Europhys. Lett., 2005, 71, 304 Search PubMed.
- D. Matulis, I. Rouzina and V. A. Bloomfield, J. Mol. Biol., 2000, 296, 1053–1063 CrossRef CAS.
- H. Mayama, T. Iwataki and K. Yoshikawa, Chem. Phys. Lett., 2000, 318, 113–117 CrossRef CAS.
- N. Makita and K. Yoshikawa, Biophys. Chem., 2002, 99, 43–53 Search PubMed.
- N. Makita and K. Yoshikawa, FEBS Lett., 1999, 460, 333–337 CrossRef CAS.
- A. Diguet and D. Baigl, Langmuir, 2008, 24, 10604–10607 Search PubMed.
- A. L. M. Le Ny and C. T. Lee, J. Am. Chem. Soc., 2006, 128, 6400–6408 CrossRef CAS.
- M. Geoffroy, D. Faure, R. Oda, D. M. Bassani and D. Baigl, ChemBioChem, 2008, 9, 2382–2385 CrossRef CAS.
- A.-L. M. Le Ny and C. T. Lee, Jr, Biophys. Chem., 2009, 142, 76–83 Search PubMed.
- Y.-C. Liu, A.-L. M. Le Ny, J. Schmidt, Y. Talmon, B. F. Chmelka and C. T. Lee, Langmuir, 2009, 25, 5713–5724 CrossRef CAS.
- T. Shang, K. A. Smith and T. A. Hatton, Langmuir, 2003, 19, 10764–10773 CrossRef CAS.
- K. Yoshikawa and Y. Matsuzawa, J. Am. Chem. Soc., 1996, 118, 929–930 CrossRef.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- A. González-Pérez, J. Carlstedt, R. S. Dias and B. Lindman, Colloids Surf., B, 2010, 76, 20–27 CrossRef CAS.
- S. Bhattacharya and S. S. Mandal, Biochemistry, 1998, 37, 7764–7777 CrossRef CAS.
- R. S. Dias, B. Lindman and M. G. Miguel, J. Phys. Chem. B, 2002, 106, 12608–12612 CrossRef CAS.
- R. S. Dias, J. Innerlohinger, O. Glatter, M. G. Miguel and B. Lindman, J. Phys. Chem. B, 2005, 109, 10458–10463 CrossRef CAS.
- E. J. Richards and S. C. R. Elgin, Cell, 2002, 108, 489–500 Search PubMed.
- D. F. Browning and S. J. W. Busby, Nat. Rev. Microbiol., 2004, 2, 57–65 CrossRef CAS.
- S. C. Dillon and C. J. Dorman, Nat. Rev. Microbiol., 2010, 8, 185–195 Search PubMed.
- I. Baeza, P. Gariglio, L. M. Rangel, P. Chavez, L. Cervantes, C. Arguello, C. Wong and C. Montanez, Biochemistry, 1987, 26, 6387–6392 CrossRef CAS.
- K. Tsumoto, F. Luckel and K. Yoshikawa, Biophys. Chem., 2003, 106, 23–29 Search PubMed.
- A. Tsuji and K. Yoshikawa, J. Am. Chem. Soc., 2010, 132, 12464–12471 Search PubMed.
- A. A. Zinchenko, F. Luckel and K. Yoshikawa, Biophys. J., 2007, 92, 1318–1325 Search PubMed.
- A. Estevez-Torres, C. Crozatier, A. Diguet, T. Hara, H. Saito, K. Yoshikawa and D. Baigl, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12219–12223 CrossRef CAS.
- S. Rudiuk, H. Saito, T. Hara, T. Inoue, K. Yoshikawa and D. Baigl, submitted.
- M. Suwalsky, W. Traub, U. Shmueli and J. A. Subirana, J. Mol. Biol., 1969, 42, 363 Search PubMed.
- T. Maniatis, J. H. Venable and L. S. Lerman, J. Mol. Biol., 1974, 84, 37 CrossRef CAS.
- D. C. Rau and V. A. Parsegian, Biophys. J., 1992, 61, 246–259 CrossRef CAS.
- J. A. Schellman and N. Parthasarathy, J. Mol. Biol., 1984, 175, 313–329 CrossRef CAS.
- M. Spotheimmaurizot, S. Ruiz, R. Sabattier and M. Charlier, Int. J. Radiat. Biol., 1995, 68, 571–577 Search PubMed.
- G. L. Newton, J. A. Aguilera, J. F. Ward and R. C. Fahey, Radiat. Res., 1996, 145, 776–780 CAS.
- M. Suzuki, C. Crozatier, K. Yoshikawa, T. Mori and Y. Yoshikawa, Chem. Phys. Lett., 2009, 480, 113–117 Search PubMed.
- T. M. Klein, E. D. Wolf, R. Wu and J. C. Sanford, Nature, 1987, 327, 70–73 Search PubMed.
- A. Perl, H. Kless, A. Blumenthal, G. Galili and E. Galun, MGG, Mol. Gen. Genet., 1992, 235, 279–284 Search PubMed.
- E. Sivamani, R. K. DeLong and R. D. Qu, Plant Cell Rep., 2008, 28, 213–221 Search PubMed.
- B. Brune, P. Hartzell, P. Nicotera and S. Orrenius, Exp. Cell Res., 1991, 195, 323–329 Search PubMed.
- A. U. Khan, Y. H. Mei and T. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 11426–11427 CAS.
- I. Nayvelt, M. T. Hyvonen, L. Alhonen, I. Pandya, T. Thomas, A. R. Khomutov, J. Vepsalainen, R. Patel, T. A. Keinanen and T. J. Thomas, Biomacromolecules, 2010, 11, 97–105 Search PubMed.
- S. A. Vanapalli, S. L. Ceccio and M. J. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16660–16665 Search PubMed.
- L. Cinque, Y. Ghomchi, Y. Chen, A. Bensimon and D. Baigl, ChemBioChem, 2010, 11, 340–343 Search PubMed.
- D. Kaiser, C. W. Tabor and H. Tabor, J. Mol. Biol., 1963, 6, 141 Search PubMed.
- W. W. Cai, H. Aburatani, V. P. Stanton, D. E. Housman, Y. K. Wang and D. C. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5164–5168 CrossRef CAS.
- R. T. Kovacic, L. Comai and A. J. Bendich, Nucleic Acids Res., 1995, 23, 3999–4000 Search PubMed.
- A. Mizuno and S. Katsura, Journal of Biological Physics, 2002, 28, 587–603 Search PubMed.
- H. A. Becerril and A. T. Woolley, Chem. Soc. Rev., 2009, 38, 329–337 RSC.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775–778 CrossRef CAS.
- A. Ongaro, F. Griffin, L. Nagle, D. Iacopino, R. Eritja and D. Fitzmaurice, Adv. Mater., 2004, 16, 1799 CrossRef CAS.
- H. Nakao, H. Shiigi, Y. Yamamoto, S. Tokonami, T. Nagaoka, S. Sugiyama and T. Ohtani, Nano Lett., 2003, 3, 1391–1394 CrossRef CAS.
- G. Wei, H. L. Zhou, Z. G. Liu, Y. H. Song, L. Wang, L. L. Sun and Z. Li, J. Phys. Chem. B, 2005, 109, 8738–8743 CrossRef CAS.
- Y. Hatakeyama, M. Umetsu, S. Ohara, F. Kawadai, S. Takami, T. Naka and T. Adschiri, Adv. Mater., 2008, 20, 1122 CrossRef CAS.
- T. C. Preston and R. Signorell, Langmuir, 2010, 26, 10250–10253 Search PubMed.
- Y. Z. You, Z. Q. Yu, M. M. Cui and C. Y. Hong, Angew. Chem., Int. Ed., 2010, 49, 1099–1102 CrossRef CAS.
- A. Ongaro, F. Griffin, P. Beeeher, L. Nagle, D. Iacopino, A. Quinn, G. Redmond and D. Fitzmaurice, Chem. Mater., 2005, 17, 1959–1964 CrossRef CAS.
- A. A. Zinchenko, K. Yoshikawa and D. Baigl, Adv. Mater., 2005, 17, 2820 CrossRef CAS.
- J. Richter, R. Seidel, R. Kirsch, M. Mertig, W. Pompe, J. Plaschke and H. K. Schackert, Adv. Mater., 2000, 12, 507 CrossRef CAS.
- R. Seidel, L. C. Ciacchi, M. Weigel, W. Pompe and M. Mertig, J. Phys. Chem. B, 2004, 108, 10801–10811 CrossRef CAS.
- C. F. Monson and A. T. Woolley, Nano Lett., 2003, 3, 359–363 CrossRef CAS.
- A. Rotaru, S. Dutta, E. Jentzsch, K. Gothelf and A. Mokhir, Angew. Chem., Int. Ed., 2010, 49, 5665–5667 CrossRef CAS.
- H. A. Becerril, P. Ludtke, B. M. Willardson and A. T. Woolley, Langmuir, 2006, 22, 10140–10144 CrossRef CAS.
- Q. Gu, C. D. Cheng and D. T. Haynie, Nanotechnology, 2005, 16, 1358–1363 CrossRef CAS.
- D. Nyamjav and A. Ivanisevic, Biomaterials, 2005, 26, 2749–2757 CrossRef CAS.
- A. Houlton, A. R. Pike, M. A. Galindo and B. R. Horrocks, Chem. Commun., 2009, 1797–1806 RSC.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS.
- M. Z. Liu, H. Asanuma and M. Komiyama, J. Am. Chem. Soc., 2006, 128, 1009–1015 CrossRef CAS.
- A. Rotaru and A. Mokhir, Angew. Chem., Int. Ed., 2007, 46, 6180–6183 CrossRef CAS.
- S. Shimizu-Sato, E. Huq, J. M. Tepperman and P. H. Quail, Nat. Biotechnol., 2002, 20, 1041–1044 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |