
End-to-end differentiable construction of molecular mechanics force fields†
 *acf
Josh
Fass,
‡ab
Benjamin
Kaminow,
ab
John E.
Herr,
a
Dominic
Rufa,
ad
Ivy
Zhang,
ab
Iván
Pulido,
a
Mike
Henry,
a
Hannah E.
Bruce Macdonald,
§a
Kenichiro
Takaba
ae
and
John D.
Chodera
*a
*acf
Josh
Fass,
‡ab
Benjamin
Kaminow,
ab
John E.
Herr,
a
Dominic
Rufa,
ad
Ivy
Zhang,
ab
Iván
Pulido,
a
Mike
Henry,
a
Hannah E.
Bruce Macdonald,
§a
Kenichiro
Takaba
ae
and
John D.
Chodera
*a
Abstract
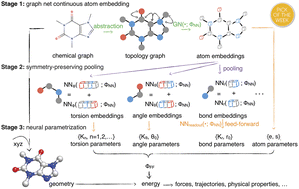
Molecular mechanics (MM) potentials have long been a workhorse of computational chemistry. Leveraging accuracy and speed, these functional forms find use in a wide variety of applications in biomolecular modeling and drug discovery, from rapid virtual screening to detailed free energy calculations. Traditionally, MM potentials have relied on human-curated, inflexible, and poorly extensible discrete chemical perception rules (atom types) for applying parameters to small molecules or biopolymers, making it difficult to optimize both types and parameters to fit quantum chemical or physical property data. Here, we propose an alternative approach that uses graph neural networks to perceive chemical environments, producing continuous atom embeddings from which valence and nonbonded parameters can be predicted using invariance-preserving layers. Since all stages are built from smooth neural functions, the entire process—spanning chemical perception to parameter assignment—is modular and end-to-end differentiable with respect to model parameters, allowing new force fields to be easily constructed, extended, and applied to arbitrary molecules. We show that this approach is not only sufficiently expressive to reproduce legacy atom types, but that it can learn to accurately reproduce and extend existing molecular mechanics force fields. Trained with arbitrary loss functions, it can construct entirely new force fields self-consistently applicable to both biopolymers and small molecules directly from quantum chemical calculations, with superior fidelity than traditional atom or parameter typing schemes. When adapted to simultaneously fit partial charge models, espaloma delivers high-quality partial atomic charges orders of magnitude faster than current best-practices with low inaccuracy. When trained on the same quantum chemical small molecule dataset used to parameterize the Open Force Field (“Parsley”) openff-1.2.0 small molecule force field augmented with a peptide dataset, the resulting espaloma model shows superior accuracy vis-á-vis experiments in computing relative alchemical free energy calculations for a popular benchmark. This approach is implemented in the free and open source package espaloma, available at https://github.com/choderalab/espaloma.
- This article is part of the themed collections: 2022 ChemSci Pick of the Week Collection and 2022 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

