
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges for reliable analysis of uranium in natural waters using laser-induced fluorimetry/LED-fluorimetry in the presence of fluoride and diverse humic substances in hot arid regions and future advances – review
D. P. S.
Rathore
 *a,
P. K.
Tarafder
b and
V.
Balaram
c
*a,
P. K.
Tarafder
b and
V.
Balaram
c
aAtomic Minerals Directorate for Exploration and Research, Department of Atomic Energy, Jaipur 302033, Rajasthan, India. E-mail: dpsr2002@yahoo.com
bAtomic Minerals Directorate for Exploration and Research, Department of Atomic Energy, Jamshedpur 831002, Jharkhand, India. E-mail: pktarafder1954@gmail.com
cCSIR-National Geophysical Research Institute, Hyderabad 500 007, Telangana, India. E-mail: balaram1951@yahoo.com
First published on 1st February 2024
Abstract
The presence of fluoride and diverse humic substances in arid regions results in changes in the content of uranium, major cations and anions if there is a time interval between sample collection and analysis, and this disrupts the reliable analysis of uranium in natural waters using laser/LED-fluorimetry, and this is discussed in this review. Complete and unequivocal preservation of samples, whether domestic waste water or natural water, is a practical impossibility. The physico-chemical and biological changes continue inevitably after sample collection due to changes in dynamic equilibrium. Thus, the use of a mobile geochemical laboratory for on the spot/quick analysis of water samples, preferably on the same day, is required. Laser-induced fluorimetry/pulsed LED-fluorimetry is a well documented, highly sensitive, and versatile technique for the determination of uranium in water samples at μg L−1 levels. This is made more challenging due to the wide variety of types of water samples, which differ in total dissolved salts found, and these include saline water, diverse humic substances and fluoride content, especially in hot arid regions as well as due to the practical impossibility of preserving natural water samples. Therefore, it is the time interval between sample collection and analysis that is the most critical factor for the reliable analysis of uranium in hot arid regions. A high level of total dissolved solids (TDS) in water samples results in greater variation in the major cations and anions, and uranium content with respect to the time interval between sample collection and analysis. Moreover, there is no availability of certified reference materials for such matrices to cross-check/ensure the reliability of the results.
 D. P. S. Rathore | , Dr D. P. S. Rathore FICS, FRSC, M-ACS, IUPAC-Affiliate Member, LM-ICC PhD Chemistry (1981), A.M.U., Aligarh, India. He is a Former Scientific Officer-G at the Atomic Minerals Directorate for Exploration and Research, Department of Atomic Energy, India. Dr Rathore has served in different capacities for 34 years, as Incharge, in different Regional Chemical Laboratories, Coordinator, mobile geochemical laboratory in various parts of the country and was involved in providing quick feedback of analytical data support for the various on-going uranium exploration projects of his Directorate. His field of research interest/expertise is metal complexation, spectrophotometric determinations and metallochromic indicator synthesis, chelating polymeric resins, optical fluorimetry, laser-induced/LED fluorimetry, reference measurement procedures and Geostandard certifications. He is actively involved as a reviewer for more than 30 international journals. |
 P. K. Tarafder | Dr P. K. Tarafder is an Indian Nuclear Scientist who served in the Atomic Minerals. Directorate for Exploration & Research of Department of Atomic Energy, Government of India for more than 33 years in different capacities. He has vast experience in the field of Analytical Chemistry and research. He has published more than 100 research papers in various international journals of repute, and authored a technical book. In addition, he has supervised about a dozen MSc dissertations, and three PhD thesis works. He is the recipient of a few National awards and a few international accolades. He has reviewed more than 50 research papers so far. He is an Editorial Board Member of a few international journals. |
 V. Balaram | Dr V. Balaram, is a Former Emeritus Scientist at the Geochemistry Division, CSIR – National Geophysical Research Institute, Hyderabad – 500 007, India. His research areas include geochemistry, mineral exploration, spectroscopy, environmental chemistry, and climate change. He has over 335 publications in international peer-reviewed journals, 25 book chapters with ∼8000 citations (h-index 45 & i10-index 145), and guided 5 PhD students. He has received several prestigious national and international awards such as the “National Geoscience Award” from the Government of India, New Delhi (2000). He represented India at the United Nations/IAEA Meeting on “ICP-MS Applications in Water Resources Management” in Oct. 2022 at the IAEA Headquarters, Vienna, Austria. His name is included in Stanford University's new list of the top 2% of Scientists in the World in 2020, 2021, 2022 & 2023. |
Environmental significanceDetermination of uranium in natural water samples is a great challenge due to diverse nature of water samples including saline water, humic substances and fluoride content especially in hot arid regions as well as due to the practical impossibility of preserving natural water samples. Various factors responsible for reliable analysis of uranium in natural waters using laser/LED-fluorimetry are discussed. It is mandatory to use a mobile geochemical laboratory for quick analysis of water samples, preferably on the same day. The standard additions method is the best for standardising the instrument response by using deionised water as a blank sample using 1, 5 and 10 ppm U standards. These independent standards serve as reference standards for water samples and ensures the traceability of the analytical results. Pre-calibrated future generation LED-fluorimeters can be designed for uranium measurement. |
1. Introduction
Uranium is important due to its main use as a fuel for the nuclear power program1 as well as because of its chemical toxicity.2–4 Uranium is present in the environment as a result of leaching from natural deposits, release in mill tailings, emissions from the nuclear industry, the combustion of coal and other fuels and the use of phosphate fertilizers that contain uranium. Uranium is nearly ubiquitous,1,2 due to its polyvalence (+4, +6), large atomic radius (0.97 Å), high chemical reactivity, relative solubility of U(VI) compounds in aqueous solution, and relative insolubility of the U(IV) compounds. Uranium forms many compounds, and enters into the structure of many minerals and disperses readily. Under oxidizing conditions, hexavalent uranium appears as the uranyl ion, UO22+(VI), which is linear and polar in nature.5–7 The uranyl ion is very stable, and maintains its identity through many chemical transformations, as found in many secondary uranium minerals. Cations in these secondary minerals are loosely held, added or removed by base exchange reactions without touching the UO2 keyed layer structures in solids represented by the general compositions, such as, A[(UO2)(RO4)]XH2O; B[(UO2)(RO4)2]XH2O; B[(UO2)(CO3)2]XH2O; where A = K(I), Na(I), H(I); B = Ca(II), Ba(II), Mg(II), Cu(II), Fe(II), Pb(II); R = P(V), As(V), V(V). These secondary minerals are variably soluble in aqueous solutions. Thus, uranium in significant concentrations may be transported as various uranyl complexes throughout a wide range of Eh and pH conditions. In this way, uranium is a relatively mobile element in many surface or near-surface environments in natural water systems.3–5 It is only the hexavalent uranium, U(VI), present in the uranyl ion, UO22+ which phosphoresces with a relatively long lifetime of μs when using a phosphate buffer mixture as a fluorescence enhancing reagent (pH ∼ 7), whereas uranium of other valences is essentially non-luminescent at the wavelengths of excitation used for its determination by fluorimetry.10 A luminescence study of tetravalent uranium in aqueous solution have shown fluorescence properties observed in the UV-vis region with a lifetime < 20 ns at room temperature by excitation at 245 nm, which corresponds to the 5f–5f electronic transition (=40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 cm−1) of this element in this oxidation state. Thus, uranyl compounds have a distinct well-known characteristic, long-lived green luminescence which can be isolated by optical filters and measured as intensity with a photodetector to give an indication of the uranium concentration in a sample.10
820 cm−1) of this element in this oxidation state. Thus, uranyl compounds have a distinct well-known characteristic, long-lived green luminescence which can be isolated by optical filters and measured as intensity with a photodetector to give an indication of the uranium concentration in a sample.10
Laser-induced fluorimetry/pulsed LED-fluorimetry is a well documented, highly sensitive and versatile technique for the determination of uranium in water samples at μg L−1 levels,4,7–9 and at the same time it is much more challenging due to wide variety of water samples which differ in their concentrations of total dissolved salts including saline water, and fluoride contents as well, due to practical difficulty in preserving natural water samples.11–21 Therefore, the time interval between sample collection and analysis is the most critical factor responsible for reliable analysis of water samples especially in hot arid regions. Detection limits for uranium measured by different analytical techniques are given in our earlier publications.4,9 In contrast, mineralized rock sample solutions are comparatively easy to analyse using the application of the differential technique of laser-induced fluorimetry/pulsed LED-fluorimetry for their uranium content due to their easy preservation over a period of time and their instability problems. Recently, we have presented at the IAEA conference, URAM-2023 (ref. 22) and later published a critical review23 called the “application of a differential technique in laser-induced fluorimetry/pulsed LED-fluorimetry: simple and reliable analysis of uranium raw materials in nuclear fuel cycle – mini-review”. The application of a differential technique in laser-induced fluorimetry/pulsed LED-fluorimetry is useful for the analysis of uranium in ores, certification of reference materials, borehole core assay, beneficiation product, and other diverse applications in the entire nuclear fuel cycle worldwide.
A literature survey indicated that so far, no attempt has been made to present a comprehensive review of the application of laser/LED-fluorimetry in the analysis of uranium in different water samples. Here an attempt is made to summarize the various factors responsible for the reliable analysis of uranium in natural waters using laser/LED-fluorimetry.
2. Experimental
2.1 Apparatus
2.2 Reagents fluorescence enhancing reagent buffer solution (Fluran),25,26 pH ∼ 7
First, 570 g of Na3PO4·12H2O and 33 g of H4P2O7·10H2O were placed in a graduated 3 L beaker. Approximately 1.5 L of deionized water and about 40 mL phosphoric acid (H3PO4) were added, and heated gently to dissolve. After cooling, and diluting to 2 L with deionized water, the pH was adjusted to ∼7.0 with H3PO4. Alternatively, a 5% solution of sodium pyrophosphate in distilled water was prepared and then the pH of the solution was adjusted to ∼7.0 by drop-wise addition of phosphoric acid (AR) Merck 85% wt in H2O, (99.99% trace metal).3. Results and discussion
It is believed that experience is a wonderful thing. It enables us to recognize a mistake when we make it again. The use of a primary method or principle of the method in analytical measurement per se does not guarantee a reliable or metrologically traceable result. It has now been well recognized that the quality of the measurement results are of the highest importance and depend on the realization of the method (strict adherence to the number of steps) into practice.31 The average uranium concentration in water is given in Table 1.| Type of water sample | Concentration (μg L−1) |
|---|---|
| Seawater | 3 |
| Groundwater | ≤0.2–4918 |
| Surface water | ≤0.2–22 |
The various steps or phases of hydro-geochemical reconnaissance survey programme for uranium exploration are well documented in the literature.7,32–34 There are advantages to exploration methods with measurements which may be made directly in the field conditions with limited facilities to cover a large area quickly and economically.7 The physicochemical and biological changes continue inevitably after sample collection35 and truly, more pronounced in the presence of fluoride and diverse humic substances in hot arid regions,21 having temperature variations/fluctuations ∼10 °C. This justifies the need and use of Mobile Geochemical Laboratory for quick analysis of water samples.32–35 The water sample collected should be a true water sample i.e., free from suspended or particulate materials, and should be analysed preferable on same day.
The water samples received were analysed in mobile geochemical laboratory while working in the areas of Meghalaya, Rajasthan and Haryana. One set of samples were treated with HNO3 to prevent the adsorption of uranium and other cations on the container walls (the pH of the samples was maintained below pH ∼ 1, whereas the other samples were left untreated). The UA-3 uranium analyser (Scintrex Limited, Concord, Ontario, Canada) was used for uranium estimation using 5% sodium pyrophosphate solution in distilled water adjusted to pH-7.0–7.2 by the drop-wise addition of orthophosphoric acid (pH ∼ 7 buffer) as a fluorescence enhancing reagent. Standard methods, as described in the literature, were followed for the determination of the contents of the major cations and anions.35,36 The sodium and potassium were determined by flame photometry, and the calcium and total calcium plus magnesium were determined by EDTA titrimetric methods using the Patton–Reeder indicator, and Eriochrome Black T, respectively. The difference between the two titrations gave the magnesium content of the sample.11 Calcium and magnesium contents were also verified by using a new indicator developed in our laboratory.37,38 Bicarbonate and carbonate contents were determined by titration with hydrochloric acid using methyl orange and phenolphthalein as indicators. The chloride content was determined by titration with silver nitrate using potassium chromate as an indicator. Sulfate was measured by visual comparison of turbidity with closely matching standards using barium chloride as precipitant, and the pH and conductivity were measured using a pH-meter and conductivity meter, respectively.11
3.1 The following observations were made
(1) Conductance (changed by 4% to 40% in 15–20 days), and the pH, carbonate, bicarbonate also changed significantly. (2) There were more variations in the uranium contents in water samples containing suspended or particulate materials. Variations in uranium values were increased up to 50% and more depending upon the amount of suspended or particulate materials present. However, in some samples, the variation was very high especially from areas in Rajasthan. (3) Calcium values were found to be reduced by 6–60% in 15–20 days but the magnesium contents did not vary significantly in general, except for a few samples. (4) Nearly all the water samples were found to contain suspended or particulate material.3.2 Interpretation of the variations of the chemical contents of samples
Although, variations in the contents of water samples due to the time interval between collection and analysis are well known,35 it is impossible to state exactly, how much elapsed time may be allowed between sample collection and its analysis. This depends on many factors: (1) Nature of the sample i.e., hydrogeochemical environment of the area, (2) analysis to be made, (3) the conditions of storage, and (4) the amount of suspended material present in the sample. All these points were discussed in depth in an earlier publication.113.3 Changes in pH-alkalinity-carbon dioxide balance and calcium content
The pH may change significantly in a matter of minutes, and dissolved gases may be lost (oxygen and carbon dioxide). With changes in the pH-alkalinity-carbon dioxide balance, precipitation of calcium carbonate takes place (solubility product of CaCO3 = 4.8 × 10−9), thereby causing a decrease in the values for calcium and total calcium plus magnesium contents thereby resulting in a change in the conductance values. Besides this, the fluoride concentration which ranges from 1 to 60 mg L−1 levels, particularly in areas of Rajasthan, probably significantly reduces the calcium content by precipitation of calcium fluoride on storage. Calcium fluoride is insoluble and settles down (solubility product of calcium fluoride = 4.0 × 10−11, magnesium fluoride = 6.5× 10−9). A close examination of the calcium and magnesium contents in sets of samples (received from the district of Barmer, Rajasthan and Haryana State) showed that some samples has a lower calcium content than that of magnesium. But, on comparing the results of the samples with the usual trend of calcium to magnesium ratio, i.e., average calcium and magnesium content of ground waters from different rock types (waters from granites – Ca, 27 mg L−1; Mg, 6.2 mg L−1; serpentinite, Ca, 9.5 mg L−1; Mg, 51 mg L−1; shale, Ca, 227 mg L−1; Mg, 29 mg L−1), it appears that the surrounding rocks are magnesium rich (maybe ultramafic rocks, and so on) and should have comparatively less uranium, sodium and potassium contents. In contrast, sodium and potassium were also high and uranium concentrations were in the range of 14 to 49 ppb. On careful visual examination, a whitish deposit/suspended material was noticed in the sample bottles and this was verified experimentally by performing chemical analysis after acidification of a sample with hydrochloric acid. The acidified samples were analysed for calcium and total calcium plus magnesium contents. Calcium contents were found to be significantly much higher in same samples in comparison to the un-acidified samples, i.e., the calcium to magnesium ratio changed significantly. Calcium may also be precipitates as calcium sulfate (solubility product of calcium sulfate = 9.1 × 10−6) thereby resulting in a change in conductance values. The losses of calcium content by precipitation do not follow a uniform pattern. However, in some samples, an increase in calcium and magnesium values have been found, and this increase is presumed to occur when suspended particulate material with comparatively high calcium and magnesium levels slowly equilibrates with the water. A similar observation has been reported,14 quote “The high mean ratio >1 of Mg2+/Ca2+ in groundwater indicated …”. Rathore further stated: “In my opinion and based on my observations as stated above, this conclusion is incorrect and highly misleading. This interpretation in the manuscript, based on the unreliable measurement results obtained (the high mean ratio (>1) of Mg2+/Ca2+ in groundwater), is simply due to variations in the contents of calcium and magnesium contents with time interval between collection of water samples.”3.4 Changes in the uranium contents of samples
Uranium levels also changed considerably, probably due to adsorption, co-precipitation and so on. In the presence of fluoride, a fine suspension or precipitation of calcium fluoride may occur, which is a well known co-precipitant of uranium.39,40 There is a well-known method for detection and determination of uranium at a ppt level: “Detection of ultra-trace levels of uranium in aqueous samples by laser-induced fluorescence spectrometry” by Perry et al.,39 and also, “determination of ultra-trace levels of uranium by selective laser excitation of precipitates” by Johnston and Wright.40 In this work, selective excitation of ion probe luminescence is applied to the analysis of uranium co-precipitation into calcium fluoride. Thus, this may result in abnormal variations in uranium levels, if, samples are not properly acidified and stored. A high acidity is essential to keep uranium in solution in the presence of fluoride. The abnormal behavior of fluoride needs further investigation and to be discussed in depth.11 It is necessary that before acidification of a water sample, it should be free from suspended particulate materials, if not, it should be filtered, otherwise, uranium levels may not be a true value and may increase or decrease. A gain is presumed to occur when a suspended particulate material with comparatively high uranium levels slowly equilibrates with the water. Losses probably occur to the container walls. In general, for the preservation of samples for the determination of metal ions at trace levels, the water samples should only be acidified after filtration. A high total of dissolved salts in water samples, results in a larger variation in major cations and anions and uranium contents, with respect to the time-interval between collection and analysis. This variation in these contents depends on the total dissolved salts. A percentage change in conductance in 22 days ranged from 4% to 40% while a further change in conductance in 27 days is in the range from 0.3 to 3.4, indicating a stabilization in their total dissolved salt contents. Simply stating that the uranium content was determined by using a laser fluorimetric technique is not correct.41,42 Thus, direct methods for the analysis of uranium in natural water samples are desirable. Any additional chemical preparation of a sample will introduce contamination, is time consuming and unreliable results are obtained due to the high blank value.413.5 Choice of an appropriate fluorescence enhancing reagent
Choosing an appropriate fluorescence enhancing reagent for different types of sample matrices is essential.9,22,23,26–28 For water samples, the best fluorescence enhancing reagent is 5% sodium pyrophosphate solution in distilled water adjusted to pH 7.0–7.2 by drop-wise addition of orthophosphoric acid (as per the manual of the instrument). In truth, the reliability of the analytical results depends on strict adherence to the various steps of the validated method and not on the laser fluorimetric technique or the laboratory or the person performing the test.11–213.6 Effect of hydrochloric acid and miscellaneous errors in uranium measurement
The chloride is a fluorescence quencher in water and hence, hydrochloric acid should never be used for maintaining the pH of water samples16,18 or cleaning the glassware. It was stated in the reply that 0.1 M HCl, and 0.1 M NaOH was used for adjusting pH of the samples, which is incorrect. It was clearly evident from the figure, where the error in the measurement (error bars) should have presumably depended on the concentration of uranium—while it is not.16,18 In another, published paper by Mukesh et al.,43 hydrochloric acid was used and moreover, 10 references in the manuscript were incorrect. Nitric acid is recommended for the cleaning of glassware and maintaining the water samples at a pH of ∼ 7.3.7 Method of measurement
There are three methods of measurement in an instrumental technique,23,44 namely, (1) calibration method, (2) standard additions, and (3) differential technique. All these methods have a significant bearing on the reliability, cost-effectiveness and traceability of the measurement data. The applications of all these methods remain good when a linear relationship between signal response and concentration of the analyte is established. In most samples, there is some quenching of fluorescence relative to distilled water or deionised water.29 The standard addition method is recommended for analysis of water samples, where the quenching is recognized and corrected by means of a ‘standard addition’ to the sample. The increase in concentration in the cell on the addition of a standard addition is given by:
For example, two additions of 5 μL of a 1 ppm standard in 7 mL is equivalent to 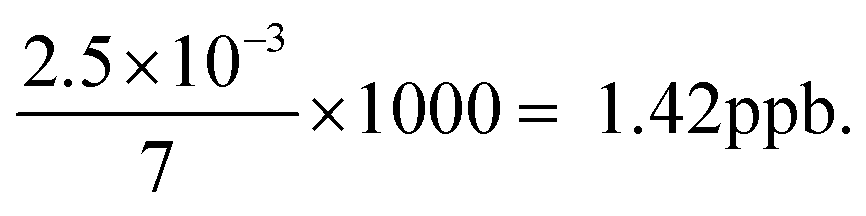
This method is the best for standardising the instrument response by using deionised water as a blank sample. Calibration of the instrument used by this method avoids the need for preparation of very dilute standards with their associated stability problems. Because, certified reference material is unavailable for such matrices to cross-check/ensure the reliability of the results, the 1 ppm, 5 ppm and 10 ppm U standards serve as reference standards for water samples and ensures the traceability of the analytical results. All these standard solutions should be prepared from independent standard stock solutions to cross-check them and their fluorescence response should be in agreement with each other (Fig. 1 and 2).
 | ||
| Fig. 1 UA-3 uranium analyzer (SCINTREX, Canada). | ||
 | ||
| Fig. 2 LED fluorimeter (model LF-2) (Quantalase, India). | ||
3.8 Effect of humic acid in water samples on uranium measurement and steps to take care of
Humic substances are complex and heterogeneous mixtures of polydisperse materials formed in soils, sediments, and natural waters by biochemical and chemical reactions during the decay and transformation of plant and microbial remains (a process called humification). Thus, humic substances are a fraction of natural organic matter and are involved in many processes in soils and natural waters: e.g., soil weathering, plant nutrition, pH buffering, trace metal mobility and toxicity, bioavailability, degradation and transport of hydrophobic organic chemicals, formation of disinfection by-products during water treatment, and heterotrophic production in black water ecosystems.45 Organic material, especially humic acids (HA) and fulvic acids (FA), also play an important role because of the complexation as well as the sorption processes.46,47 As stated and observed by Riggle and Wandruszka in their work on the stability of uranium(VI) complexes of humates and fulvates in biphasic systems, slight momentary heating of aqueous solutions (by ∼10 °C), containing humic substances (HSs) and uranyl ions (UO22+) yielded humic flocs that carried the bulk of the ions down with them.48 These biphasic systems were at a near-neutral pH, and after returning to ambient temperatures they remained intact. The formation of flocs due to organic matter were simply due to a ∼10 °C change in temperature, and calcium fluoride co-precipitation11 are also most interesting observations and may result in large variations in the uranium contents in samples with varying time intervals between sample collection and analysis in hot arid regions of India and worldwide. A detailed survey of dissolved organic carbon in natural waters in hot arid regions of India is still required. The authors strongly recommend a detailed survey of dissolved organic carbon to understand its role and correlation with the high values of uranium found in the hot arid regions of India. Humic acid has been shown to play a most significant role in the environmental behavior of many metal cations as it can function as both a complexant and a redox agent.49 A number of models have been proposed to explain their complexation role, but most use quite different chemical descriptions of the metal-humate interactions. Two of these models which have been applied to humic acid interaction with actinide cations are briefly discussed. In one model in which the humics are treated as anionic polyelectrolytes, cations can bind to specific anionic donor sites (site binding) as well as being attracted by the net anionic charge of the macromolecules (the polyelectrolyte model). In the second model (the charge neutralization model), the binding for each cation is assumed to be associated with a number of carboxylate groups equal to the cationic charge. It was concluded that the charge neutralization model is more useful in geochemical calculation codes, whereas the polyelectrolyte model can provide more insight into the chemical behavior of the humic acids. Although, a laser fluorimeter is designed to determine selectively a long lived uranyl phosphorescence signal in the presence of dissolved organic matter, there was still a time delayed phosphorescence signal due to the competing complexing behavior between organic carbon and polyphosphates (used as a fluorescence enhancing reagent) with uranium was observed. The response of the instrument after addition of polyphosphate as fluorescence enhancing reagent to a uranium bearing solution is usually rapid, except in case of some organic-rich samples, when the extraction of uranium from the organic species appears to be rather slow.7 A similar effect has been noted in laboratory samples containing high humic acid concentrations,26 in which more than 80% of the final uranium value is reached after only 60 seconds or so. The time required per sample in these cases is correspondingly increased, but the waters could be treated with polyphosphate some minutes prior to analysis to avoid delay. Alternatively,26 such water samples can be analysed after the removal of dissolved organic compounds by evaporating with concentrated nitric acid. While using Fluran or pyrophosphate buffer the pH of the test solution should be ∼7.0. However, while using sodium dihydrogen phosphate buffer (pH ∼ 2.5),26,27 there is no need of pH adjustment.The reliability/quality of the measurement results of the water samples depend on strict adherence to each step of sampling, preservation of samples, time-interval between sampling and analysis for filtered but non-acidified water samples, and on the methodology adopted.11 However, of the direct methods, the fission track method seems to be the nearest competitor.12,503.9 Uranium determination in saline water samples
Chloride ions at high concentrations in aqueous solution depress uranyl fluorescence. If the uranium concentrations to be determined are relatively high, an appropriate dilution of the samples can be used to minimize the interference effects. If, however, the uranium level is very low, in such cases, as per the recommended procedure, potassium persulfate29 is added to the sample which is then heated to dryness to drive off chloride ions as chlorine. The residue is taken up in water and made up to volume and analysed with laser/LED fluorimetry using acidic sodium dihydrogen phosphate buffer, pH ∼ 2.5 as a fluorescence enhancing reagent using the standard addition method. Alternately, after adjusting the pH of the treated sample to pH ∼ 7, the sample is analysed by using sodium pyrophosphate, pH ∼ 7 buffer as fluorescence enhancing reagent (Fig. 2).3.10 LED-fluorimeter for diverse applications and advances
Recent advances in narrow and broadband LED light sources from the far UV to the NIR wavelength regions have also opened up the flexibility for additional designs, electronics, and instrumentation. With the addition of new fluorescence reagents51 or probes/sensing materials,52–54 advancements in the technologies have continued with high performance qualification, portable instrumentation and appear to show promise for future applications.55 Previous researchers have investigated the use of LEDs in arrays to produce a wider spectrum of light with a higher spectral output.56–58 The drawbacks of early LED-based systems include insufficient light output, instability, poor lifetime, expense and, until recently, lack of lower wavelength LEDs, so limiting their application to long wavelength ultra-violet, visible and near infrared excitable fluorescent compounds.56 However, recent step change advances in manufacturing processes are now making LEDs an attractive alternative to conventional white-light sources. LEDs offer a highly energy-efficient means of producing monochromatic light.59 They also provide a concentrated, small, cool emitter ideal for miniature analytical devices because of their reduced power of operation, size, and longer life time and also provide spectral control without the need for high overhead optics. Recently, the availability and reductions in cost of UV-LEDs that emit to 270 nm and below, have broadened the horizons for their use in LED-based fluorescent devices for new applications. Characterising the organic and microbial matrix of water are key issues in ensuring a safe potable water supply. The current technique only confirms water quality retrospectively via laboratory analysis of discrete samples. Whilst such analysis is required for regulatory purposes, it would be highly beneficial to monitor water quality in situ in real time, thus, enabling rapid water quality assessment and facilitating proactive management of the water supply system. The scope for LED based instrumentation for in situ, real time assessment of the organic and microbial matrix of potable water is the most interesting application.59With the advent of brighter and more stabilized LED lights, a pulsed LED-fluorimeter based on this excitation source has become available from Quantalase Enterprises Pvt Ltd., Indore, India (https://quantalase.in/services).60
The excitation source was a bank of pulsed LEDs emitting at 400 nm, model LF-2,60Tables 1 and 2. Several innovations were added to the instrument to make it more reliable, more versatile and easier to operate than the nitrogen laser fluorimeter. With LED in the visible range (400 nm) replacing the laser (337 nm) in the UV range, the tolerance levels of many associated interfering elements improved almost 10 times in this new LED-based technique. Tolerance to pre-filters like Fe(III) was found to be high (10 fold increase as compared to laser-induced fluorimetry). This is the same as the case with post filters like Mn, Cu, Cr, etc. It was found that the tolerance to many ions have been enhanced at least 10 times in LED fluorimetry in comparison to laser-induced fluorimetry.
| Pulsed UV LEDs (light emitting diodes) emitting at a 400 nm wavelength and with a suitable filter. 20 μJ or higher less than 1%, 5 years |
| Detector – photomultiplier tube with precision multilayer optical filter |
| Analyte volume – 6 mL cuvette, made from ultra-low fluorescence fused silica |
| Dynamic range: 0.1–1000 ppb (0.1–1000 μg L−1) |
| Accuracy: better than 10%. Can be significantly better if the instrument is used in a temperature controlled environment |
| Reproducibility: better than 5% |
| Averaging: the average of 1280 measurements is used for the calculation of uranium concentration |
| LED fluorimeter: Quantalase enterprises private limited, Indore, India (https://quantalase.in/services). |
Pradhan and Ambade61 developed a solid phase extraction procedure based on the absorption of uranium(VI)-2,3-dihydroxynapthalene complex on microcrystalline naphthalene for the extraction of uranium in water samples. The solid mass consisting of uranium(VI)-2,3-dihydroxynapthalene complex was ignited in a furnace (700 °C) and dissolved in dilute HNO3. This solution was used to determine uranium by LED fluorimetry using the fluorescence enhancing pyrophosphate buffer. The detection limit of the method is 0.025 ng mL−1 and is useful in the determination of very low concentrations of uranium in a variety of water samples including seawater. Sahoo et al.62 used LED fluorimetry to determine uranium in 55554 surface water as well as groundwater samples collected across India for mapping uranium content in drinking water sources across the nation in order to identify high uranium concentration zones, to enable the regulatory authorities to take proper remedial measures.
In the present LED fluorimeter, an average of 1280 measurements were used for calculation of uranium concentration and moreover, the pulsed UV LEDs emitting at a 400 nm wavelength with suitable filter, 20 μJ or higher less than 1%, with a lifetime of five years, this can be exploited practically in the design of a pre-calibrated LED-fluorimeter for future generations, Table 1. In this way, with the advancement in LEDs, digitization and computer controlled electronic signal processing, the instrument can be practically pre-calibrated to read the concentration of uranium in the ppb range directly in test samples. A LED-fluorimeter coupled with unique features of flow-injection analysis, can be exploited to enable us to design and develop microchemielectronic devices. Such portable microchemielectronic devices will be more compatible with the tools of the computer age and also help in handling of radioactive solutions in chemical laboratories of uranium processing industries at various stages of the nuclear fuel cycle and discussed in depth in our earlier publication.28
3.11 Comparison of laser fluorimetry/LED-fluorimetry versus ICP-MS techniques for uranium determination in water
Both laser fluorimetry and LED fluorimetry have been extensively used for the determination of uranium in the mobile geochemical laboratory of the Atomic Minerals Directorate for Exploration and Research for hydrogeochemical reconnaissance surveys. Shenoy et al.63 reported a comparative analysis of uranium in potable waters using laser fluorimetry and ICP-MS techniques64 and also reported a comparative study65 of LED vs. laser fluorimetry for the determination of uranium in natural waters. Mehra et al.66 reported a comparative study of uranium concentration using two different analytical techniques, LED fluorimetry and ICP-MS, for the assessment of physicochemical parameters in groundwater. The results of the uranium determination in water by laser fluorimetry/LED fluorimetry compared favourably with the ICP-MS technique and are well documented in the literature. Moreover, a portable pulsed LED-fluorimeter is an excellent substitute for the UA-3 laser-induced fluorimeter available in the market for uranium determination in diverse matrices in the nuclear fuel cycle including hydrogeochemical reconnaissance surveys infield conditions as well as for base laboratory investigations. This also fulfills the essential criteria of reliability, applicability and practicability (RAPs).9 A comparison of the salient features of commonly used analytical techniques for uranium determination in water is given in Table 3.| Method | Advantages | Limitations |
|---|---|---|
| ICP-MS | Large linear dynamic range | Low tolerance to high TDS in solution |
| Very sensitive (ppt-ppb) | Isobaric interference | |
| Isotopes (U-235/U238) can be quantified | Polyatomic ion interference | |
| High vacuum requirement | ||
| Base laboratory technique | ||
| Needs expertise, is costly and not eco-friendly | ||
| Pellet fluorimetry | Very sensitive | Separation is a must |
| After separation of uranium, less interference | Large volume of solvents required | |
| Simple and stable instrument | Aluminum nitrate is a toxic chemical | |
| Low instrument cost | Corrosive flux (NaF) is used | |
| High temperature fusion | ||
| Costly platinum is involved | ||
| Tedious | ||
| Not eco-friendly | ||
| LED fluorimetry | Very sensitive (ppb level of U) | Chloride is a fluorescence quencher above 300 ppm in the presence of a pyrophosphate buffer, pH ∼ 7 |
| Only reagent is a fluorescence enhancing buffer and a variety of reagents are available | ||
| Simple protocols | ||
| Environmentally friendly | ||
| Low cost instrument with a stable LED source | ||
| A versatile technique for U determination in various matrices | ||
| Low interferences | ||
| Easy calibration | ||
| Instrument runs on 12 V DC supply and hence can be battery operated | ||
| Can be employed in field labs | ||
| High tolerance to fluorescence quenchers | ||
| A simple room AC is enough for temperature equilibration | ||
| Fast results | ||
| Three modes of calibration and measurement | ||
| Even uranium in sea water can be determined |
3.12 The following are the steps for the self-evaluation of data of water samples
(1) pH, conductance and temperature and so on should be measured at the site of sample collection. (2) Attention should also be given to the types of containers used and to the manner of transport and storage. Water samples should be collected in polyethylene bottles, using a rapid sampling system after filtration of the sample under pressure. (3) A periodic re-assessment should be made of methods with respect to the sensitivity precision and accuracy of sample collection and its analysis. Intra-laboratory as well as inter-laboratory evaluations should be carried out. (4) Control samples should be prepared. Control samples can be natural water samples or samples with standard additions (natural water with one or more chemicals added e.g., tap water) that have been assayed by several “referee” laboratories preferably by several different methods providing good agreement, so that a “known” value can be assigned to the sample. Synthetic control samples can also be prepared to assess the accuracy of the results. Control charts prepared from the data obtained by re-cycling about 20% of the samples (duplicate samples) for duplicate determinations along with each of set of samples, can be used to check the precision of the determination. (5) The expression of analytical results in milligram-equivalents permits one to check, to a certain degree, the accuracy of the analysis performed, because in this case, the sum of milligram equivalents of the cations must obviously equal the sum of the milliequivalents of the anions. This relationship will hold good, when the analysis is performed promptly within 2–3 days, and then the error will be within the 2–3% limit, which is an indication of satisfactory analysis. (6) The analytical data of the hydrogeochemical samples should be tabulated in the form of maps for future developments. The following points will be more useful: (i) The time interval between the collection of the sample and analysis undertaken should be mentioned. (ii) The exact sample location together with the rock types of the area should be mentioned. (iii) The level of seasonal variations, if any, should also be recorded. (iv) The analytical data of the samples should be reflected in maps together with the other analytical data obtained by other geochemical surveys. (v) The methods adopted for the chemical analysis of samples should be reflected in the final report. The previous points/suggestions will be highly useful to uncover weaknesses in the analytical chain and permit improvement to be instituted without delay. The results can disclose whether the trouble stems from faulty sample collection/treatment, poor calibration practices, impure or incorrectly standardized reagents.4. Conclusions
It is the time interval between sample collection and analysis that is the most critical factor for the reliable analysis of uranium in hot arid regions. A high TDS in the water samples results in a greater variation in the major cations and anions, and uranium content with respect to the time interval between sample collection and analysis. The standard addition method is recommended for analysis of the water samples, where the quenching is recognized and corrected by means of a ‘standard addition’ to the sample. This method is the best for standardising the instrument response by using deionised water as a blank sample. Calibration of the instrument by this method avoids the need for the preparation of very dilute standards with their stability problems. Because, there is no certified reference material available for use with such matrices to cross-check/ensure reliability of results, the 1 ppm, 5 ppm and 10 ppm U standards serve as reference standards for water samples and ensures the traceability of the analytical results. All these standard solutions should be prepared from independent standard stock solutions to cross-check them and their fluorescence response should be in agreement with each other. It is without question that LED-fluorimetry is a portable device more suitable for field conditions, with its easy equipment calibration/standardization, high sensitivity, better reproducibility, cost-effectiveness, eco-friendliness, traceability, and comparability.Author contributions
All authors have contributed to their best.Conflicts of interest
There are no conflicts of interest to declare.References
- R. Chidambaram and C. Ganguly, Curr. Sci., 1996, 70, 21–35 CAS
.
- D. Misra and S. Gupta, Eur. Chem. Bull., 2023, 12(S2), 2550–2558, DOI:10.31838/ecb/2023.12.s2.318
- Uranium in Drinking-Water, Background Document for Development of WHO Guidelines for Drinking-Water Quality, WHO/SDE/WSH/03.04/118, https://www.who.int/water_sanitation_health Search PubMed.
- V. Balaram, A. Rani and D. P. S. Rathore, Geosyst. Geoenviron., 2022, 1, 100043, DOI:10.1016/j.geogeo.2022.100043
-
E. Staff, Significance of Mineralogy in the Development of Flowsheets for Processing Uranium Ores, Technical Report Series No. 196, IAEA, 1980 Search PubMed
-
B. Mason and C. B. Moore, Principles of Geochemistry, Wiley Eastern Limited, New Delhi, India, 4th edn, 1985, pp.46–47 Search PubMed
- J. C. Robbins, CIM Bull., 1978, 71, 61–67 CAS
-
J. C. Robbins and J. D. Kinrade, US pat. 4239964, 1980 Search PubMed
- D. P. S. Rathore, Talanta, 2008, 77, 9–20, DOI:10.1016/j.talanta.2008.06.019
- A. Kirishima, T. Kimura, O. Tochiyama and Z. Yoshida, Chem. Commun., 2003, 7, 910–911 RSC
- D. P. S. Rathore, Explor. Res. At. Miner., 2013, 23, 207–215 CAS
- D. P. S. Rathore, J. Radioanal. Nucl. Chem., 2013, 298, 717–719, DOI:10.1007/s10967-013-2432-z
- D. P. S. Rathore, Hum. Ecol. Risk Assess., 2013, 19, 1147–1149, DOI:10.1080/10807039.2013.791148
- D. P. S. Rathore, J. Geol. Min. Res., 2013, 5, 108–113 Search PubMed
- D. P. S. Rathore, J. Radioanal. Nucl. Chem., 2013, 298, 721–723, DOI:10.1007/s10967-013-2445-7
- D. P. S. Rathore and V. K. Garg, Int. J. Occup. Environ. Med., 2014, 5, 169–171 Search PubMed
- D. P. S. Rathore, Curr. Sci., 2014, 106, 792 Search PubMed
- D. P. S. Rathore, J. Radioanal. Nucl. Chem., 2014, 302, 745–746, DOI:10.1007/s10967-014-3392-7
- D. P. S. Rathore, Chem. Sci. J., 2017, 8, 154, DOI:10.4172/2150-3494.1000154
- D. P. S. Rathore, Adv. Recycl. Waste Manage., 2017, 2, 146, DOI:10.4172/2475-7675.1000146
- D. P. S. Rathore, Environ. Sci. Technol. Lett., 2018, 5, 591–592, DOI:10.1021/acs.estlett.8b00382
-
D. P. S. Rathore, P. K. Tarafder, V. Balaram, M. Mishra, J. Pari, A. G. Bhujle and D. D. Bhawalkar, Simple and reliable analysis of uranium in mineralised rocks, ores, beneficiation products and other diverse matrices: application of differential technique in laser-induced fluorimetry/pulsed led-fluorimetry-a green methodology, e-poster presented, track-3, in International Symposium on Uranium Raw Material for the Nuclear Fuel Cycle: Innovation for Sustaining Future Resources and Production (URAM-2023) (Abstract ID#040), IAEA Headquarters, Vienna, Austria, 2023 Search PubMed
- D. P. S. Rathore, P. K. Tarafder, V. Balaram, M. Mishra, J. Pari, A. G. Bhujle and D. D. Bhawalkar, Environ. Sci. Adv., 2023, 2, 1340–1350, 10.1039/D3VA00134B
- V. Balaram and S. S. Sawant, Minerals, 2022, 12, 394, DOI:10.3390/min12040394
- Analytical Techniques in Uranium Exploration and Ore Processing, Technical Reports Series No. 341, IAEA, Vienna, 1992, Chapters 3–4 and references cited there in Search PubMed.
- P. K. Tarafder, R. K. Mondal and G. Chakrapani, Anal. Methods, 2020, 12, 830–838, 10.1039/c9ay02225b
- D. P. S. Rathore, P. K. Tarafder, M. Kayal and M. Kumar, Anal. Chim. Acta, 2001, 434, 201–208, DOI:10.1016/S0003-2670(01)00850-9
- D. P. S. Rathore and M. Kumar, Talanta, 2004, 62, 343–349, DOI:10.1016/j.talanta.2003.08.002
-
J. C. Robbins, C. Castledine and W. Kostiak, Analytical Procedures for UA-3 Uranium Analysis — Applications Manual, Scintrex Ltd, Ontario, Canada, 1985 Search PubMed
- W. Davies and W. Gray, Talanta, 1964, 11, 1203–1211, DOI:10.1016/0039-9140(64)80171-5
- D. P. S. Rathore, M. Kumar and P. K. Tarafder, Accredit. Qual. Assur., 2012, 17, 75–84, DOI:10.1007/s00769-011-0838-2
- Geochemical Exploration for Uranium, IAEA, Vienna, 1988, TRS No. 284 Search PubMed.
-
Uranium exploration methods, IAEA, Proceedings of a Panel on Uranium Exploration Methods Held in Vienna during 10-14 April, 1972, 1973 Search PubMed
-
E. M. Cameron and C. C. Durhan, Further Studies of Hydro Geochemistry Applied to Mineral Exploration in the Northern Canadian Shield, Report of Activities, Part C, Geological Survey of Canada, 1975, pp. 233–238 Search PubMed
-
American Public Health Association (APHA), Standard Methods for the Examination of Water and Waste Water, American Public Health Association, Washington, D.C., 15th edn, 1980 Search PubMed
-
A. I. Vogel, A Textbook of Quantitative Inorganic Analysis, Longman, London, 4th edn., 1985, pp. 257–278, 325–327 Search PubMed
- D. P. S. Rathore, P. K. Bhargava, M. Kumar and R. K. Talra, Anal. Chim. Acta, 1993, 281, 173–177 CrossRef CAS
- D. P. S. Rathore, M. Kumar and P. K. Bhargava, Chem. Anal., 1995, 40, 805–813 CAS
- D. L. Perry, S. M. Klainer, H. R. Bowman, F. P. Milanovich, T. Hirschfeld and S. Miller, Anal. Chem., 1981, 53, 1048–1950 CrossRef CAS
- M. V. Johnston and J. C. Wright, Anal. Chem., 1981, 53, 1050–1054 CrossRef CAS
- N. S. Shenoy, A. Verma, S. A. Kumar, S. Pandey, S. D. Kumar and A. V. R. Reddy, J. Radioanal. Nucl. Chem., 2012, 294, 413–417 CrossRef CAS
- S. P Prabhu, P. D. Sawant, S. S. Raj, A. Kumar, R. M. Tripathi and P. K. Sarkar, J. Radioanal. Nucl. Chem., 2012, 294, 443–446 CrossRef
- M. Prasad, P. Semwal, P. Panwar, G. S. Gusain and R. C. Ramola, J. Radioanal. Nucl. Chem., 2022, 331, 1933–1940, DOI:10.1007/s10967-021-08128-6
- D. P. S. Rathore, Res. At. Miner., 2007, 17, 145–149 Search PubMed
- International Humic Substances Society (IHSS), https://humic-substances.org/ Search PubMed.
- N. Labonne-Wall, V. Moulin and J. P. Vilarem, Radiochim. Acta, 1997, 79, 7–49 CrossRef
- Y. H. Shen, Chemosphere, 1999, 38, 1505–1515 CrossRef CAS
- J. Riggle and R. V. Wandruszka, Ann. Environ. Sci., 2008, 2, 1–6 CAS
- G. R. Choppin and N. Labonne-Wall, J. Radioanal. Nucl. Chem., 1997, 221, 67–71, DOI:10.1007/BF02035244
- D. P. S. Rathore, J. Public Health Epidemiol., 2013, 5(3) DOI:10.5897/JPHE2013.0517
- M. Krishnakumar, G. Johnson and A. A. Patwardhan, J. Radioanal. Nucl. Chem., 2023, 332, 1795–1804, DOI:10.1007/s10967-023-08897-2
- W. Liu, D. Xing, Z. Bai, Y. Wang, Z. YangYanlong, L. Zhang, L. Xu, L. Zhang, L. Chen, Y. Li, D. Gui, J. Diwu, J. Wang, R. Zhou, Z. Chai and S. Wang, Environ. Sci. Technol., 2017, 51, 3911–3921, DOI:10.1021/acs.est.6b06305
- W. Liu, Y. Wang, L. Song, M. A. Silver, J. Xie, L. Zhang, L. Chen, J. Diwu, Z. Chai and S. Wang, Talanta, 2019, 196, 515–522, DOI:10.1016/j.talanta.2018.12.088
- Z. Zhang, D. Zhang, C. Shi, W. Liu, L. Chen, M. Yu, J. Diwu, J. Li and S. Wang, Environ. Sci. Nano, 2019, 6, 1457–1465, 10.1039/C9EN00148D
- Fluorescence Spectrophotometry, https://www.thermoscientific.com/MaterialsScience Search PubMed.
- S. J. Hart and R. D. Jiji, Analyst, 2002, 127, 1693–1699, 10.1039/B207660H
- A. E. Moe, S. Marx, N. Banani, M. Liu, B. Marquardt and D. M. Wilson, Sens. Actuators B, 2005, 111–112, 230–241, DOI:10.1016/j.snb.2005.01.057
- J. E. Dickens, M. S. Vaughn, M. Taylor and M. Ponstingl, Sens. Actuators B, 2011, 158, 35–42, DOI:10.1016/j.snb.2011.04.077
- J. Bridgeman, A. Baker, D. Brown and J. B. Boxall, Sci. Total Environ., 2015, 524–525, 338–346, DOI:10.1016/j.scitotenv.2015.04.050
- Quantalase, Quantalase Enterprises Pvt Ltd., Indore, India, https://quantalase.in Search PubMed.
- S. K. Pradhan and B. Ambade, Anal. Sci., 2020, 36, 207–211, DOI:10.2116/analsci.19P216
- S. K. Sahoo, S. K. Jha, V. N. Jha, A. C. Patra and M. S. Kulkarni, Curr. Sci., 2021, 120, 1482–1490 CrossRef CAS
- N. S. Shenoy, A. Verma, S. A. Kumar, S. Pandey, S. D. Kumar and A. V. R. Reddy, J. Radioanal. Nucl. Chem., 2012, 294, 413–417, DOI:10.1007/s10967-012-1705-2
- V. Balaram, Geol. J., 2021, 56, 2300–2359, DOI:10.1002/gj.4005
-
N. Shenoy, H. Parab, S. Sounderajan, K. Kumar, S. D. Kumar and A. V. R. Reddy, CTAC-2015: BRNS-AEACI Symposium on Current Trends in Analytical Chemistry, Association of Environmental Analytical Chemistry of India, Mumbai, India, 2015, p. 100, 262 Search PubMed
- R. Mehra, K. M. Anamika and M. Praveen, J. Rad. Nucl. Appl., 2018, 3, 149–156, DOI:10.18576/jrna/030304
| This journal is © The Royal Society of Chemistry 2024 |