
Rapid self-healing and superior toughness in ionically crosslinked polymer ionogels and strain sensing applications†
Anil Kumar
Padhan‡
a,
Diksha
Sharma‡
a,
Tino S.
Thomas
a,
Aayushi Prakash
Sinha
a,
Adarsha Narayan
Mallick
b and
Debaprasad
Mandal
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Punjab 140001, India. E-mail: dmandal@iitrpr.ac.in
bDepartment of Biomedical Engineering, Indian Institute of Technology Ropar, Punjab 140001, India
First published on 12th March 2024
Abstract
Self-healing polymeric ionogel/hydrogel materials are mostly viscoelastic and lack high mechanical toughness; however, simultaneous rapid self-healing and robust mechanical properties are essential for various applications. Herein, an ionically crosslinked polymer ionogel comprising poly(vinylimidazol) [PVIM] and polyphosphoric acid [PPA] having a dual interpenetrating network of ionic and hydrogen bonding interactions is reported. The designed ionogel PVIM–PPA shows rapid and repeatable self-healing within a few seconds without any external stimulus, realizes exceptional mechanical properties such as high tensile strength (30 ± 1 MPa), superior toughness (43.3 ± 0.5 MJ m−3), and fracture energy (4100 J m−2) with ∼90% of healing efficiency and possesses intrinsic flexibility, transparency, and fire-resistant properties. In addition, the mechanical and self-healing properties of the polymer were also investigated by incorporating Ca-ions into the PVIM–PPA ionogels. The PVIM–PPA–Ca ionogel shows an enhancement of the tensile strength to 39 ± 1 MPa and fracture energy of 4700 J m−2 with ≥87% self-healing efficiency. The ionic conductivity of the PVIM–PPA–Ca polymer membrane increases almost five times (5.93 × 10−5 S cm−1) w.r.t PVIM–PPA (1.25 × 10−5 S cm−1) and it can retain ∼90% of the conductivity after self-healing. The recovery efficiency remains almost constant from the second cycle up to the sixth subsequent failure–recovery cycle for these ionogels. Furthermore, a wearable strain sensor was designed using the designed ionogels to detect various human motions accurately and reliably.
Introduction
The advancement in polymeric hydrogel/ionogel materials having new functionalities with tunable structural and mechanical properties as well as responsiveness, which exhibit rapid and autonomous self-healing have attracted great attention due to their applications in various fields, viz., soft robotics,1 neural prostheses, implantable biosensors, electrostimulated drug release devices, artificial skin, tissue engineering, etc.2–4 Autonomous self-healing in polymeric materials depends on the recovery of dynamic bonds during the healing process; however, the slow dynamics of polymer chains often prevent dynamic bonds from reforming.5–7 Even the kinetics of dynamic bond recovery play an important role since some of these bonds are formed within a cracked surface and reshuffling of the bonded pairs is required for efficient bridging between the fractured surfaces during the healing process. Thus, fast, autonomous, and complete healing is limited to polymers containing highly dynamic bonds with good molecular mobility, like hydrogels and soft elastomers.8–11 However, high molecular mobility generally contradicts the mechanical strength of polymer materials.Ionogels/hydrogels based on intrinsic ionic interactions and H-bonding exhibit notable advantages owing to their fast and autonomous self-healing ability without any external stimuli or energy input, and involve a simple preparation method. Conventional self-healing polymer ionogels/hydrogels are considered to be weak and brittle (low toughness) with a fracture energy of <1000 J m−2, which largely restricts their widespread application, particularly in demanding robust mechanical properties. An ionogel is a 3D polymer matrix network containing interpenetrating chemical and physical crosslinking via polymer chain entanglement and ionic interactions. Ionogels haveadvantages over conventional hydrogels in superior environmental stability, as they are highly resistant to dehydration, freezing, and other environmental factors. Despite recent progress in the development of ionogels, existing self-healing polymeric materials generally possess either rapid self-healing ability or mechanical robustness with tensile strengths of <1 MPa, modulus < 0.1 MPa and toughness < 1000 J m−2. Therefore, the development of a mechanically robust material capable of autonomous rapid self-healing on a time scale of seconds is of great demand.12–16
Herein, we have developed ionically crosslinked polymeric materials associated with a hydrogen bond containing a protonated polyvinyl imidazolium cation [PVIMH]n+ and polyphosphate anion [PPA]n−. This crosslinked material intrinsically takes ∼10 wt% water and forms an ionogel with rapid self-healing ability within a few seconds under ambient conditions without any external stimulus.
The dynamic and reversible process of hydrogen bonding contributed to energy dissipation and network reconstruction in the ionogel, which provided ionogel with fast self-healing ability, high tensile strength, and good ionic conductivity.17–21 We have also explored the effect of Ca-ion incorporation within the polymer matrix on self-healing and mechanical properties, which resulted in an increase in ionic conductivity but a slight decrease in rigidity and healing performance. NaCl or LiCl was avoided to prevent the ionogel from absorbing moisture from air. These outstanding properties make zwitterionic nanocomposite gels promising for skin strain sensors to monitor body movement.22–25
Results and discussion
Synthesis of ionically crosslinked polymer ionogels
The PVIM–PPA crosslinked ionic network was prepared by mixing the polymer PVIM (1) and PPA solutions in deionized water under sonication at 60 °C for 1 h as shown in Fig. 1a. A white viscous polymer gel was formed which was isolated and dried. The resulting ionically crosslinked network of the PVIM–PPA (2) polymer remains insoluble in most nonpolar solvents, but swells in polar solvents like DMF, DMSO, methanol, and ethanol and forms an ionogel in water. The synthesized PVIM–PPA crosslinked polymer forms a thin transparent and flexible film as shown in Fig. S1 (ESI†). Similarly, Ca-ion incorporating polymer ionogels PVIM–PPA–Ca (3a–c) were prepared by mixing PVIM, PPA, and various concentrations of Ca(NO3)2 (9, 18, and 27 wt% PPA) as depicted in Fig. 1. These PVIM–PPA–Ca (3a–c) polymers also form ionogels in water and slightly swell in polar solvents similar to 2. Surprisingly, when phosphoric acid (>85%) was used during synthesis instead of PPA, no gel formation was observed even after standing for one day under similar experimental conditions. Microwave plasma atomic emission spectroscopy (MPAES) analysis revealed 0.12, 0.50, and 1.56 wt% of Ca-ions in polymer ionogels 3a, 3b, and 3c, respectively (Fig. S2, ESI†). Furthermore, the presence of Ca-ions was also confirmed by using the energy dispersive X-ray spectra (EDS) of PVIM–PPA–Ca (3a–c), as shown in Fig. S3 (ESI†). | ||
| Fig. 1 (a) Synthesis of crosslinked polymer ionogels PVIM–PPA (2), and PVIM–PPA–Ca (3a, 3b, and 3c) with added Ca-ions of 9, 18, and 27 wt% of PPA. (b) FT-IR spectra of PPA and ionogels 1, 2, and 3a–3c. (c) DSC curve of ionogels and (d) water uptake of ionogels. | ||
FT-IR analysis
The crosslinked ionogels PVIM–PPA (2) and PVIM–PPA–Ca (3a–c) were primarily assessed by FT-IR spectroscopy. The ionic interactions associated with PVIM–PPA (2) and PVIM–PPA–Ca (3a–c) were supported by FT-IR spectra, in which PVIM–PPA–Ca exhibit quite similar FT-IR spectra to those of PVIM–PPA. As observed in Fig. 1b, the νC–H(stretch) peak at 3100 cm−1 of PVIM was shifted by 30 cm−1 to 3130 cm−1 in PVIM–PPA (2) and PVIM–PPA–Ca (3a–c). The C–N–C(stretch) peak at 1498 cm−1 also shifted to 1540 cm−1 in PVIM–PPA (2) and PVIM–PPA–Ca (3a–c). Both shifts to a higher frequency in all the cross-linked polymers support ion pair formation. The distinct phosphate peak at 920 cm−1 was also found to shift to a lower frequency of ∼890 cm−1 in all the cross-linked polymers. The broadening of the peak at ∼890 cm−1 for PVIM–PPA–Ca (3a–c) was consistent at various concentrations of Ca-ions, suggesting successful crosslinking of the polymers.Thermal analysis
Differential scanning calorimetry (DSC) was performed at temperatures ranging from −50 to 80 °C at a rate of 10 °C min−1 for all the ionogels. DSC was also conducted from 20 to 200 °C at a rate of 40 °C min−1 after freeze-drying the polymers. As shown in Fig. 1c, ionogels 2, 3a, 3b, and 3c (without freeze-drying) exhibit glass transition temperatures (Tg) of −10.2, −3.8, 1.8, and 8.2 °C, respectively. The DSC analysis of the freeze-dried polymers, presented in Fig. S4a and S13–S18 (ESI†), reveals only Tg without any crystallization or melting. Homopolymer PVIM (1) and PPA exhibit Tg values of 172.6, and −11.3 °C, respectively. For PVIM–PPA (2), Tg is 126.2 °C, which progressively increases to 135.6, 143.1, and 152.3 °C for 3a–3c, respectively. The higher Tg values observed in freeze-dried polymers resulted from increased rigidity due to the removal of water molecules during the freeze-drying process. Moreover, the increase in Tg values observed in both ionogels and freeze-drying polymers, correlates well with the increase in Ca-ion concentration, indicating a gradual increase in the rigidity and constraint of chain mobility within the polymer matrix.Water uptake and effect of salts and urea on ionogels
The water uptake study is essential for ionic polymers where water uptake was calculated using the following equation:26| Water uptake (%) = (Ws − Wd)/Wd × 100 |
The saline stabilities of ionogels 2, 3a, 3b, and 3c were investigated by immersing these gels in 3 M NaCl solution for 48 h. The photographs and corresponding swelling ratio data are shown in Fig. S5 and S6 (ESI†). The swelling percentage was determined by using the equations:
| Swelling ratio (%) = (Vs − Vd)/Vd × 100 |
All the ionogels show good saline stability as their shapes remained unchanged and only slight swelling was observed in the range of 118–128 (%). When the solution was heated to 60 °C, the ionogel started dissociating within 3 h and completely dissolved within 10 h as shown in Fig. S6,† due to the weakening of ionic bonds in the presence of NaCl solution. Similarly, the effect of 3 M urea solution was also investigated and is shown in Fig. S7 (ESI†). The ionogels are stable for up to 10–12 h, after which the shape of ionogels distorted possibly due to disturbances of H-bonding and some extent of ionic bonding. The disturbance of the network structure by saline water and urea solution proves the presence of interpenetrating ionic and H-bond interactions in these ionogels.
Self-healing performance
Initially the self-healing ability of the PVIM–PPA ionogel was evaluated by cutting a test sample into two pieces using a sharp surgical blade as shown in Fig. 2A, followed by contacting the freshly created cut surfaces at room temperature without any external force. As shown in Fig. 2A, all the samples rejoined the interface of these two pieces completely within a few seconds. Rapid damage repair occurs due to the molecular motion created from both interionic attraction and the hydrogen bonding network between the polymeric cation [PVIMH]+ and [PPA]− anion. The healed ionogel could withstand certain mechanical forces such as pulling to a large extent without failure at the interface as shown in Fig. 2A. A similar set of experiments was further performed for 3b and 3c, and are presented in Fig. 2. Furthermore, clear visualization of the healing performance was demonstrated by taking pieces of fresh ionogel 3c and methylene blue dye-doped ionogel 3c and joining them as shown in Fig. 2B–E and S8 (ESI†). The healed ionogel shows remarkable load-bearing capacity as shown in Fig. 2D and E, and movie clip M1.† The self-healing efficiency of these crosslinked polymers was further investigated qualitatively based on rheology, tensile properties and conductivity measurement. As calcium-ion content was lower (9 wt%) in ionogel 3a, it was not included in further studies. | ||
| Fig. 2 (A) Room temperature self-healing of 2, and 3b virgin ionogel, cut sample, self-healed ionogel at ambient temperature, and stretched after healing; (B) pieces of fresh ionogels 3c and methylene blue dye-doped ionogel 3c (left, dark brown) (C) self-healing, (D and E) load bearing after healing. | ||
Rheology analysis and self-healing performance
The healing performance was also evaluated by rheology analysis. The stress–strain profile from oscillatory rheology measurements at 25 °C illustrates the tensile nature and shear-thinning behaviour of ionogels. The yield points of ∼1000, 3500, and 5000 Pa for 2, 3b, and 3c, respectively in Fig. 3a, demonstrate the enhanced mechanical strength with 3.5 and 5 times Ca-ion incorporation. The storage modulus (G′) and loss modulus (G′′) of 2, 3b, and 3c as a function of oscillatory strain amplitude are shown in Fig. 3b. The large value of critical strain with a ∼50% linear viscoelastic regime could be ascribed to the stretchability of the polymer ionogel. The observed G′ and G′′ were higher for PVIM–PPA–Ca (3b and 3c) than that for PVIM–PPA (2), as the formation of calcium phosphate improves the mechanical properties. The G′ and G′′ for 2 and 3b, 3c as a function of angular frequency (ω) obtained from rheology measurements are shown in Fig. 3c, wherein it was observed that G′ and G′′ increase with increasing frequency.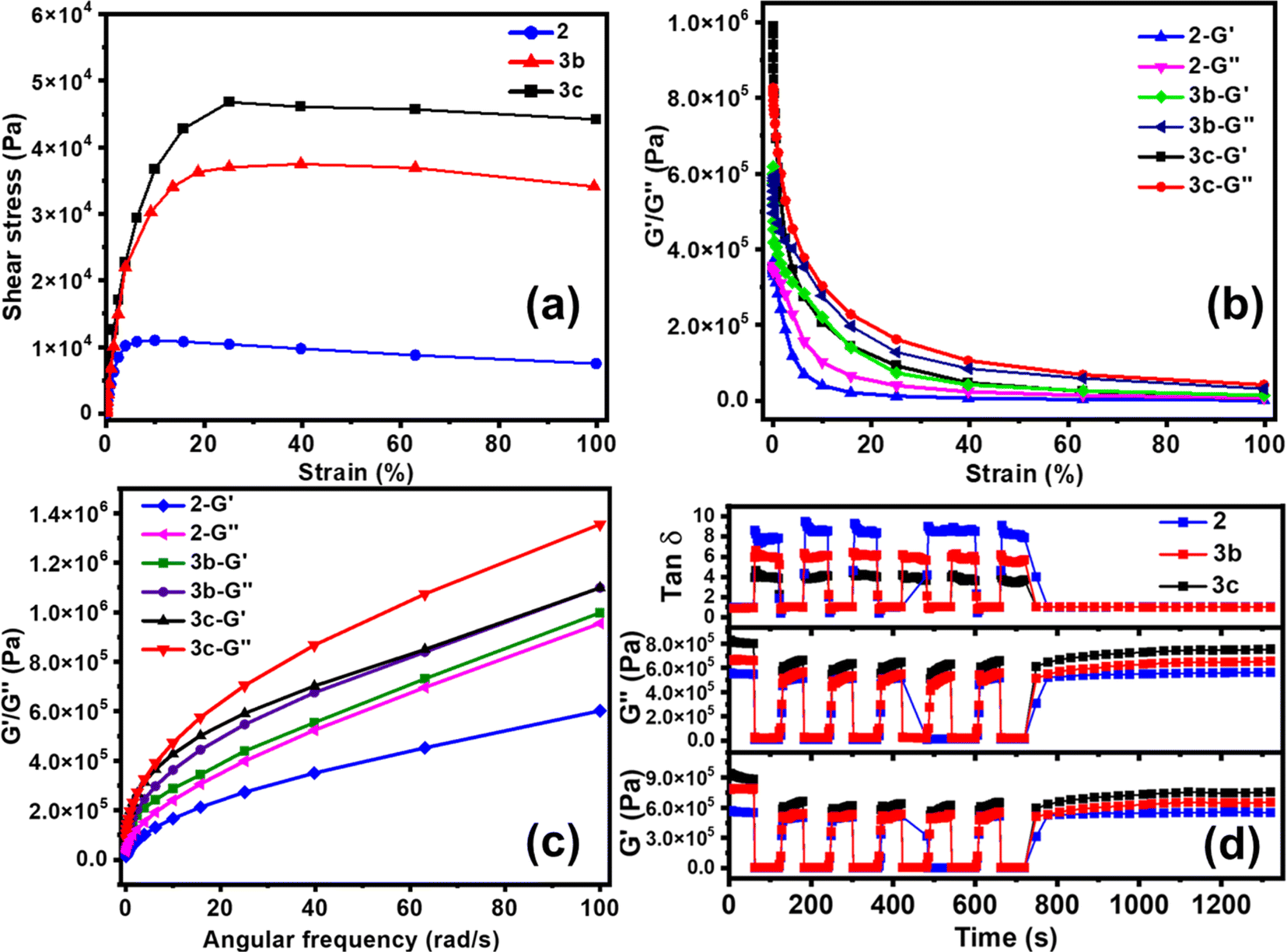 | ||
Fig. 3 Viscoelastic measurements of (2) and (3b–c); (a) shear stress–strain profile, (b) amplitude sweep, (c) frequency sweep at 25 °C, and (d) self-healing efficiency from rheology failure–recovery tests where gel failure was induced by a sudden increase in 200% strain at 25 °C and a constant angular frequency of 10 rad s−1. Recovery of the storage modulus (G′), loss modulus (G′′), and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ for each cycle (d) comparison of 2, 3b, and 3c. δ for each cycle (d) comparison of 2, 3b, and 3c. | ||
The self-healing efficiency was evaluated by oscillatory rheology measurements. A high strain (∼200%) was applied by a sudden increase in the damping factor to cause gel network failure, accompanied by a decrease in both storage and loss modulus. Self-healing efficiency was obtained from the failure recovery of G′, G′′ and loss factors (tanδ = G′′/G′). When the strain was brought to its initial value, a very fast recovery was observed for each of these three variables G′, G′′ and tanδ. As can be seen from Fig. 3d, PVIM–PPA (2) shows 88 ± 2% recovery efficiency as obtained from G′ in the first failure cycle. A small decrease in the recovery efficiency of 85 ± 2% and 81 ± 2% was observed for 3b and 3c (PVIM–PPA–Ca) in the first failure cycle (Fig. S10 (ESI†)). The failure–recovery cycles were repeated 6 times and no noticeable loss of the self-healing ability was observed from the 2nd cycle onwards, and the recovery efficiency remains almost the same for all crosslinked polymers. These findings suggest the complete recovery of the crosslink network after disruption and a repeatable self-healing ability of these ionogels. With the introduction of Ca-ions (3c, 27 wt% w.r.t PPA), a slight decrease in the healing efficiency with a 7% loss as compared to that of 2 was observed. Moreover, mechanical properties and self-healing efficiency were analyzed under different environmental conditions at different temperatures (−25, 80 and 120 °C) for 24 h and at 80% humidity for 3 days. As shown in Fig. S23 and Table S4 (ESI†). ionogel 3c shows good mechanical characteristics at −25 amd 80 °C and under humid conditions, and it healed within 2 min with a high healing efficiency of 96%. However, when the temperature was increased to 120 °C, due to the partial evaporation of water, the ionogels become brittle, and mechanical characteristics and self-healing efficiency decreased.
Mechanical properties and self-healing
Stress–strain tensile studies were performed to quantitatively evaluate the self-healing performance by investigating the mechanical properties using a universal testing machine (UTM) with dog-bone shaped samples of polymers 2, 3b and 3c before and after the healing process. As depicted in Fig. 4a and Table 1, 2 shows a very high ultimate tensile stress of 30 ± 1 MPa, elongation at break of 155 ± 2%, and Young's modulus of 3.1 ± 0.1 MPa. After a cut with a sharp surgical blade, the PVIM–PPA (2) sample was allowed to self-heal at room temperature for 3 minutes, and furthermore, the mechanical properties were evaluated which shows >90% retention of the initial ultimate tensile strength (28 ± 2 MPa), Young's modulus (2.7 ± 0.1 MPa) and the initial elongation at break (146 ± 2%). The tensile strength and Young's modulus were found to increase by ∼15% in 3b and ∼30% in 3c with respect to those of 2. However, the elongation at break decreases to the same extent (∼15 and 30%) upon increasing the Ca-ion concentration. Interestingly, 3b and 3c also retained a healing efficiency similar to that of ionogel 2, while after the healing process, the healed networks of 3c retained ∼87% of the initial ultimate tensile strength (34 ± 2 MPa), and ∼90% of their initial elongation at break. These results demonstrate that Ca-ion incorporation has a minimal effect on reducing the healing performance but greatly improves mechanical properties. Self-healing repeatability of 3c was also monitored from the tensile test up to 5 cycles by joining two fracture parts of the sample with a healing time of ∼1 min. The stress–strain curves in Fig. S24 (ESI†) demonstrate a good healing cycle repeatability. | ||
| Fig. 4 (a) Tensile properties before and after healing and (b) toughness of polymers 2, 3b, and 3c. (c) Healing efficiency and (d) force–extension curves obtained from the tearing test for polymer ionogels 2, 3b, and 3c; inset shows fracture energy. | ||
| Ionogels | Young's modulus (MPa) | Ultimate stress (MPa) | Elongation at break (%) | Toughness (MJ m−3) |
|---|---|---|---|---|
| PVIM–PPA (2) (virgin) | 3.1 ± 0.1 | 30 ± 1 | 155 ± 2 | 43.3 ± 1 |
| PVIM–PPA (2) (healed) | 2.7 ± 0.1 | 28 ± 2 | 146 ± 2 | 37.9 ± 1 |
| PVIM–PPA–Ca (3b) (virgin) | 3.6 ± 0.1 | 34 ± 1 | 128 ± 2 | 41.3 ± 1 |
| PVIM–PPA–Ca (3b) (healed) | 2.9 ± 0.1 | 31 ± 2 | 117 ± 2 | 33.9 ± 2 |
| PVIM–PPA–Ca (3c) (virgin) | 4.1 ± 0.2 | 39 ± 1 | 110 ± 2 | 41.4 ± 2 |
| PVIM–PPA–Ca (3c) (healed) | 3.3 ± 0.2 | 34 ± 2 | 98 ± 2 | 31.3 ± 2 |
The toughness is the ability of a material to absorb energy before breaking and is measured from the area under the stress–strain curve. The polymer ionogels 2, 3b, and 3c exhibited a high toughness of 43.3 ± 1, 41.3 ± 1, and 41.4 ± 2 MJ m−3, respectively, which are much higher than those of related self-healing ionogels as can be seen from Table S1 (ESI†). These ionogels possess robust mechanical properties and high toughness simultaneously with rapid autonomous self-healing. This can be explained by the synergistic contribution of intrinsic interionic interactions and hydrogen bonding, which create a crosslinked interpenetrating polymer network (IPN) structure. After healing, the toughness for 2, and 3b, 3c dropped to 37.9 ± 1, 33.9 ± 2 and 31.3 ± 2 MJ m−3 respectively (Fig. 4c). Toughness values of 2 are slightly higher than that of 3b and 3c (PVIM–PPA–Ca), suggesting a better self-healing nature in terms of toughness. It can be seen that the ionogels require small strains to reach yield points, after which the stress remains almost constant against strain until the breaking point, which demonstrates an improved performance, such as a higher Young's modulus, and they were stiffer and also stronger, so that they can bear higher load (actual stress) without breaking.
Tearing energy/fracture energy
The tearing energy (fracture energy) T was calculated at a constant tearing force F using the relation T = 2F/w, where w is the thickness of the sample. As shown in Fig. 4d, by applying a constant stretching force, a steady state of crack propagation is obtained. The tearing energies (T) are 4140, 4535, and 4730 J m−2 for polymer ionogels 2, 3b, and 3c, respectively. Most synthetic ionogels are brittle, with fracture energies of ∼10 J m−2 as compared to 1000 J m−2 for cartilage and 10000 J m−2 for natural rubber.
Flame-retardant behaviour
Besides self-healing and physical, chemical, and mechanical properties, the flame-retardant property is highly important as it enhances the safety and sustainability while maintaining longer durability.27,28 The flame-retardant properties were demonstrated by exposing the polymer films to a direct flame from a butane torch for 2 minutes of ignition. Fig. 5 illustrates the ignition test of pure cotton, cotton coated with the polymer, and direct ignition of the polymer in the presence of a butane torch. Upon exposure to a direct flame, the polymer PVIM–PPA exhibited fire resistance properties witnessed from its minimum ignition due to the ionic nature of polymer PPIM–PPA and phosphate. The UL-94 experiment in Fig. S25 (ESI†) demonstrates an excellent flame-resistant performance up to 20 seconds. | ||
| Fig. 5 Photograph of the flame-retardant test of (a) cotton without the polymer and (b) a small film of 2 wrapped over cotton, and (c) 2 direct ignitions for 2 min. | ||
Ionic conductivity and self-healing performance
The ionic conductivities of polymer membranes 2, 3b, and 3c were measured by four probe conductivity measurements at room temperature before and after the self-healing experiment, as shown in Fig. 6a–c, using the following equation:29| σ = L/(Rwt) |
 | ||
| Fig. 6 Images of (a) fresh, (b) cut and (c) healed polymer membranes of 2 used for ionic conductivity measurement. (d) Ionic conductivity of fresh and healed polymers of 2, 3b, and 3c. | ||
Strain sensing performance
Excellent mechanical properties, rapid self-healing, and high ionic conductivity demonstrated by the PVIM–PPA–Ca (3c) ionogel led us to explore its potential application as a wearable sensor for fast response to diverse human movements in real time, despite its limited stretchability. To assess the reliability and reproducibility of the sensing performances, cycles of loading/unloading experiments were performed on the ionogel under ambient conditions. The relative fluctuation in resistance (ΔR/R0) of the ionogel was calculated by using the following equation:| ΔR/R0 = (R − R0)/R0 |
As shown in Fig. 7a, the motion of a finger in response to relative resistance (ΔR/R0) can be detected while bending the finger from 0° to 90°, indicating that the ionogel-based sensor has high sensitivity. Furthermore, to validate the practicability of the sensor, the ionogel membranes were placed at the main joints of the human body, such as the arm and knees, to sense human movements accurately in real time, as shown in Fig. 7b and c. The relative resistance signal consistently increases as the arm and knee are bent and promptly returns to the initial state when straightened. Moreover, the designed sensor could instantly detect the speed of knee motion during walking, wherein the relative resistance signal is greater when knee is bent slower and lower when knee is bent faster due to a change in the angle, demonstrating excellent sensitivity.
 | ||
| Fig. 7 Real-time monitoring of the PVIM–PPA–Ca (3c) ionogel-based wearable strain sensor for various human activities. Signals of the relative resistance change (ΔR/R0) of 3c as a function of (a) finger bending at various angles, (b) arm bending, (c) knee bending, and (d) speaking different words; (e) photograph of 3c at various applied strains; (f) corresponding relative resistance (ΔR/R0) and gauge factors. | ||
In addition to simple motions, the sensor responded precisely to tiny yet complex muscle movements such as vocal movements during speaking. The ionogel was fixed to a volunteer's throat and signals were recorded for various pronounced words such as “good morning” and “beautiful” where a distinct and repeatable signal can be observed for each phrase spoken as shown in Fig. 7d and S26 (ESI†). The response to relative resistance (ΔR/R0) remains constant over the cycles and responds swiftly to all the deformations and stable responses with little drift or fluctuation in multiple cycles, indicating the repeatability of the ionogel sensors. These results confirm the high sensitivity of the PVIM–PPA–Ca (3c) ionogel-based sensor for practical application. Furthermore, a good strain response was observed as shown in Fig. 7e and f, where the relative resistance of the ionogel (ΔR/R0) precisely changes with increasing tensile strain applied to the ionogel (20%, 40%, 60%, 80%, 100%, 120%, and 140%).
The gauge factor (GF), a crucial parameter for demonstrating the sensitivity of the sensor was calculated from the fitting curve of the relative resistance change against the applied strain. The GF values were 2.18 at less than 80% strain, and 5.54 at 140% strain, indicating the excellent strain sensitivity of ionogel 3c at lower as well as higher stretching as shown in Fig. 7f. The excellent mechanical and self-healing properties of the PVIM–PPA–Ca ionogel sensor can aid in monitoring human body motions with longer lifetimes and adhesion of the ionogel provides intimate contact for a fast response to amplify signals with high stretchability and sensitivity.
Biocompatibility test
The pH of ionogel 3c is between 6.6 and 7.0, and therefore the biocompatibility test was performed. The cell viability of 2 and 3c was examined in a murine fibroblast cell line (L929). L929 cells had a vitality of more than 100%. The L929 cells were incubated with 2 and 3c for 24 h, while the untreated cells served as a control. Ionogels 2 and 3c demonstrated 78% and 82% cell viability, respectively (Fig. S27, ESI†).Conclusions
We have developed an ionically crosslinked polymer ionogel network comprising poly(vinylimidazolium) [PVIMH]n+ and a polyphosphate anion [PPA]n−. The synthesized ionogel shows rapid and repetitive self-healing at room temperature on a time scale of a few seconds without any external stimulus. Self-healing of the crosslinked polymer ionogel was achieved by the contribution of dual interpenetrating ionic bonds and a hydrogen bonding network. Rheology analysis revealed a constant recovery efficiency up to 6 subsequent failure–recovery cycles of 85% of the original storage and loss modulus, and the ionogel demonstrated a high tensile strength of 30 ± 1 MPa, superior toughness of 43.3 ± 0.5 and fracture energy of 4100 J m−2 with ∼90% healing efficiency.The incorporation of Ca-ions enhances the storage modulus (G′), loss modulus (G′′), and tensile strength of 39 ± 1 MPa, a superior toughness of 41.0 MJ m−3 with ≥87% healing efficiency. Moreover, the ionic conductivity of PVIM–PPA–Ca (3c) increases almost 5 times (5.93 × 10−5 S cm−1) w.r.t PVIM–PPA (1.25 × 10−5 S cm−1), and the self-healed membrane retains ∼90% efficiency. The designed ionogel has been shown to be an efficient strain sensor for measuring various human activities with precision and reliability, even for small motions of the human body (e.g., finger, wrist, and elbow), indicating its tremendous potential for use as a wearable device.
Experimental section
Synthesis
Preparation of poly(vinylimidazol) (1) (under microwave heating): a 30 mL microwave glass vial was charged with vinyl imidazole (0.941 g, 10.00 mmol), AIBN (0.013 g, 0.08 mmol), and anhydrous DMSO (3.0 mL). The reaction mixture was degassed by evacuating at 0.1 mbar followed by purging with argon for 15 min. The reaction mixture was stirred at ambient temperature for 10 min and then heated at 140 °C in a ‘Monowave 200’ instrument programmed with a 2 min ramp, 40 min hold at 140 °C. The progress of the reaction was monitored by 1H NMR and FT-IR spectroscopy. Upon completion, the polymer was precipitated in acetone (30 mL). The residue was filtered, washed with acetone (20 mL × 4), and dried under vacuum (2 × 10−3 mbar) at 60 °C to afford 1 (0.673 g, 71 wt%) as a white powder. Polymer 1 is soluble in water, methanol, and DMSO.(Under conventional heating): a Schlenk tube was charged with vinylimidazole (0.941 g, 10.00 mmol), AIBN (0.013 g, 0.08 mmol), and anhydrous toluene (4.0 mL). The mixture was degassed in three freeze–thaw cycles under vacuum and purged with argon for 15 min. The reaction mixture was heated in an 80 °C oil bath for 24 h to give a precipitate. The obtained solid was purified by precipitation in diethyl ether and dried under vacuum to afford 1 as a white powder (0.750 g, 79.7 wt%). Polymer 1 is soluble in water, methanol, and DMSO but insoluble in chloroform and toluene; 1HNMR (400 MHz, D2O. δ): 7.06–6.64 (br, 3H, NCHCHN and NCHN), 3.74–2.57 (br, 1H, CH(N)CH2), 2.12–1.99 (br, 2H, CH(N)CH2) (Fig. S11 (ESI†)). 1H (DMSO-d6) δ ppm: 7.03–6.74 (br, 3H, NCHCHN & NCHN) 3.73–2.59 (br, 1H, CH(N)CH2), 2.30–1.50 (br, 2H, CH(N)CH2). 13C {1H, DMSO-d6}: 136.28 (NCHN), 129.25 (NCHCHN), 116.77 (NCHCHN), 50.85 (CH(N)CH2) (Fig. S12 (ESI†)). FT-IR (ATR, cm−1): 3358.6, 3110.2, 2960.2, 2199.5, 1647.5, 1495.7, 1451.5, 1414.2, 1212.9, 1227.0, 1108.4, 1081.6, 1019.8, 915.2, 819.3, 739.9, 661.3, 634.6. Mn (GPC): 11100 (Fig. S9 (ESI†)).
Preparation of PVIM–PPA (2)
Poly(vinyl imidazole) 1 (2.000 g, 21.26 mmol considering monomer mol. wt) and PPA (83% P2O5 basis) (2.500 g, 21.26 mmol) were dissolved separately in deionized water. These solutions were mixed in a beaker under sonication at 60 °C. A white milky emulsion was formed instantly and was kept under sonication for 1 h at 60 °C to form a viscous crosslinked polymer ionogel. The resulting PVIM–PPA polymer was washed with deionized water and dried under vacuum (2 × 10−3 mbar) to obtain 2 as a yellowish-white solid (3.76 g, 83.5 wt%).FT-IR (ATR, cm−1): 3375.9, 3122.2, 1642.5, 1571.8, 1538.9, 1416.7, 1177.1, 1069.8, 966.5, 877.2, 750.4, 698.7, 646.9.
Synthesis of PVIM–PPA–Ca (3a, 3b, and 3c)
Poly(vinyl imidazole) (1.400 g, 14.88 mmol) and Ca(NO3)2·4H2O (0.157 g, 0.67 mmol, 9 wt% of PPA) were mixed in 10.0 mL deionized water. PPA (83% P2O5 basis) (1.750 g, 14.88 mmol) was freshly prepared in deionized water (10.0 mL) and was added to the above solutions under sonication at 60 °C, and a white emulsion suspension was formed instantly. The mixture was sonicated for 1 h at 60 °C and isolated as a viscous polymer ionogel. The resulting PVIM–PPA–Ca polymer gel was washed with deionized water (3 × 20 mL) and dried under vacuum resulting in a yellowish-white solid (2.71 g, 82 wt%). Similar experiments were performed varying Ca(NO3)2·4H2O, (0.315 g, 1.33 mmol, 18 wt% w.r.t PPA and 0.472 g, 2.00 mmol, 27 wt% w.r.t PPA) to obtain 82 wt% of 3b and 85 wt% of 3c, respectively.FT-IR (ATR, cm−1) 3a: 3357.9, 3122.2, 1648.5, 1574.2, 1538.9, 1416.7, 1238.2, 1183.4, 1069.7, 966.5, 877.2, 750.4, 698.7, 647.1. 3b: 3357.9, 3122.0, 1652.4, 1572.3, 1538.6, 1416.2, 1238.1, 1183.7, 1069.8, 966.5, 877.2, 750.4, 698.6, 646.8. 3c: 3357.8, 3122.1, 1657.3, 1571.6, 1538.7, 1416.4, 1238.2, 1183.7, 1069.8, 966.5, 877.2, 750.4, 698.6, 646.9.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Mandal acknowledges CSIR (02(0436/21/EMR-II)) and DGNSM/DMSRDE/PROC(PRR)/22-23/013 for financial support. AK Padhan, D. Sharma and A. N. Mallick thank IIT Ropar for fellowships. A. P. Sinha thanks DST for the Inspire Fellowship (DST/INSPIRE/03/2019/000968) and T. S. Thomas thanks UGC for the fellowship (DEC18-114672). We thank V. Chawla and Dr Yashveer Singh (IIT Ropar) for the biocompatibility test.References
- D. Jiao, Q. L. Zhu, C. Y. Li, Q. Zheng and Z. L. Wu, Acc. Chem. Res., 2022, 55, 1533–1545 CrossRef CAS PubMed.
- Y. Cao, Y. J. Tan, S. Li, W. W. Lee, H. Guo, Y. Cai, C. Wang and B. C. K. Tee, Nat. Electron., 2019, 2, 75–82 CrossRef.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- F. Li, X. Cai, G. Liu, H. Xu and W. Chen, Adv. Funct. Mater., 2023, 33, 2300701 CrossRef CAS.
- A. K. Padhan and D. Mandal, in Self-Healing Polymer-Based Systems, ed. S. Thomas and A. Surendran, Elsevier, 2020, pp. 17–73, DOI:10.1016/B978-0-12-818450-9.00002-7.
- A. K. Padhan and D. Mandal, Polym. Chem., 2018, 9, 3248–3261 RSC.
- J. Mo, Y. Dai, C. Zhang, Y. Zhou, W. Li, Y. Song, C. Wu and Z. Wang, Mater. Horiz., 2021, 8, 3409–3416 RSC.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- D. Zhang, Y. Tang, Y. Zhang, F. Yang, Y. Liu, X. Wang, J. Yang, X. Gong and J. Zheng, J. Mater. Chem. A, 2020, 8, 20474–20485 RSC.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- Y. Ou, T. Zhao, Y. Zhang, G. Zhao and L. Dong, Mater. Horiz., 2022, 9, 1679–1689 RSC.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- D. Sharma and D. Mandal, J. Mater. Chem. A, 2023, 11, 25683–25691 RSC.
- W. Li, L. Li, Z. Liu, S. Zheng, Q. Li and F. Yan, Adv. Mater., 2023, 35, 2301383 CrossRef CAS PubMed.
- M. Wang, J. Hu and M. D. Dickey, JACS Au, 2022, 2, 2645–2657 CrossRef CAS PubMed.
- M. Wang, P. Zhang, M. Shamsi, J. L. Thelen, W. Qian, V. K. Truong, J. Ma, J. Hu and M. D. Dickey, Nat. Mater., 2022, 21, 359–365 CrossRef CAS PubMed.
- Z. Yu and P. Wu, Adv. Funct. Mater., 2021, 31, 2107226 CrossRef CAS.
- X. Qu, W. Niu, R. Wang, Z. Li, Y. Guo, X. Liu and J. Sun, Mater. Horiz., 2020, 7, 2994–3004 RSC.
- J. Tie, Z. Mao, L. Zhang, Y. Zhong and H. Xu, Adv. Funct. Mater., 2023, 2307367 CrossRef CAS.
- D.-L. Zhang, X. Ju, L.-H. Li, Y. Kang, X.-L. Gong, B.-J. Li and S. Zhang, Chem. Commun., 2015, 51, 6377–6380 RSC.
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef CAS PubMed.
- T. Q. Trung, T. M. L. Dang, S. Ramasundaram, P. T. Toi, S. Y. Park and N.-E. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 2317–2327 CrossRef CAS PubMed.
- J. Sun, Y. Yuan, G. Lu, L. Li, X. Zhu and J. Nie, J. Mater. Chem. C, 2019, 7, 11244–11250 RSC.
- S. Wu, K. Moody, A. Kollipara and Y. Zhu, ACS Appl. Mater. Interfaces, 2023, 15, 1798–1807 CrossRef CAS PubMed.
- X. Li and F. Sun, ACS Appl. Mater. Interfaces, 2023, 15, 37717–37727 CrossRef CAS PubMed.
- G. Singh, M. Kumar, T. S. Thomas, T. C. Nagaiah and D. Mandal, ACS Appl. Energy Mater., 2021, 4, 14689–14699 CrossRef CAS.
- J. H. Cho, V. Vasagar, K. Shanmuganathan, A. R. Jones, S. Nazarenko and C. J. Ellison, Chem. Mater., 2015, 27, 6784–6790 CrossRef CAS.
- Y. Du, Y. Xie, X. Liu, H. Jiang, F. Wu, H. Wu, Y. Mei and D. Xie, ACS Sustainable Chem. Eng., 2023, 11, 4498–4508 CrossRef CAS.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07277k |
| ‡ A. K. Padhan and D. Sharma contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |