
Exploring how cation entropy influences electric double layer formation and electrochemical reactivity†
Beichen
Liu
 ,
Wenxiao
Guo
,
Seth R.
Anderson
,
Samuel G.
Johnstone
,
Siqi
Wu
,
Megan C.
Herrington
and
Matthew A.
Gebbie
*
,
Wenxiao
Guo
,
Seth R.
Anderson
,
Samuel G.
Johnstone
,
Siqi
Wu
,
Megan C.
Herrington
and
Matthew A.
Gebbie
*
Department of Chemical and Biological Engineering, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA. E-mail: gebbie@wisc.edu
First published on 1st December 2023
Abstract
Electric double layers are crucial to energy storage and electrocatalytic device performance. While double layer formation originates in electrostatic interactions, electric double layer properties are governed by a balance of both electrostatic and entropic driving forces. Favorable ion–surface electrostatic interactions attract counterions to charged surfaces to compensate, or “screen,” potentials, but the confinement of these same ions from a bulk reservoir to the interface incurs an entropic penalty. Here, we use a dicationic imidazolium ionic liquid and its monovalent analogue to explore how cation valence and entropy influence double layer formation and electrochemical reactivity using CO2 electroreduction as a model reaction. We find that divalent and monovalent cations display similar CO2 reduction kinetics but differ vastly in steady-state reactivity due to rapid electrochemically induced precipitation of insulating dicationic (bi)carbonate films. Using in situ surface-enhanced Raman scattering spectroscopy, we find that potential-dependent cation reorientation occurs at similar potentials between the two ionic liquids, but the introduction of a covalent link in the divalent cation imparts a more ordered double layer structure that favors (bi)carbonate precipitation. In mixed monovalent-divalent electrolytes, we find that the divalent cations dominate interfacial properties by preferentially accumulating at surfaces even at very low relative concentrations. Our findings confirm that ion entropy plays a key role in modulating local electrochemical environments. Furthermore, we highlight how double layer properties are sensitive to the properties of counterions that pay the lowest entropic penalty to accumulate at interfaces. Overall, we illustrate that ion entropy provides a new knob to tune reaction microenvironments and unveil how entropy plays a major role in modulating electrochemical reactivity in mixed ion electrolytes.
Introduction
Electrochemical interfaces are a core area of research in the soft matter community1–3 for advanced energy storage4,5 and electrocatalytic systems6,7 that provide opportunities to offset anthropogenic CO2 emissions.8–10 For example, converting CO2 into useful chemical products through electrocatalysis offers a way to not only mitigate the effects of CO2 emissions, but also to sustainably produce chemicals for industrial processes.10,11 However, achieving this vision requires considerable advancements in fundamental electrolyte and interface science as well as overall electrochemical systems design.To date, the electrocatalysis community has demonstrated that CO2 can be reduced to a variety of potentially useful products by judiciously tuning catalyst materials12–15 and electrolyte properties.12,16–18 The formation of CO is typically the first reduction step for the production of more complex C2+ products.19,20 However, reducing CO2 to CO requires significant energy input due to the relative stability of the CO2 double bonds,21,22 making electrocatalytic reduction of CO2 to CO an especially relevant model reaction for fundamental studies.
In most cases, CO2 reduction to CO proceeds through a two-electron, two-proton reduction process (eqn (1)), where the intermediate has been reported to be a negatively-charged radical, CO−2.21,22 The formation of this intermediate, as well as the transformation of CO2 from a nonpolar molecule to the polar CO molecule, means that this reaction is particularly sensitive to potential gradients and therefore presents a unique avenue for the study of electric double layers.
| CO2 + 2H+ + 2e− → CO + H2O | (1) |
Notably, CO2 electroreduction is carried out at high cathodic potentials, often exceeding −1 V below open circuit potential.23 These intense electrode polarizations induce the formation of nanostructured electric double layers,24–29 where ion–ion correlations and excluded volume effects play defining roles in determining local potential gradients and reaction microenvironments.30,31 Hence, electric double layer formation at electrocatalytic interfaces often hinges on collective ion self-assembly, which is not considered in classical electric double layer theories such as the Gouy–Chapman–Stern model, where ion size, ion shape, and ion–ion interactions are neglected.32–35
In this work, we aim to expand our understanding of how cation entropy influences ion assembly, electric double layer formation, and ultimately local reaction microenvironments under conditions of high applied bias that are inherent to CO2 electrocatalytic reduction. We selected ionic liquid-mediated CO2 reduction to CO on Ag electrodes as a model system, as ionic liquid molecular structures can be systematically modulated via synthetic protocols.36
Ionic liquids are salts that are often composed of weakly coordinating organic cations and anions and have been heavily studied as excellent promoters of CO2 reduction in both aqueous37–40 and non-aqueous electrolytes.30,41,42 Imidazolium-based salts are the prototypical class of ionic liquids used for mediating CO2 electroreduction due to their propensity to donate protons and coordinate CO2.37 Increasingly, however, studies are highlighting how other organic cations that cannot donate protons or form coordination interactions can also effectively promote CO2 electroreduction,43,44 leaving many open questions surrounding how and why organic cations can facilitate electrocatalytic reactions.
We hypothesize that cation entropy provides an important and underexplored element in explaining why ionic liquids can facilitate efficient CO2 electroreduction. Electric double layer formation hinges on both entropic and electrostatic interactions,45 where the large size and irregular shape of ionic liquid ions can lead to additional dielectric fluctuations and responses which can contribute to electrostatic screening and enhance interfacial potential gradients.32,46,47 An ion confined to the double layer not only experiences electrostatic interactions, but also incurs entropic penalties.48 However, the degree to which the entropic penalty influences double layer formation, and subsequently electrostatic screening, remains poorly understood.
Here, we systematically isolate the influence of entropy on electric double layer formation by comparing electric double layer properties and electrocatalytic CO2 reduction rates in two ionic liquids that are structurally identical except for a covalent linkage that forms a dimer analogue of a common monovalent ionic liquid. Specifically, we examine the role of entropy in double layer formation by comparing the interfacial behavior and reactivity of 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIm][TFSI]) and its dicationic analogue, 1,8-bis(3-methylimidazolium-1-yl)octane bis(trifluoromethanesulfonyl)imide ([BisMImO][TFSI]2) (Fig. 1).
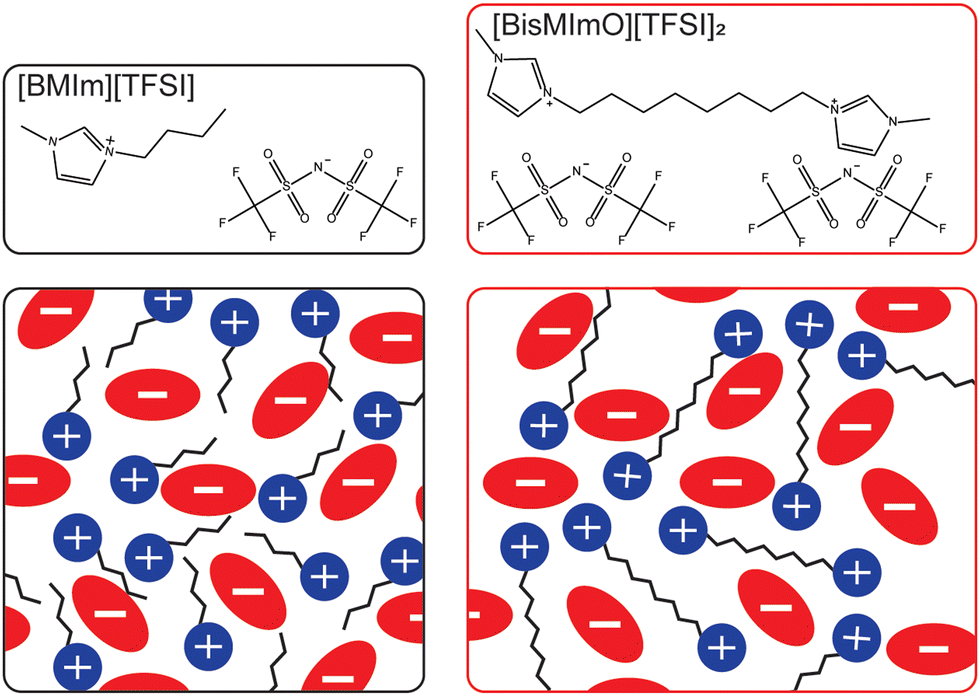 | ||
| Fig. 1 Chemical structures of the ionic liquids (top) and cartoon schematic (bottom) of the neat ionic liquids. The 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIm][TFSI], black box) ionic liquid contain monovalent cations and anions. The dicationic ionic liquid, 1,8-bis(3-methylimidazolium-1-yl)octane bis(trifluoromethanesulfonyl)imide ([BisMImO][TFSI]2, red box), consists of a divalent cation and monovalent anions. The introduction of a single covalent bond on the N3 substituents of the [BMIm]+ cation yields the [BisMImO]+ cation, which has lower entropy of confinement within the double layer. Thus, at the same total concentration of ionic liquid, the [BisMImO][TFSI]2 solution has twice the number of charges as the [BMIm][TFSI] solution. | ||
Ions confined to the interface must pay an entropic penalty due to the loss of translational motion of 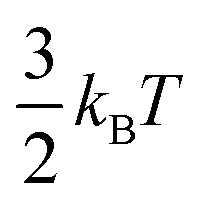 , where kB and T represent the Boltzmann constant (J/K) and temperature (K), respectively. As this entropic penalty pertains to any single molecule regardless of size or shape, [BisMImO]2+ pays an entropic penalty of
, where kB and T represent the Boltzmann constant (J/K) and temperature (K), respectively. As this entropic penalty pertains to any single molecule regardless of size or shape, [BisMImO]2+ pays an entropic penalty of  while [BMIm]+ must pay an entropic penalty of
while [BMIm]+ must pay an entropic penalty of  to confine two positive charges to the interface. Therefore, it is more thermodynamically favorable to confine the [BisMImO]2+ cation to the double layer than 2 [BMIm]+ ions for the same amount of charge screening.
to confine two positive charges to the interface. Therefore, it is more thermodynamically favorable to confine the [BisMImO]2+ cation to the double layer than 2 [BMIm]+ ions for the same amount of charge screening.
We hypothesized that salts with lower entropic penalties per charge carried, such as [BisMImO][TFSI]2, will preferentially accumulate at interfaces, generally leading to thinner and more efficiently-screening double layers. However, the effects of a thinner double layer may be tempered by the additional electrostatic interactions between the imidazolium rings of the [BisMImO]2+ cation, as well as the imposed constraint on the number of positions the imidazoliums can adopt relative to each other in [BisMImO]2+ when compared to [BMIm]+. We evaluated the electrochemical reactivity and interfacial structure of [BisMImO][TFSI]2 and [BMIm][TFSI] in acetonitrile to avoid the competing hydrogen evolution reaction49 and on Ag working electrodes for its selectivity for CO.50
Our findings demonstrate that entropic contributions to double layer formation can dominate interfacial ion assembly and govern reaction rates. Using cyclic voltammetry (CV), we show that [BisMImO][TFSI]2 generally exhibits lower current densities than [BMIm][TFSI], but that current densities begin to converge in more concentrated electrolytes. We then use chronoamperometry (CA) and product analyses such as gas chromatography (GC) to show that CO2 electroreduction in [BisMImO][TFSI]2-containing electrolytes lead to noticeable precipitation of the bicarbonate salt at the electrode surface, which compromises steady state reactivity, while [BMIm][TFSI] does not.
To better understand the structure within the double layer and how it is related to the reaction rate, we performed in situ electrochemical surface-enhanced Raman scattering (SERS) spectroscopy and found that [BisMImO][TFSI]2 more readily forms ordered double layers, especially under inert gas-sparging conditions. Furthermore, in a mixture of the two ionic liquids, we provide evidence that electrocatalytic performance and interfacial structuring are dominated by the ionic liquid with the lower entropic penalty of de-mixing from the bulk electrolyte.
Experimental methods
Chemicals
1-Butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIm][TFSI], >98% purity, Iolitec) was used as purchased.The dicationic ionic liquid, 1,8-bis(3-methylimidazolium-1-yl)octane bis(trifluoromethanesulfonyl)imide ([BisMImO][TFSI]2) was synthesized according to established literature procedures.51,52 Briefly, 1-methylimidazole (99%, Thermo Scientific) and 1,8-dibromooctane (98%, Sigma-Aldrich) were added to a round-bottom flask in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and allowed to react for 24 hours at room temperature to make 1,8-bis(3-methylimidazolium-1-yl)octane dibromide ([BisMImO][Br]2). The [BisMImO][Br]2 was then washed with acetone and dried, resulting in a dry and white powder.
:1 molar ratio and allowed to react for 24 hours at room temperature to make 1,8-bis(3-methylimidazolium-1-yl)octane dibromide ([BisMImO][Br]2). The [BisMImO][Br]2 was then washed with acetone and dried, resulting in a dry and white powder.
To conduct the anion exchange, [BisMImO][Br]2 and [Li][TFSI] (>98%, TCI) in a 1:2 molar ratio were added to a round bottom flask and fully dissolved in MilliQ H2O. The mixture was stirred at room temperature for 3 hours. The water layer was decanted off and the resulting ionic liquid was washed with MilliQ H2O. After several washes, the water layer was tested with a silver nitrate halide test to confirm the removal of halides.
The crude [BisMImO][TFSI]2 was then purified by first dissolving it in acetonitrile and then stirring it with activated charcoal (Sigma-Aldrich) for 2–3 days. After filtering out the activated charcoal, the solution was dried using a rotary evaporator (BUCHI R-300M Rotavapor) followed by a vacuum drying oven (Yamato ADP200C) at 80 °C for 2 days. The resulting [BisMImO][TFSI]2 was a viscous and colorless oil that slowly began to solidify upon storage. A more detailed method is presented in ESI.†
The water content of HPLC grade acetonitrile (>99.9%, Sigma-Aldrich) was adjusted to be 900–1000 ppm according to our previous protocol to ensure that protons were available for the reaction.30 Potassium bromide was purchased from MP Biomedicals and silver nitrate was from Alfa Aesar.
Electrochemical methods
A VSP potentiostat from BioLogic was used for all electrochemical experiments. In electrochemical measurements, a Ag/Ag+ non-aqueous reference electrode containing 0.1 M [TBA][BF4] and 0.01 M [Ag][NO3] in acetonitrile was prepared,53 and a coiled platinum wire was used as the counter electrode.For cyclic voltammetry and steady-state electrolysis, we used a 3 mm polycrystalline Ag electrode (geometric surface area of 0.071 cm2) purchased from BASi as the working electrode, which was polished to a mirror finish using 15 μm, 3 μm, and 1 μm diamond polish followed by 0.05 μm alumina (BASi) before ultrasonication in water. All electrolytes were purged with either Ar or CO2 prior to electrochemical measurements. CV measurements were conducted in one-compartment borosilicate electrochemical cells with a 3D-printed cap to ensure consistent electrode placement. All presented CV measurements were carried out at a scan rate of 100 mV s−1 and the results are presented in their raw form, as IR-corrections do not change the conclusions of this study, since the current passed is low enough to negate the minor changes in solution resistance that occur as the ion concentration is altered in the reported electrolytes (Tables S1, S2 and Fig. S1, S2, ESI†).
Product analysis
CA measurements were conducted in a custom two-compartment H-cell separated by a Nafion 117 membrane. The Nafion 117 membranes were activated according to prior established protocols54,55 and stored in a solution of 0.1 M [TBA][BF4] in acetonitrile. They were rinsed thoroughly with acetonitrile prior to use. Potentials of −2.5 V vs. Ag/Ag+ were held for 1 hour, where steady state was typically reached after 10 minutes. Ar-sparged controls were conducted to assess the background current density (Fig. S3, ESI†) and to ensure that the ionic liquid did not decompose at these applied potentials (Fig. S4 and S5, ESI†).The cells were continuously stirred with Teflon stir bars (VWR) and CO2 was continuously purged through the cathode compartment at 10 standard cubic centimeters per minute (sccm). The resulting gas stream outlet fed directly into a gas chromatograph (Multiple Gas Analyzer #5, SRI Instruments), which was equipped with a flame-ionization detector (FID) coupled with a methanizer and a thermal conductivity detector (TCD). The FID was connected with a HayeSep D and a Molesieve 5A column (Restek) while the TCD was connected to a HayeSep D column. The carrier gas was ultrahigh purity grade Ar (Airgas).
Product analysis was conducted every 6 minutes and faradaic efficiencies were calculated using the following equation:56
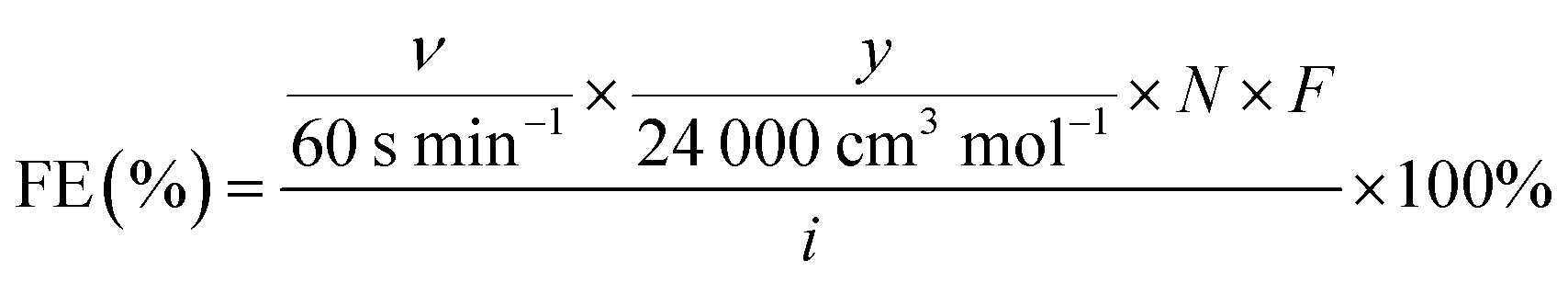 | (2) |
500 C mol−1 is the Faraday constant, and i is the total current passing through the working electrode in the unit of ampere. Reported steady-state results and standard deviations were calculated by averaging current densities and faradaic efficiencies after 8 minutes of applying the potential.
We also used NMR spectroscopy to identify products in the catholyte after electrolysis. The NMR (400 MHz, Bruker Avance) was equipped with a BBFO probe.
SERS measurements
Electrochemical surface-enhanced Raman scattering (SERS) spectroscopy measurements were carried out in a single-compartment PTFE electrochemical cell. A polycrystalline L-shaped Ag working electrode (Shanghai Fanyue Electronic Technology Co. Ltd) was first polished and then electrochemically roughened following reported methods in literature with modifications.57Briefly, the electrode was submerged in an aqueous solution of 0.1 M [K][Br] under continuous Ar purging. The electrode was then cycled between −0.3 V and +0.3 V vs. open-circuit potential (OCP) for 4 cycles. The electrode was then held at −0.3 V vs. OCP for 10 seconds to desorb bromide from the silver surface. The electrode was thoroughly rinsed with water after roughening and dried prior to use. A mechanically polished and coiled Pt wire was the counter electrode and a non-aqueous Ag/Ag+ (0.01 M [Ag][NO3]) reference electrode was used.
The electrolytes were purged with either Ar or CO2 for 20 minutes at 10 sccm prior to measurement. SERS spectra were collected using a Thermo-Fisher Scientific DXRxi Raman Imaging Microscope with 532 nm excitation laser at 10× magnification. The laser power was 8.0 mW, the acquisition time was 0.125 seconds, and 25 scans were collected for each spectrum. The potential was swept from −0.5 V to −2.5 V vs. Ag/Ag+ and then back to −0.7 V vs. Ag/Ag+ at intervals of 0.2 V. Only the forward scans are reported in the main text as the trends were reversible.
Results and discussion
Cation entropy influences reaction rate and selectivity
Using a polycrystalline Ag working electrode, we systematically conducted CV scans on a series of ionic liquid concentrations in acetonitrile under continuous CO2 sparging. The current density at −2.5 V vs. Ag/Ag+ was used for comparison among different concentrations.We found that the [BisMImO][TFSI]2 electrolytes display a maximum current density at similar salt concentrations as [BMIm][TFSI] but that the current densities of [BisMImO][TFSI]2-containing electrolytes are lower than those containing [BMIm][TFSI] at all concentrations (Fig. 2(A)). More interestingly, after accounting for the total number of charge-carrying imidazoliums in each cation, we observe that the discrepancies in current densities between the two ionic liquids are larger at lower concentrations (<1 M) but converge to similar current densities as concentration increases (Fig. 2(B)).
 | ||
| Fig. 2 Electrochemical behavior of [BisMImO][TFSI]2 and [BMIm][TFSI]. CV experiments show that [BisMImO][TFSI]2–acetonitrile electrolytes (red) always yield lower current densities than [BMIm][TFSI] (black). (A) Accounting for total ionic liquid concentration, we find that [BisMImO][TFSI]2 electrolytes reach a maximum current density at a lower concentration than [BMIm][TFSI]. (B) If total imidazolium concentrations are considered, however, we find that current densities begin to converge at higher concentrations. Datapoints and standard deviations were calculated by averaging the current density at −2.5 V vs. Ag/Ag+ for 2 cycles of CV at 100 mV s−1 for 3 separate electrolyte samples at each concentration. | ||
We attribute the discrepancy in CO2 reduction rates at lower imidazolium concentrations to the reorientation of the cations in the double layer and their corresponding role in the reaction. The imidazolium cation is thought to stabilize the CO−2 intermediate through either complexing with the C2 position,37,58 or through hydrogen bonding between CO−2 and the C4 and C5 protons.42,59 The covalent link between two imidazolium rings in the [BisMImO]2+ cation serves to further constrain the imidazolium ring orientation relative to the CO−2 intermediate. As such, the imidazoliums in [BisMImO]2+ have fewer degrees of freedom and hence fewer stabilizing conformations compared to the [BMIm]+ cation. Additionally, at low concentrations, the [BMIm]+ cations remain fully solvated and “free” while the imidazoliums in the [BisMImO]2+ cations experience enhanced interactions due to the covalent linkage, which could promote the formation of electrode-blocking precipitates containing (bi)carbonate anions. The imposed constraint and corresponding entropic limitations are likely the reason behind the attenuated reactivity of the [BisMImO][TFSI]2 at concentrations lower than 1 M of imidazolium cation.
As concentration increases past 1 M, however, the emergence of bulk ion–ion correlations and the formation of multi-ion aggregates makes screening less efficient than the predictions of classical double layer theory.60 At these concentrations, ion–ion interactions between [BMIm]+ cations also decrease screening efficiency such that correlations within [BMIm]+ electrolytes become similar to those in [BisMImO]2+ electrolytes. As a result, increasing concentration towards the neat limit yields converging current densities for the two ionic liquids as double layer screening efficiency decreases and the double layer thickness increases.
We then conducted chronoamperometry (CA) studies combined with product analysis techniques to evaluate the two ionic liquids for steady-state electrolysis performance. CA experiments were conducted using polycrystalline Ag working electrodes in a well-mixed H-cell with continuous CO2 sparging at 10 sccm. The outlet gas was directed into the sample loop of a gas chromatograph to identify gaseous products.
We notice that within 10–15 minutes of applying a potential of −2.5 V vs. Ag/Ag+, the current density and faradaic efficiency of CO2 conversion to CO for all [BisMImO][TFSI]2 electrolytes drop and remain at a low level for the remainder of the experiment (Fig. 3(A)). In contrast, the [BMIm][TFSI] electrolytes maintain a relatively constant current density and faradaic efficiency (Fig. 3(B)). Upon inspection of the working electrode after electrolysis with the dicationic analogue, we observed that a solid precipitate had formed on the surface and completely occluded the working electrode. This precipitate was not observed after electrolysis in [BMIm][TFSI], suggesting that covalently joining the two imidazoliums together has the effect of inducing precipitate formation.
 | ||
| Fig. 3 Representative CA and GC results show that within a few minutes of applying a −2.5 V vs. Ag/Ag+ potential, the current density and faradaic efficiencies drop for 125 mM [BisMImO][TFSI]2 (A) but remain relatively stable for 250 mM [BMIm][TFSI] (B). This is due to the formation of (bi)carbonate species at the interface, leading to the growth of a solid precipitate. | ||
Next, we performed CA at incremented potentials following the reaction onset (Fig. S7, ESI†) and observed this precipitate formation at potentials near the onset potential of CO2 reduction but did not observe the precipitate under Ar-sparged conditions (Fig. S8, ESI†). Further, even low concentrations (0.0625 M) of [BisMImO][TFSI]2 displayed this behavior, as a decrease in current density was observed even between different cycles of CV (Fig. S9, ESI†).
The formation of the obstructing precipitate on the electrode under CO2-sparged and its absence under Ar-sparged conditions indicates that the formation of byproducts during CO2 reduction is the likely cause of the precipitate. Indeed, (bi)carbonate formation and subsequent precipitation of (bi)carbonate salts (Fig. S10, ESI†), which is caused by the reaction between interface-generated [OH]− and dissolved CO2, is a common issue in CO2 electrocatalysis.10,61 The precipitate that is formed lowers the current density and the subsequent concentration of gaseous products, which can approach or fall below the detection limits of our instrumentation and makes rigorous analysis of product distributions challenging. Analysis of the carbonate precipitate suggests that the precipitate can trap products like formate at the electrode–electrolyte interface, which can further decrease the faradaic efficiency for CO (Fig. S11, ESI†). Carbonate salt precipitate formation is also observed in inorganic electrolytes and remains a common source of diminishing reactor performance in steady state CO2 electrolysis.62,63 Our observations are also consistent with our prior work,30,31 where we previously reported the formation of a solid precipitate in electrolytes with 1-ethyl-2,3-dimethylimidazolium tetrafluoroborate, in which the bicarbonate anion paired with the imidazolium cation was insoluble in acetonitrile.
Hence, simply joining two imidazolium rings together dramatically alters the steady-state electrocatalytic behavior. It is difficult to overstate the importance of ion molecular structure and how it impacts both double layer formation and reactivity. Prior works have demonstrated significant cation effects in both aqueous64 and non-aqueous systems,65 which often focus on how changing the cation structure or size can enhance local concentration gradients66 or the availability of protons.67 Here, we show that cation entropy can have similarly drastic effects on CO2 electroreduction by preserving the inherent structure of the imidazolium cation but introducing an entropic constraint. To further investigate the double layer that is formed when the cation is entropically-constrained, we used surface-enhanced Raman scattering (SERS) spectroscopy to examine the structure of the double layer at various applied potentials.
Electrochemical reactions disrupt interfacial ion organization
We collected in situ SERS spectra at low and intermediate concentrations of the two ionic liquids to probe the structure of the double layer. Our experiments were conducted in one-compartment SERS cells with surface-roughened Ag working electrodes.57 All solutions were sparged with either Ar or CO2 for 20 minutes prior to data collection. Our group has previously used this method to interrogate the orientation of the imidazolium ring in the double layer as cathodic potentials were applied.30,31In the alkyl substituent region, we observed the rise of a peak at 1003 cm−1, which is particularly pronounced in [BisMImO][TFSI]2 electrolytes and is observed primarily in Ar-sparged conditions (Fig. 4). Upon the reverse potential scan in Ar-sparged electrolytes, the intensity of the 1003 cm−1 peak diminishes. Interestingly, we found that there was a marked difference in the growth of the 1003 cm−1 peak when the electrolyte had been sparged with Ar compared to CO2 (Fig. 4). Additionally, while we do observe peaks that may be assigned to Raman-active modes of carbonate, these peaks overlap with those associated with the alkyl chain region of the cation (Fig. S12, ESI†) and definitive assignment of these peaks in CO2-sparged solutions is therefore challenging.
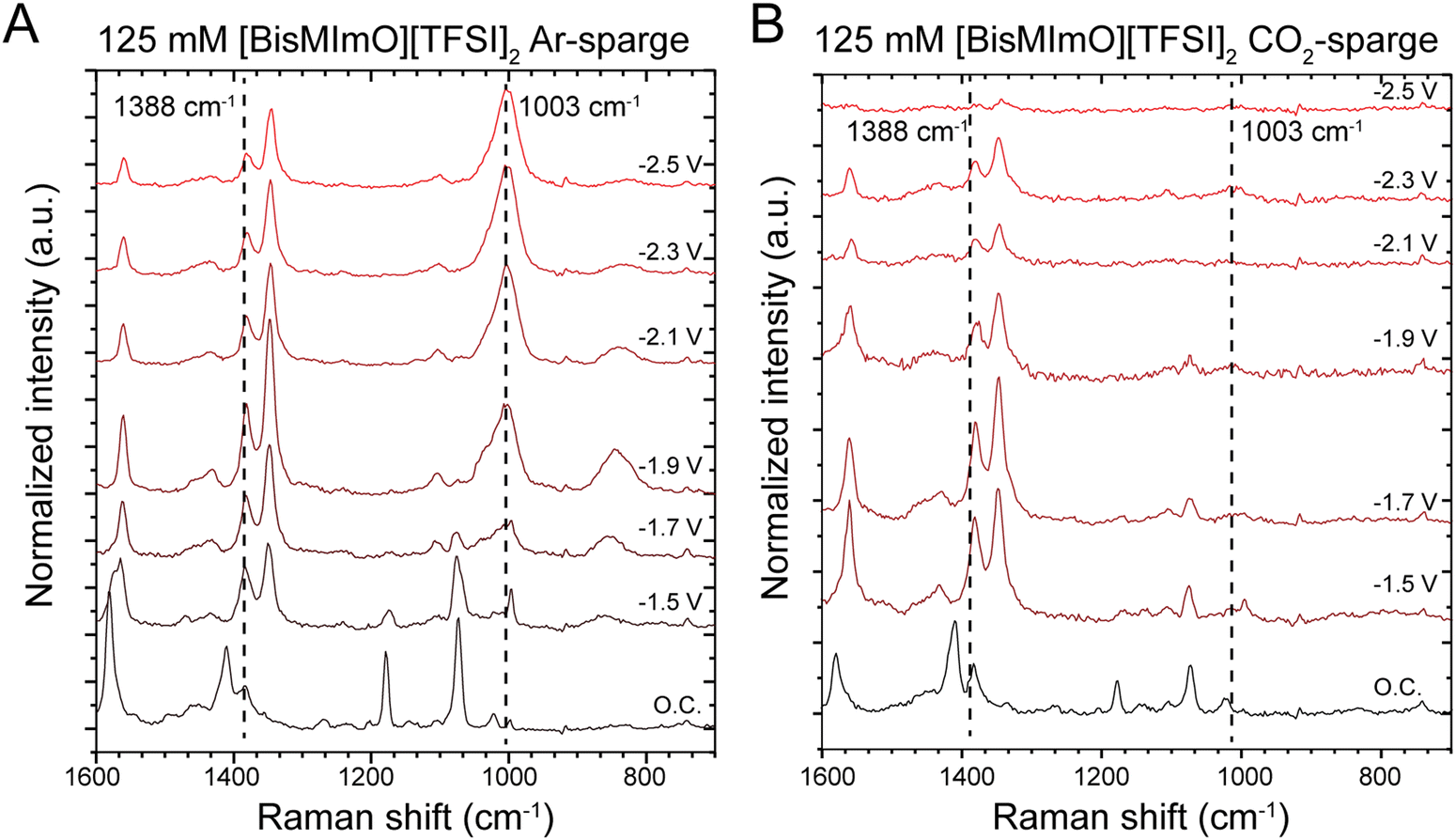 | ||
| Fig. 4 In situ SERS spectra of 125 mM [BisMImO][TFSI]2 under Ar- and CO2-sparged conditions highlight the differences in double layer structure in a capacitive and reactive double layer, respectively. In the presence of Ar (A), the rise of a peak ca. 1003 cm−1 associated with the alkyl chain substituents that appears at −1.7 V demonstrates potential-dependent restructuring of the alkyl domains of the cation. When CO2 is present (B), however, the 1003 cm−1 peak is not observed. We hypothesize that this could be due to the transport of reactants and products through the double layer and the formation of negatively charged byproducts that disrupt double layer ordering. The formation of a precipitate at the surface at −2.5 V vs. Ag/Ag+ hinders Raman spectra collection under CO2-sparged conditions. | ||
To analyze the growth of the 1003 cm−1 peak, we took the ratio of the peak intensity of the 1003 cm−1 peak relative to the peak intensity of the 1388 cm−1 peak, which is associated with the stretching mode of the imidazolium ring structure and remains largely unchanged with applied potential. We find that the peak intensity of the 1003 cm−1 peak surpasses that of the 1388 cm−1 peak at −1.9 V vs. Ag/Ag+ (Fig. S13, ESI†). After reaching this potential, the intensity of the 1003 cm−1 peak becomes several times larger than the 1388 cm−1 peak in Ar-sparged solutions. A similar trend is observed in [BMIm][TFSI] solutions, but the growth of the 1003 cm−1 peak in Ar-sparged solutions is much more subdued (Fig. S14, ESI†).
The 1003 cm−1 peak is associated with the alkyl substituents on the imidazolium and has been previously shown to correspond to C–C stretching.68,69 The pronounced 1003 cm−1 peaks likely arise from interfacial segregation of the alkyl chains from the imidazolium ring structures given that the alkyl chains, especially for [BisMImO]2+, are less polar than the positively charged imidazolium ring. Additionally, the formation of segregated alkyl chain domains have previously been observed in monovalent imidazolium ionic liquid systems.28,70 There was also a greater enhancement of the 1003 cm−1 signal in [BisMImO]2+, suggesting that the connecting alkyl chain imparts more organization to the double layer. Notably, the peak at 1003 cm−1 disappeared altogether after −1.9 V vs. Ag/Ag+ under CO2-sparged conditions, which is approximately the onset potential for CO2 electroreduction.
The CO2 reduction process requires the transport of molecules across the double layer and forms primarily CO but can also generate unwanted byproducts such as [OH]− (eqn (3)) and, by recombination of the [OH]− with CO2, (bi)carbonate.31,61
| CO2 + H2O + 2e− → CO + 2OH− | (3) |
Transporting CO2 towards the electrode and transporting products and byproducts away from the electrode can disrupt the structure that would have formed in the absence of a reaction in the double layer. Furthermore, the formation of negatively charged products like the CO−2 intermediate or (bi)carbonates can electrostatically balance the charge of the cations already in the double layer, therefore negating the screening effect of the cation. As a result, more cations are recruited to the double layer as the neutral clusters of ions diffuse away from the surface, thus creating a more dynamic and disrupted double layer structure.
We find that the increase in the intensity of the 1003 cm−1 peak under Ar-sparged conditions was also observed in N2-sparged electrolytes (Fig. S15, ESI†). Combined with the CO2-sparged results, this suggests that the double layer under inert conditions is fundamentally different from double layer under reactive conditions, in which the transport of reactants and (by)products creates a more dynamic interface. This finding has far-reaching implications, the most significant of which is that double layer models developed for inert conditions, or capacitive double layers, are not directly applicable to dynamic reactive double layers.
Cation reorientation occurs at similar potentials for mono- and dicationic imidazolium species
Imidazolium rings have been shown to adopt an orientation parallel to the electrode surface at high cathodic potentials, leading to enhanced cation packing and therefore more efficient screening.30,71 To determine if concentration had a significant impact on cation orientation when CO2 was present, we collected SERS data for low concentrations (0.125 M [BisMImO][TFSI]2 and 0.250 M [BMIm][TFSI]) and concentrated electrolytes (0.825 M [BisMImO][TFSI]2 and 1.65 M [BMIm][TFSI]). These concentrations were chosen to keep the total number of imidazoliums consistent between the two ionic liquids.The imidazolium ring region of 1344 to 1580 cm−1 can be used to qualitatively understand the reorientation of the imidazolium ring due to the development of an electric field as a potential is applied to the electrode. In SERS spectroscopy, the vibration modes with tensors perpendicular to the electrode plane are enhanced so trends in signal intensities can be used to gauge the strength of the Raman mode. We observed the red-shifting of the 1580 cm−1 peak to 1561 cm−1, which is indicative of increased proximity between the C4 and C5 protons with the surface72–74 (Fig. 5). We also observed a decrease in the intensity of the 1415 cm−1 peak, which is associated with the in-plane stretching mode of the imidazolium,74,75 and a corresponding increase in the intensity of the 1344 cm−1 peak, associated with CH2(N) stretching,74–76 as the potential becomes more cathodic for both [BisMImO][TFSI]2 and [BMIm][TFSI] electrolytes. Taken together, these trends in ring Raman modes as a function of applied potential suggests that the imidazolium ring begins to adopt an orientation parallel to the electrode surface as applied potential increases,73,75,76 likely with the alkyl substituents roughly also parallel to the electrode.
 | ||
| Fig. 5 In situ SERS spectra of 0.125 M [BisMImO][TFSI]2 (A) and 0.250 M [BMIm][TFSI] (B) under CO2-sparged conditions. The potential at which the 1344 cm−1 peak intensity surpasses the 1415 cm−1 peak is bolded. The relative intensities of the 1344 and 1415 cm−1 peaks are used to determine the extent of imidazolium reorientation (C). The normalized peak intensity of the 1344 cm−1 peak surpasses that of the 1415 cm−1 peak at −1.1 V vs. Ag/Ag+ for both samples. Comparing the ratios of the peak intensities of the 1344 to 1415 cm−1 peaks, we find that the 1.65 M [BMIm][TFSI] inversion potential is more negative than the other ionic liquid concentrations. The inversion potential for the 0.825 M [BisMImO][TFSI]2 remains similar to the lower concentration electrolytes, which suggests that the covalent link provides a “pre-structuring” effect between the two imidazoliums of the [BisMImO]+ cation. | ||
We used the ratio of the intensity of the 1344 and 1415 cm−1 peaks as a metric for assessing the strength of the electric field, as stronger electric fields can reorient an imidazolium ring at less cathodic potentials than weaker electric fields (Fig. 5(B)). Although we observe the same general trend of diminishing 1415 cm−1 peak intensities between the two ionic liquids, the potentials at which the ratio of the peak intensities become greater than 1 are quite different at various concentrations.
For example, the intensity inversion for 0.125 M [BisMImO][TFSI]2 and 0.250 M [BMIm][TFSI] are very similar and occur at approximately −1.0 V vs. Ag/Ag+. At higher concentrations, however, we observed a delayed inversion potential of −1.6 V vs. Ag/Ag+ for the 1.65 M [BMIm][TFSI] electrolyte. The inversion potential for 0.825 M [BisMImO][TFSI]2 occurs at approximately −1.0 V vs. Ag/Ag+, indicating little difference between the low and intermediate concentrations of [BisMImO][TFSI]2.
The imidazolium ring reorientation inversion potential allows us to gauge how bulk ion–ion correlations impact double layer formation. For the lower concentrations of [BisMImO][TFSI]2 and [BMIm][TFSI], the intensity inversion occurs at similar potentials, as ion–ion correlations have not yet begun to dominate. In the 1.65 M [BMIm][TFSI] electrolyte, we observed a delayed inversion potential, which can be explained by ion–ion interactions, as well as the formation of clusters and aggregates in the bulk electrolyte that decrease screening efficiency and thus lower the strength of the interfacial electric field. Although there are the same number of charges, 0.825 M [BisMImO][TFSI]2 did not have such a delay and the potential of peak intensity inversion was more similar to lower concentration electrolytes, indicating a stronger electric field in 0.825 M [BisMImO][TFSI]2 than 1.65 M [BMIm][TFSI].
One explanation for this is that the linkage between the two imidazoliums in [BisMImO]2+ can help with the potential-dependent reorientation through a decrease in accessible orientations of imidazolium rings relative to [BMIm]+, hence imparting a “pre-structure” to the double layer. The ratio of the 1344 cm−1 and 1415 cm−1 peaks, however, was smaller than that of the lower concentrations especially at potentials more cathodic than −1.5 V vs. Ag/Ag+. Thus, the SERS results indicate that the extent of this reorientation is smaller in more concentrated electrolytes and is likely hindered by ion–ion interactions.
Divalent cations are surface active
We also aimed to understand how lower entropic penalties impacted ion availability in the double layer using mixtures of [BisMImO][TFSI]2 and [BMIm][TFSI]. Additionally, we expected that mixing ionic liquids would help mitigate the precipitation issue, as having multiple types of cations present could disrupt the ion packing needed to make the solid (bi)carbonate salt. We evaluated the mixtures of the two ionic liquids using CA, GC, and SERS spectroscopy. For simplicity, we kept the total ionic liquid concentration at 125 mM.First, we find that by adding 5 mM [BisMImO][TFSI]2 to 120 mM [BMIm][TFSI], the steady-state current density drops to 25% of the current density observed for the 125 mM [BMIm][TFSI] electrolyte. As the ratio of [BisMImO][TFSI]2 increases, the steady state current density decreases until it becomes smaller than −2.5 mA cm−2 in a mixture of 25 mM [BisMImO][TFSI]2 and 100 mM [BMIm][TFSI]. Notably, faradaic efficiency for CO decreases gradually until it reaches approximately 50% at 125 mM [BisMImO][TFSI]2 (Fig. 6).
 | ||
| Fig. 6 Results of mixed electrolytes demonstrate the preferential accumulation of the [BisMImO]2+ cation. Steady state electrolysis (A), where current density (black) and faradaic efficiency for CO (red) in mixtures of [BisMImO][TFSI]2 and [BMIm][TFSI] show that the addition of 5 mM [BisMImO][TFSI]2 to 120 mM [BMIm][TFSI] results in a decrease in current density and the formation of an occluding phase on the electrode surface. The faradaic efficiency for CO decreases steadily with increasing concentration of [BisMImO][TFSI]2. SERS spectra of 10 mM [BisMImO][TFSI]2 and 115 mM [BMIm][TFSI] under Ar-sparged conditions (B) reveal the presence and growth of the 1003 cm−1 peak that was observed in [BisMImO][TFSI]2 electrolytes. Due to the drop in current density and the formation of an occluding phase on the electrode surface, our interpretation is that the [BisMImO]2+ cation accumulates within the double layer and thus has an outsized effect on reactivity. | ||
We observed a pronounced decrease in current density with the addition of relatively small amounts of [BisMImO][TFSI]2 as well as precipitate formation, which together indicates that a distinct [BisMImO]-enriched phase forms on the electrode surface. Interestingly, the potential applied to the electrode surface dictated the physical appearance of the precipitate in solutions containing only [BisMImO][TFSI]2. For instance, a solid white crystal nucleated and propagated on the surface when a potential of −2.5 V vs. Ag/Ag+ was held under CO2-sparged conditions (Fig. S3, ESI†). If the potential was quickly stepped to −2.7 V vs. Ag/Ag+ and held, however, a dynamic precipitate formation process was observed that occurred in two steps. Evidence of liquid–liquid phase separation was observed as islands of a liquid phase formed on the electrode surface and began to coalesce. Within these liquid droplets, solid precipitates began to form, ultimately resulting in the formation of an orange gel-like film spanning the entire electrode surface, likely composed of the (bi)carbonate salt and decomposed imidazolium (Fig. S16, Supplemental Video, ESI†). These same trends were observed in mixtures of [BisMImO][TFSI]2 and [BMIm][TFSI].
Hence, we find that decreasing the entropic penalty of constraining cations within electric double layers appears to amplify driving forces for electrochemically induced phase separation. Electrochemically induced phase separation has been previously demonstrated by Leibler and coworkers to be possible in binary mixtures of silicone oil and paraffin oil,77 which was followed up with a theoretical model for electric field gradient-induced phase separation in ion-containing mixtures.78 In experiments by Lahiri and coworkers, the mechanism for potential-dependent phase separation in ionic liquid-containing electrolytes was through the formation of unstable bonds, resulting in the eventual decomposition of the electrolyte into two phases.79
Even more interestingly, electrolytes containing only 10 mM [BisMImO][TFSI]2 (with 115 mM [BMIm][TFSI]) formed a clear liquid phase on the electrode surface that was distinguishable from the bulk electrolyte under CO2-sparged conditions but not in Ar-sparged conditions. We hypothesize that this is due to the formation of the [BisMImO]2+ (bi)carbonate salt, where the concentration of the salt is not sufficient to induce precipitation. Additionally, the presence of [BMIm]+ could disrupt the formation of a solid crystal since the (bi)carbonate salt of [BMIm]+ does not precipitate out of solution, leading to the metastable liquid–liquid phase separation process.
To the best of our knowledge, liquid–liquid phase separation due to the active electrocatalytic processes in ionic liquid electrolytes has not been reported in literature. This separate liquid phase was fluid and moved when the cell was lightly disturbed before reverting to its original state (Supplemental Video, ESI†). A white crystal began to nucleate and grow after more than 30 minutes of applied potential of −2.7 V vs. Ag/Ag+. Further increases in the ratio of [BisMImO][TFSI]2 to [BMIm][TFSI] in the electrolyte mixtures began to yield precipitates that were indistinguishable from that of electrolytes containing only [BisMImO][TFSI]2. Hence, we conclude that the formation of solid precipitates in these mixed electrolytes appears to pass through a non-classical multistep nucleation and growth process, as explained by De Yoreo and colleagues for carbonate precipitation in biological systems.80
The low concentration required to observe this phase separation indicates that the [BisMImO][TFSI]2 preferentially accumulates in the double layer but crystal formation is suppressed or delayed due to the presence of [BMIm]+ cations with lower propensity to form solid (bi)carbonate crystals. Although the full mechanism is the subject of ongoing work, the potential-dependent liquid–liquid phase separation we observe in these mixed ionic liquids appears to have intriguing connections to the process of coacervate formation in biology, where changes in pH and ionic strength drive formation of distinct liquid phases.80 We therefore expect that the interface would contain higher concentrations of [BisMImO][TFSI]2 than the bulk.
To investigate the composition of the double layer, we collected SERS spectra of the electrolyte mixtures, focusing on the electrolyte compositions with low ratios of [BisMImO][TFSI]2:[BMIm][TFSI]. Given that even mixtures of low concentrations of [BisMImO][TFSI]2 exhibited electrochemical behavior more like that of [BisMImO][TFSI]2-only electrolytes, we expected that the SERS spectra of the interface would reflect an enhanced abundance of interfacial [BisMImO][TFSI]2 in these mixed electrolytes.
Under Ar-sparged conditions, we indeed found that the electrolytes containing small amounts of the [BisMImO][TFSI]2 behaved more like the electrolyte containing primarily [BisMImO][TFSI]2. In short, the 1003 cm−1 peak outgrew the 1388 cm−1 peak in intensity in all solutions containing [BisMImO][TFSI]2 regardless of whether [BMIm][TFSI] was also present (Fig. S17, ESI†). We observed the appearance of the 1003 cm−1 peak under Ar-sparged conditions in a mixture of 10 mM [BisMImO][TFSI]2 and 115 mM [BMIm][TFSI] (Fig. 6(B)). This suggests there is organization of the alkyl chain domains of the cations and an enhanced presence of the [BisMImO]+ cation at the interface. In other mixtures, the ratio of the peak intensities in the mixed electrolytes began to approach that of the 125 mM [BisMImO][TFSI]2 solution as the concentration of [BisMImO][TFSI]2 increased (Fig. S18, ESI†), suggesting that the [BisMImO]2+ concentrates within the double layer. Otherwise, the low ratio of [BisMImO][TFSI]2 to [BMIm][TFSI] should yield SERS spectra more like an electrolyte containing only [BMIm][TFSI].
These findings, combined with the electrolysis data, point to the preferential accumulation of the lower entropic penalty ionic liquid, [BisMImO][TFSI]2, at the electrode/electrolyte interface. In short, the electrochemical behavior of a mixed electrolyte is dictated by the salt with the lowest entropic penalty for double layer formation (Fig. 7). Intriguingly, our results on entropic considerations of double layer formation may also help explain observations made in aqueous studies of mixed alkali metal salts, where the reactivity of mixed electrolytes containing mixtures of Li+ and larger alkali metal cations is dominated by the reactivity of the larger alkali metal cation,81 or the enhancement of CO2 electroreduction in the presence of multivalent ions.61
 | ||
| Fig. 7 Ionic liquids with higher entropic penalties of confinement within the double layer form fluid interfaces (left column) while those with lower entropic penalties preferentially accumulate at the interface, whichcan lead to electrode-blocking precipitation formation (right column). Mixtures of ionic liquids exhibit electrocatalytic behavior that depend on the ion with the lower entropic penalty and can yield liquid–liquid phase separated interfaces (middle column). | ||
Cation entropy governs interfacial reactivity
Our findings comparing [BisMImO][TFSI]2 and [BMIm][TFSI] highlight the importance of cation entropy in dictating interfacial electroreduction in a manner rivaling that of changing the overall cation structure. Although the core imidazolium structure remains constant, imposing an entropic constraint through the covalent bond leads to decreases in CO2 reduction rates due to the formation of a precipitate layer on the electrode surface. By tethering two [BMIm]+ ions together to make the [BisMImO]2+ ion, the primary change is in the orientational degrees of freedom of the cation, as the basic [BMIm]+ structure is preserved.When confined within the double layer, the [BMIm]+ ion has more orientational degrees of freedom. The imidazoliums in the [BisMImO]2+ ion, on the other hand, have a reduced number of available orientations available relative to one another. The covalent bond in [BisMImO]2+ also serves to impose an additional layer of ion correlations, resulting in smaller current densities at lower concentrations due to the formation of precipitates. These effects become less pronounced at high concentrations, however, as the emergence of ion–ion correlations in [BMIm]+ attenuates the difference between CO2 reduction in [BisMImO]2+ and [BMIm]+, leading to converging reaction rates in the highly concentrated ionic liquid limit.
Analysis of SERS results in [BisMImO][TFSI]2 electrolytes also point towards a fundamental difference in the structure of the double layer under inert or reactive conditions. We found that under inert conditions, the long alkyl chains of the [BisMImO]2+ formed segregated domains or local nanostructures. When CO2 is present, however, the double layer structure becomes disrupted. This can be attributed to the dynamic nature of a reactive double layer, in which the transport of reactants and products disturbs the interfacial structure, and formation of charged byproducts disrupts the local electric field. This difference in double layer structure under two different gas sparging conditions highlights the complex nature of double layers and suggests that conventional models of double layers, which assume no faradaic reactions, may not be directly applicable to reactive double layers.
In [BisMImO][TFSI]2 electrolytes, we observed solid precipitate formation, which passivated the electrode surface and prevented further CO2 electroreduction from occurring. These phase separations only occurred under CO2-sparged conditions, suggesting that byproducts of the CO2 reduction reaction facilitate the phase separation. Additionally, the overall appearance of the solid precipitate differs at different applied potentials, indicating that the presence of other electrochemical products, such as those made by the deactivation of imidazolium, can also modify for surface passivation processes.
The inclusion of an ion like [BMIm]+ in mixed electrolytes disrupted the solid [BisMImO]–(bi)carbonate precipitate crystal structure, thus leading to the liquid–liquid phase separation observed in the 10 mM [BisMImO][TFSI]2 115 mM [BMIm][TFSI] solutions. Precipitate formation poses a serious hurdle in the adoption of CO2 electrolysis cells, as formation of solid (bi)carbonate salts creates a passivating layer on the electrode surface, ultimately leading to electrolytic cell failure.10,61 Our mixed electrolyte experiments show that this precipitation process occurs via a multistep mechanism, in which liquid–liquid phase separation first occurs and is followed by precipitate nucleation. Multistep nucleation processes have been demonstrated to have much lower energy barriers than classical nucleation,80 which may explain the prevalence of the precipitate problem and also offer methods for prevention. We have shown that mixing two salts can have a stabilizing effect on the metastable liquid formed at the electrode surface, but higher compositional complexity may be a more suitable approach for completely suppressing nucleation.
Conclusion
We compared a dicationic ionic liquid, [BisMImO][TFSI]2, with its monovalent counterpart [BMIm][TFSI] to investigate the role of entropy in double layer formation. We found that differences in cation entropy can fundamentally alter reaction rates and selectivity during steady-state electrolysis. SERS spectroscopy results show a pronounced difference between double layers under reactive and non-reactive conditions for both the monocationic and dicationic species. We also found that when CO2 was present, the reorientation of the imidazolium rings of [BisMImO]2+ and [BMIm]+ were similar at low concentrations but had more pronounced differences at higher concentrations. In mixtures of the two ionic liquids, we concluded that the cation that pays the lowest entropic penalty accumulates within the double layer and dictates reactivity.Our findings highlight the key role entropy plays in determining interfacial potential gradients and electrochemical reactivity. We also demonstrate that entropy–reactivity relationships offer a new approach to tuning electrocatalytic reactions, in which the design of an electrochemical system must account for both ion entropy of confinement to the double layer, as well as the ability of ions to reorient at the interface. Double layer models must take both entropic considerations and reactive conditions into account to understand the dynamic nature of the reactions occurring in double layers. Ions at the interface can facilitate or hinder a reaction, as evidenced by the onset of liquid–liquid phase separation followed by solid precipitate formation in [BisMImO][TFSI]2 electrolytes.
Our observation of multi-step nucleation processes of solid precipitates in mixed electrolytes also warrants reexamination of electrochemically induced phase separation and its pertinence for electrolyte engineering. Multistep nucleation mechanisms, akin to those described by De Yoreo and coworkers for biological carbonate precipitation,80 have much lower energy barriers than classical nucleation. It is possible that (bi)carbonate salts of [BMIm]+ undergo classical nucleation with high energy barriers, while [BisMImO]2+ salts tend to undergo multi-step nucleation pathways and are therefore more prone to precipitate formation. Thus, understanding how entropy governs double layer formation can offer fundamentally new ways to leverage entropy as an additional parameter for double layer engineering and can offer an exciting opportunity to reexamine precipitate nucleation and growth mechanisms.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This material is based upon work supported by the National Science Foundation under award no. CBET-2237311. Acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this reasearch. M. A. G. acknowledges support from the Michael F. and Virginia H. Conway Assistant Professorship. B. L. acknowledges support from the Fenton-May Graduate Fellowship. The authors gratefully acknowledge use of facilities and instrumentation at the UW-Madison Wisconsin Centers for Nanoscale Technology (wcnt.wisc.edu) partially supported by the NSF through the University of Wisconsin Materials Research Science and Engineering Center (DMR-1720415). The Bruker AVANCE 400 NMR spectrometer was supported by NSF grant CHE-1048642.References
- A. C. Maggs and R. Podgornik, General theory of asymmetric steric interactions in electrostatic double layers, Soft Matter, 2016, 4, 1219–1229 RSC
.
- E. F. Silkina, T. Y. Molotilin, S. R. Maduar and O. I. Vinogradova, Ionic equilibria and swelling of soft permeable particles in electrolyte solutions, Soft Matter, 2020, 16(4), 929–938, 10.1039/c9sm01602c
- R. Kjellander, The intimate relationship between the dielectric response and the decay of intermolecular correlations and surface forces in electrolytes, Soft Matter, 2019, 15(29), 5866–5895, 10.1039/c9sm00712a
- C. H. Ryu, H. Lee, H. Lee and H. Ren, Learning from the Heterogeneity at Electrochemical Interfaces, J. Phys. Chem. Lett., 2022, 13(33), 7838–7846, DOI:10.1021/acs.jpclett.2c02009
- H. Shin, J. M. Yoo, Y.-E. Sung and D. Y. Chung, Dynamic Electrochemical Interfaces for Energy Conversion and Storage, JACS Au, 2022, 2(10), 2222–2234, DOI:10.1021/jacsau.2c00385
- A. Groß, Grand-Canonical Approaches To Understand Structures And Processes At Electrochemical Interfaces From An Atomistic Perspective, Curr. Opin. Electrochem., 2021, 27, 100684, DOI:10.1016/j.coelec.2020.100684
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Energy and Fuels from Electrochemical Interfaces, Nat. Mater., 2017, 16, 57–69, DOI:10.1038/nmat4738
- L. D. Chen, M. Urushihara, K. Chan and J. Nørskov, Electric Field Effects in Electrochemical CO2 Reduction, ACS Catal., 2016, 6(10), 7133–7139, DOI:10.1021/acscatal.6b02299
- H. Su, W. Zhou, H. Zhang, W. Zhou, X. Zhao, Y. Li, M. Liu, W. Cheng and Q. Liu, Dynamic Evolution of Solid–Liquid Electrochemical Interfaces over Single-Atom Active Sites, J. Am. Chem. Soc., 2020, 142(28), 12306–12313, DOI:10.1021/jacs.0c04231
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels, Nat. Energy, 2019, 4, 732–745, DOI:10.1038/s41560-019-0450-y
- P. Zhu and H. Wang, High-Purity and High-Concentration Liquid Fuels Through CO2 Electroreduction, Nat. Catal., 2021, 4, 943–951, DOI:10.1038/s41929-021-00694-y
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Rational Catalyst and Electrolyte Design for CO2 Electroreduction Towards Multicarbon Products, Nat. Catal., 2019, 2, 198–210, DOI:10.1038/s41929-019-0235-5
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Potential Link between Cu Surface and Selective CO2 Electroreduction: Perspective on Future Electrocatalyst Designs, Adv. Mater., 2020, 32(17), 1908398, DOI:10.1002/adma.201908398
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, Hierarchical Copper with Inherent Hydrophobicity Mitigates Electrode Flooding for High-Rate CO2 Electroreduction to Multicarbon Products, J. Am. Chem. Soc., 2021, 143(21), 8011–8021, DOI:10.1021/jacs.1c01190
- W. Ge, Y. Chen, Y. Fan, Y. Zhu, H. Liu, L. Zong, Z. Liu, C. Lian, H. Jiang and C. Li, Dynamically Formed Surfactant Assembly at the Electrified Electrode–Electrolyte Interface Boosting CO2 Electroreduction, J. Am. Chem. Soc., 2022, 144(14), 6613–6622, DOI:10.1021/jacs.2c02486
- K. Yang, R. Kas and W. A. Smith, In Situ Infrared Spectroscopy Reveals Persistent Alkalinity near Electrode Surfaces during CO2 Electroreduction, J. Am. Chem. Soc., 2019, 141, 15891–15900, DOI:10.1021/jacs.9b07000
- R. Kas, R. Kortlever, H. Yılmaz, M. T. M. Koper and G. Mul, Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions, ChemElectroChem, 2014, 2(3), 354–358, DOI:10.1002/celc.201402373
- A. Xu, N. Govindarajan, G. Kastlunger, S. Vijay and K. Chan, Theories for Electrolyte Effects in CO2 Electroreduction, Acc. Chem. Res., 2022, 55(4), 495–503, DOI:10.1021/acs.accounts.1c00679
- R. Kortlever, J. Shen, J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6(20), 4073–4082, DOI:10.1021/acs.jpclett.5b01559
- Y. Hori, A. Murata and Y. Yoshinami, Adsorption of CO, Intermediately Formed in Electrochemical Reduction of CO2, at a Copper Electrode, J. Chem. Soc., Faraday Trans., 1991, 87(1), 125–128, 10.1039/FT9918700125
- K. Chadrasekaran and J. O. M. Bockris,
In situ Spectroscopic Investigation of Adsorbed Intermediate Radicals in Electrochemical Reactions: CO2− on Platinum, Surf. Sci., 1987, 185(3), 495–514, DOI:10.1016/S0039-6028(87)80173-5
- J. O. M. Bockris and J. C. Wass, On the Photoelectrocatalytic Reduction of Carbon Dioxide, Mater. Chem. Phys., 1989, 22(3–4), 249–280, DOI:10.1016/0254-0584(89)90001-1
-
Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, 2008, vol. 42, pp.89–189 Search PubMed
- R. G. Mariano, M. Kang, O. J. Wahab, I. J. McPherson, J. A. Rabinowitz, P. R. Unwin and M. W. Kanan, Microstructural Origin of Locally Enhanced CO2 Electroreduction Activity on Gold, Nat. Mater., 2021, 20, 1000–1006, DOI:10.1038/s41563-021-00958-9
- C. Merlet, D. T. Limmer, M. Salanne, R. van Roij, P. A. Madden, D. Chandler and B. Rotenberg, The Electric Double Layer Has a Life of Its Own, J. Phys. Chem. C, 2014, 118(32), 18291–18298, DOI:10.1021/jp503224w
- Y. Zhang, T. Ye, M. Chen, Z. A. H. Goodwin, G. Feng, J. Huang and A. A. Kornyshev, Enforced Freedom: Electric-Field-Induced Declustering of Ionic-Liquid Ions in the Electrical Double Layer. Energy & Environmental, Materials, 2020, 3(3), 414–420, DOI:10.1002/eem2.12107
- J. P. de Souza, Z. A. H. Goodwin, M. McEldrew, A. A. Kornyshev and M. Z. Bazant, Interfacial Layering in the Electric Double Layer of Ionic Liquids, Phys. Rev. Lett., 2020, 125(11), 116001, DOI:10.1103/PhysRevLett.125.116001
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Self-assembly in the electrical double layer of ionic liquids, Chem. Commun., 2011, 47(23), 6572–6574, 10.1039/c1cc11322d
- Z. A. H. Goodwin, M. McEldrew, J. P. de Souza, M. Z. Bazant and A. A. Kornyshev, Gelation, clustering, and crowding in the electrical double layer of ionic liquids, J. Chem. Phys., 2022, 157(9), 094106, DOI:10.1063/5.0097055
- B. Liu, W. Guo and M. A. Gebbie, Tuning Ionic Screening To Accelerate Electrochemical CO2 Reduction in Ionic Liquid Electrolytes, ACS Catal., 2022, 12(15), 9706–9716, DOI:10.1021/acscatal.2c02154
- W. Guo, B. Liu and M. A. Gebbie, Suppressing Co-Ion Generation via Cationic Proton Donors to Amplify Driving Forces for Electrochemical CO2 Reduction, J. Phys. Chem. C, 2023, 127(29), 14243–14254, DOI:10.1021/acs.jpcc.3c04004
- A. A. Kornyshev, Double-Layer in Ionic Liquids: Paradigm Change?, J. Phys. Chem. B, 2007, 111(20), 5545–5557, DOI:10.1021/jp067857o
- M. V. Fedorov and A. A. Kornyshev, Towards Understanding the Structure and Capacitance of Electrical Double Layer in Ionic Liquids, Electrochim. Acta, 2008, 53(23), 6835–6840, DOI:10.1016/j.electacta.2008.02.065
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Double Layer in Ionic Liquids: Overscreening versus Crowding, Phys. Rev. Lett., 2011, 106(4), 046102, DOI:10.1103/PhysRevLett.106.046102
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, A. A. Lee, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Long range electrostatic forces in ionic liquids, Chem. Commun., 2017, 53(7), 1214–1224, 10.1039/c6cc08820a
- S. Koutsoukos, J. Becker, A. Dobre, Z. J. Fan, F. Othman, F. Philippi, G. J. Smith and T. Welton, Synthesis of Aprotic Ionic Liquids, Nat. Rev. Methods Primers, 2022, 2(1), 49, DOI:10.1038/s43586-022-00129-3
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Ionic Liquid-Mediated Selective Conversion of CO2 to CO at Low Overpotentials, Science, 2011, 334(6056), 643–644, DOI:10.1126/science.1209786
- N. G. Rey and D. D. Dlott, Structural Transition in an Ionic Liquid Controls CO2 Electrochemical Reduction, J. Phys. Chem. C, 2015, 119(36), 20892–20899, DOI:10.1021/acs.jpcc.5b03397
- N. G. Rey and D. D. Dlott, Effects of water on low-overpotential CO2 reduction in ionic liquid studied by sum-frequency generation spectroscopy, Phys. Chem. Chem. Phys., 2017, 19(16), 10491–10501, 10.1039/c7cp00118e
- X. H. Yang, M. Papasizza, A. Cuesta and J. Cheng, Water-In-Salt Environment Reduces the Overpotential for Reductionof CO(2)to CO2-in Ionic Liquid/Water Mixtures, ACS Catal., 2022, 12(11), 6770–6780, DOI:10.1021/acscatal.2c00395
- L. Y. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Switching the Reaction Course of Electrochemical CO2 Reduction with Ionic Liquids, Langmuir, 2014, 30(21), 6302–6308, DOI:10.1021/la5009076
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, New Insights Into the Role of Imidazolium-Based Promoters for the Electroreduction of CO2 on a Silver Electrode, J. Am. Chem. Soc., 2016, 138(25), 7820–7823, DOI:10.1021/jacs.6b03366
- S. F. Zhao, M. Horne, A. M. Bond and J. Zhang, Is the Imidazolium Cation a Unique Promoter for Electrocatalytic Reduction of Carbon Dioxide, J. Phys. Chem. C, 2016, 120(42), 23989–24001, DOI:10.1021/acs.jpcc.6b08182
- E. E. L. Tanner, C. Batchelor-McAuley and R. G. Compton, Carbon Dioxide Reduction in Room-Temperature Ionic Liquids: The Effect of the Choice of Electrode Material, Cation, and Anion, J. Phys. Chem. C, 2016, 120(46), 26442–26447, DOI:10.1021/acs.jpcc.6b10564
- J. Theodoor and G. Overbeek, The Role of Energy and Entropy in the Electrical Double Layer, Colloids Surf., 1990, 51, 61–75, DOI:10.1016/0166-6622(90)80132-N
- Y. F. Wang, L. Adhikari, G. A. Baker and G. J. Blanchard, Cation structure-dependence of the induced free charge density gradient in imidazolium and pyrrolidinium ionic liquids, Phys. Chem. Chem. Phys., 2022, 24(32), 19314–19320, 10.1039/d2cp01066f
- K. Ma, R. Jarosova, G. M. Swain and G. J. Blanchard, Modulation of an Induced Charge Density Gradient in the Room-Temperature Ionic Liquid BMIM + BF4−, J. Phys. Chem. C, 2018, 122(13), 7361–7367, DOI:10.1021/acs.jpcc.8b02345
- J. Lindner, F. Weick, F. Endres and R. Schuster, Entropy Changes upon Double Layer Charging at a (111)-Textured Au Film in Pure 1-Butyl-1-Methylpyrrolidinium Bis[(trifluoromethyl)sulfonyl]imide
Ionic Liquid, J. Phys. Chem. C, 2020, 124(1), 693–700, DOI:10.1021/acs.jpcc.9b09871
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile, ACS Catal., 2016, 6(4), 2382–2392, DOI:10.1021/acscatal.5b02543
- A. A. Peterson and J. K. Nørskov, Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. Journal of Physical, Chem. Lett., 2012, 3(2), 251–258, DOI:10.1021/jz201461p
- Q. B. Liu, F. van Rantwijk and R. A. Sheldon, Synthesis and Application of Dicationic Ionic Liquids, J. Chem. Technol. Biotechnol., 2006, 81(3), 401–405, DOI:10.1002/jctb.1411
- J. L. Anderson, R. F. Ding, A. Ellern and D. W. Armstrong, Structure and properties of high stability geminal dicationic ionic liquids, J. Am. Chem. Soc., 2005, 127(2), 593–604, DOI:10.1021/ja046521u
- V. V. Pavlishchuk and A. W. Addison, Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C, Inorg. Chim. Acta, 1999, 298(1), 97–102, DOI:10.1016/S0020-1693(99)00407-7
- K. J. Chae, M. Choi, F. F. Ajayi, W. Park, I. S. Chang and I. S. Kim, Mass Transport through a Proton Exchange Membrane (Nafion) in Microbial Fuel Cells, Energy Fuels, 2008, 22(1), 169–176, DOI:10.1021/ef700308u
- H. Sheng, E. D. Hermes, X. Yang, D. Ying, A. N. Janes, W. Li, J. R. Schmidt and S. Jin, Electrocatalytic Production of H2O2 by Selective Oxygen Reduction Using Earth-Abundant Cobalt Pyrite (CoS2), ACS Catal., 2019, 9, 8433–8442, DOI:10.1021/acscatal.9b02546
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate, Nat. Commun., 2018, 9(1), 1320, DOI:10.1038/s41467-018-03712-z
- D. P. dos Santos, M. L. A. Temperini and A. G. Brolo, Mapping the Energy Distribution of SERRS Hot Spots from Anti-Stokes to Stokes Intensity Ratios, J. Am. Chem. Soc., 2012, 134(32), 13492–13500, DOI:10.1021/ja305580t
- B. A. Rosen, J. L. Haan, P. Mukherjee, B. Braunschweig, W. Zhu, A. Salehi-Khojin, D. D. Dlott and R. I. Masel, In Situ Spectroscopic Examination of a Low Overpotential Pathway for Carbon Dioxide Conversion to Carbon Monoxide, J. Phys. Chem. C, 2012, 116(29), 15307–15312, DOI:10.1021/jp210542v
- K. Motobayashi, Y. Maeno and K. Ikeda, In Situ Spectroscopic Characterization of an Intermediate of CO2 Electroreduction on a Au Electrode in Room-Temperature Ionic Liquids, J. Phys. Chem. C, 2022, 126(29), 11981–11986, DOI:10.1021/acs.jpcc.2c03012
- A. M. Smith, A. A. Lee and S. Perkin, The Electrostatic Screening Length in Concentrated Electrolytes Increases with Concentration, J. Phys. Chem. Lett., 2016, 7(12), 2157–2163, DOI:10.1021/acs.jpclett.6b00867
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Electrolyte Effects on CO2 Electrochemical Reduction to CO, Acc. Chem. Res., 2022, 55(14), 1900–1911, DOI:10.1021/acs.accounts.2c00080
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, Zero-Gap Electrochemical CO2 Reduction Cells: Challenges and Operational Strategies for Prevention of Salt Precipitation, ACS Energy Lett., 2023, 8(1), 321–331, DOI:10.1021/acsenergylett.2c01885
- B. C. Kash, R. J. Gomes and C. V. Amanchukwu, Mitigating Electrode Inactivation during CO2 Electrocatalysis in Aprotic Solvents with Alkali Cations, J. Phys. Chem. Lett., 2023, 14(4), 920–926, DOI:10.1021/acs.jpclett.2c03498
- X. Qin, T. Vegge, H. A. Hansen and C. O. Cation-Coordinated Inner-Sphere, 2) Electroreduction at Au–Water Interfaces, J. Am. Chem. Soc., 2023, 145(3), 1897–1905, DOI:10.1021/jacs.2c11643
- A. T. Chu and Y. Surendranath, Aprotic Solvent Exposes an Altered Mechanism for Copper-Catalyzed Ethylene Electrosynthesis, J. Am. Chem. Soc., 2022, 144(12), 5359–5365, DOI:10.1021/jacs.1c12595
- W. Ren, A. Xu, K. Chan and X. Hu, A Cation Concentration Gradient Approach to Tune the Selectivity and Activity of CO2 Electroreduction, Angew. Chem., Int. Ed., 2022, 61(49), e202214173, DOI:10.1002/anie.202214173
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment, Nat. Commun., 2022, 13(1), 7596, DOI:10.1038/s41467-022-35415-x
- T. Moumene, E. H. Belarbi, B. Haddad, D. Villemin, O. Abbas, B. Khelifa and S. Bresson, Vibrational Spectroscopic Study of Imidazolium Dicationic Ionic Liquids: Effect of Cation Alkyl Chain Length, J. Appl. Spectrosc., 2016, 83(2), 165–171, DOI:10.1007/s10812-016-0264-7
- V. H. Paschoal, L. F. O. Faria and M. C. C. Ribeiro, Vibrational Spectroscopy of Ionic Liquids, Chem. Rev., 2017, 117(10), 7053–7112, DOI:10.1021/acs.chemrev.6b00461
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, Monolayer to Bilayer Structural Transition in Confined Pyrrolidinium-Based Ionic Liquids, J. Phys. Chem. Lett., 2013, 4(3), 378–382, DOI:10.1021/jz301965d
- S. Baldelli, Surface Structure at the Ionic Liquid−Electrified Metal Interface, Acc. Chem. Res., 2008, 41(3), 421–431, DOI:10.1021/ar700185h
- M. Zhang, S. Duan, S. Luo, Y. Zhong, J. Yan, G. Liu, B. Mao and Z. Tian, Structural Exploration of Multilayered Ionic Liquid/Ag Electrode Interfaces by Atomic Force Microscopy and Surface-Enhanced Raman Spectroscopy, ChemElectroChem, 2020, 7(24), 4936–4942, DOI:10.1002/celc.202001294
- V. O. Santos Jr., M. B. Alves, M. S. Carvalho, P. A. Suarez and J. C. Rubim, Surface-enhanced Raman scattering at the silver electrode/ionic liquid (BMIPF6) interface, J. Phys. Chem. B, 2006, 110(41), 20379–20385, DOI:10.1021/jp0643348
- J. Kiefer, J. Fries and A. Leipertz, Experimental Vibrational Study of Imidazolium-Based Ionic Liquids: Raman and Infrared Spectra of 1-Ethyl-3-methylimidazolium Bis(Trifluoromethylsulfonyl)imide and 1-Ethyl-3-methylimidazolium Ethylsulfate, Appl. Spectrosc., 2007, 16(12), 1306–1311, DOI:10.1366/00037020778329200
- H. S. Schrekker, M. A. Gelesky, M. P. Stracke, C. M. Schrekker, G. Machado, S. R. Teixeira, J. C. Rubim and J. Dupont, Disclosure of the imidazolium cation coordination and stabilization mode in ionic liquid stabilized gold(0) nanoparticles, J. Colloid Interface Sci., 2007, 316(1), 189–195, DOI:10.1016/j.jcis.2007.08.018
- N. Nanbu, T. Kato, Y. Sasaki and F. Kitamura, In Situ SEIRAS Study of Room-Temperature Ionic Liquid | Gold Electrode Interphase, Electrochemistry, 2005, 73(8), 610–613, DOI:10.5796/electrochemistry.73.610
- Y. Tsori, F. Tournilhac and L. Leibler, Demixing in simple fluids induced by electric field gradients, Nature, 2004, 430(6999), 544–547, DOI:10.1038/nature02758
- Y. Tsori and L. Leibler, Phase-separation in ion-containing mixtures in electric fields, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(18), 7348–7350, DOI:10.1073/pnas.0607746104
- A. Lahiri, N. Behrens, G. Pulletikurthi, A. Yochelis, E. Kroke, T. Cui and F. Endres, Electrochemically induced phase separation and in situ formation of mesoporous structures in ionic liquid mixtures, Sci. Adv., 2018, 4(10), eaau9663, DOI:10.1126/sciadv.aau9663
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Crystallization by particle attachment in synthetic, biogenic, and geologic environments, Science, 2015, 349(6247), aaa6760, DOI:10.1126/science.aaa6760
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2017, 139(32), 11277–11287, DOI:10.1021/jacs.7b06765
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: https://doi.org/10.1039/d3sm01302b |
| This journal is © The Royal Society of Chemistry 2024 |