
Upcycling of plastic waste to atomic nickel site-decorated carbon for efficient electrochemical CO2 conversion†
Lan-Hui
Feng
,
Zhi-Hui
Lv
,
Yi-Jie
Kong
and
Xin-Ming
Hu
 *
*
Environment Research Institute, Shandong University, Qingdao 266237, China. E-mail: huxm@sdu.edu.cn
First published on 6th May 2024
Abstract
Excessive CO2 emission and plastic waste pollution constitute two pressing threats to our environment. Effective tactics are urgently demanded to address these issues. Herein, we report that the thermal treatment of plastic wastes in the presence of nickel chloride, melamine, and KCl/LiCl salt mixture produces atomic nickel and nitrogen-doped carbon materials (Ni–N–C), which are proven to be effective for electrocatalytic CO2 conversion. We disclose that the treating temperature and the activation of molten KCl/LiCl play critical roles in fabricating Ni–N–C with high porosity, abundant Ni sites, and good charge transfer ability. As a result, Ni–N–C-800 prepared from polyethylene terephthalate bottles exhibits good performance in electrochemical CO2-to-CO conversion. Importantly, a large current density of 400 mA cm−2 and a high faradaic efficiency of 90% for CO production can be achieved in the flow cell, which endows Ni–N–C-800 with great prospects for industrial applications. Furthermore, we show that the thermal treatment of other plastic wastes such as polyethylene, polypropylene, and polyvinyl chloride can also yield effective Ni–N–C catalysts, demonstrating good universality of upcycling plastic wastes to efficient catalysts. This study puts forward a sustainable solution for the simultaneous valorization of both CO2 and plastic wastes.
1 Introduction
With the excessive exploitation and consumption of fossil fuels since the industrial revolution, a large amount of CO2 has been discharged into the atmosphere, causing a series of ecological and environmental problems such as global warming, sea level rise, and climate change.1,2 These issues can be addressed by two typical technological routes: (1) CO2 capture and storage; (2) CO2 conversion and utilization. Compared to traditional CO2 capture and storage, CO2 conversion technology can not only alleviate the excessive CO2 emission but also produce carbon monoxide (CO), formic acid, ethylene, ethanol, and other valuable chemicals/fuels.3–5 Among the multiple products coming from CO2 conversion, CO is of great importance for its widespread downstream utilization, for instance, in the Fischer–Tropsch process6 and synthesis of various chemicals.7The heat-, light-, and electricity-driven CO2 reduction are all promising technologies for the conversion of CO2 to CO. In particular, the electrocatalytic reduction of CO2 is becoming a hot research field as it can take place under mild conditions (i.e., room temperature and ambient pressure) with high efficiency and be well controlled by tuning the applied potentials.8,9 Unfortunately, this process is often accompanied by the competitive reaction of hydrogen evolution and suffers from sluggish reaction kinetics.10 Therefore, the development of efficient electrocatalysts for CO2-to-CO conversion has become the key to this technology.11
Up to now, electrocatalysts that can effectively catalyze the reduction of CO2 to CO mainly include metal complex molecular catalysts represented by Fe/Co/Ni porphyrin/phthalocyanine,12–14 precious metals such as Au and Ag nanomaterials,15,16 and single-atom catalysts (SACs) that have been developed rapidly in recent years.17,18 SACs comprise atomic metal sites that are usually coordinated with nitrogen atoms and embedded in carbon supports, which are also called metal- and nitrogen-doped carbon (M–N–C) materials. The M–N–C materials have been considered as one type of promising electrocatalysts for CO2 reduction due to their advantages of maximal utilization of metal atoms and high selectivity for CO formation.19–22
The M–N–C materials are usually prepared by thermal treatment of the selected precursors at high temperatures. To achieve the effective anchoring of single metal atoms, the choice of carbon source is critical. Metal–organic frameworks (MOFs) such as ZIF-8, equipped with high crystallinity and ultra-fine porosity, have been widely used as precursors to prepare M–N–C materials.23 For example, Feng et al. prepared Ni–N–C by thermal treatment of ZIF-8 with adsorbed Ni2+ ions.24 The Ni–N–C exhibits a large current density of 726 mA cm−2 and high faradaic efficiency for CO production (FECO > 90%) in a flow cell. In two of our recent reports, we successfully synthesized Zn–N–C and In–N–C catalysts through pyrolysis of MOFs made of Zn/In ions and benzene-1,3,5-tricarboxylic acid in the presence of dicyandiamide.25,26 Notably, Zn–N–C can achieve a large current density of 1 A cm−2 and a high FECO of 95% in a flow cell. A variety of polymers, such as polycarbazole and polydopamine, have also been utilized as carbon and nitrogen sources to prepare Fe–N–C and Ni–N–C materials, which exhibit good activity for electrochemical CO2-to-CO conversion.27,28 In addition, carbon nanotubes (CNTs) possessing a high surface area and a well-defined channel structure and graphene oxide (GO) exhibiting abundant defective sites have been proven to be promising carbon substrates for supporting single metal atoms.29,30 For example, Wu et al. used N-doped carbon nanotubes (N-CNTs) to trap isolated Ni2+ at high temperatures, achieving up to 98% of FEco at low potentials.31 Zhang et al. used GO as a carbon source to support Ni single atoms.32 The above results reveal that MOFs, polymers, CNTs, and GO can all serve as carbon sources to fabricate M–N–C materials with good-to-excellent electrocatalytic activity for CO2 reduction. However, these carbon sources all need to be synthesized through tedious and costly processes. Therefore, it is highly desired to search for abundant and easily accessible carbon sources for sustainable synthesis of M–N–Cs.
Plastic production has developed rapidly in recent years, and global demand for plastic products is expected to be triple of the present by 2050.33 However, it should be noted that polyethylene terephthalate (PET) accounts for 12% of the world's solid wastes,34 which represents the main municipal plastic wastes together with polyethylene (PE), polyvinyl chloride (PVC), and polypropylene (PP).35 Due to the difficulty in biodegradation, these plastic wastes are usually treated by incineration or landfill.36 Even worse, such treatment will emit CO2 and/or cause secondary pollution to the environment. Considering that the carbon content in plastic wastes is considerably high (for example, PET and PE contain ∼60% and ∼85% carbon, respectively), some researchers have started to utilize plastic wastes as precursors to prepare various carbon materials for CO2 capture,37,38 electrochemical energy storage and conversion,39,40 and removal of organic pollutants.41 Inspired by the previous research studies, we believe that plastic wastes can also be employed as a carbon source to prepare M–N–C materials for electrocatalytic CO2 reduction as long as the single metal atoms can be properly incorporated. This would open a new avenue for the recycling of plastic waste, which has been rarely studied so far.
In this work, we successfully prepare a series of Ni–N–C materials through the thermal treatment of plastic wastes in molten salt in the presence of nickel chloride and melamine. The addition of the KCl/LiCl mixture, which formed molten salt at high temperatures, is found helpful in creating high porosity and bringing abundant Ni sites in the resulting materials. The optimal material, Ni–N–C-800, made from a used PET bottle at 800 °C, exhibits large current density and high faradaic efficiency for the electrocatalytic reduction of CO2 to CO in both H-cell and flow cell reactors. Importantly, Ni–N–C materials with high catalytic activity can also be prepared using a variety of plastic wastes, including PE bags, PP cups, and PVC hoses, showing the universality of the strategy for upcycling plastic wastes to efficient CO2 reduction catalysts. Hence, we demonstrate a viable tactic to accomplish the valorization of both the solid plastic waste and the gaseous CO2 waste.
2 Experimental
2.1 Chemicals
Nickel chloride hexahydrate (NiCl2·6H2O), melamine (C3H6N6), potassium hydroxide (KOH), and polytetrafluoroethylene suspension (PTFE, 60 wt%) were purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd. Lithium chloride (LiCl) and potassium chloride (KCl) were obtained from Shanghai Maclin Biochemical Technology Co. Ltd. Nafion solution (5 wt%) was from Sigma-Aldrich Chemical Reagent Co. Ltd. Potassium bicarbonate (KHCO3) and methanol were acquired from Sinopharm Chemical Reagent Co. Ltd. The PET plastic waste is the used mineral water bottles of Nongfu Spring brand. The PE and PP plastic wastes are the disposable bags and cups of Miaojie brand, respectively. The PVC is the hose waste of Lehua plastic. Ultrapure water (18.2 MΩ cm) was used in all experiments. All chemicals were used as received without further purification.2.2 Material synthesis
1.5 g NiCl2·6H2O, 3.0 g melamine, 8.25 g KCl, and 6.75 g LiCl were ground in a mortar to form a uniform mixture. 3.0 g PET plastic of the used mineral water bottles was cut into pieces with the size of ca. 0.5 × 0.5 cm and then added to the above mixture. The resulting mixture was transferred into a quartz boat and pyrolyzed in a tube furnace at 800 °C for 2 h under an Ar atmosphere. After cooling down to room temperature, the material was washed in 1.0 M HCl at 60 °C, washed in ultrapure water and methanol, and dried at 60 °C. Eventually, 0.89 g Ni–N–C-800 was obtained after a secondary carbonization under the same conditions as the first pyrolysis, corresponding to a yield of 30% relative to the mass of the starting PET plastic. Ni–N–C-700 and Ni–N–C-900 were prepared by performing the pyrolysis at temperatures of 700 and 900 °C, respectively, with other experimental procedures kept the same. The yields for these two materials were 40% and 26%, respectively. Ni–N–C-800(NS), Ni–C-800, and N–C-800 were synthesized under the same conditions as for Ni–N–C-800 except without adding the KCl/LiCl salt mixture, melamine, and NiCl2·6H2O, respectively. Ni–N–C-800-PE, Ni–N–C-800-PVC, and Ni–N–C-800-PP were prepared under the same conditions as for Ni–N–C-800 except that the PET was replaced with PE, PVC, and PP plastic wastes, respectively. The yields for these three materials were 8%, 19%, and 7% relative to the mass of their starting plastics, respectively.2.3 Characterization
Brunauer–Emmett–Teller (BET) specific surface areas and pore size distributions of the materials were measured at −196 °C with a physical sorption apparatus (Micromeritics ASAP 2460) using N2 as the adsorbate. The X-ray diffraction patterns were acquired on an X-ray diffractometer (XRD, Japan Rigaku Miniflex 600) using Cu Kα radiation. Raman spectra were recorded on the Thermo Fisher Scientific DXR2 Raman spectrometer with a laser excitation of 532 nm. The nickel content of the materials was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Thermo Scientific ICAP 7400). X-ray photoelectron spectra (XPS) were recorded on the Thermo Scientific Nexsa X-ray photoelectron spectroscope. The C 1s binding energy of 284.8 eV was used for energy referencing. The observation of Ni single atoms in the material was done on a high angle annular dark field scanning transmission electron microscopy (HAADF-STEM, Thermofisher Spectra 300, probe-corrected TEN, 300 kV, convergence angle of 21 mrad, detected range of 50–200 mrad), and the probe used for elemental mapping was Super EDS detector.The X-ray absorption spectra (XAS) of Ni K-edge were collected at the beamline of TPS44A1 in the National Synchrotron Radiation Research Center (NSRRC), Taiwan. This beamline adopted 4-bounce channel-cut Si (111) monochromator for X-ray absorption spectroscopy. The end station is equipped with three ionization chambers and a Lytle/SDD detector after the focusing position of the KB mirror for transmission mode X-ray absorption spectroscopy. The photon flux on the sample ranged from 1 × 1011–3 × 109 photon per s for X-ray energy from 6–27 keV. The X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were analyzed using Athena and Artemis softwares.42
For wavelet transform analysis, the χ(k) exported from Athena was imported into the Hama Fortran code.43 The parameters were listed as follows: R range of 0–6 Å, k range of 0–12.0 Å−1, k weight of 2, and Morlet function of κ = 10, as well as σ = 1. These parameters were used as the mother wavelet to provide the overall distribution.
2.4 Electrochemical measurements in the H-cell
The working electrodes were prepared according to the following procedure. 6.0 mg of the material, 450 μL of ultrapure water, 90 μL of isopropyl alcohol, and 60 μL of 5 wt% Nafion were mixed and sonicated for 30 min. Then, 100 μL of the suspension was drop-cast onto both sides of the carbon paper (TGP-H-060) and dried in the air. The effective area of the electrode was 2 cm2, and the catalyst loading was 0.5 mg cm−2.The catalytic performance of the materials for electrochemical CO2 reduction was evaluated in an H-type three-electrode cell with two compartments segregated by a Nafion-117 proton exchange membrane in 0.5 M KHCO3 electrolyte using platinum mesh as the counter electrode and saturated Ag/AgCl as the reference electrode.
Before the beginning of electrolysis, the cathodic electrolyte was saturated with CO2. In the course of electrolysis, CO2 was purged into the catholyte at a constant rate controlled by the gas mass flowmeter. The outlet of the gas in the cathodic chamber was connected to the inlet of the gas chromatograph (Thermofisher Trace 3000), and the gas products were analyzed online.
The faradaic efficiency of CO and H2 products (FEco and FEH2) were calculated using eqn (1).
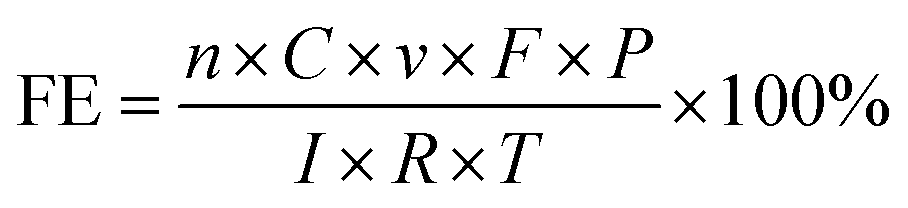 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1; P is the atmospheric pressure, 101325 Pa; I is the current at the given sampling time; R is the ideal gas constant, 8.314 J mol−1 K−1; T is the room temperature.
485 C mol−1; P is the atmospheric pressure, 101325 Pa; I is the current at the given sampling time; R is the ideal gas constant, 8.314 J mol−1 K−1; T is the room temperature.
All potentials were reported without iR correction. The potential against Ag/AgCl was converted to the potential against reversible hydrogen electrode (RHE) according to eqn (2).
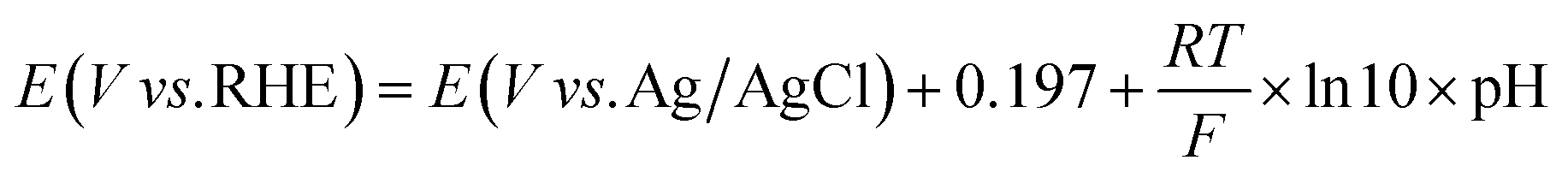 | (2) |
485 C mol−1). Note that the pH values of a CO2-saturated 0.5 M KHCO3 aqueous solution and an Ar-saturated 0.5 M KHCO3 aqueous solution are 7.3 and 8.4, respectively.
Linear sweep voltammetric (LSV) curves were recorded in Ar and CO2-saturated 0.5 M KHCO3 solution between 0 and −1.2 V (vs. RHE) at a scan rate of 10 mV s−1. The electrochemical active surface area (ECSA) was analyzed by measuring the electrochemical double layer capacity (Cdl). Cdl was calculated based on the double layer current in the cyclic voltammograms (CVs) in the potential ranges from −0.6 to −0.8 V (vs. RHE) at different scan rates (10, 20, 30, 40, and 50 mV s−1). Electrochemical impedance spectroscopy (EIS) was implemented at an AC voltage amplitude of 5 mV over a frequency ranging from 100000 to 0.1 Hz.
2.5 Electrochemical measurements in the flow cell
The preparation method for the gas diffusion electrode was as follows. 40 mg of the material, 9 mL of anhydrous ethanol, 1 mL of 5 wt% PTFE, and 40 μL of 5 wt% Nafion were mixed and sonicated for 60 min. The suspension was drop-cast onto the gas diffusion layer electrode (YLS-30T). The effective area was 1 cm2, and the catalyst loading was 2 mg cm−2.Electrochemical CO2 reduction at a high current density was conducted in a flow electrolytic cell (Gauss Union). The cathodic and anodic chambers were separated by anion exchange membrane (Fumatech, FAA-3-PK-130). A platinum sheet was used as the counter electrode and saturated Hg/HgO as the reference electrode. The circulation of electrolytes (1.0 M KOH) in the cathodic and anodic chambers was controlled by a peristaltic pump and a gas–liquid mixed flow pump, respectively. The gas outlet of the cathodic chamber was connected to the inlet of gas chromatography for online detection of gaseous products.
3 Results and discussion
The synthesis protocol for plastic wastes to Ni–N–C materials is shown in Fig. 1. The wasted bottle made from PET was first cut into small pieces and mixed with melamine, NiCl2·6H2O, and the KCl/LiCl salt mixture in a certain proportion, and then heated at different temperatures (i.e., 700, 800, and 900 °C). The PET plastic served as a carbon source and melamine as a nitrogen source for anchoring Ni atoms coming from NiCl2·6H2O. The molar ratio of KCl/LiCl was set to be 0.41:0.59, which helped in creating porosity in the catalyst during thermal treatment, as the eutectic point of the salt mixture at this ratio was 353 °C, lower than the major weight loss temperature range of PET (400–500 °C).44 Subsequently, the KCl/LiCl salt mixture and Ni nanoparticles formed during thermal treatment were removed by washing with dilute hydrochloric acid and water. Since the acid washing would leave some defective structures behind,45 the materials were subjected to a secondary thermal treatment, which resulted in a higher carbonization degree of the material that would be beneficial for electrocatalysis. The resulting materials are named Ni–N–C-T, where T represents the temperature of the thermal treatment.
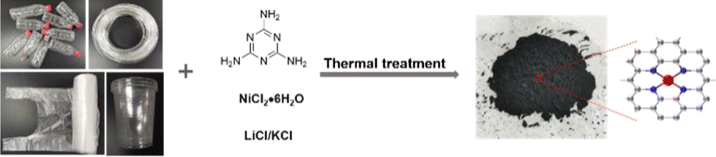 | ||
| Fig. 1 Protocol for the synthesis of Ni–N–C using plastic wastes. | ||
The performance of Ni–N–C-T (T = 700, 800, or 900 °C) for electrochemical CO2 reduction reaction (eCO2RR) was first assessed by electrochemical tests in a classical three-electrode H-type electrolytic cell in 0.5 M KHCO3 solution. Fig. S1† displays the linear sweep voltammograms recorded in Ar- and CO2-saturated KHCO3. Compared to the case under Ar atmosphere, all three materials showed enhanced current density under CO2 atmosphere within the usual potential range for CO2 reduction between −0.6 V and −0.9 V vs. reversible hydrogen electrode (RHE), indicating that the dominant reaction of the electrolytic process became the CO2 reduction rather than hydrogen evolution reaction (HER). The eCO2RR performance of the materials was further evaluated through controlled electrolysis at different potentials. The products were analyzed by gas chromatography and 1H nuclear magnetic resonance (1H NMR). It can be seen from the 1H NMR that no product was detected in the electrolyte (Fig. S2†), and gas chromatography showed that only CO and H2 were produced in the electrolysis. In general, the three materials exhibit comparably high selectivity for the CO2-to-CO conversion (Fig. 2a). The similar selectivity can be explained by the same Ni active sites in these three materials. Nevertheless, Ni–N–C-800 prepared through thermal treatment at 800 °C exhibited the highest absolute cathodic current density (|j|) and partial current density for CO production (|jco|) in a wide range of potentials (Fig. 2b and S3†).
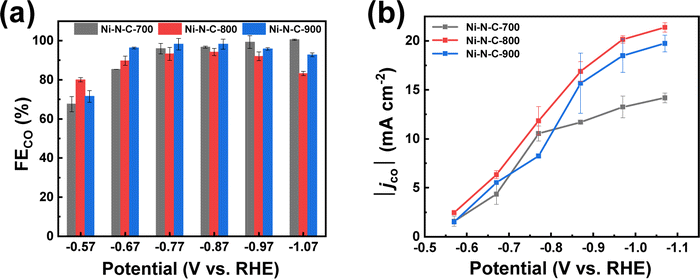 | ||
| Fig. 2 (a) FECO and (b) |jco| measured after 20 min electrolysis at varying potentials on Ni–N–C-T (T = 700, 800, and 900 °C). | ||
A series of structural characterizations were conducted to uncover the origin of the different eCO2RR activities among the three materials. Firstly, X-ray photoelectron spectroscopy (XPS) was used to analyze the surface chemical composition of each material. The presence of C 1s, N 1s, O 1s, and Ni 2p signals in the survey spectra suggested that Ni–N–C-T samples were mainly composed of C, N, O, and Ni elements (Fig. S4†). The N 1s spectra of all materials could be deconvoluted to pyridinic, Ni coordinated, pyrrolic, graphitic, and oxidized N species, respectively (Fig. 3a). Thus, we can infer that the Ni in the Ni–N–C materials are mainly coordinated with N atoms to form Ni–N active sites. In the Ni 2p spectra, the binding energy of Ni 2p3/2 in the three materials is around 855 eV, which were between Ni0 from metallic Ni and Ni2+ from nickel porphyrin (Fig. 3b), indicating that Ni atoms were likely to be at a low valence state in the materials.46 The Ni contents of Ni–N–C-700, Ni–N–C-800, and Ni–N–C-900, which were further determined by ICP-OES, were 3.21, 2.54, and 1.32 wt%, respectively, being lower than the surface Ni content measured by XPS (Table S1†). These results suggest that Ni atoms mainly exist on the surface of the materials, providing abundant active sites for electrocatalysis. Notably, the Ni and N contents decreased gradually with the rise of treating temperature, indicative of a higher carbonization degree at higher temperatures.47 This also explained the lower yield of the catalyst synthesized at higher temperatures. Nevertheless, the selectivity for CO production was almost the same while the |jCO| first increased and then decreased with rising temperature, indicating that the difference in eCO2RR performance over the three materials cannot be simply attributed to their varying content of Ni sites.
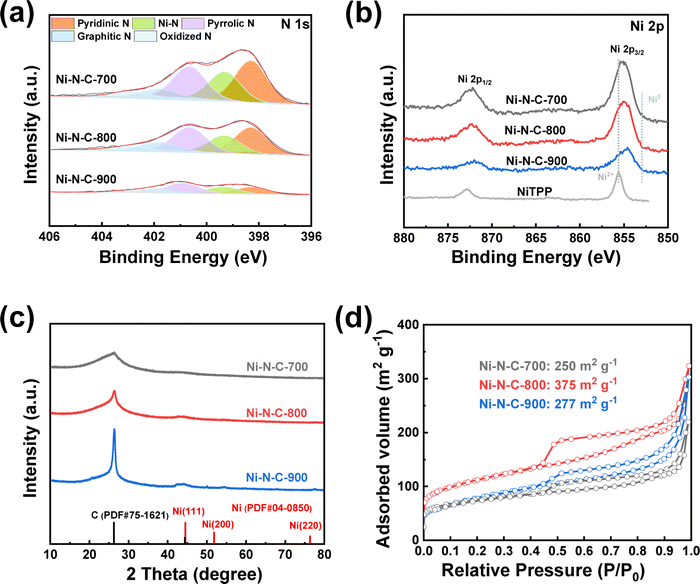 | ||
| Fig. 3 (a) XPS N 1s spectra, (b) XPS Ni 2p spectra, (c) XRD patterns, and (d) N2 adsorption–desorption isotherms of Ni–N–C-T (T = 700, 800, and 900 °C). | ||
The X-ray diffraction (XRD) patterns do not show any diffraction peak attributed to nickel particles (Fig. 3c), which further supports our inference that Ni in the materials is atomically dispersed. Notably, the diffraction peak at around 26° corresponds to the graphitic carbon.48 This peak intensified with a higher treating temperature, confirming the higher degree of graphitization when the temperature goes up. For this reason, some minor peaks associated with graphitic carbon are also observed for Ni–N–C-900. In the Raman spectra, two typical broad peaks around 1350 and 1580 cm−1 were observed (Fig. S5†), which were attributed to the D (defective) and G (graphitic) bands of carbon, respectively. The intensity ratio of the two peaks (ID/IG) that reflects the defect degree of the material decreased with the higher treating temperature, in good agreement with the XPS and XRD results, which show that higher temperature led to a higher degree of graphitization. The degree of graphitization would have an impact on the charge transfer ability of the materials.
To analyze the charge transfer ability of different materials during the process of CO2 reduction, electrochemical impedance spectroscopy (EIS) was conducted at the polarization state (−0.57 V vs. RHE), and the data was fitted by the equivalent circuit displayed in Fig. S6.† To our surprise, the variation of charge transfer ability of different materials does not follow the order of the degree of graphitization. Such a phenomenon suggests that the impedance value in the polarization state is not only related to the conductivity of the material itself but also depends on the charge transfer between the catalyst and CO2 at the electrode interface. It can be seen from Fig. S7 and Table S2† that Ni–N–C-800 has the strongest interfacial charge transfer ability in the process of CO2 reduction, which is one of the reasons that explains its highest current density for eCO2RR.
Furthermore, we performed N2 adsorption–desorption experiments to analyze the specific surface area and pore size distribution of the materials (Fig. 3d and Table S3†). They all show type IV isotherms and H4 hysteresis loops.49 Notably, Ni–N–C-800 exhibits a specific surface area of 375 m2 g−1, which is higher than the materials of the other two temperature series. This suggests that the treating temperature also has a significant effect on the porosity of the materials. Low treating temperatures would lead to incomplete carbonization, while too high temperature may cause the pore collapse, both producing materials with smaller specific surface area. For this reason, Ni–N–C-800 possessed more micropores and mesopores than Ni–N–C-700 and Ni–N–C-900 (Fig. S8†).
Although a higher specific surface area is conducive to exposing more active sites, the actual surface area involved in the electrochemical catalytic reaction needs to be taken into account during the reaction process. We then analyzed the electrochemical active surface area (ECSA) of each material by determining their double-layer capacity (Cdl, Fig. S9†). Ni–N–C-800 shows the largest double-layer capacity (Cdl = 19.5 mF cm−2). The Cdl of the three materials actually follows the same order as their specific surface area. Taken together, Ni–N–C-800 exhibits the highest specific surface area and ECSA as well as the best charge transfer capability among the three materials, which facilitates the CO2 transport and electron transfer during eCO2RR, resulting in an optimal |jco|. Therefore, it is very important to choose the appropriate treatment temperature for Ni–N–C synthesis.
To understand the role of each reagent in Ni–N–C-800 synthesis, we prepared controlled materials, Ni–N–C-800(NS), Ni–C-800 and N–C-800 in the absence of KCl/LiCl salt mixture, melamine, and NiCl2·6H2O, respectively. Linear sweep voltammograms and controlled potential electrolysis both revealed that leaving out any reagent in the synthesis produced materials with poorer activity for eCO2RR (Fig. S10†). Specifically, N–C-800 exhibits a FECO of 60% and a |jco| of 0.84 mA cm−2 for eCO2RR at −0.87 V vs. RHE due to the lack of isolated Ni sites, manifesting the crucial role of Ni atoms as active sites for eCO2RR. Even worse, the dominant reaction over Ni–C-800 was no longer the reduction of CO2 to CO but HER. This was also attributed to the lack of effective atomic Ni sites in the material due to the absence of a nitrogen source in the material synthesis. Instead, the Ni in Ni–C-800 exists in the form of metallic particles that are wrapped by carbon layers, as we observed intense diffraction peaks for metallic Ni particles in XRD but no Ni signals in XPS (Fig. S11 and S12†). This points to the fact that nitrogen atoms are indispensable for the formation of atomically dispersed Ni sites in the course of material synthesis.
Ni–N–C-800(NS), which was prepared without adding the KCl/LiCl salt mixture, exhibited similar FECO but significantly lower |jCO| than Ni–N–C-800 at −0.87 V vs. RHE. The lower activity of Ni–N–C-800(NS) can be explained by the following aspects originating from the absence of the KCl/LiCl salt mixture during the material synthesis. First, Ni–N–C-800(NS) exhibited a smaller specific surface area and ECSA than Ni–N–C-800 (Fig. S13 and S14†). Second, Ni atoms tended to agglomerate to form metallic Ni particles without the addition of KCl/LiCl salt mixture (Fig. S12†), leading to a smaller content of atomically dispersed Ni sites on the surface of Ni–N–C-800(NS) as determined by XPS, despite its larger overall Ni content than Ni–N–C-800 as determined by ICP-OES (Table S1†). These results highlight the importance of the KCl/LiCl salt mixture for synthesis of high-performance Ni–N–C electrocatalysts, not only creating more porosity in the material but also making the metal atoms more dispersive, as the molten salt can dissolve the Ni ions and serve as porogen (Fig. 4). All in all, the results revealed that the metal source, nitrogen source, and the salt mixture are all crucial to prepare high-performance Ni–N–C electrocatalysts for eCO2RR.
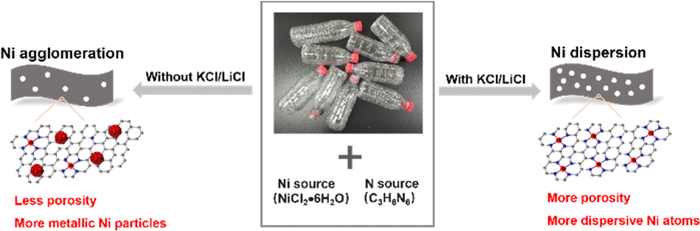 | ||
| Fig. 4 The impact of molten salt (KCl/LiCl) on the structure of the resulting carbon materials. | ||
Based on the above results, we selected Ni–N–C-800 for more thorough characterizations. The morphology of Ni–N–C-800 was analyzed by a high angle annular dark field scanning transmission electron microscope (HAADF-STEM), and the results are shown in Fig. 5. The elemental mapping reveals that C, N, and Ni in Ni–N–C-800 were uniformly distributed. In the enlarged images acquired by aberration-corrected HAADF-STEM, uniformly distributed bright spots can be clearly seen, which confirms that Ni exists in the form of isolated atoms rather than nanoclusters in Ni–N–C-800.
 | ||
| Fig. 5 (a–d) The STEM image of Ni–N–C-800 with corresponding elemental mapping images of C, N, and Ni; (e and f) aberration-corrected HAADF-STEM images of Ni–N–C-800. | ||
X-ray absorption fine spectroscopy (XAFS) was performed to further analyze the valence state and coordination environment of Ni single atoms in Ni–N–C-800. We found that the Ni K-edge energy of Ni–N–C-800 was between those of Ni foil and NiO through the comparison of their X-ray absorption near-edge spectra (XANES) (Fig. 6a), indicating that the valence state of Ni atoms in Ni–N–C-800 was between 0 and +2, in good agreement with the XPS results. The Fourier transform (FT) of the extended X-ray absorption fine spectroscopy (EXAFS) is presented in Fig. 6b. Ni–N–C-800 clearly showed a dominant radial distance at ∼1.4 Å, which could be attributed to the Ni–N coordination structure.50 The EXAFS spectra were subjected to wavelet transform (WT) analysis in both R and k spaces (Fig. 6c).51 The maximum intensity of the WT contour plots for Ni–N–C was closer to that for nickel phthalocyanine (NiPc), which further proved the Ni–N coordination structure in Ni–N–C-800. Through fitting the EXAFS data of k space, we obtained a coordination structure of Ni–N4 for the Ni atoms in Ni–N–C-800 (Fig. 6d, e, S15, and Table S4†).
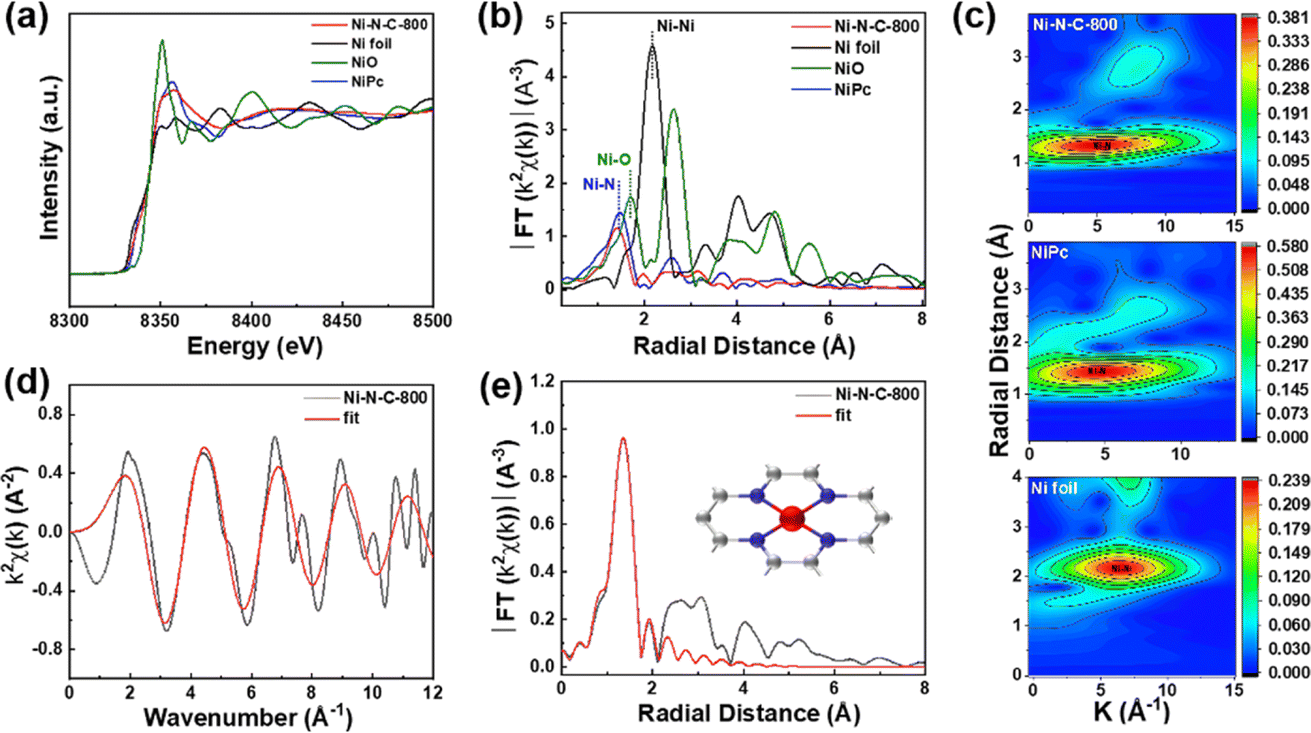 | ||
| Fig. 6 (a) Ni K-edge XANES spectra of Ni–N–C-800, Ni foil, NiO, and NiPc; (b) Fourier transform of the EXAFS spectra of Ni foil, NiO, NiPc, and Ni–N–C-800; (c) wavelet transforms (WT) of Ni foil, NiPc, and Ni–N–C-800; (d) EXAFS fitting of Ni–N–C-800 at K space; (e) Fourier transform-EXAFS fitting results of Ni–N–C-800 (inset: the corresponding coordination structure model). | ||
To investigate the potential for practical applications, we carried out electrolysis experiments at large current densities using Ni–N–C-800 in a flow cell. An alkaline electrolyte (1.0 M KOH) was used to suppress the competitive HER. It can be seen from Fig. 7a and S16† that the FECO remained high (>90%) when the current density increased from 50 to 400 mA cm−2. In order to determine the stability of Ni–N–C-800, we conducted electrolysis at 100 mA cm−2 for 12 h. It maintained a high selectivity of 90% for CO production throughout the 12 h electrolysis, with the working potential fluctuating around −0.6 V vs. RHE (Fig. 7b). Such potential fluctuation could be attributed to the flooding and salting out that have often occurred on the working electrode during large current density electrolysis, regardless of the catalyst used.25,52 This suggests that Ni–N–C-800 could work stably in the flow cell at high current densities and thus showed good prospects for practical applications. It also needs to be pointed out that the performance of Ni–N–C is comparable to or even higher than many other Ni-based electrocatalysts made from different carbon sources (Table S5†).
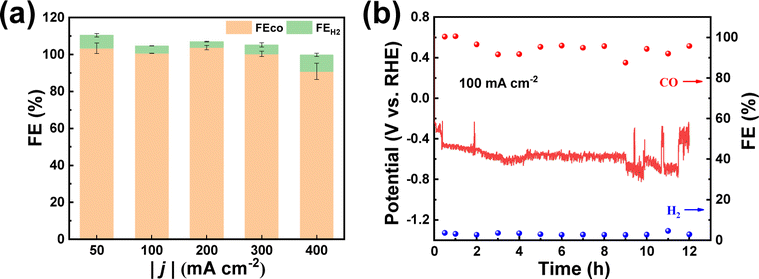 | ||
| Fig. 7 Recording of FE on Ni–N–C-800 with (a) |j| varying from 50–400 mA cm−2 for 15 min electrolysis and with (b) |j| = 100 mA cm−2 for 12 h electrolysis of CO2 using a gas-fed flow-cell reactor in 1.0 M KOH. | ||
Finally, we synthesized a family of Ni–N–C-800-PE, Ni–N–C-800-PP, and Ni–N–C-800-PVC under the same conditions except replacing the PET plastic waste with other common plastic wastes, such as PE bags, PP cups, and PVC hoses. The catalyst yield varied when using different carbon sources, following an order of PET > PVC > PE ≈ PP at the same synthesis temperature (800 °C), which was attributed to the different composition and structure of the plastics. The highest yield for the catalyst derived from PET could be due to the presence of aromatic rings in PET, while the other plastics consisted of only aliphatic chains. Furthermore, the presence of heteroatoms seems to favour the production of carbon materials, as shown by the higher yield of Ni–N–C-800-PVC than Ni–N–C-800-PE and Ni–N–C-800-PP. This provides guidance for the choice of plastic waste for the high-yield synthesis of Ni–N–C catalysts.
As can be seen from Fig. 8 and S17,† these Ni–N–Cs also exhibit similarly high CO selectivity and current density, regardless of using different types of plastic waste. The small differences in the activity and selectivity could be attributed to the slightly different composition and structure of these Ni–N–C materials originating from the use of different plastic precursors (Fig. S18–S22 and Table S1–S3†). Nonetheless, these results demonstrate the good universality of upcycling various plastic wastes to high-performance Ni–N–C catalysts for eCO2RR.
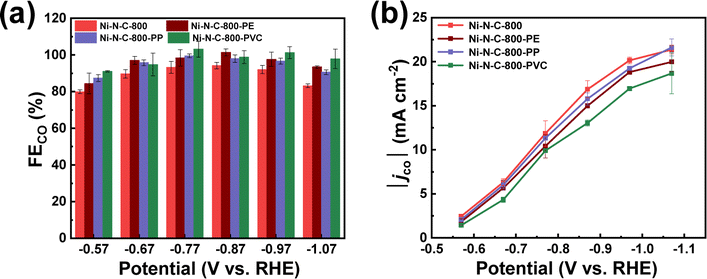 | ||
| Fig. 8 (a) FECO and (b) |jco| measured after 20 min electrolysis at varying potentials on Ni–N–C-800, Ni–N–C-800-PE, Ni–N–C-800-PP, and Ni–N–C-800-PVC. | ||
4 Conclusions
We have demonstrated that the thermal treatment of various plastic wastes, including PET, PE, PP, and PVC, in the presence of nickel source, nitrogen source, and KCl/LiCl salt mixture can yield a family of carbon materials containing isolated Ni sites coordinated with nitrogen atoms. The control of the treating temperature and the use of KCl/LiCl molten salt are crucial for generating effective Ni–N–C catalysts, which possess high porosity, abundant Ni sites, and good charge transfer ability. The resulting Ni–N–Cs thus exhibit good performance for electrochemical CO2 conversion to CO in both H-cell and flow cell, indicating their great prospect for industrial applications. This work demonstrates the concept of recycling solid plastic waste to valorize gaseous CO2 waste, opening a new avenue to address the environmental problems of climate change and plastic pollution.Author contributions
Lan-Hui Feng: conceptualization, methodology, investigation, writing – original draft, formal analysis. Zhi-Hui Lv: writing – review & editing. Yi-Jie Kong: writing – review & editing. Xin-Ming Hu: conceptualization, supervision, writing – review & editing, project administration, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Shandong Provincial Natural Science Foundation (ZR2021QB100), National Natural Science Foundation of China (22109089, 22376120), Shandong Provincial Science Foundation for Excellent Young Scholars Overseas (2022HWYQ-002), and Taishan Scholars program from Shandong Province (tsqn202103021).References
- T. Geng, F. Jia, W. J. Cai, L. X. Wu, B. Gan, Z. Jing, S. J. Li and M. J. McPhaden, Increased occurrences of consecutive La Niña events under global warming, Nature, 2023, 619, 774–781 CrossRef CAS PubMed.
- S. Koseki, J. Tjiputra, F. Fransner, L. R. Crespo and N. S. Keenlyside, Disentangling the impact of Atlantic Niño on sea-air CO2 flux, Nat. Commun., 2023, 14, 3649 CrossRef CAS PubMed.
- D. U. Nielsen, X. M. Hu, K. Daasbjerg and T. Skrydstrup, Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- K. Fernández-Caso, G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, Electroreduction of CO2: Advances in the continuous production of formic acid and formate, ACS Energy Lett., 2023, 8, 1992–2024 CrossRef.
- S. A. Farooqi, A. S. Farooqi, S. Sajjad, C. L. Yan and A. B. Victor, Electrochemical reduction of carbon dioxide into valuable chemicals: A review, Environ. Chem. Lett., 2023, 21, 1515–1553 CrossRef CAS.
- Y. L. Hu, C. C. Lee, M. Grosch, J. B. Solomon, W. Weigand and M. W. Ribbe, Enzymatic Fischer-Tropsch-type reactions, Chem. Rev., 2022, 123, 5755–5797 CrossRef PubMed.
- S. Fujimori and S. Inoue, Carbon monoxide in main-group chemistry, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Electrochemical approaches for CO2 conversion to chemicals: A Journey toward practical applications, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS PubMed.
- M. Zhuansun, T. Wang, J. Wang, G. Han, X. Wang and Y. Wang, Reactors for electro-upgrading carbon dioxide into value-added chemicals, Mater. Today Sustain., 2022, 19, 100185 CrossRef.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and challenges for the electrochemical reduction of CO2 to CO: From fundamentals to industrialization, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- M. Rybacki, A. V. Nagarajan and G. Mpourmpakis, Ligand removal energetics control CO2 electroreduction selectivity on atomically precise, ligated alloy nanoclusters, Environ. Sci.: Nano, 2022, 9, 2032–2040 RSC.
- X. M. Hu, M. H. Ronne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Enhanced catalytic activity of cobalt porphyrin in CO2 electroreduction upon immobilization on carbon materials, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- X. Z. Lv, Q. Liu, H. Yang, J. H. Wang, X. J. Wu, X. T. Li, Z. F. Qi, J. H. Yan, A. J. Wu, T. Cheng and H. B. Wu, Nanoconfined molecular catalysts in integrated gas diffusion electrodes for high-current-density CO2 electroreduction, Adv. Funct. Mater., 2023, 33, 2301334 CrossRef CAS.
- M. R. Smith, C. B. Martin, S. Arumuganainar, A. Gilman, B. E. Koel and M. L. Sarazen, Mechanistic elucidations of highly dispersed metalloporphyrin metal-organic framework catalysts for CO2 electroreduction, Angew. Chem., Int. Ed., 2023, 62, e202218208 CrossRef CAS PubMed.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. X. Zhao, L. Shang, C. Zhou, L. Jiang and T. R. Zhang, Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- A. Thevenon, A. Rosas-Hernández, A. M. Fontani Herreros, T. Agapie and J. C. Peters, Dramatic HER suppression on Ag electrodes via molecular films for highly selective CO2 to CO reduction, ACS Catal., 2021, 11, 4530–4537 CrossRef CAS.
- P. F. Yao, J. W. Zhang, Y. L. Qiu, Q. O. Zheng, H. M. Zhang, J. W. Yan and X. F. Li, Atomic-dispersed coordinated unsaturated nickel-nitrogen sites in hollow carbon spheres for the efficient electrochemical CO2 reduction, ACS Sustainable Chem. Eng., 2021, 9, 5437–5444 CrossRef CAS.
- H. Wang, Y. Li, M. Wang, S. Chen, M. Yao, J. L. Chen, X. L. Liao, Y. W. Zhang, X. Lu, E. Matios, J. M. Luo, W. Zhang, Z. X. Feng, J. C. Dong, Y. Q. Liu and W. Y. Li, Precursor-mediated in situ growth of hierarchical N-doped graphene nanofibers confining nickel single atoms for CO2 electroreduction, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2219043120 CrossRef CAS PubMed.
- Y. Li, X. F. Lu, S. Xi, D. Luan, X. Wang and X. W. D. Lou, Synthesis of N-doped highly graphitic carbon urchin-like hollow structures loaded with single-Ni atoms towards efficient CO2 electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202201491 CrossRef CAS PubMed.
- Y. L. Jia, Z. Q. Xue, J. Yang, Q. L. Liu, J. H. Xian, Y. C. Zhong, Y. M. Sun, X. X. Zhang, Q. H. Liu, D. X. Yao and G. Q. Li, Tailoring the electronic structure of an atomically dispersed zinc electrocatalyst: Coordination environment regulation for high selectivity oxygen reduction, Angew. Chem., Int. Ed., 2022, 61, e202110838 CrossRef CAS PubMed.
- X. Li, K. Hu, Y. Z. Huang, Q. Q. Gu, Y. W. Chen, B. Yang, R. L. Qiu, W. H. Luo, B. M. Weckhuysen and K. Yan, Upcycling biomass waste into Fe single atom catalysts for pollutant control, J. Energy Chem., 2022, 69, 282–291 CrossRef CAS.
- M. Wen, N. N. Sun, L. Jiao, S. Q. Zang and H. L. Jiang, Microwave-assisted rapid synthesis of MOF-based single-atom Ni catalyst for CO2 electroreduction at ampere-level current, Angew. Chem., Int. Ed., 2024, 63, e202318338 CrossRef CAS PubMed.
- Y. Q. Chen, J. R. Zhang, J. Y. Tian, Y. Guo, F. F. Xu, Y. Zhang, X. Z. Wang, L. J. Yang, Q. Wu and Z. Hu, Hierarchical Ni/N/C single-site catalyst achieving industrial-level current density and ultra-wide potential plateau of high CO Faradic efficiency for CO2 electroreduction, Adv. Funct. Mater., 2023, 33, 2214658 CrossRef CAS.
- Y. Li, N. M. Adli, W. T. Shan, M. Y. Wang, M. J. Zachman, S. Hwang, H. Tabassum, S. Karakalos, Z. X. Feng, G. F. Wang, Y. G. C. Li and Y. G. Wu, Atomically dispersed single Ni site catalysts for high-efficiency CO2 electroreduction at industrial-level current densities, Energy Environ. Sci., 2022, 15, 2108–2119 RSC.
- S. M. Li, S. Q. Zhao, X. Y. Lu, M. Ceccato, X. M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Low-valence Znδ+ (0<δ<2) single-atom material as highly efficient electrocatalyst for CO2 reduction, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- S. M. Li, X. Y. Lu, S. Q. Zhao, M. Ceccato, X. M. Hu, A. Roldan, M. Liu and K. Daasbjerg, p-Block indium single-atom catalyst with low-coordinated In-N motif for enhanced electrochemical CO2 reduction, ACS Catal., 2022, 12, 7386–7395 CrossRef CAS.
- X. M. Hu, D. Mendoza, M. R. Madsen, D. Joulié, B. Lassalle-Kaiser, M. Robert, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Achieving near-unity CO selectivity for CO2 electroreduction on an Iron-decorated carbon material, ChemSusChem, 2020, 13, 6360–6369 CrossRef CAS PubMed.
- Y. J. Kong, A. C. Zheng, Z. Z. Wu, Q. Chen and X. M. Hu, Hollow carbon spheres with isolated Ni atoms on both external and internal surfaces for efficient CO2 electroreduction, Mater. Today Chem., 2023, 28, 101386 CrossRef CAS.
- J. Sheng and Y. Li, Applications of carbon nanotubes in oxygen electrocatalytic reactions, ACS Appl. Mater. Interfaces, 2022, 14, 20455–20462 CrossRef CAS PubMed.
- A. Hasani, M. A. Teklagne, H. H. Do, S. H. Hong, Q. Van Le, S. H. Ahn and S. Y. Kim, Graphene-based catalysts for electrochemical carbon dioxide reduction, Carbon Energy, 2020, 2, 158–175 CrossRef CAS.
- S. D. Wu, F. Yi, D. Ping, S. G. Huang, Y. F. Zhang, L. F. Han, S. W. Wang, H. Wang, X. Z. Yang, D. J. Guo, G. L. Liu and S. M. Fang, Constructing single-atomic nickel sites in carbon nanotubes for efficient CO2 electroreduction, Carbon, 2022, 196, 1–9 CrossRef CAS.
- K. W. Mou, Z. P. Chen, X. X. Zhang, M. Y. Jiao, X. P. Zhang, X. Ge, W. Zhang and L. C. Liu, Highly efficient electroreduction of CO2 on nickel single-atom catalysts: Atom trapping and nitrogen anchoring, Small, 2019, 15, 1903668 CrossRef CAS PubMed.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Plastic futures and their CO2 emissions, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Machine learning-aided engineering of hydrolases for PET depolymerization, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- J. D. Chea, K. M. Yenkie, J. F. Stanzione and G. J. Ruiz-Mercado, A generic scenario analysis of end-of-life plastic management: Chemical additives, J. Hazard. Mater., 2023, 441, 129902 CrossRef CAS PubMed.
- G. Kwon, D. W. Cho, J. Park, A. Bhatnagar and H. Song, A review of plastic pollution and their treatment technology: A circular economy platform by thermochemical pathway, Chem. Eng. J., 2023, 464, 142771 CrossRef CAS.
- W. A. Algozeeb, P. E. Savas, Z. Yuan, Z. Wang, C. Kittrell, J. N. Hall, W. Y. Chen, P. Bollini and J. M. Tour, Plastic waste product captures carbon dioxide in nanometer pores, ACS Nano, 2022, 16, 7284–7290 CrossRef CAS PubMed.
- X. Z. Yuan, N. M. Kumar, B. Brigljević, S. J. Li, S. Deng, M. Byun, B. Lee, C. S. K. Lin, D. C. W. Tsang, K. B. Lee, S. S. Chopra, H. Lim and Y. S. Ok, Sustainability-inspired upcycling of waste polyethylene terephthalate plastic into porous carbon for CO2 capture, Green Chem., 2022, 24, 1494–1504 RSC.
- J. Xu, S. M. Dou, W. Zhou, C. Yang, I. Manke, P. P. Zhang, Z. H. Yan, Y. H. Xu, Q. H. Yuan, Y. L. Zhang, W. D. Liu, R. J. Chen and Y. N. Chen, Scalable waste-plastic-derived carbon nanosheets with high contents of inbuilt nitrogen/sulfur sites for high performance potassium-ion hybrid capacitors, Nano Energy, 2022, 95, 107105 Search PubMed.
- J. H. Zhao, Q. Niu, J. J. Zhang and P. F. Zhang, Core–shell construction of metal@carbon by mechanochemically recycling plastic wastes: Towards an efficient oxygen evolution reaction, Green Chem., 2023, 25, 8047–8056 RSC.
- M. Priyadarshini, A. Ahmad and M. M. Ghangrekar, Efficient upcycling of iron scrap and waste polyethylene terephthalate plastic into Fe3O4@C incorporated MIL-53(Fe) as a novel electro-fenton catalyst for the degradation of salicylic acid, Environ. Pollut., 2023, 322, 121242 CrossRef CAS PubMed.
- B. Ravel and M. Newville, Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- H. Funke, A. C. Scheinost and M. Chukalina, Wavelet analysis of extended x-ray absorption fine structure data, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094110 CrossRef.
- X. Yuan, S. Li, S. Jeon, S. Deng, L. Zhao and K. B. Lee, Valorization of waste polyethylene terephthalate plastic into N-doped microporous carbon for CO2 capture through a one-pot synthesis, J. Hazard. Mater., 2020, 399, 123010 CrossRef CAS PubMed.
- X. M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M. M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Selective CO2 reduction to CO in water using earth-abundant metal and nitrogen-doped carbon electrocatalysts, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
- H. B. Yang, S. F. Hung, S. Liu, K. D. Yuan, S. Miao, L. P. Zhang, X. Huang, H. Y. Wang, W. Z. Cai, R. Chen, J. J. Gao, X. F. Yang, W. Chen, Y. Q. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Atomically dispersed Ni(I) as the active site for electrochemical CO2 reduction, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- H. H. Peng, J. Cao, W. P. Xiong, Z. H. Yang, M. Y. Jia, S. W. Sun, Z. Y. Xu, Y. R. Zhang and H. C. Cai, Two-dimension N-doped nanoporous carbon from KCl thermal exfoliation of Zn-ZIF-L: Efficient adsorption for tetracycline and optimizing of response surface model, J. Hazard. Mater., 2021, 402, 123498 CrossRef CAS PubMed.
- W. Ren, X. Tan, C. Jia, A. Krammer, Q. Sun, J. T. Qu, S. C. Smith, A. Schueler, X. L. Hu and C. Zhao, Electronic regulation of nickel single atoms by confined nickel nanoparticles for energy-efficient CO2 electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202203335 CrossRef CAS PubMed.
- H. Wang, H. Y. Chuai, X. Y. Chen, J. L. Lin, S. Zhang and X. B. Ma, Self-supported porous carbon nanofibers decorated with single Ni atoms for efficient CO2 electroreduction, ACS Appl. Mater. Interfaces, 2023, 15, 1376–1383 CrossRef CAS PubMed.
- H. P. Yang, Q. Lin, C. Zhang, X. Y. Yu, Z. Cheng, G. D. Li, Q. Hu, X. Z. Ren, Q. L. Zhang, J. H. Liu and C. X. He, Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- C. M. Lv, K. Huang, Y. Fan, J. Xu, C. Lian, H. L. Jiang, Y. Z. Zhang, C. Ma, W. M. Qiao, J. T. Wang and L. C. Ling, Electrocatalytic reduction of carbon dioxide in confined microspace utilizing single nickel atom decorated nitrogen-doped carbon nanospheres, Nano Energy, 2023, 111, 108384 CrossRef CAS.
- Q. Chen, X. Q. Wang, Y. Z. Zhou, Y. Tan, H. M. Li, J. W. Fu and M. Liu, Electrocatalytic CO2 reduction to C2+ products in flow cells, Adv. Mater., 2024, 36, 2303902 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00244j |
| This journal is © The Royal Society of Chemistry 2024 |