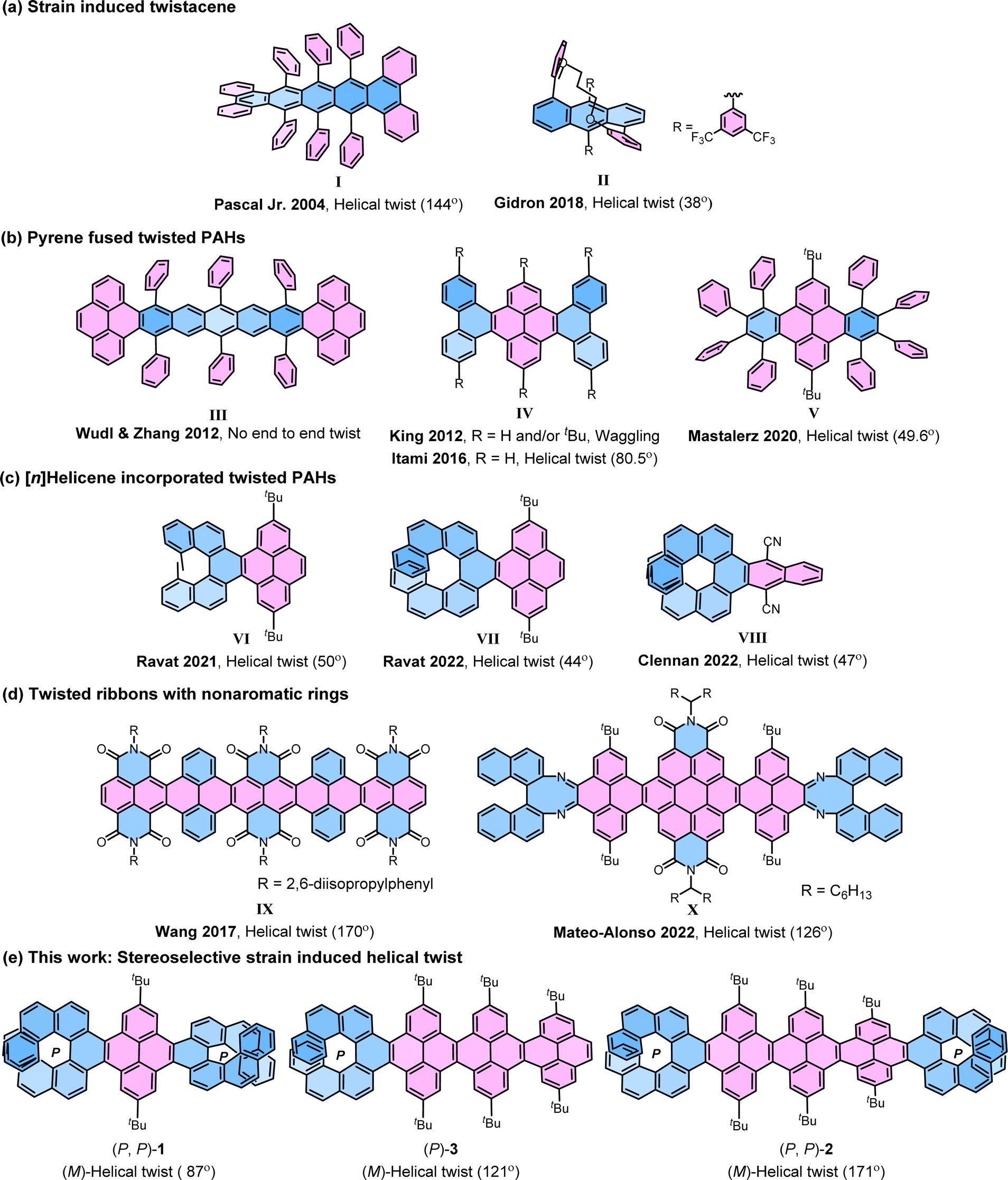DOI:
10.1039/D4SC01814A
(Edge Article)
Chem. Sci., 2024,
15, 11737-11747
Helically twisted nanoribbons via stereospecific annulative π-extension reaction employing [7]helicene as a molecular wrench†
Received
18th March 2024
, Accepted 15th May 2024
First published on 23rd May 2024
Abstract
Over the past decade, significant progress has been made in synthesizing atomically precise carbon nanostructures, particularly graphene nanoribbons (NRs), employing advanced synthetic methodologies. Despite these advancements, achieving control over the stereochemistry of twisted NRs has proven to be a formidable challenge. This manuscript presents a strategic approach to achieve absolute control over the single-handed helical conformation in a cove-edged NR. This strategy leverages enantiopure helicenes as a molecular wrench, intricately influencing the overall conformation of the NR. [7]helicenes stitched to the terminal K-regions of a conjugated pyrene NR through a stereospecific annulative π-extension reaction to produce a helically twisted NR with an end-to-end twist of 171°. Furthermore, a detailed investigation of the impact of twisting on the conformational population was studied by quantum chemical calculations.
Introduction
The electronic properties of graphene nanoribbons (NRs)1–8 composed of linearly fused polycyclic aromatic hydrocarbons (PAHs) depend on their size, shape, and most importantly, edge structures. Based on the edges NRs can be classified as cove-edge, armchair-edge, and zigzag-edge NRs.9–13 The cove-edge NRs14 are of special interest as they have the potential to be chiral as a result of the non-planarity arising from the steric hindrance in the cove regions. Cove-edged NRs can adopt a twisted configuration, whether helical or waggling (randomly twisted), contingent upon specific steric congestion along their edges.15–18 However, they suffer from low configurational stability and the minimal relative energy difference between helical and waggling conformers due to the rapid flipping of the inner cove's chirality.14,19,20 The NRs with fjord regions—such as supertwistacene21 by Wang et al. and triply conjugated HBC (hexa-peri-hexabenzocoronene)22 by Campana et al.—exhibit a slightly higher barrier, allowing room temperature chiral resolution. The nanographenes with bay regions are relatively difficult to twist, as the majority of the rings lie flat in the orthogonal plane with limited options for substitution. Chalifoux et al. achieved a 35° end-to-end twist by substituting only one side of the bay region,23 while Würthner et al. substituted all positions in the bay region, resulting in an enhanced twist of 76° in quaterrylene bisimide with an enantiomerization barrier of 30 kcal mol−1.24
The strategy of strain-induced twist has been widely applied to achieve longitudinally twisted acenes (‘twistacenes’), the narrowest NRs, which can be manipulated into helical or waggling conformations through the strategic application of crowded substitutions or benzyl annulations (Fig. 1).4,25–27 In the late 1990s, Pascal Jr et al. pioneered the twisting of acenes by employing bulky phenyl substitutions reaching an astonishing end-to-end twist of 144° (I).28–30 This record was surpassed by Kilway et al. in 2018 with the hexacene derivative, achieving an end-to-end twist of 184°.31 In twistacenes, the strain-induced helical twist propagates along the aromatic ring planes. However, often twistacenes owing to the low configurational stability cannot be resolved into enantiomers. Gidron et al. engineered helically locked tethered anthracenes (II), which can be isolated in an enantiopure form, achieving an end-to-end twist of up to 38°.32,33 Several attempts were made to incorporate promising π-electron-rich cores, such as pyrenes34 into twisted acenes. In most cases, the attachment takes place at the K-region of the pyrene, as it offers greater accessibilities compared to other reaction sites of pyrene. Wudl and Zhang developed a molecule (III) containing a pentacene core terminally locked by two pyrenes.35 However, it failed to propagate a uniform twist from one end to the other as the twist reverses at the central benzene ring due to the absence of steric groups at crucial positions in the central acene. King et al. prepared a K-region phenanthrene annulated pyrene exhibiting waggling conformation (IV).36 In contrast, later Itami et al. achieved the helical conformation of IV with an unsubstituted core.37 Lately, eight phenyl substituted dibenzo[e, l]pyrene (V) by Mastalerz et al. showed the helical structure with an end-to-end twist of 49.6°.38 Recently, we introduced [n]helicene as a strain-inducing tensor for generating twisted acene core within a pyrene-fused [n]helicene moiety, achieving end-to-end twist of 50° by incorporating methyl substituted [5]helicene at the K-region of pyrene (VI) and a 44° end-to-end twist upon using [7]helicene (VII).39–41 The helicity of twisted core was dictated by the helicity of the attached [n]helicene, which was opposite to each other. This approach was then followed by Clennan to produce a 47° end-to-end twist in a [7]helicene-incorporated anthracene (VIII).42 Mateo-Alonso et al. consequently employed this concept in making a pyrene-coronene cored helical nanoribbon (X), where enantiopure 1,1′-binaphthyl-2,2′-diamine was fused to form [5]helicenoid at both terminal ends containing distorted octagonal rings with four cove region [4]helicene subunits achieving an end-to-end twist of 126° for the central core.43 It is worth mentioning that, similar to X, Wang et al. in 2017 reported a helically twisted decatwistacene (IX) with a much higher end-to-end torsion twist of 170° which was achieved solely through steric hindrance between imide groups and benzene rings in the cove region, without the use of any external steric tensor such as [n]helicinoids, as shown in X.44 The axially chiral binaphthyl moiety within the ribbon X is not fully conjugated and possesses multiple nonaromatic rings with two nitrogen atoms, as well as two imide moieties. Furthermore, in this ribbon, the cove-region gained additional configurational stability from the buttressing effect45 of imide moieties. The conformation of X was susceptible to temperature fluctuations because of the dynamic nature of the cove regions. Hence, the challenge of developing a sturdy cove-edged hydrocarbon nanoribbon with a precisely stable conformation remains unaddressed.
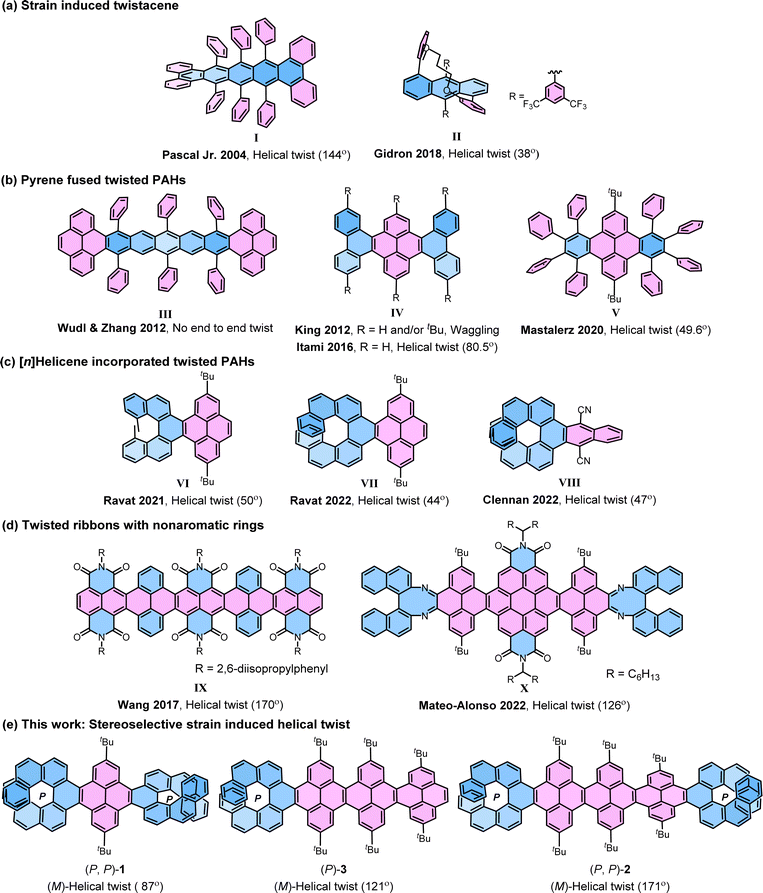 |
| | Fig. 1 Selected previous examples of twisted molecules (a)–(d) and newly synthesized helically twisted NRs (e). The helical end-to-end twist is reported in parenthesis. | |
Earlier investigations into strain-induced twisting suffered from limitations in controlling stereochemistry and low configurational stability, making it a challenging endeavor to synthesize helically twisted chiral hydrocarbon NRs in a stereospecific manner. To address this challenge, we aimed to exert precise control over the twist of cove-edged NRs by integrating configurationally stable enantiopure [n]helicenes46–49 at the terminals preceding our earlier work where we attain 99.5% conformational stability for VII. The incorporation of enantiopure [n]helicene on both ends of NRs gives rise to three potential stereoisomers: left-handed, right-handed, or waggling. In this study, we demonstrate that the overall conformation and twist of cove-edged NRs can be systematically adjusted by employing terminal helicene moieties, effectively acting as a molecular wrench. This article presents a strategic methodology focused on achieving helically twisted chiral nanoribbons, featuring a central pyrene core NR, securely anchored at terminal K-regions through one or two [7]helicenes via stereospecific APEX reactions (Fig. 1e). The influence of [7]helicene on stabilizing the conformers was explored using DFT-optimized structures and single-crystal structure analysis. The (chir)optical properties were comprehensively examined through UV-Vis absorption, emission, electronic circular dichroism (ECD), and circularly polarized luminescence (CPL) spectroscopies. Furthermore, the inversion barrier of compound 1 was estimated through kinetic measurements. The experimental findings were effectively correlated with quantum chemical calculations, providing a comprehensive understanding of the synthesized helically twisted chiral nanoribbons and their intriguing properties.
Results and discussion
Synthesis and characterization
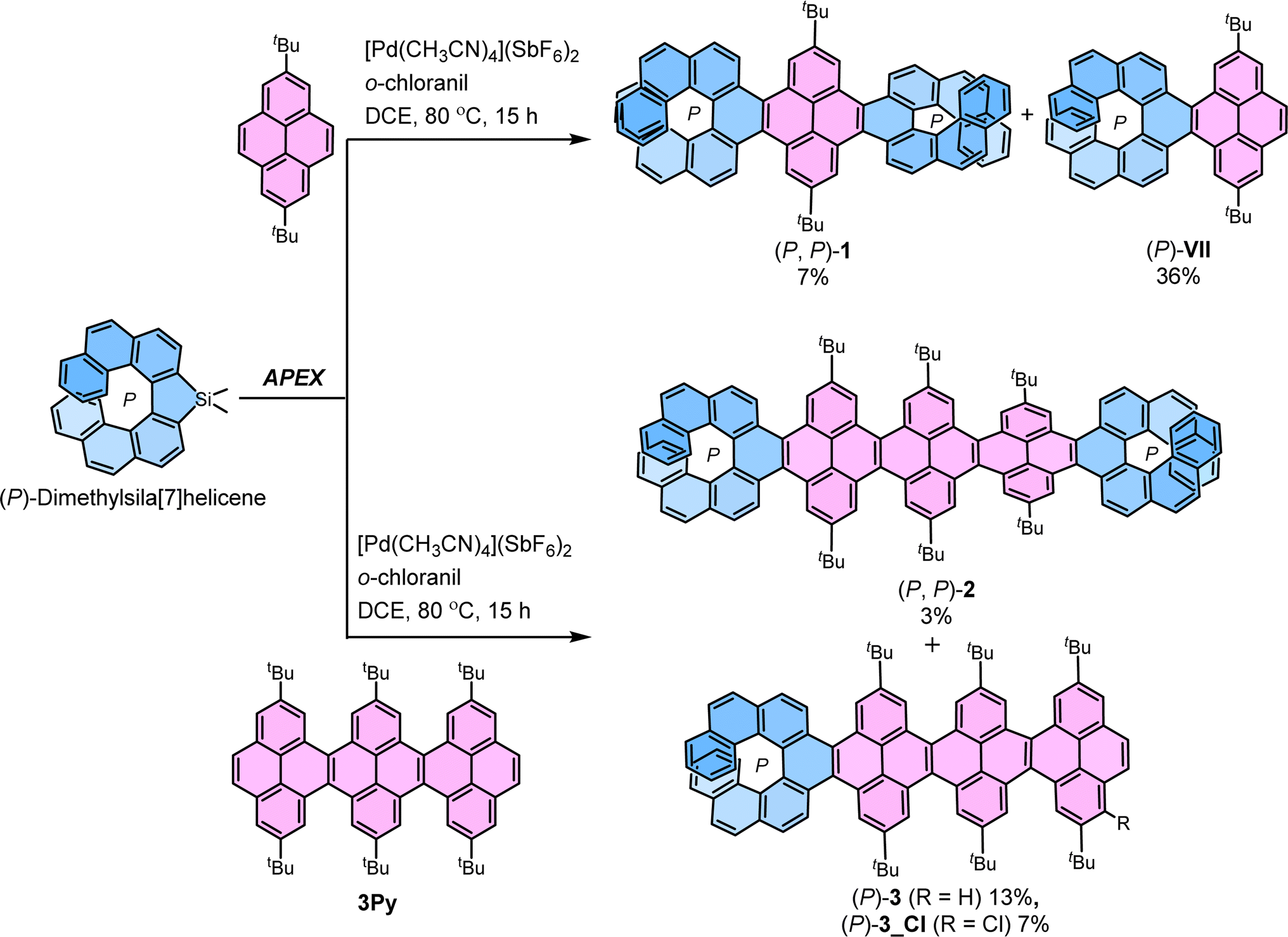
The most straightforward instance of cove-edged nanoribbons (NRs) involves a linearly fused pyrene connected at the K-region. In this study, a central NR comprising three linearly fused pyrene units (3Py) was selected. The peri-fused benzene rings in pyrene served to anchor essential points necessary for transmitting twists from one end to the other. Recently, we developed a stereospecific method for incorporating [n]helicene at the K-region of pyrene through a one-pot Suzuki coupling – C–H activation process.39–41 Initially, we applied the same strategy to synthesize (P, P)-1 by conducting a one-pot Suzuki coupling – C–H activation reaction between (S)-3,3-dibromo-4,4′-biphenanthrene40 (ee > 99%) and 2-(2,7-di-tert-butylpyren-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane.50 (P, P)-1 was successfully obtained with a moderate 35% yield, considering two Suzuki coupling and two C–H activation reaction steps (ESI Section S2†). However, this approach proved unsuitable for synthesizing 2 and 3 due to inaccessibility of required pyrene-NR (3Py) with boronic ester group. To surmount these obstacles, we adopted the K-region specific annulative π-extension (APEX) reaction developed by Ito and Itami (Scheme 1).51
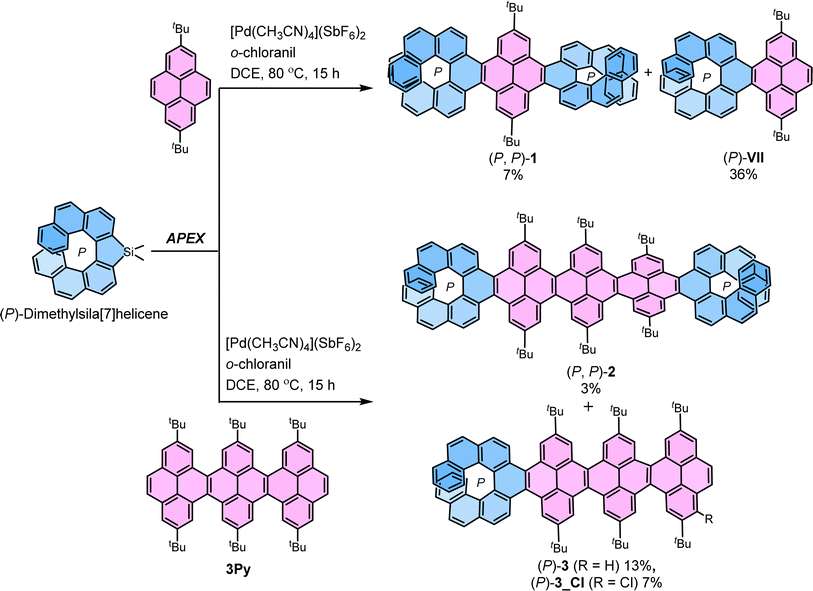 |
| | Scheme 1 Synthesis of (P, P)-1, (P, P)-2 and (P)-3via stereospecific APEX reaction. | |
(P, P)-1 and (P, P)-2 were synthesized from 2,7-di-tert-butylpyrene and 3Py,52 and (P)-dimethylsila[7]helicene (ee > 99%),53 through a palladium-catalyzed double C–H/C–Si coupling (K-region APEX reaction).51 A mixture of the respective pyrene precursor and (P)-dimethylsila[7]helicene was heated at 80 °C for 15 hours in the presence of [Pd(CH3CN)4](SbF6)2 and o-chloranil, producing the double annulative (P, P)-1 and (P, P)-2 with stereospecificity. The second competitive addition of [7]helicene occurs selectively at the K-region of the pyrene possibly due to its high olefinic character,54 compared to the two K-regions of the first annulated [7]helicene where aromatic character is dominant.51 This selectivity can be rationalized by the presence of neighboring conjugated Clar's sextet. In the K-region of pyrene, four peri-fused benzene rings shared two Clar's sextets, whereas in the K-region of [7]helicene, it conjugates with two neighboring Clar's sextets. The APEX reactions also produced single annulated (P)-3 and (P)-VII (ref. 40) with moderate yield. Similarly, (M, M)-1 and (M, M)-2 were synthesized from (M)-dimethylsila[7]helicene in comparable yields and enantiopurity. The identical enantiopurity of 1 (ee > 99%) in APEX reaction and Suzuki coupling–C-H activation confirms the stereospecificity of the APEX reaction (ESI Fig. S1†). To the best of our knowledge, these are the first examples of site-selective and stereospecific APEX reactions, as the enantiomeric excess (ee) was retained throughout the reaction scheme.55 The structures of 1, 2 and 3 were confirmed by unambiguous assignment of 1H and 13C peaks to the respective atoms by COSY, NOESY, HSQC, and HMBC NMR measurements (ESI Fig. S10†). Additionally, the structure of 1 and 3Py was determined by single-crystal X-ray diffraction (ESI Fig. S9†).
Pyrene bridged double [7]helicene
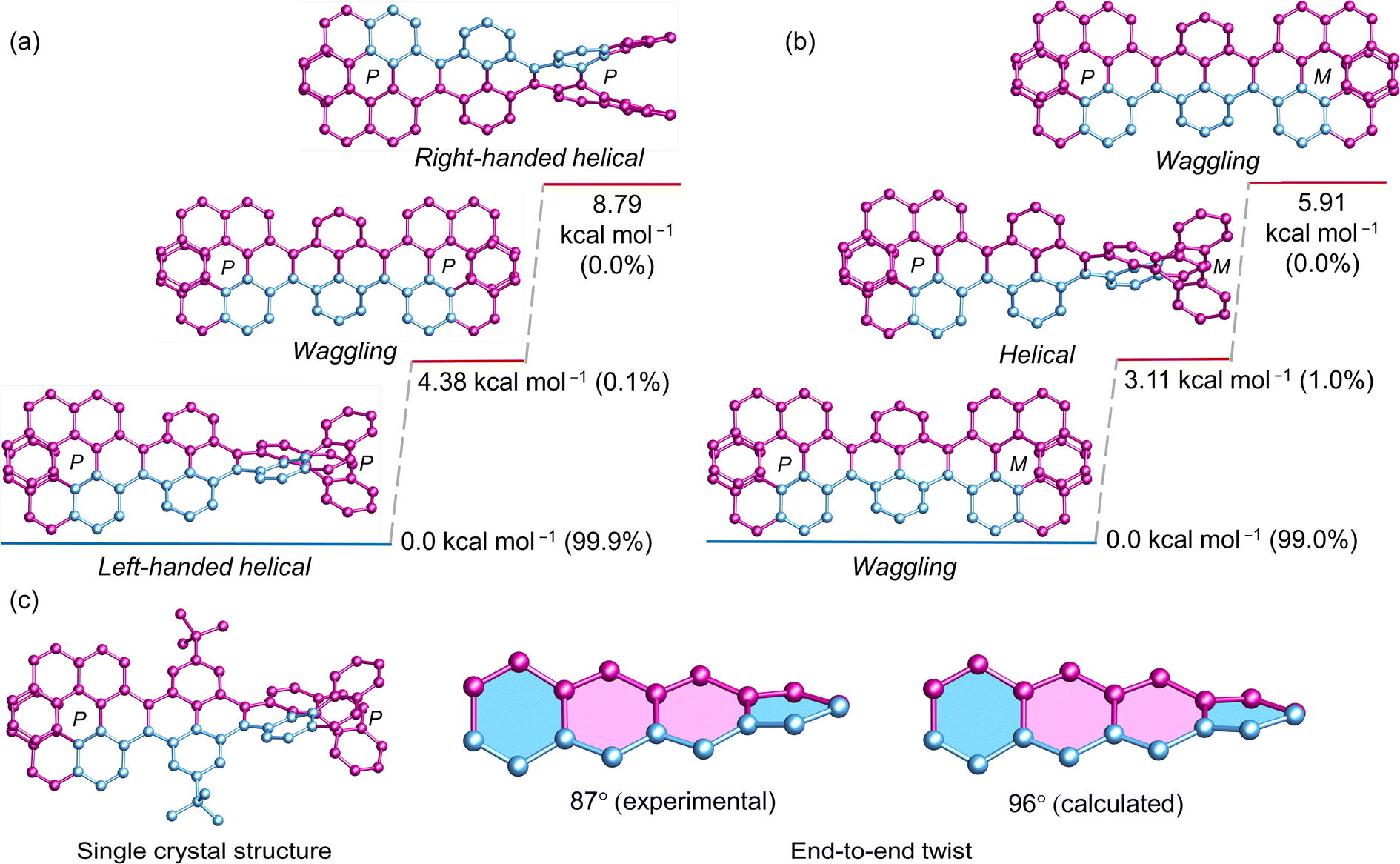
Before exploring longer fused pyrene NRs, an extensive study was conducted on conformational stability of pyrene bridged double [7]helicene (1). DFT optimized geometry and energy level analyses of (P, P)-1 revealed that the diastereomer with an opposite chirality in the cove region to [7]helicene (left-handed helical conformation) is notably most stable, constituting a relative Boltzmann population of 99.9% (Fig. 2a). The remaining 0.1% encompasses the higher energy waggling conformation of (P, P)-1. The theoretical calculations were substantiated by the obtained single crystal structure of (P, P)-1 (Fig. 2c), where all four [4]helicene subunits adopt the M conformation. The resemblance between the experimental (87°) and calculated (96°) end-to-end twists confirm the precision of the DFT optimized structures. In the case of (P, M)-1, pyrene bridged by two [7]helicenes with opposite chirality, the scenario reverses, just the opposite of (P, P)-1. The waggling conformation with opposite helicity to [7]helicene in the cove is the most stable, representing 99% of the relative population (Fig. 2b). The remaining 1% comprises the helical conformation. Notably, in both (P, P)-1 and (P, M)-1, the conformer with similar helicity in the cove and adjacent [7]helicene represents the highest energy conformation, with an almost negligible relative population.
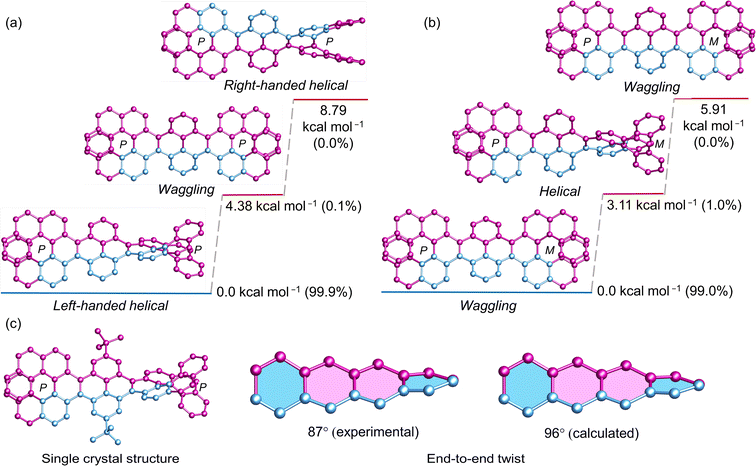 |
| | Fig. 2 DFT (ωB97XD/6-31G(d,p)) calculated conformational isomers of (a) (P, P)-1, (b) (P, M)-1 with relative total energy and population at 298 K. (c) Single crystal structure of (P, P)-1. Hydrogen atoms in all and tert-butyl groups in calculated structures are omitted for clarity. | |
The 99.9% conformational stability of (P, P)-1 allow us to assess the configurational stability, unlike the dynamic nanoribbon (VII) reported by Mateo-Alonso et al. where mixture of conformations exists at variable temperature for fixed helicity of the [5]helicenoid.43 (P, P)-1 was heated at elevated temperature to observe formation of other isomers. Upon heating two new peaks appeared in HPLC, which were assigned to (M, M)-1 and (P, M)-1 (ESI Fig. S2a†). Using Eyring equation the ΔH and ΔS values were calculated to be 44.1 kcal mol−1 and 21.4 cal K−1 mol−1, respectively (ESI Fig. S3b†). Accordingly, the Gibbs activation energy ΔG‡ (298 K) for diastereomerization of 1 was calculated to be 37.7 kcal mol−1, lower than the enantiomerization barrier of pristine [7]helicene (41.2 kcal mol−1).49,56,57 It should be noted that, both ΔH and ΔS for the diastereomerization of 1 are significantly higher than those for the enantiomerization of the [7]helicene (ΔH = 40.4 kcal mol−1, ΔS = −2.8 cal K−1 mol−1).49 Hence the decreased ΔG‡ for 1 can be rationalized for the much higher and positive ΔS value—an entropically favored process. ΔG‡ (438 K) of 1 (35.3 kcal mol−1) is slightly higher than that of VII (34.1 kcal mol−1)40 indicating a more ordered conformation upon locking the structure with the second [7]helicene.58 Upon prolonged heating (P, P)-1 reaches to a diastereomeric equilibrium of (P*, P*)-1 to meso (P, M)-1 at a ratio of 3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI Fig. S1 and S2†), similar to the ratio (3.8:1) obtained from racemic reaction (ESI Fig. S1†). This is well in accordance with the calculated relative population of two diastereomers (3.8:1) by DFT.
:1 (ESI Fig. S1 and S2†), similar to the ratio (3.8:1) obtained from racemic reaction (ESI Fig. S1†). This is well in accordance with the calculated relative population of two diastereomers (3.8:1) by DFT.
Conformation isomers of conjugated cove-edged NR (3Py)
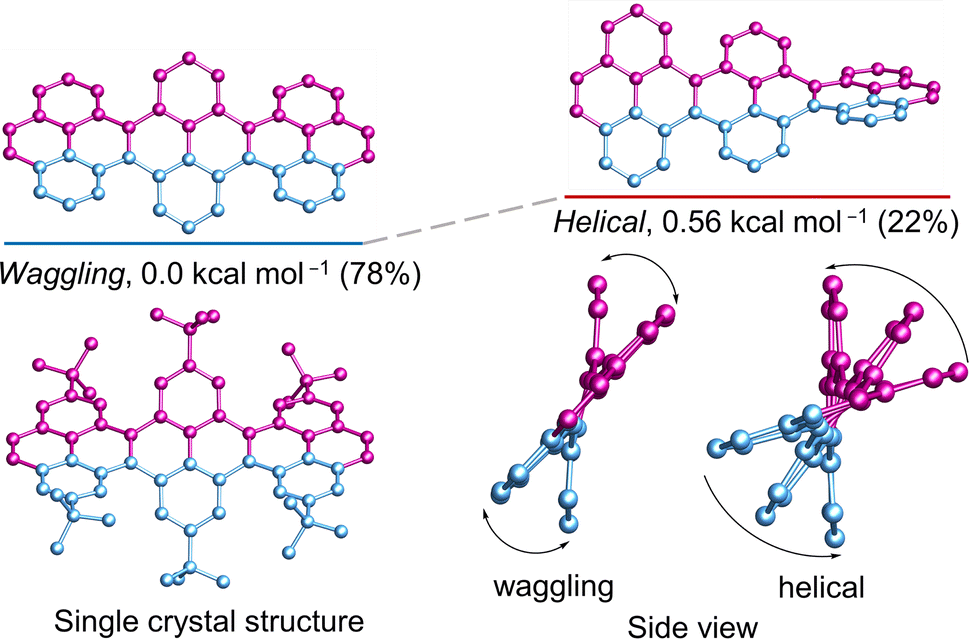
The cove-edged nanoribbons showcase a nonplanar geometry primarily attributed to steric repulsion between two hydrogen atoms positioned at the inner core of recurring [4]helicene units, resulting in tilted upward and downward topologies. The conformational stability of 3Py was evaluated through DFT calculations (Fig. 3). At room temperature, the waggling conformer of 3Py prevails as the most stable, constituting 78% of the population, while the remaining portion corresponds to the helical conformer. The calculations were substantiated by the obtained single crystal of 3Py, revealing a waggling twist. Notably, there is no net end-to-end twist observed, as the twist from one terminal to the other end does not continue consistently but instead reverses the twist direction in the middle pyrene. The rapid flipping of adjacent pyrene in waggling twist is evident from the 1H NMR experiment, where all tert-butyl groups exhibit only one distinct sharp singlet at 1.63 ppm (ESI Fig. S15†).
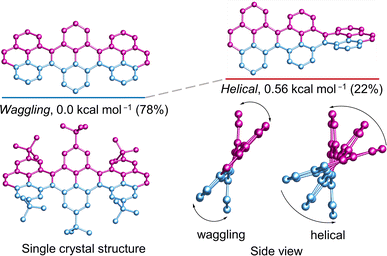 |
| | Fig. 3 DFT (ωB97XD/6-31G(d,p)) calculated conformational isomers and single crystal structure of 3Py with relative total energy and population at 298 K. Hydrogen atoms in all and tert-butyl groups in calculated structures are omitted for clarity. | |
Effect of one terminal [7]helicene on conformational stability of cove-edged NR
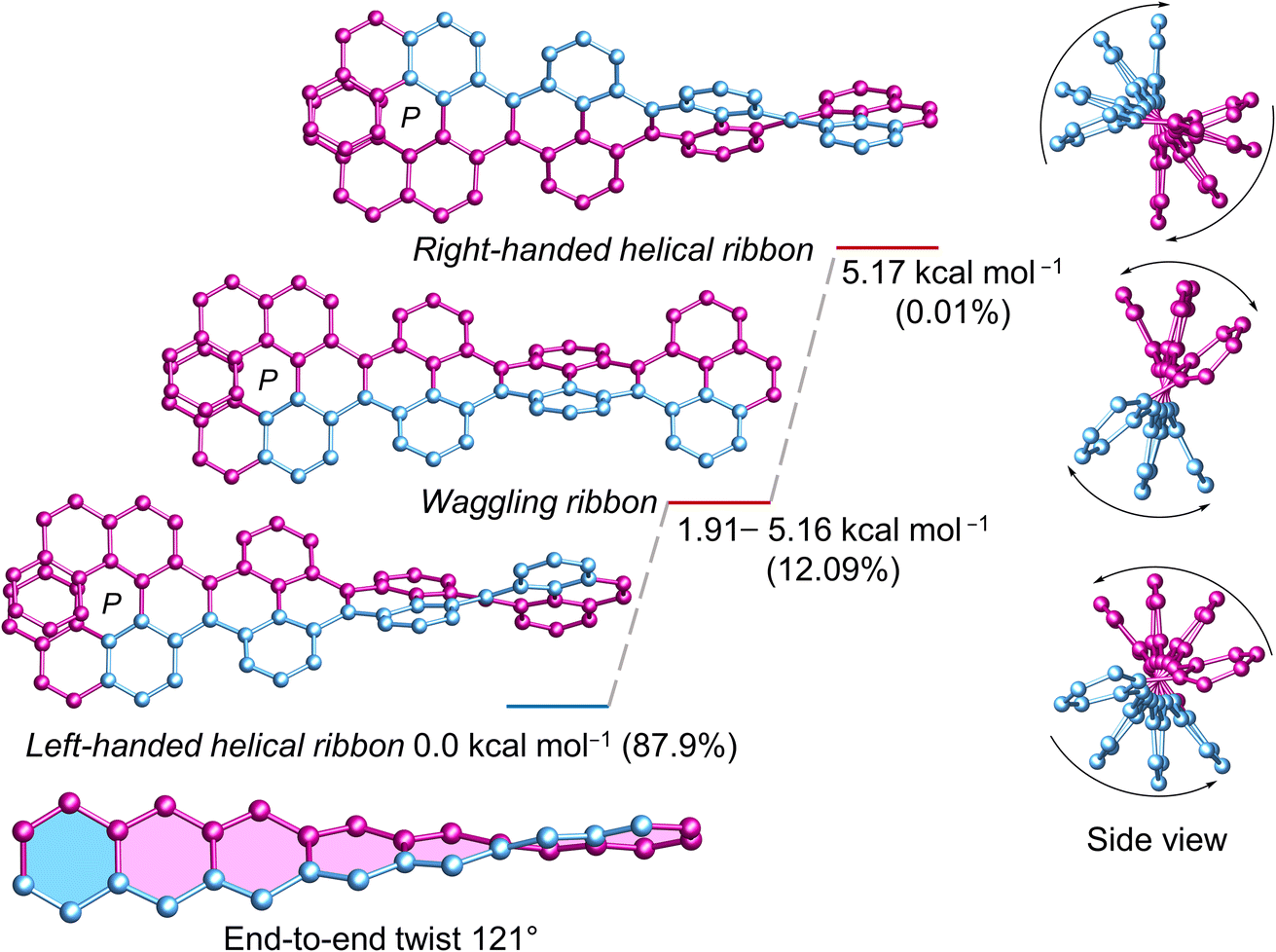
Next, we investigated the influence of one (P)-[7]helicene unit on the conformation of 3Py. Although (P)-3 appears as flexible as 3Py, the left-handed helical conformation emerges as the most stable, constituting a relative population of 87.9% (Fig. 4). Conversely, the lowest waggling conformer, slightly higher in energy (1.91 kcal mol−1), accounts for a relative population of 6.3%, while combined population of all four waggling conformers amounts to 12.09%. The highest energy right-handed helical conformer (5.17 kcal mol−1) comprises only 0.01% of the population. Despite numerous attempts in various solvent combinations and at low temperatures, obtaining a single crystal of (P)-3 remained elusive. However, the twisted structure of the C2 symmetric (P)-3 is discernible in the 1H NMR, where all three sets of tert-butyl groups exhibit three distinct singlet signals, each corresponding to 18 protons (ESI Fig. S32†). Consequently, rapid flipping of adjacent pyrenes in the waggling conformation just as 3Py is not applicable to (P)-3. The end-to-end twist of (P)-3 measures 121°, which is 34° higher than that of (P, P)-1.
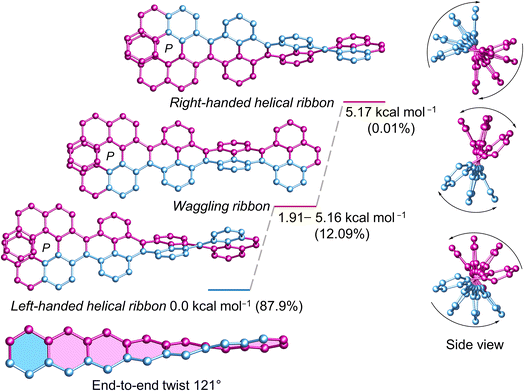 |
| | Fig. 4 DFT (ωB97XD/6-31G(d,p)) calculated conformational isomers of (P)-3 with relative total energy and population at 298 K. The lowest energy waggling conformation of (P)-3 is depicted here. Refer to ESI Section S6† for additional waggling conformations. Hydrogen atoms and tert-butyl groups are omitted for clarity. | |
Effect of two terminal [7]helicenes on conformational stability of cove-edged NR
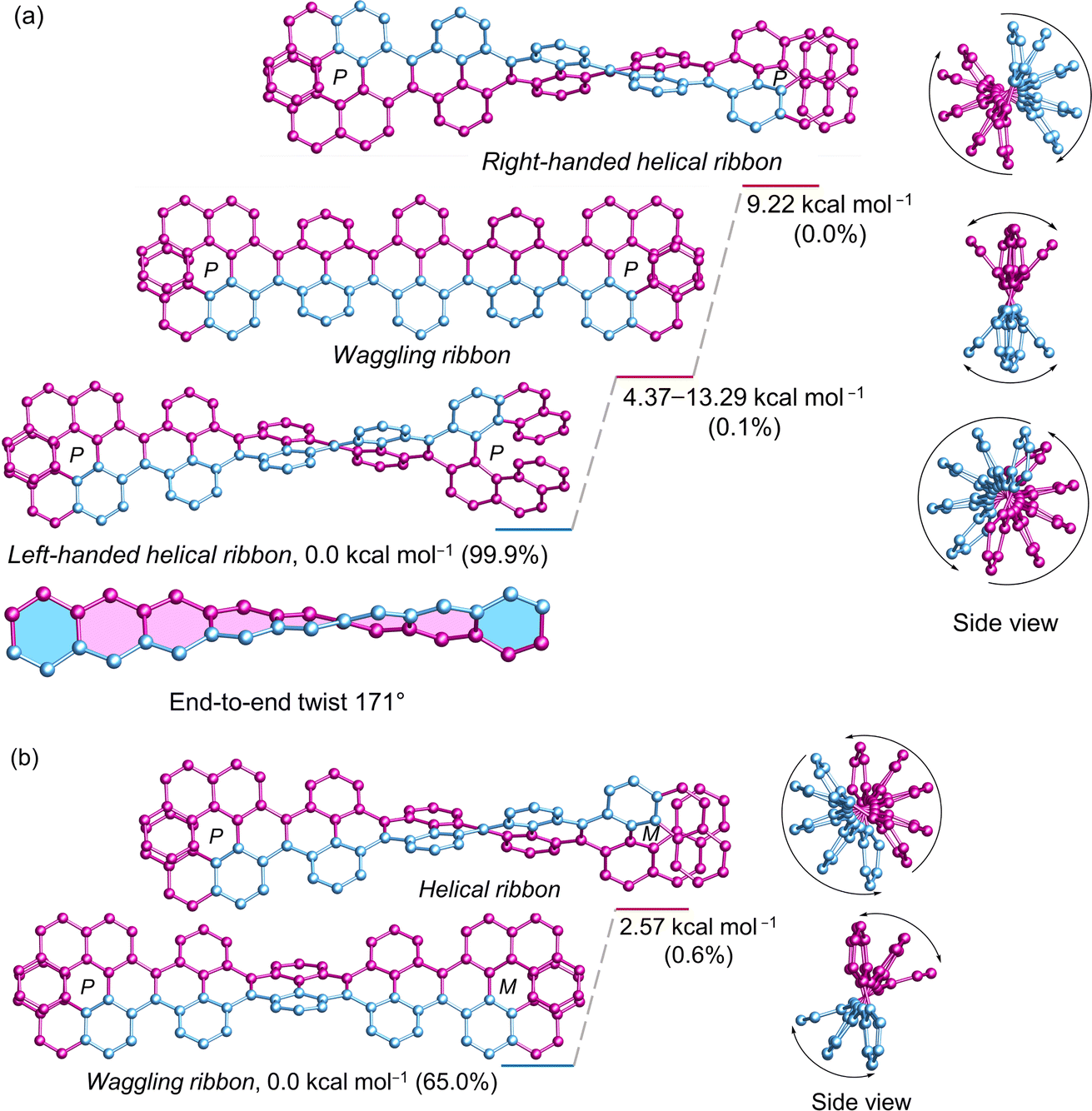
The (P)-[7]helicene-locked nanoribbon (P, P)-2 follows a similar trend as (P)-3 and (P, P)-1 established by DFT calculated energies. The left-handed helical nanoribbon represents the most stable conformer with a relative population of 99.9%, while the waggling conformers account for the remainder (Fig. 5a). The right-handed helical ribbon represents the highest energy conformer (9.22 kcal mol−1) with a negligible population. Notably, upon locking both terminal K-regions by (P)-[7]helicene in 2,7-di-tert-butyl pyrene and 3Py, the left-handed helical ribbon becomes stabilized by 4.37–4.38 kcal mol−1 from the lowest waggling conformer and by 8.79–9.22 kcal mol−1 from the right-handed helical ribbon. In the case of (P, M)-2, where helicenes of opposite chirality attached to the terminal of NR, the waggling conformer is stable by 2.57 kcal mol−1 from the helical conformer, constituting a relative population of 65% (Fig. 5b). Although (P, P)-2 possesses twice the number of labile [4]helicene subunits in comparison to the pyrene-coronene nanoribbon (X) described by Mateo-Alonso et al.,43 its left-handed helical conformer shows significantly greater stability than the waggling conformers, comprising a relative population of 99.9% versus 93.8%. The end-to-end twist of (P, P)-2 measures 171° making it as the second-most twisted central acene core following the twisted hexacene reported by Kilway et al.31 The end-to-end twist in (P, P)-2 is nearly double that of (P, P)-1 and increased significantly from (P)-3 demonstrating that the terminal [7]helicenes act as a molecular wrench in maintaining the twist. The average torsion angle per benzene ring4 measures 21.75° and 21.38° in (P, P)-1 and (P, P)-2, respectively, in contrast to 17.3° in (P)-3 and 14.8° in (P)-VII, which highlights the increased strain resulting from the conformational lock imposed by the second [7]helicene. The distance between the terminal benzene centroids of [7]helicene substructure decreased significantly from 4.13 Å in (P, P)-1 to 3.77 Å in (P, P)-2, while the torsional twist slightly increased from 22.4° (1) to 22.9° in 2. This highlights the additional strain imposed by the [7]helicene in (P, P)-2 to maintain such a high level of twist in the central core. Regardless of its size (∼3.1 nm), and the extensive aromatic core comprising 100 carbon atoms, (P, P)-2 demonstrates excellent solubility in a wide range of organic solvents, owing to the highly twisted structure.
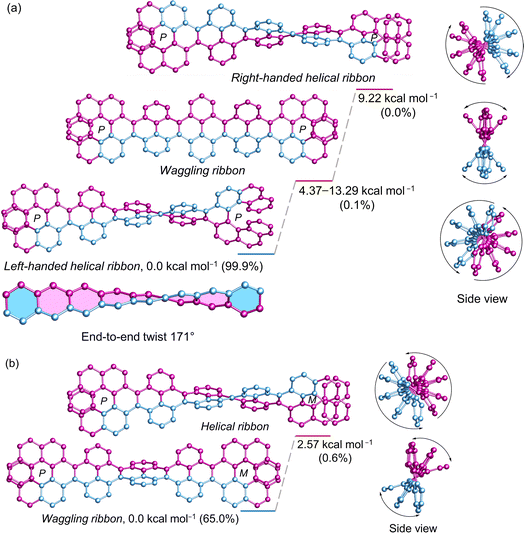 |
| | Fig. 5 DFT (ωB97XD/6-31G(d,p)) calculated conformational isomers of (a) (P, P)-2 and (b) (P, M)-2 with relative total energy and population at 298 K. The lowest energy waggling conformation of 2 is depicted here. Refer to ESI Section S6† for additional waggling conformations. Hydrogen atoms and tert-butyl groups are omitted for clarity. | |
Nucleus-independent chemical shift calculations
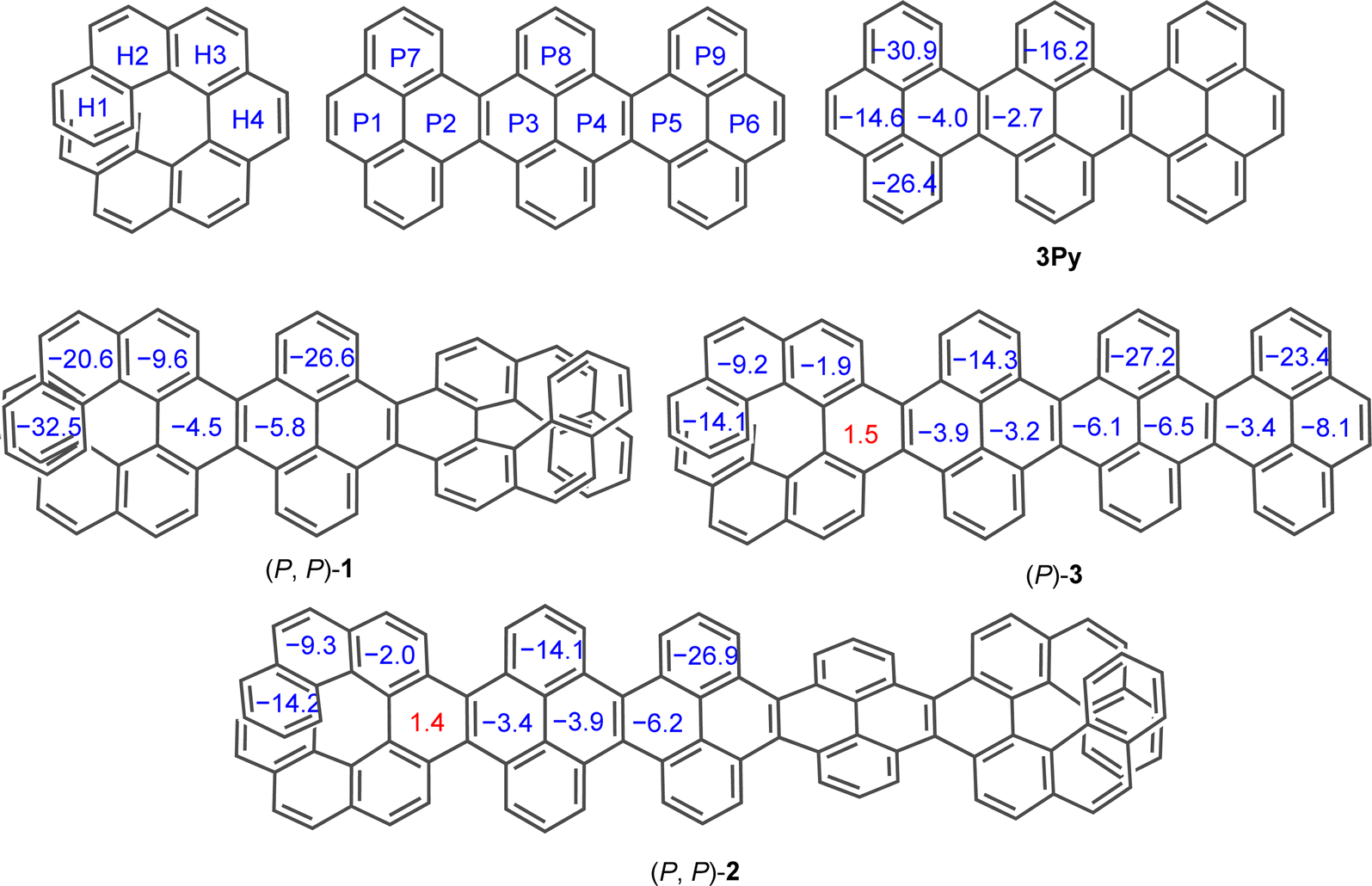
To evaluate the effect of twisting on the aromaticity, the nucleus-independent chemical shift (NICS) values were computed for all discussed molecules (Fig. 6). All benzene rings exhibited negative NICS(1)ZZ values, indicative of their aromaticity, except for H4 ring in (P)-3 and (P, P)-2. The marginally positive value NICS(1)ZZ for ring H4 in (P)-3 and (P, P)-2 corroborates to its highly distorted structure compared to other rings. In the [7]helicene subunit of NRs, the aromaticity increased on moving away from the central rings (H4 to H1) similar to pristine [7]helicene.59,60 Aromaticity within the acene core (P1 to P6) decreases on moving away from terminal rings into the central core in 3Py. However, this trend reverses in (P, P)-1 and (P, P)-2 as terminal rings (P1 or P6) show smaller NICS(1)zz values with a gradual increase to the inner core. Away from the centrally tethered twisting, the P7–P9 rings in the pyrene exhibit relatively higher negative NICS(1)ZZ values, as do the H1 and H2 rings of the [7]helicene. In general, the NICS(1)ZZ values of the waggling conformer of 1 and 2 are significantly higher than those of the left-handed helical conformer of (P, P)-1 and (P, P)-2, likely due to absence of an end-to-end twist (ESI Table S6†).
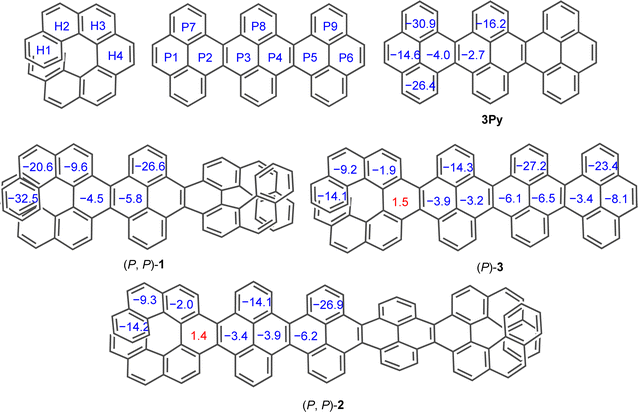 |
| | Fig. 6 Calculated (GIAO-B3LYP/6-311+G(2d,p)) NICS(1)ZZ values for 3Py, (P, P)-1, (P, P)-2 and (P)-3. tert-Butyl groups are omitted for clarity. | |
Chiroptical properties
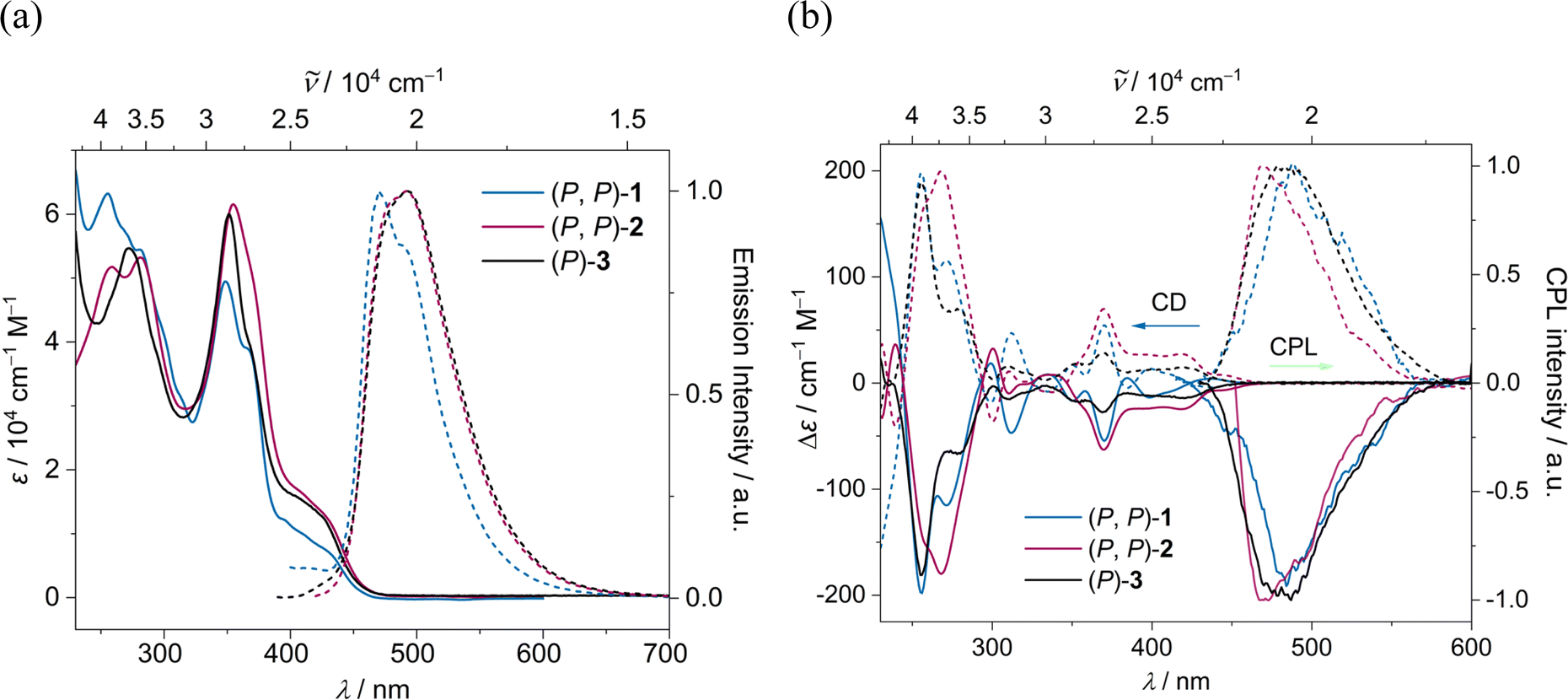
The absorption and emission spectra of (P, P)-2 and (P)-3 closely resemble each other, with a slight red shift compared to (P, P)-1, due to an increase in π-conjugation (Fig. 7a). The optical energy gap for (P, P)-1, (P, P)-2, and (P)-3 falls within the range of 2.79–2.83 eV, notably lower than that of [7]helicene (3.06 eV)61 and 3Py (2.92 eV) (ESI Table S2†). TD-DFT (B3LYP/6-31g(d,p)) calculations revealed that for all three compounds the lowest energy absorption band mainly stemming from HOMO → LUMO transition with similar oscillator strength of 0.13–0.14 (ESI Table S5†). The emission maximum for (P, P)-1, (P, P)-2 and (P)-3 were recorded at 473, 492 and 493 nm respectively with fluorescence quantum yields (FQYs) of 0.05, 0.15 and 0.19 in dichloromethane. The decrease in FQY of (P, P)-2 compared to (P)-3 can be attributed to twist-enhanced inter system crossing as observed in twisted acenes.62,63 The fluorescence decay lifetimes of (P, P)-1, (P, P)-2, and (P)-3 range between 5.07–5.55 ns, significantly shorter than that of [7]helicene (13.8 ns).64
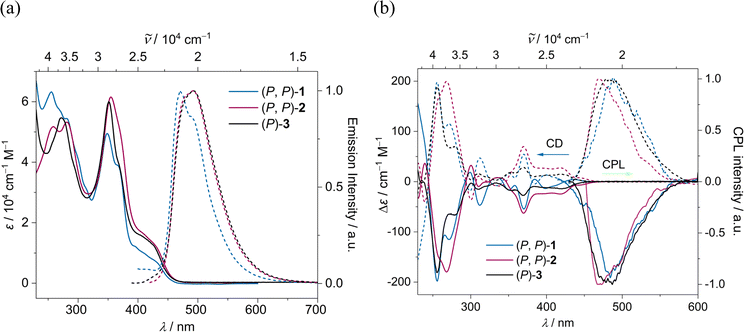 |
| | Fig. 7 (a) UV-Vis absorption (solid line) and emission (dashed line) spectra and (b) ECD and CPL spectra of (P, P) (solid line) and (M, M) (dashed line) of 1 (blue), 2 (red) and 3 (black) in DCM (c ∼ 10−5 M). | |
The absolute configuration of the enantiomers of 1, 2, and 3 was assigned comparing experimental and TD-DFT calculated CD spectra (ESI Fig. S8†). The lowest energy CD signals are stronger for 2, followed by 1 and 3 respectively (Fig. 7b). The experimentally obtained luminescence dissymmetry factor (glum) of 2 is 1.54 × 10−3, nearly three times that of 3 (0.63 × 10−3) and 1 (0.54 × 10−3) (ESI Fig. S7†). The increase in chiroptical response of 2 can be attributed to the increased twist in the NR core, resulting in macro chirality. This was further supported by TD-DFT calculations, which showed enhanced excited state magnetic transition dipole moment and higher cosθ values for 2 compared to those for 1 and 3 (ESI Table S3†). In contrast to dynamic pyrene-coronene NR (X) reported by Mateo-Alonso et al.,43 the variable temperature (278–333 K) ECD and CPL measurements showed minimal change (ESI Fig. S6f†), indicating the robust chirality of the (P, P)-2, corroborating its higher glum/gabs ratio of 0.81.65
Conclusion
In summary, we have succeeded in achieving unparalleled control over the conformation of cove-edged graphene NRs. Leveraging enantiopure [7]helicenes as molecular wrenches, we have integrated these helical motifs into the terminal K-regions of a conjugated pyrene NR through a stereospecific APEX reaction. Among these acene-cored NRs, (P, P)-2 displayed an impressive end-to-end twist of 171°. These robust chiral NRs exhibited exceptional configurational and conformational stability with a relative population of helical NR amounting to 99.9% at room temperature. The progressive increase in chiroptical responses from (P)-3 to (P, P)-2 highlights the emergence of macro chirality resulting from the overall twist of the nanoribbon. Our study not only addresses the longstanding challenge of controlling the stereochemistry of twisted hydrocarbon NRs but also sheds light on the impact of twisting on the conformational population. Through extensive quantum chemical calculations, we have provided a detailed understanding of the energetically favored conformers and their relative populations. The outlined strategy, allowing the late-stage introduction of helicene units by APEX reaction, holds promise for facilitating the synthesis of diverse cove edge NR variants with desired conformations.
Data availability
The experimental procedures, characterizations, spectral analysis and DFT computational details are available in the ESI.†
Author contributions
A. S. performed the experimental work, analyzed the data, and wrote the manuscript. K. R. and H. B. analyzed the crystallographic data. P. R. conceived the project, supervised the research, and prepared the final draft of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We dedicate this paper to Professor Frank Würthner (University of Würzburg) on the occasion of his 60th birthday. P. R. thanks Julius-Maximilians-Universität Würzburg for “Excellent Ideas” program. A. S. and P. R. sincerely thank Prof. Christoph Lambert (University of Würzburg) for generously supporting our research. Funding: this project received funding from Deutsche Forschungsgemeinschaft (DFG) (Project No. 448604676). The CPL/CD hybrid spectrometer was funded by the DFG (Project No. 444286426). We thank the reviewers for their valuable suggestions to improve the manuscript.
References
- X.-Y. Wang, A. Narita and K. Müllen, Precision synthesis versus bulk-scale fabrication of graphenes, Nat. Rev. Chem., 2017, 2, 0100 CrossRef.
- J. M. Fernández-García, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Chiral Molecular Carbon Nanostructures, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed.
- Y. Segawa, H. Ito and K. Itami, Structurally uniform and atomically precise carbon nanostructures, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- M. Rickhaus, M. Mayor and M. Juríček, Strain-induced helical chirality in polyaromatic systems, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- J. R. Sanchez-Valencia, T. Dienel, O. Gröning, I. Shorubalko, A. Mueller, M. Jansen, K. Amsharov, P. Ruffieux and R. Fasel, Controlled synthesis of single-chirality carbon nanotubes, Nature, 2014, 512, 61 CrossRef CAS PubMed.
- S. Eigler and A. Hirsch, Chemistry with Graphene and Graphene Oxide—Challenges for Synthetic Chemists, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
-
E. Clar, Polycyclic Hydrocarbons, Springer, 1964 Search PubMed.
- W. Niu, Y. Fu, Z.-L. Qiu, C. J. Schürmann, S. Obermann, F. Liu, A. A. Popov, H. Komber, J. Ma and X. Feng, π-Extended Helical Multilayer Nanographenes with Layer-Dependent Chiroptical Properties, J. Am. Chem. Soc., 2023, 145, 26824–26832 CrossRef CAS PubMed.
- A. Narita, X. Feng and K. Müllen, Bottom-Up Synthesis of Chemically Precise Graphene Nanoribbons, Chem. Rec., 2015, 15, 295–309 CrossRef CAS PubMed.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, New advances in nanographene chemistry, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- S. Dutta and S. K. Pati, Novel properties of graphene nanoribbons: a review, J. Mat. Chem., 2010, 20, 8207–8223 RSC.
-
J. Liu, R. Berger, K. Müllen and X. Feng, in From Polyphenylenes to Nanographenes and Graphene Nanoribbons, Springer, 2017, vol. 278, pp. 1–32 Search PubMed.
- J. Li, S. Sanz, N. Merino-Díez, M. Vilas-Varela, A. Garcia-Lekue, M. Corso, D. G. de Oteyza, T. Frederiksen, D. Peña and J. I. Pascual, Topological phase transition in chiral graphene nanoribbons: from edge bands to end states, Nat. Commun., 2021, 12, 5538 CrossRef CAS PubMed.
- J. Liu, B.-W. Li, Y.-Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, Toward Cove-Edged Low Band Gap Graphene Nanoribbons, J. Am. Chem. Soc., 2015, 137, 6097–6103 CrossRef CAS PubMed.
- Y. Gu, R. Muñoz-Mármol, S. Wu, Y. Han, Y. Ni, M. A. Díaz-García, J. Casado and J. Wu, Cove-Edged Nanographenes with Localized Double Bonds, Angew. Chem., Int. Ed., 2020, 59, 8113–8117 CrossRef CAS PubMed.
- Y. Gu, V. Vega-Mayoral, S. Garcia-Orrit, D. Schollmeyer, A. Narita, J. Cabanillas-González, Z. Qiu and K. Müllen, Cove-Edged Hexa-peri-hexabenzo-bis-peri-octacene: Molecular Conformations and Amplified Spontaneous Emission, Angew. Chem., Int. Ed., 2022, 61, e202201088 CrossRef CAS PubMed.
- X. Wang, J. Ma, W. Zheng, S. Osella, N. Arisnabarreta, J. Droste, G. Serra, O. Ivasenko, A. Lucotti, D. Beljonne, M. Bonn, X. Liu, M. R. Hansen, M. Tommasini, S. De Feyter, J. Liu, H. I. Wang and X. Feng, Cove-Edged Graphene Nanoribbons with Incorporation of Periodic Zigzag-Edge Segments, J. Am. Chem. Soc., 2022, 144, 228–235 CrossRef CAS.
- W. A. Chalifoux, The Synthesis of Non-planar, Helically Coiled Graphene Nanoribbons, Angew. Chem., Int. Ed., 2017, 56, 8048–8050 CrossRef CAS PubMed.
- M. Daigle, D. Miao, A. Lucotti, M. Tommasini and J.-F. Morin, Helically Coiled Graphene Nanoribbons, Angew. Chem., Int. Ed., 2017, 56, 6213–6217 CrossRef CAS PubMed.
- W. Niu, J. Ma, P. Soltani, W. Zheng, F. Liu, A. A. Popov, J. J. Weigand, H. Komber, E. Poliani, C. Casiraghi, J. Droste, M. R. Hansen, S. Osella, D. Beljonne, M. Bonn, H. I. Wang, X. Feng, J. Liu and Y. Mai, A Curved Graphene Nanoribbon with Multi-Edge Structure and High Intrinsic Charge Carrier Mobility, J. Am. Chem. Soc., 2020, 142, 18293–18298 CrossRef CAS PubMed.
- S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, Supertwistacene: A Helical Graphene Nanoribbon, J. Am. Chem. Soc., 2020, 142, 16887–16893 CrossRef CAS PubMed.
- S. Castro-Fernández, C. M. Cruz, I. F. A. Mariz, I. R. Márquez, V. G. Jiménez, L. Palomino-Ruiz, J. M. Cuerva, E. Maçôas and A. G. Campaña, Two-Photon Absorption Enhancement by the Inclusion of a Tropone Ring in Distorted Nanographene Ribbons, Angew. Chem., Int. Ed., 2020, 59, 7139–7145 CrossRef PubMed.
- W. Yang, R. R. Kazemi, N. Karunathilake, V. J. Catalano, M. A. Alpuche-Aviles and W. A. Chalifoux, Expanding the scope of peropyrenes and teropyrenes through a facile InCl3-catalyzed multifold alkyne benzannulation, Org. Chem. Front., 2018, 5, 2288–2295 RSC.
- B. Mahlmeister, M. Mahl, H. Reichelt, K. Shoyama, M. Stolte and F. Würthner, Helically Twisted Nanoribbons Based on Emissive Near-Infrared Responsive Quaterrylene Bisimides, J. Am. Chem. Soc., 2022, 144, 10507–10514 CrossRef CAS PubMed.
- R. A. Pascal, Twisted Acenes, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS PubMed.
- R. S. Walters, C. M. Kraml, N. Byrne, D. M. Ho, Q. Qin, F. J. Coughlin, S. Bernhard and R. A. Pascal Jr, Configurationally Stable Longitudinally Twisted Polycyclic Aromatic Compounds, J. Am. Chem. Soc., 2008, 130, 16435–16441 CrossRef CAS PubMed.
- I. I. Schuster, L. Craciun, D. M. Ho and R. A. Pascal, Synthesis of a strained, air-sensitive, polycyclic aromatic hydrocarbon by means of a new 1,4-benzadiyne equivalent, Tetrahedron, 2002, 58, 8875–8882 CrossRef CAS.
- J. Lu, D. M. Ho, N. J. Vogelaar, C. M. Kraml and R. A. Pascal, A Pentacene with a 144° Twist, J. Am. Chem. Soc., 2004, 126, 11168–11169 CrossRef CAS PubMed.
- X. Qiao, M. A. Padula, D. M. Ho, N. J. Vogelaar, C. E. Schutt and R. A. Pascal, Octaphenylnaphthalene and Decaphenylanthracene, J. Am. Chem. Soc., 1996, 118, 741–745 CrossRef CAS.
- Y. Xiao, J. T. Mague, R. H. Schmehl, F. M. Haque and R. A. Pascal Jr, Dodecaphenyltetracene, Angew. Chem., Int. Ed., 2019, 58, 2831–2833 CrossRef CAS PubMed.
- R. G. Clevenger, B. Kumar, E. M. Menuey and K. V. Kilway, Synthesis and Structure of a Longitudinally Twisted Hexacene, Chem.–Eur. J., 2018, 24, 3113–3116 CrossRef CAS PubMed.
- A. Bedi, L. J. W. Shimon and O. Gidron, Helically Locked Tethered Twistacenes, J. Am. Chem. Soc., 2018, 140, 8086–8090 CrossRef CAS.
- A. Bedi and O. Gidron, The Consequences of Twisting Nanocarbons: Lessons from Tethered Twisted Acenes, Acc. Chem. Res., 2019, 52, 2482–2490 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Pyrene-Based Materials for Organic Electronics, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- J. Xiao, H. M. Duong, Y. Liu, W. Shi, L. Ji, G. Li, S. Li, X.-W. Liu, J. Ma, F. Wudl and Q. Zhang, Synthesis and Structure Characterization of a Stable Nonatwistacene, Angew. Chem., Int. Ed., 2012, 51, 6094–6098 CrossRef CAS PubMed.
- B. Kumar, C. E. Strasser and B. T. King,
t-Butyl Biphenylation of o-Dibromoarenes: A Route to Soluble Polycyclic Aromatic Hydrocarbons, J. Org. Chem., 2012, 77, 311–316 CrossRef CAS PubMed.
- Y. Yano, H. Ito, Y. Segawa and K. Itami, Helically Twisted Tetracene: Synthesis, Crystal Structure, and Photophysical Properties of Hexabenzo[a,c,fg,j,l,op]tetracene, Synlett, 2016, 27, 2081–2084 CrossRef CAS.
- S. M. Elbert, K. Baumgärtner, J. A. Esteves, L. Weber, F. Rominger and M. Mastalerz, Pyrene-Based Diarynes as Precursors for Twisted Fused Polycyclic Aromatic Hydrocarbons: A Comparison of Two Routes, Org. Mater., 2020, 02, 358–361 CrossRef CAS.
- A. K. Swain, K. Kolanji, C. Stapper and P. Ravat, C2- and C1-Symmetric Configurationally Stable Pyrene-Fused [5]Helicenes Connected via Hexagonal and Heptagonal Rings, Org. Lett., 2021, 23, 1339–1343 CrossRef CAS PubMed.
- A. K. Swain, K. Radacki, H. Braunschweig and P. Ravat, Pyrene-Fused [7]Helicenes Connected Via Hexagonal and Heptagonal Rings: Stereospecific Synthesis and Chiroptical Properties, J. Org. Chem., 2022, 87, 993–1000 CrossRef CAS PubMed.
- A. Swain and P. Ravat, Pyrene bridged double[7]helicene embedded with a heptagonal ring, Org. Chem. Front., 2023, 10, 3714–3725 RSC.
- J. A. Weber, E. L. Clennan and N. Arulsamy, A Computational Physical Organic Study of a Torque, Lock, and Propagate Approach and Validation with the Synthesis of Configurationally Stable First-Generation Helically Twisted Acenes, Eur. J. Org Chem., 2022, e202101533 CrossRef CAS.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, Inducing Single-Handed Helicity in a Twisted Molecular Nanoribbon, J. Am. Chem. Soc., 2022, 144, 2765–2774 CrossRef CAS PubMed.
- W. Fan, T. Winands, N. L. Doltsinis, Y. Li and Z. Wang, A Decatwistacene with an Overall 170° Torsion, Angew. Chem., Int. Ed., 2017, 56, 15373–15377 CrossRef CAS PubMed.
- W. Theilacker and H. Böhm, Optically Active 2,2′-Dimethylbiphenyl, the Simplest Atropisomeric Hydrocarbon, Angew. Chem., Int. Ed., 1967, 6, 251 CrossRef CAS.
- R. H. Martin, The Helicenes, Angew. Chem., Int. Ed., 1974, 13, 649–660 CrossRef.
- Y. Shen and C. F. Chen, Helicenes: synthesis and applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, G. Félix and R. Peresutti, One hundred years of helicene chemistry. Part 2: stereoselective syntheses and chiral separations of carbohelicenes, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- P. Ravat, Carbo[n]helicenes Restricted to Enantiomerize: An Insight into the Design Process of Configurationally Stable Functional Chiral PAHs, Chem.–Eur. J., 2021, 27, 3957–3967 CrossRef CAS PubMed.
- L. Ji, I. Krummenacher, A. Friedrich, A. Lorbach, M. Haehnel, K. Edkins, H. Braunschweig and T. B. Marder, Synthesis, Photophysical, and Electrochemical Properties of Pyrenes Substituted with Donors or Acceptors at the 4- or 4,9-Positions, J. Org. Chem., 2018, 83, 3599–3606 CrossRef CAS PubMed.
- K. Ozaki, K. Kawasumi, M. Shibata, H. Ito and K. Itami, One-shot K-region-selective annulative π-extension for nanographene synthesis and functionalization, Nat. Commun., 2015, 6, 6251 CrossRef CAS PubMed.
- F. Liu, X. Shen, Y. Wu, L. Bai, H. Zhao and X. Ba, Synthesis of ladder-type graphene ribbon oligomers from pyrene units, Tetrahedron Lett., 2016, 57, 4157–4161 CrossRef CAS.
- H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Facile Synthetic Route to Highly Luminescent Sila[7]helicene, Org. Lett., 2013, 15, 2104–2107 CrossRef CAS PubMed.
- T. Hasegawa, M. Sekine, W. P. Schaefer and H. Taube, Preparation and characterization of a bis(pentaammineosmium(II)) pyrene complex, Inorg. Chem., 1991, 30, 449–452 CrossRef CAS.
- H. Ito, K. Ozaki and K. Itami, Annulative π-Extension (APEX): Rapid Access to Fused Arenes, Heteroarenes, and Nanographenes, Angew. Chem., Int. Ed., 2017, 56, 11144–11164 CrossRef CAS PubMed.
- R. H. Martin and M.-J. Marchant, Thermal racemisation of [6], [7], [8] and [9] helicene, Tetrahedron Lett., 1972, 13, 3707–3708 CrossRef.
- J. Barroso, J. L. Cabellos, S. Pan, F. Murillo, X. Zarate, M. A. Fernandez-Herrera and G. Merino, Revisiting the racemization mechanism of helicenes, Chem. Commun., 2018, 54, 188–191 RSC.
- J. M. Fernández-García, P. Izquierdo-García, M. Buendía, S. Filippone and N. Martín, Synthetic chiral molecular nanographenes: the key figure of the racemization barrier, Chem. Commun., 2022, 58, 2634–2645 RSC.
- G. Portella, J. Poater, J. M. Bofill, P. Alemany and M. Solà, Local Aromaticity of [n]Acenes, [n]Phenacenes, and [n]Helicenes (n = 1–9), J. Org. Chem., 2005, 70, 2509–2521 CrossRef CAS PubMed.
- L. Rulíšek, O. Exner, L. Cwiklik, P. Jungwirth, I. Starý, L. Pospíšil and Z. Havlas, On the Convergence of the Physicochemical Properties of [n]Helicenes, J. Phys. Chem. C, 2007, 111, 14948–14955 CrossRef.
- Y. Nakai, T. Mori and Y. Inoue, Theoretical and experimental studies on circular dichroism of carbo[n]helicenes, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS.
- P. Malakar, V. Borin, A. Bedi, I. Schapiro, O. Gidron and S. Ruhman, The impact of twisting on the intersystem crossing in acenes: an experimental and computational study, Phys. Chem. Chem. Phys., 2022, 24, 2357–2362 RSC.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Enhanced intersystem crossing in core-twisted aromatics, Chem. Sci., 2017, 8, 1776–1782 RSC.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. Vander Donckt, Fluorescence of the higher helicenes, Chem. Phys. Lett., 1976, 43, 33–36 CrossRef CAS.
- H. Tanaka, Y. Inoue and T. Mori, Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Krzysztof
Radacki
a,
Krzysztof
Radacki