
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dioxide affinity (“carboxophilicity”) of trivalent light metal pyrazolates†
Felix
Kracht
,
Philipp
Rolser
,
Klaus
Eichele
 ,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
*
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen (EKUT), Auf der Morgenstelle 18, 72076, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 23rd August 2024
Abstract
Trivalent group 3 and 13 light metal pyrazolates were synthesised and their reactivity towards CO2 was investigated. The homoleptic complex Al(pztBu2)3 reversibly inserts two molecules of CO2 to afford Al(CO2·pztBu2)2(pztBu2), exhibiting CO2 release only at elevated temperatures (>100 °C). In contrast, donor-stabilised Sc(pztBu2)3(thf) forms mono-inserted species [Sc(μ-CO2·pztBu2)(pztBu2)2]2, which already releases CO2 at ambient temperature and pressure and hence is isolable only at low temperature. For the yttrium complex Y(pztBu2)3(thf)2, insertion of CO2 is not observable at ambient temperature. The new homoleptic aluminium diisopropyl pyrazolate complex [Al(pziPr2)3]2 shows exhaustive CO2 insertion, while dimethyl pyrazolate could be isolated as the separated ion pair [Al(N,N′,N′′-Al{pzMe2}3Me)2][Al(pzMe2)3Me]. The scandium complex Sc(pztBu2)3(thf) performed best in the catalytic cycloaddition reaction of CO2 and epoxides, unveiling an inverse correlation of carboxophilicity ( ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO2 affinity) and catalytic activity. Carboxophilicity is assessed using CO2-release temperature (via VT 1H NMR spectroscopy and thermogravimetric analysis).
CO2 affinity) and catalytic activity. Carboxophilicity is assessed using CO2-release temperature (via VT 1H NMR spectroscopy and thermogravimetric analysis).
Introduction
Anthropogenic greenhouse gases, in particular carbon dioxide, are the main reason for global climate change. Consequently, combating CO2 build-up has developed into the most important and compelling research field in the chemical sciences.1–11 Carbon dioxide capture and storage (CCS) from flue gas or directly from air (DAC) are industrially realised with aqueous amine scrubbers; this technology, however, is affected by low capacities (<15 wt% CO2), sensitivity to oxygen and high regeneration energies (high “energy penalty”).8,9 Academic research has focused on amino-functionalized porous materials such as mesoporous silica, zeolites or metal–organic frameworks, with the latter showing promising results under simulated flue gas.5,12–17 Moreover, a detailed molecular understanding of metal–ligand interactions with carbon dioxide seems crucial for developing efficient (catalytic) processes for its chemical valorization.18 Here, metal–N(ligand) cooperativity has proven particularly effective in CO2 transformations together with a synergistic interplay to reversibly insert this heteroallene.18 Any reversible insertion/de-insertion process will rely on a fine balance of metal-centred oxophilicity and ligand-nitrogen nucleophilicity, which we have termed carboxophilicity (Fig. 1, top).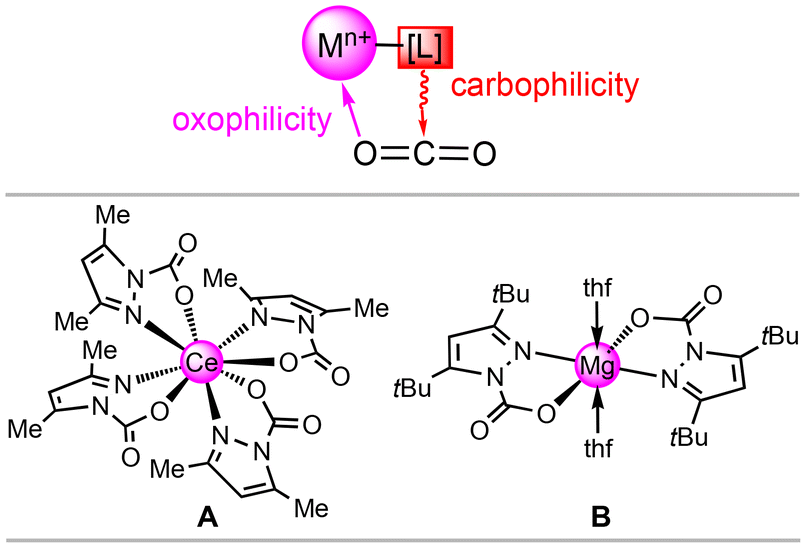 | ||
| Fig. 1 Top: Carboxophilicity of metal–ligand entities as driven by the oxophilicity of the metal centre and nucleo(carbo)philicity of the ligand. Bottom: Examples of tetravalent (cerium, A)19 and divalent (magnesium, B) carbamate complexes,20 which feature fully reversible CO2 insertion. | ||
Recently, we found that CO2 reversibly inserts into metal–pyrazolato bonds involving oxophilic metal centres. It was revealed that tetravalent dimeric [Ce(pzMe2)4]2 and the trivalent cluster [Ce(pzMe2)3]4 can accommodate 25 wt% CO2.19 Adapting this approach to the environmentally friendlier earth-abundant light metal magnesium, parent pyrazolate [Mg(pz)2]n achieved an exceptionally high reversible CO2 uptake of up to 35.7 wt%.20 Structurally authenticated complexes include tetravalent Ce(CO2·pzMe2)4 (A)19 and divalent Mg(CO2·pztBu2)2(thf)2 (B),20 showing exclusive κ2(N,O) carbamato coordination (Fig. 1, bottom). Moreover, all pyrazolate complexes displayed moderate catalytic activity in the conversion of CO2 and epoxides to their corresponding cyclic carbonates. In this work, we report on the reactivity of trivalent pyrazolates of highly oxophilic group 3 and 13 light metals aluminium, scandium and yttrium towards CO2. In addition, we discuss CO2 release temperature as a quantifiable measure for assessing carboxophilicity.
The reactivity of organoaluminium complexes towards carbon dioxide was described by Ziegler in 1960 and expanded on later in 1970 by Weidlein.21,22 Accordingly, CO2 inserts irreversibly into trialkyl aluminium compounds to afford tertiary alkoxides or carboxylates, which upon hydrolysis, produced their corresponding alcohols or carboxylic acids. Dimeric [Al(μ-CO2·N(iPr)2)(CO2·N(iPr)2)2]2 features the first structurally characterised aluminium carbamate complex, accessed from AlBr3/HN(iPr)2/CO2.23 Moreover, dimeric heteroleptic carbamate [Me2Al(μ-CO2·N(iPr)2)]2 was obtained (among others) via CO2 insertion into [Me2Al(μ-N(iPr)2)2Mg(μ-Me)]n.24 More recently, compound [Al(κ2-N,N-2-{methylamino}pyridine)2R] (R = Et, iBu) was shown to insert CO2 not only into the Al–C(alkyl) bond but also in one of the two Al–N bonds of each pyridine ligand.25,26 Furthermore, the frustrated Lewis acid pair 9,9-dimethylxanthene(PPh2-AlMe2Cl-AlMeCl) engaged in reversible CO2 insertion.27 In addition, aluminium compounds supported by a rigid ligand backbone, such as porphyrinato, salen or amino-tris(phenolato), are active in the cycloaddition of CO2 with epoxides, with the latter displaying a promising performance with low catalyst loads.28–36 The porphyrinate complex (TPP)Al(OMe) showed reversible insertion behaviour towards CO2 in the presence of 1-methylimidazole.28
For rare-earth metals, a rich CO2-insertion chemistry is already existent.19b However, for most metal–ligand entities, CO2 insertion turned out to be irreversible, forming carboxylates/formiate from alkyls/hydrides,37–42 carbonates from alkoxide/aryloxide43,44 or carbamates from amide complexes.45–48 Scandocene [(C5Me5)2Sc(CO2·p-tolyl)] features the first structurally characterised organoscandium CO2 insertion complex.37 Evans et al. described the first yttrium carbamate complex, [(C5Me5)2Y(CO2·ε-caprolactam)]2, obtained by CO2 insertion into the Y–N bond of (C5Me5)2Y(O,N-ε-caprolactam).47 Rare-earth-metal complexes also gained attention for their catalytic performance in the co-polymerisation of CO2 and epoxides to polycarbonates,43,49–52 as well as the cycloaddition of both compounds to the cyclic carbonates with amino-tris(phenolates) as the most promising catalysts.52–59 Particularly thanks to the works of the groups of Deacon, Junk, and Winter, there exists a vast variety of rare-earth-metals pyrazolates, featuring distinct coordination modes.60–64 We were mostly interested in diamagnetic scandium and yttrium pyrazolates, such as Sc(pztBu2)3,61 [Y(pzMe2)3(thf)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 62 and Y(pztBu2)3(do)2.60
62 and Y(pztBu2)3(do)2.60
Results and discussion
Selection and CO2 insertion of group 13 pyrazolates
:3 of both compounds was used, only a single methane elimination occurred at ambient temperature, affording the well-known [Me2AlpzMe2]2 (2a) (Scheme 1).66,67
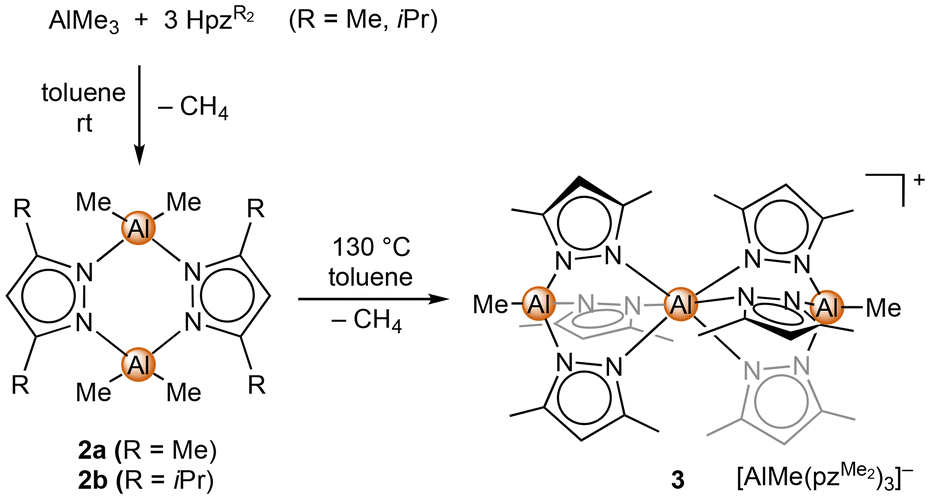 | ||
| Scheme 1 Protonolysis reactions of AlMe3 with three equivalents HpzR2 (R = Me and iPr), to form [Me2AlpzMe2]2 (3) at ambient temperature and separated ion pair [Al(N,N′,N′′-Al{pzMe2}3Me)2][MeAl(pzMe2)3] (4) by refluxing at 130 °C. | ||
Complex 2a features a recurring structural motif,68–70 which was also observed for the new diisopropyl derivative [Me2AlpziPr2]2 (2b). Refluxing a 1:3 mixture in toluene gave a more extensive but still incomplete ligand exchange. The crystalline material gained overnight revealed the solid-state structure of the separated ion pair [Al(N,N′,N′′-Al{pzMe2}3Me)2][MeAl(pzMe2)3] (3) (Fig. S102†). The mono-cationic fragment revealed an aluminium(III) centre coordinated by two mono-anionic [MeAl(pzMe2)3]− ligands. The latter are reminiscent of the tris(pyrazolyl)hydroborato ligand TpMe,Me, while the Tp analogous aluminium ligand is accessed via a protonolysis reaction of LiAlH4 with pyrazole.71–74 The separated anion in complex 3 is another [MeAl(pzMe2)3]− entity. The central aluminium atom of the cationic fragment features a slightly distorted octahedral geometry (N–Al–N angles ±3° off). The aluminium centres of the tridentate “scorpionato”-type ligand adopt a slightly distorted tetrahedral geometry with N–Al–N angles around 100° and C–Al–N angles around 117°. The respective angles in the separated [MeAl(pzMe2)3]− anion are 103° and 112° and hence are closer to tetrahedral. The three aluminium centres of the cationic fragment are almost linearly arranged (∠Al, Al, Al 1.5°), with the outer Al1/Al3–N (av. 1.874 Å and 1.893 Å) and inner Al2–N distances (av. 2.035 Å and 2.048 Å) reflecting the distinct coordination numbers. The trinuclear complex [AlMe({N,N′-3,3′-bipyrazolate}2AlMe2)2], obtained from bispyrazole and AlMe3 (1:1.5),75 shows a similar structural motif. The 27Al NMR spectrum of compound 3 revealed only two signals at δ = 75.7 ppm and 0.3 ppm, assignable to 4- and 6-coordinated aluminium centres, respectively.
Further heating a 1:3 mixture of AlMe3 and HpzMe2 in mesitylene to 180 °C, in order to achieve full methyl elimination, did not lead to any isolable product, and thus the homoleptic [Al(pzMe2)3] remains elusive.
Unlike the dimethyl derivative, potassium diisopropyl pyrazolate reacted smoothly with aluminium chloride via a salt-metathesis route (Scheme 2). The crystal structure revealed the homoleptic bimetallic complex [Al(pziPr2)3]2 (4) (Fig. 2), with two bridging pyrazolato ligands arranged in an approximate boat conformation with the aluminium atoms. Interestingly, the terminal pyrazolato ligands show different coordination modes. One aluminium centre features terminal pyrazolatos coordinated exclusively in a κ1 coordination mode, while the other one accommodates the N-ligand in both κ1 and κ2 fashion. The 1H and 13C NMR spectra of 4 show the bridging and terminal pyrazolatos as two different signal sets in a ratio of 1:2. The 27Al NMR resonance was detected at δ = 68.1 ppm. Some reactions gave 1H NMR spectra with an additional slightly shifted signal set. We were able to fractionally crystallize this side product, and the solid-state structure revealed the monomeric mixed pyrazole/pyrazolato complex Al(pziPr2)3(HpziPr2) (4a) (Fig. S104†). The structure of 4a is quite similar to that of 4 with one Al(pziPr2)2 fragment replaced by a proton, which forms a hydrogen bond with the pyrazolyl moieties. Most likely, the pyrazole originated from partial hydrolysis caused by residual moisture in the solvent THF.
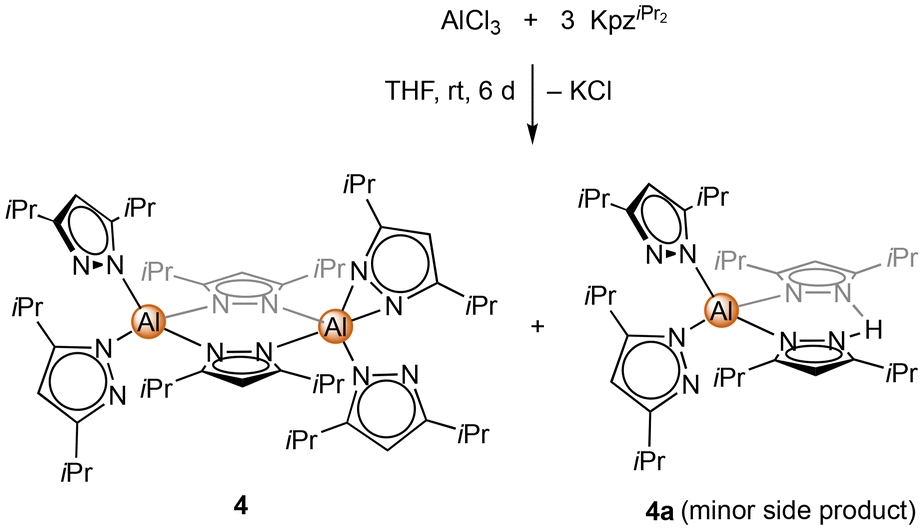 | ||
| Scheme 2 Synthesis of homoleptic [Al(pziPr2)3]2 (4) via salt metathesis of AlCl3 and KpziPr2. The side product Al(pziPr2)3(HpziPr2) (4a) is formed in the presence of residual moisture. | ||
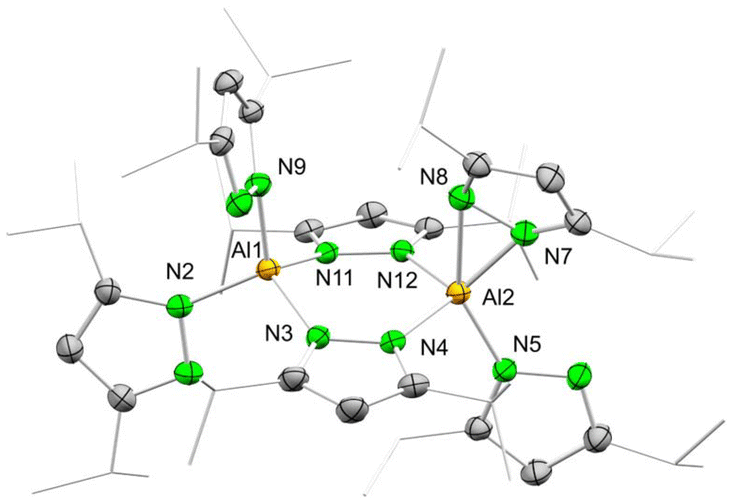 | ||
| Fig. 2 Crystal structure of [Al(pziPr2)3]2 (4). Ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
 | ||
| Scheme 3 Reaction of GaCl3 with KpztBu2 to yield [Ga(pztBu2)(μ-N,N,C-pztBu,C(CH3)2CH2)]2 (5) as a side product. | ||
An X-ray structural analysis revealed the formation of the dimeric complex [Ga(pztBu2)(μ-N,N,C-pztBu,C(CH3)2CH2)]2 (5) (Fig. 3). C–H-bond activation at one tBu moiety under release of pyrazole generated a dianionic pyrazolato ligand, bridging with both nitrogen atoms and one CH2 group between the gallium atoms.
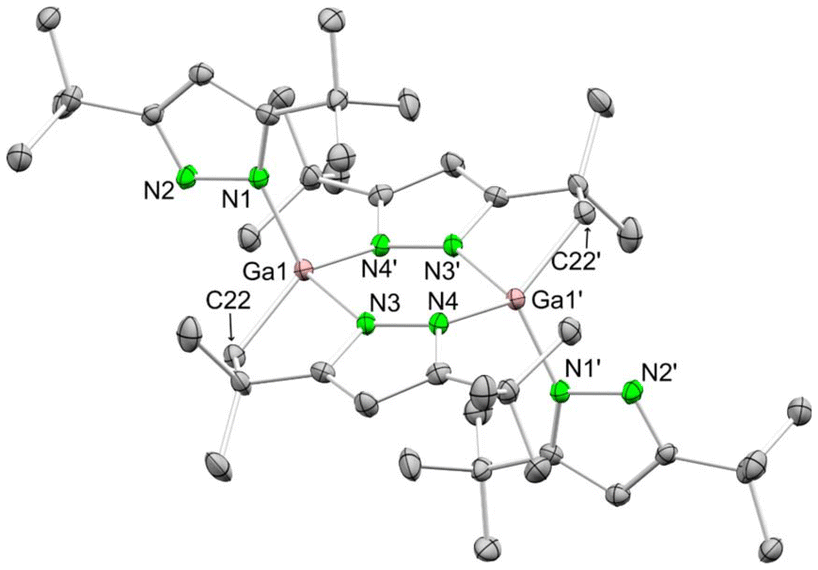 | ||
| Fig. 3 Crystal structure of [Ga(pztBu2)(μ-N,N,C-pztBu,C(CH3)2CH2)]2 (5). Ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
The molecular structure of complex 5 indicates a rather complex reactivity of gallium pyrazolates. Such distinct behaviour of aluminium and gallium in salt-metathesis reactions is not unknown, as previously shown for MCl3/LiN(SiMe3)2 or MCl3/LiN(SiHMe2)2 mixtures (M = Al, Ga). There, gallium also engaged in extensive Si–X-bond activation (X = C, H).80 Application of a protonolysis protocol employing a 1:3 mixture of GaMe3 and HpztBu2 led to the known [Me2Ga(pztBu2)]2,81 analogous to the aluminium-based reactions displayed in Scheme 1.
 | ||
| Scheme 4 Reaction of Al(pztBu2)3 (1) with excess CO2 in toluene. | ||
The crystal structure of 1-CO2 revealed a strongly distorted octahedral geometry (see Fig. 4). The aluminium centre is coordinated with two carbamato moieties in the κ2(N,O) mode and one pyrazolato ligand in the terminal κ2(N,N) mode. Because of the sterically demanding tBu moieties, all three ligands are twisted against each other, which gives the complex a propeller-like overall geometry. Both stereoisomers (Λ,Δ) can be found in the unit-cell which is further corroborated by the centrosymmetric space group Pbca. The solid-state structure might also explain the non-occurrence of an exhaustive CO2 insertion, since the bulky tBu moieties hinder a vital tilt of the third pyrazolato ligand. The inserted CO2 moieties exhibit an angle around 127.5°, along with localized C–O double (1.199–1.203 Å) and single bonds (1.285–1.288 Å). Such bond localization is also found in the pyrazolato rings. The 4C–3/5C and the 1/2N–3/5C distances of the non-inserted pyrazolato ring differ slightly by 0.005 Å, whereas, for the carbamate pyrazolato rings, the differences are significantly larger at 0.031–0.034 Å and 0.047 Å, respectively.
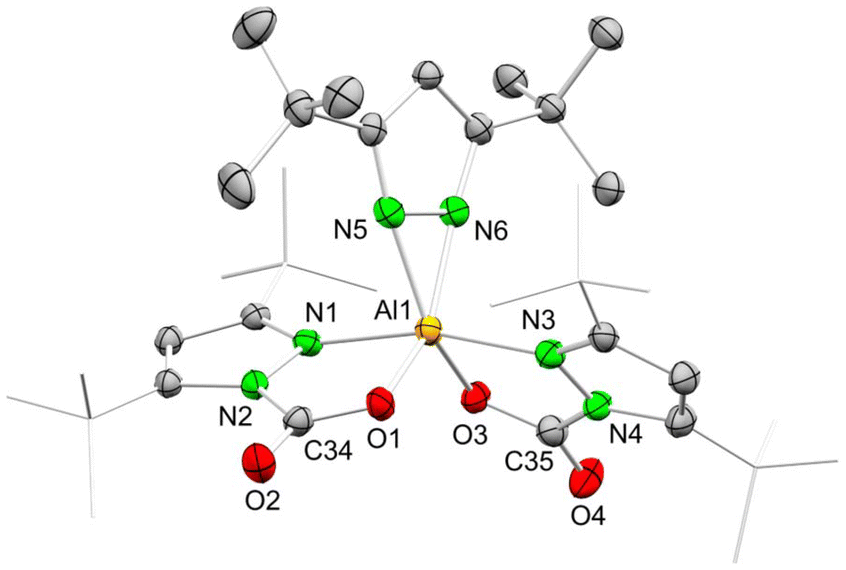 | ||
| Fig. 4 Crystal structure of Al(CO2·pztBu2)2(pztBu2) (1-CO2). Ellipsoids are set at the 50% probability level. Hydrogen atoms and disorder in the tBu moieties are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
DRIFTS measurements further validate this behaviour, displaying strong absorption bands at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1774/1763 cm−1 and = 1328 cm−1 for the CO and C–O stretching vibrations, respectively. The 1H NMR spectrum of 1-CO2 shows two singlets for the pyrazolato backbone proton at δ = 6.32 and 6.00 ppm in a 1:2 ratio. Additionally, three singlets with an integral of 18 protons each, one at δ = 1.50 and two at δ = 1.05 ppm, are detected, in accordance with three distinct tBu moieties. Further, the 13C NMR spectrum of 1-CO2 shows a signal at δ = 146.9 ppm, assignable to inserted CO2, while the 27Al resonance at δ = 17.0 ppm appears slightly shifted, broadened and less intense (due to loss of symmetry) compared to that of precursor 1 (δ = 23.3 ppm).
= 1774/1763 cm−1 and = 1328 cm−1 for the CO and C–O stretching vibrations, respectively. The 1H NMR spectrum of 1-CO2 shows two singlets for the pyrazolato backbone proton at δ = 6.32 and 6.00 ppm in a 1:2 ratio. Additionally, three singlets with an integral of 18 protons each, one at δ = 1.50 and two at δ = 1.05 ppm, are detected, in accordance with three distinct tBu moieties. Further, the 13C NMR spectrum of 1-CO2 shows a signal at δ = 146.9 ppm, assignable to inserted CO2, while the 27Al resonance at δ = 17.0 ppm appears slightly shifted, broadened and less intense (due to loss of symmetry) compared to that of precursor 1 (δ = 23.3 ppm).
Unlike the previously examined cerium and magnesium pyrazolates, solid 1-CO2 does not show any CO2 release at ambient temperature, not even under reduced pressure. This high carboxophilicity was further revealed by a variable-temperature (VT) NMR study in THF-d8 (see S15), revealing prominent CO2 release only at 90 °C. At this temperature, the formation of small amounts of the mono-inserted complex [Al(CO2·pztBu2)(pztBu2)2] can be detected, while increasing the temperature to 100 °C led to small amounts of 1. This is in contrast to the CO2-inserted magnesium pyrazolates which fully de-insert the heteroallene in this temperature range.19 Cooling the sample which was heated to 100 °C to ambient temperature shows the re-formation of 1-CO2 as the predominant species, along with small amounts of the mono-inserted complex and 1. Retreatment of this mixture with an excess of CO2 fully restores compound 1-CO2. A thermogravimetric analysis (TGA) of 1-CO2 revealed CO2 release in the temperature range between 114 °C and 196 °C, with a mass loss of 15.8% which is in good agreement with the calculated 13.5%. A single CO2-release step like in solution could not be observed.
Treatment of [Al(pziPr2)3]2 (4) with 1 bar CO2 gave no crystalline material; however, the NMR data suggest an exhaustive insertion into every pyrazolato ligand (see S16–S18†). Three signals for the pyrazolato backbone protons appear in the 1H NMR spectrum in a 1:1:1 integral ratio. Furthermore, four septets for the tertiary iPr proton are detected in a 3:1:1:1 integral ratio. This indicates the expected asymmetry of the iPr groups resulting from CO2 insertion, in accordance with distinct signals for the iPr moieties adjacent to the inserted CO2 and one signal for iPr distant to the inserted CO2. This assignment is further strengthened by the 13C NMR spectrum, which shows three signals for inserted CO2 ranging from δ = 147.0 to 146.6 ppm. A 1H–13C HMBC experiment revealed that each of the three pyrazolato backbone proton signals couples with two carbon signals in the shift region for 3/5-pyrazolato carbon atoms, lending more evidence for the new asymmetry. The isopropyl CH3 groups appear as one multiplet with 24 protons and four doublets with 3 protons. This suggests a hindered rotation of the iPr moieties as a result of the three inserted CO2. Upon CO2 insertion, the 27Al NMR signal shifted to higher field, from δ = 68.1 ppm in 4 to δ = 12.1 ppm. The putative [Al(CO2·pziPr2)3] (4-CO2) would infer a capacity of 21.6 wt% CO2.
Reactivity of group 3 metal pyrazolates towards CO2
The scandium pyrazolate Sc(pztBu2)3(thf) (7) was synthesized by applying the salt-metathesis protocol described by Deacon for aluminium pyrazolate Al(pztBu2)3 (1, vide supra). Accordingly, the mercury-free synthesis using THF-activated scandium chloride and KpztBu2 gave 7 in a good yield (see Scheme 5).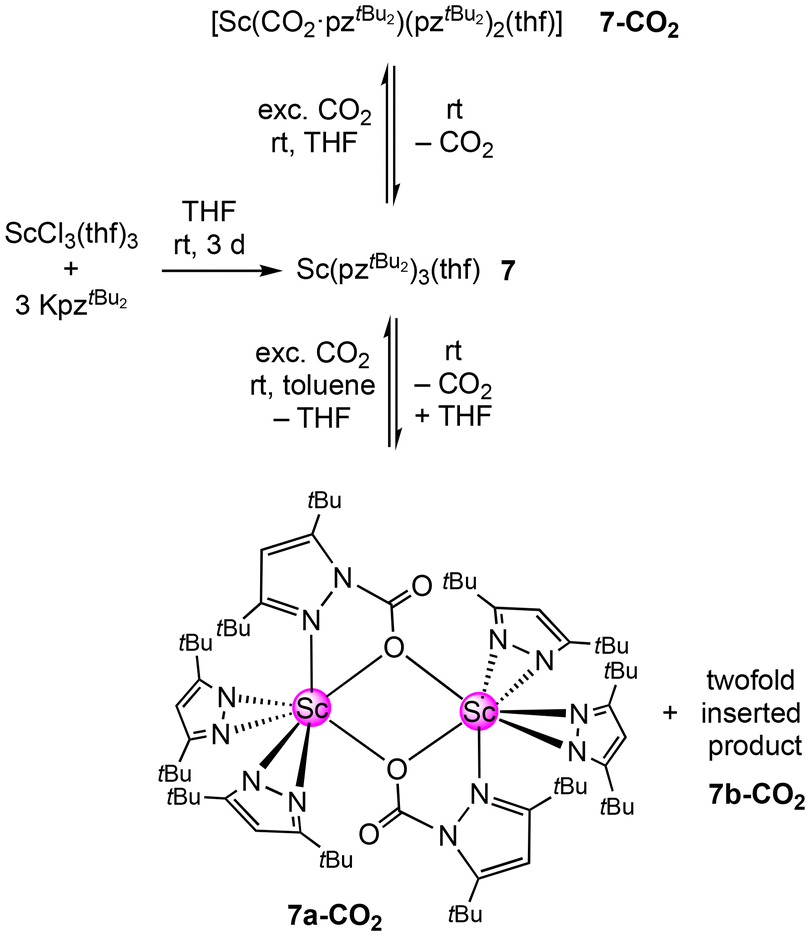 | ||
| Scheme 5 Salt-metathesis reaction of ScCl3(thf)3 and KpztBu2 to yield donor-stabilised Sc(pztBu2)3(thf) (7). Treatment with an excess of CO2 produces the donor-stabilised mono-insertion product [Sc(CO2·pztBu2)(pztBu2)2(thf)] (7-CO2) in THF and a mixture of the mono-inserted species [Sc(CO2·pztBu2)(pztBu2)2]2 (7a-CO2) and a higher inserted species 7b-CO2 in toluene. | ||
The crystal structure of 7 revealed one THF donor molecule bound to the scandium centre and three κ2(N,N)-coordinated pyrazolato ligands (see Fig. S107†). Unsurprisingly, the Sc–N distances are slightly longer than those in donor-free Sc(pztBu2)3.61 The scope of group 3 metal pyrazolates was extended to the literature known complex [Y(pzMe2)3(thf)]2 (9) obtained via protonolysis of YCp3 with HpzMe2.62 Dimeric 9 contains two bridging and four terminal pyrazolato ligands which appear in solution 1H and 13C NMR experiments as only one signal set due to fast ligand exchange. A 13C CP/MAS NMR experiment revealed separated signal sets for the terminal and bridging ligands (see S26†). The quaternary carbon atoms of the terminal pyrazolatos even appear as four well separated signals. Applying the synthesis protocol as used for scandium complex 7, monomeric [Y(pztBu2)3(thf)2] (10) was obtained. The solid-state structure of 10 revealed two THF donor molecules bound to the metal centre, isostructural to the pyridine-stabilized derivative [Y(pztBu2)3(Py)2] (see S108†).60
Treatment of Sc(pztBu2)3(thf) (7) with 1 bar CO2 in THF-d8 gave immediate CO2 insertion, as evidenced by 1H NMR spectroscopy. Three signals for tBu moieties appeared at δ = 1.51, 1.24 and 0.77 ppm, with an integral ratio of 9:36:9, respectively, as well as two signals for pyrazolato backbone protons at δ = 6.15 ppm and δ = 6.04 ppm with an integral ratio of 2:1, respectively. In addition, two signals of coordinated THF were observed. These signals can be assigned to the mono-inserted donor-stabilized complex [Sc(CO2·pztBu2)(pztBu2)2(thf)] (7-CO2). The 13C NMR spectrum shows the resonance for the inserted CO2 at δ = 149.7 ppm. The CO2 insertion can be also tracked by 45Sc NMR spectroscopy, since the sharp resonance of 7 at δ = 41.6 ppm shifts slightly towards lower field at δ = 69.1 ppm for 7-CO2, also involving signal broadening. Unfortunately, we were not able to isolate any crystalline material of 7-CO2, due to immediate complete CO2 release in the absence of solvent, already at ambient temperature and pressure. This effective and immediate reversibility of 7/7-CO2 is a stark contrast to the aluminium derivative 1-CO2, where CO2 release only occurred at elevated temperature and was not observed at reduced pressure. To further confirm CO2 insertion into scandium pyrazolate 7, in situ DRIFTS experiments were pursued. At 1 bar CO2 atmosphere, the immediate formation of a single strong band at = 1759 was observed, indicative of a CO bond stretching vibration (see S98 and S99†). Replacing the carbon dioxide atmosphere by argon resulted in complete CO2 de-insertion and back-formation of 7. A VT 1H NMR experiment uncovered a fully dynamic equilibrium between 7 and 7-CO2 in solution inside a closed vessel (see S31†). Starting with 7-CO2, even at ambient temperature, traces of 7 were detected. By gradually increasing the temperature, the conversion of 7-CO2 into 7 became more and more prominent and vice versa. Cooling the mixture to ambient temperature resulted in an almost identical 1H NMR spectrum as that at the outset of the VT NMR experiment.
Treatment of 7 with 1 bar CO2 in toluene-d8 instead of THF-d8 resulted in immediate CO2 insertion as well. However, this time, the 1H NMR spectrum revealed the formation of two distinct insertion products, 7a-CO2 and 7b-CO2. One species shows three signals for tBu moieties (9:36:9) with chemical shifts close to 7-CO2, while the second species gives four tBu proton signals at δ = 1.69, 1.49, 0.74 and 0.49 ppm with an integral ratio of 18:9:18:9. This second species appeared to be a product with a higher degree of inserted CO2. Correspondingly, the pyrazolato backbone protons appeared as two sets of two singlets with an integral ratio of 2:1. Furthermore, the 45Sc NMR spectrum showed two resonances at δ = 165.7 ppm and δ = 39.1 ppm.
This time, single-crystalline 7a-CO2 could be obtained from the solution; however, like observed for 7-CO2, solid 7a-CO2 showed immediate CO2 release at ambient temperature. Therefore, permanent cooling at −40 °C was essential for setting up and performing a successful XRD measurement. The crystal structure revealed the dimeric donor-free mono-inserted species [Sc(CO2·pztBu2)(pztBu2)2]2 (7a-CO2) bearing the two carbamato ligands in a [μ-1κ2(N,O):2κ(O)] bridging fashion (Fig. 5). Moreover, the scandium centres accommodate two terminal κ2(N,N′) pyrazolatos. As indicated by the 1H NMR spectrum, coordinated THF from the starting material was displaced.
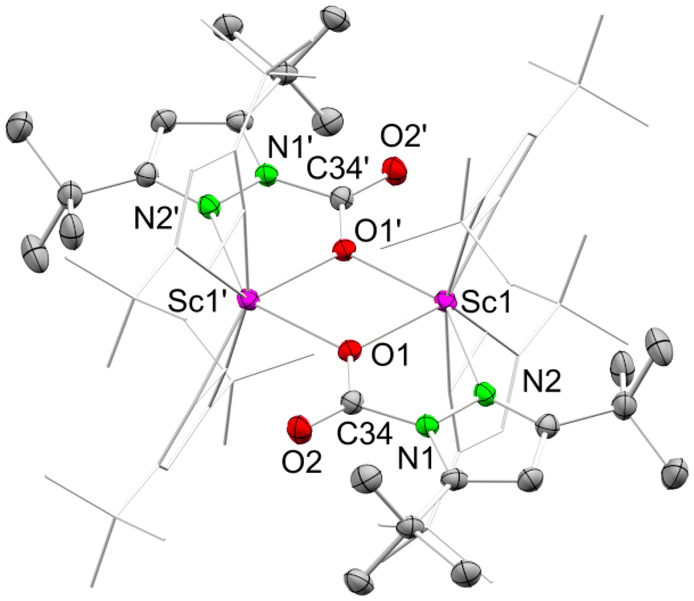 | ||
| Fig. 5 Crystal structure of [Sc(CO2·pztBu2)(pztBu2)2]2 (7a-CO2). Ellipsoids are set at the 50% probability level. Hydrogen atoms and disorder in the tBu moieties are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
The overall reversibility of the CO2 insertion of 7a-CO2 is akin to that of 7-CO2. The original NMR solution as well as isolated 7a-CO2 redissolved in toluene-d8, giving similar 1H NMR spectra, both showing small amounts of the second species 7b-CO2. In the absence of a CO2 atmosphere, 7b-CO2 seems to be unstable in solution at ambient pressure, which is why no crystalline 7b-CO2 could be isolated. Given the tBu proton signal set (integral ratio of 18:9:18:9), representing two carbamato ligands and one pyrazolato ligand, species 7b-CO2 likely features the twofold CO2-inserted compound [Sc(CO2·pztBu2)2(pztBu2)].
Our attempts to synthesize the homoleptic scandium dimethyl pyrazolate via a salt-metathesis protocol [ScCl3(thf)3 + 3KpzMe2] were unsuccessful. However, on one occasion, the trimetallic oxo-centred cluster [Sc3O(pzMe2)7(HpzMe2)2] (8) was obtained as the main product in a crystalline yield of 74% (see Scheme 6, Fig. 6).82
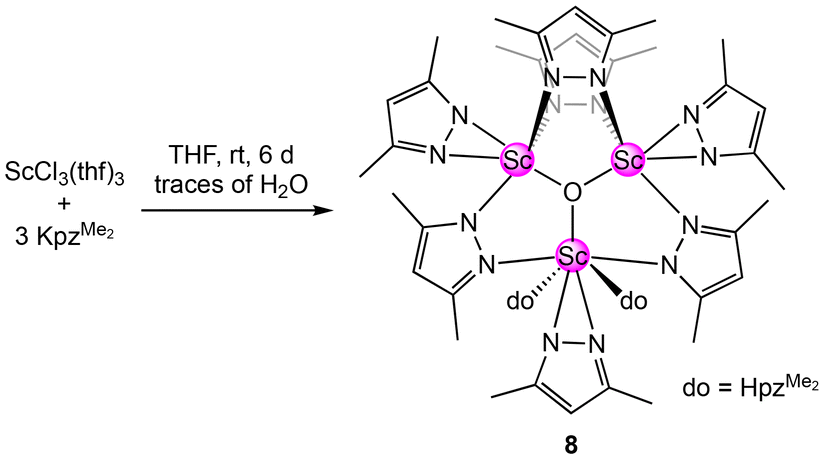 | ||
| Scheme 6 Synthesis of the oxo-centred cluster [Sc3O(pzMe2)7(HpzMe2)2] (8). | ||
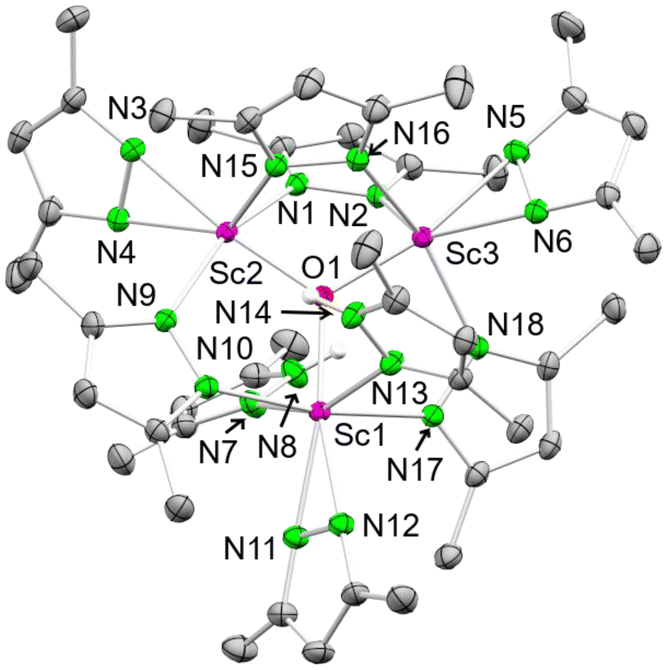 | ||
| Fig. 6 Crystal structure of [Sc3O(pzMe2)7(HpzMe2)2] (8). Ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
Two scandium centres in 8 are coordinated by four pyrazolato ligands each, and one is coordinated by three pyrazolatos and two donor pyrazoles. Each scandium centre bears one terminal κ2(N,N′) pyrazolato ligand, while two scandium centres are bridged twice by μ-1κ(N):2κ(N′) pyrazolatos. The scandium centre being single-bridged to the other two accommodates the two pyrazole donors. Overall, a total of one equivalent H2O is formally required to yield complex 8, which most likely originated from residual water in the solvent THF.
The 1H NMR spectrum of compound 8 displays the methyl groups and the backbone protons of the pyrazolato and pyrazole ligands as one signal each, at δ = 2.13 ppm and δ = 5.73 ppm, respectively, which indicates rapid exchange of the ligands in solution. The two NH-protons appear as one broad signal at δ = 11.82 ppm. As expected, the 45Sc NMR spectrum features two resonances at δ = 153.2 ppm and δ = 45.1 ppm in a ratio of 1:2. More surprisingly, treatment of 8 with 1 bar CO2 led to two well-defined products as suggested by NMR spectroscopy. One product presents the carbamic acid HO2CpzMe2 with typically broad proton and carbon signals, which we reported lately as a co-product of the reaction of mixed pyrazolato/pyrazole magnesium complexes with CO2 or as a hydrolysis product of the carbamate complexes.20 The second product 8-CO2 appeared as three sharp signals in the 1H NMR spectrum at δ = 5.95, 2.26 and 2.18 ppm. This pattern indicates exhaustive CO2 insertion into the oxo-cluster. This is further implied by the 45Sc NMR spectrum, featuring only one signal at δ = 53.6 ppm. The 13C NMR spectrum revealed two signals for inserted CO2 at δ = 151.2 ppm and δ = 150.2 ppm, assignable to the carbamic acid and the CO2-functionalized oxo cluster, respectively. However, no crystalline material of 8-CO2 could be obtained yet, and attempts to reproduce 8 by using an equimolar amount of water in the reaction depicted in Scheme 5 were unsuccessful.
The CO2-inserted product of [Y(pzMe2)3(thf)]2 (9) shows the typical splitting of the methyl groups in the 1H NMR spectrum (THF-d8), resonating at δ = 2.18 ppm and δ = 2.09 ppm, indicative of the formation of Y(CO2·pzMe2)3(thf) (9-CO2). However, several other non-assignable signals were detected along with that of putative 9-CO2 (see Fig. S45†), most likely representing a rather complex cluster species. A VT 1H NMR study revealed that 9-CO2 fully back-converts into 9 at 70 °C, while the unknown species remains unchanged (see Fig. S48†). Recently, we introduced a solvent-free procedure, in which the pyrazolate compound is reacted with CO2 neat as a solid.20 By using this procedure for 9, the resulting 9-CO2 exhibited a mass gain of 15.6 wt%. This is slightly higher than the expected value for a two-fold insertion (14.5 wt%) and lower than that for a complete insertion (20.3 wt%). Correspondingly, the 13C CP/MAS spectrum of 9-CO2 revealed two signal sets. One can be assigned to unreacted 9 and the other to putative 9-CO2. The chemical shifts are comparable to those observed for [Mg(CO2·pzMe2)2]n with a resolved signal for inserted CO2 at 152.1 ppm. A final statement on the amount of inserted CO2 in 9-CO2 cannot be made. In contrast, Y(pztBu2)3(thf)2 (10) did not show any traceable CO2 insertion into the Y–N bond at ambient temperature. Only traces of the carbamic acid HO2CpztBu2 could be detected in the 1H NMR spectrum, pointing to some HpztBu2 impurities in 10 (see Fig. S51†). Overall, this indicates that yttrium complex 10 has an even less pronounced capability to bind CO2 than the scandium congener 7. This is also in agreement with the CO2-insertion capability of homoleptic ceric pyrazolates Ce(pzRR′)4 which was found to decrease in the order of pzMe2 > pztBu,Me > pzPh2 > pztBu2.83
Synthesis and reactivity of unsubstituted “parent” pyrazolates of aluminium and scandium
Since the parent pyrazolate [Mg(pz)2]n displayed an exceptionally high reversible CO2 uptake of up to 35.7 wt%,20 it was of interest to examine such reaction behaviour for the trivalent light metals. The trivalent parent pyrazolates [Al(pz)3]n (11) and [Sc(pz)3]n (12) were accessed via transamination of Al(pztBu2)3 (1) and Sc(pztBu2)3(thf) (7) with Hpz in toluene, as indicated by the instant formation of a white precipitate. The 13C CP/MAS spectra of amorphous 11 and 12 showed the expected signal sets (Fig. S52/S54†). The 27Al CP/MAS experiment revealed a signal at 5.2 ppm (Fig. S53†) featuring the most high-field shifted signal observed for a homoleptic aluminium pyrazolate. For polymeric 12, the 45Sc resonance was resolved at 156.0 ppm (Fig. S55†). Not unexpectedly, the CO2-uptake behaviours of 11 and 12 were in line with those of the discrete substituted pyrazolates (vide supra). When exposing the solids in a very simple manner to an atmosphere of 1 bar CO2 for three hours, mass gains of 8.2 wt% for 11 and 6.9 wt% for 12 were found. However, these materials rapidly release CO2 at atmospheric pressure, as indicated by FTIR spectroscopy.Catalytic cycloaddition of epoxides and CO2
Given our previous works on metal pyrazolates, we were interested in the behaviour of trivalent metal pyrazolates as catalysts in the cycloaddition of epoxides and CO2 (Table 1). Applying the standard protocol (0.5 mol% catalyst, 1 mol% cocatalyst tetra-n-butylammonium bromide (TBAB), epoxide as solvent), the catalysis was performed under 1 bar CO2 at ambient temperature. After 24 h, the conversion was determined from the 1H NMR spectra. Pyrazolates Al(pztBu2)3 (1) [Al(pziPr2)3]2 (4), Sc(pztBu2)3(thf) (7), [Y(pzMe2)3(thf)]2 (9) and Y(pztBu2)3(thf)2 (10) were employed as catalysts in addition to tetrametallic cluster [Ce(pzMe2)3]4 (13) and the tetravalent [Ce(pzMe2)4]2 (14) for comparison. Propylene oxide, styrene oxide, 2-tert-butyloxirane and 1,2-epoxyhexane were employed as substrates. Aluminium complex 1 showed only moderate catalytic activity, converting 65% of propylene oxide to the cyclic carbonate (entry 1). The sterically more demanding epoxides were only minimally converted by complex 1, which achieved the highest conversion for 1,2-epoxyhexane (7%).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | R = Me | R = Ph | R = tBu | R = nBu |
| a Reaction conditions: 1 bar CO2, 0.5 mol% metal centre of the catalyst, 1 mol% TBAB at ambient temperature for 24 h. b Conversion determined by comparison of the integral protons at the α-position of the epoxide and the corresponding cyclic carbonate (expect for 3,3-dimethyl-1,2-butene carbonate where the integral of the tBu moieties was used). | |||||
| 1 | 1-Al | 65% | 5% | 2% | 7% |
| 2 | 4-Al | 43% | 8% | 3% | 11% |
| 3 | 7-Sc | >99% | 27% | 24% | 29% |
| 4 | 9-Y | 84% | 20% | 5% | 22% |
| 5 | 10-Y | 97% | 22% | 8% | 18% |
| 6 | 13-Ce III | 96% | 13% | 5% | 9% |
| 7 | 14-Ce IV | 88% | 23% | 13% | 25% |
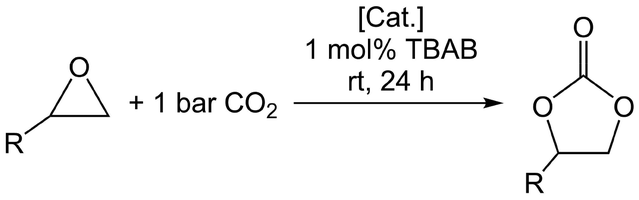
Dimeric complex 4 displayed an even lower conversion of 43% for propylene oxide, but performed slightly better in the cases of styrene oxide (8%) and 1,2-epoxyhexane (11%) (entry 2). This can be ascribed to the smaller iPr groups of 4 (versus tBu of 1) in agreement with the observations made for magnesium pyrazolates.20
Scandium complex 7 performed best in this cycloaddition reaction, affording 99% conversion of propylene oxide with only traces of the starting material left (entry 3). Complex 7 also exhibited the highest catalytic activity of the metal pyrazolates under study for the three other epoxides, ranging from 24 to 29%. The yttrium-based catalysts 9 and 10 provided similar results, with monomeric 10 achieving slightly higher conversions than dimeric 9, except for 1,2-epoxyhexane (entry 4 and 5). The results obtained for the cerium-based catalysts were in line with our earlier report, showing only slight deviations.19 Strikingly, there seems to be a negative correlation between the carboxophilicity of the metal pyrazolate and its catalytic activity (Table 2). As described above, scandium carbamate 7-CO2 releases CO2 readily in the absence of a solvent or when placed in an open vessel. Also, under the applied conditions, monomeric yttrium pyrazolate 10 did not insert any significant amount of CO2, while dimeric yttrium pyrazolate 9 did insert CO2 and displayed a CO2-releasing step at 70 °C. Since the CO2-release temperature range of complexes that reversibly insert CO2 is an approximate measure of their carboxophilicity, comparison with the respective catalytic conversions clearly show that the lower the carboxophilicity, the higher the catalytic activity (Table 2). On the other hand, the carboxophilicity does not reflect the trends in the extent of the (calculated) oxophilicity (Table 2)84 and electronegativity (Pauling scale)85 and might be affected further by the Lewis acidity of the metal centre (Al3+ > Sc3+ > Y3+), as well as the carbophilicity,86 sterics and coordination mode (terminal versus bridging) of the carbamato ligand. For further comparison, the average N–C(CO2) distance in complexes 1-Al-CO2 (1.444(3) Å), 7-Sc-CO2 (1.436(2) Å), [Ce(CO2·pzMe2)3]4 (13-CO2) (1.432(12) Å), [Ce(CO2·pzMe2)4] (14-CO2 = A, Fig. 1 bottom) (1.436(3) Å) and [Mg(CO2·pztBu2)2(thf)2] (B, Fig. 1 bottom) (1.477(1) Å) were detected in a close range.
| Catalyst | CO2-release temperature [°C] | Catalytic convers. [%] | Oxophilicity M [Θ]b | |
|---|---|---|---|---|
| 1H NMR start → end | TGA start → end | |||
| a Previous work Mg,20 Ce.19 b Ref. 84. c De-insertion not complete at this temperature, which is the upper limit of the VT NMR setup. | ||||
| 15-Mg | 70–105 | 134–233 | 56 | 0.6 |
| 1-Al | 100–120c | 114–196 | 65 | 0.8 |
| 9-Y | rt–70 | 60–154 | 84 | 0.8 |
| 14-Ce IV | 10–60a | 55–95a | 88 | 0.9 |
| 13-Ce III | 52–90a | 96 | 0.9 | |
| 10-Y | — | 97 | 0.8 | |
| 7-Sc | rt | rt | >99 | 0.8 |
Finally, the best-performing scandium complex 7 was examined for the cycloaddition of propylene oxide and CO2 under varying conditions (Table 3). The aforementioned performance using the standard procedure (conversion of >99%) corresponds to a TON of >199 (entry 1). Decreasing the catalyst concentration to 0.25 mol% lowered the overall catalytic conversion to 71, but increased the TON to 284 (entry 2). Employing only 0.01 mol% of 7 at harsher reaction conditions (10 bar CO2 and 90 °C), further increased the TON to 4600 (entry 3). The TOF of 7 was determined at 120 h−1 over the progress of the reaction applying our standard procedure (Fig. S86 and Table S1†).
| Entry | T [°C]/p[bar] | C (Cat.) [mol%] | Conversionb [%] | TONc |
|---|---|---|---|---|
| a 24 h in neat epoxide. b Conversion determined by comparison of the integrals protons at the α-position of the epoxide and the corresponding cyclic carbonate. c ((epoxide/Mg) × conversion)/100. | ||||
| 1 | 24/1 | 0.5 | >99% | 199 |
| 2 | 24/1 | 0.25 | 71% | 284 |
| 3 | 90/10 | 0.01 | 45% | 4600 |
Accordingly, complete conversion (>99%) was already detected after 12 h, but already after 6 h, the conversion was noted at 96%. Using half the amount of catalyst (entry 2) did not give similarly high conversions after 6 or 10 h, pointing to the catalyst load as the determining factor. Overall, a mechanism/catalytic cycle as described for the cerium and magnesium congeners is proposed.19,20 To put the present catalytic results into a proper perspective, even the most active catalysts under study show only moderate catalytic activity in comparison to systems described in literature. For example, the chlorinated tetraphenyl porphyrin aluminium complex (tetra(2,4-chloro)phenylporphyrin)AlCl reaches TOFs of up to 185200 h−1.36 For the rare-earth metals, the scorpionate complex [La{N(SiHMe2)2}2{κ3-(1-[2,2-bis(pzMe2)-1,1-Ph2Et]-1,3-Cp)}] was noted at TOFs of 15000 h−1 and TONs of up to 306667.57
Conclusions
The scope and feasibility of reversible metal-pyrazolate CO2-insertion chemistry is expandable to the trivalent light metals of groups 3 and 13. The reaction behaviors of monomeric aluminium(III) and scandium(III) pyrazolates are strikingly distinct under identical conditions. While homoleptic Al(pztBu2)3, bearing the comparatively small metal centre, inserts two CO2 molecules to form Al(CO2·pztBu2)2(pztBu2), Sc(pztBu2)3(thf) accommodates only one molecule CO2 per scandium, affording the dimeric complex [Sc(μ-CO2·pztBu2)(pztBu2)2]2. Both complexes feature completely reversible heteroallene insertion, as revealed by FTIR and VT 1H NMR spectroscopy and thermogravimetric analysis (TGA). Again, strikingly, the aluminium complex releases the CO2 only at temperatures >100 °C, while the scandium complex undergoes CO2 de-insertion at ambient temperature and pressure. The implications of the pyrazolato substituents for the extent of CO2 insertion was demonstrated for the new homoleptic complex [Al(pziPr2)3]2, which gave exhaustive CO2 insertion, marking a capacity of 21.6 wt% CO2. Applying the metal pyrazolates under study as catalysts in the cycloaddition of CO2 and epoxides to cyclic carbonates revealed an inverse correlation of the catalytic activity and carboxophilicity (CO2 affinity): the higher the carboxophilicity, the lower the catalytic activity. Given the reversible CO2 insertion into metal pyrazolates, the carboxophilicity can be assessed by the CO2-release temperature (via VT 1H NMR spectroscopy and TGA). The scandium complex Sc(pztBu2)3(thf) performed best, with a maximum TOF of 120 h−1 and a TON of 284 at ambient conditions.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We are grateful to the VECTOR foundation (grant P2021-0099) for generous support. We thank Dr Markus Ströbele for running the TGA experiments.References
- S. Dhakal, J. C. Minx, F. L. Toth, A. Abdel-Aziz, M. J. Figueroa Meza, K. Hubacek, I. G. C. Jonckheere, Y.-G. Kim, G. F. Nemet, S. Pachauri, X. Tan and T. Wiedmann, 2022: Emissions Trends and Drivers. In IPCC, 2022: Climate Change 2022: Climate Change 2022: Mitigation of Climate Change. Contribution of Working Group III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. P. R. Shukla, J. Skea, R. Slade, A. Al Khourdajie, R. van Diemen, D. McCollum, M. Pathak, S. Some, P. Vyas, R. Fradera, M. Belkacemi, A. Hasija, G. Lisboa, S. Luz and J. Malley, Cambridge University Press, Cambridge, UK and New York, NY, USA. DOI:10.1017/97810091579926.004.
- P. Falkowski, R. J. Scholes, E. Boyle, J. Canadell, D. Canfield, J. Elser, N. Gruber, K. Hibbard, P. Högberg, S. Linder, F. T. Mackenzie, B. Moore III, T. Pedersen, Y. Rosenthal, S. Seitzinger, V. Smetacek and W. Steffen, The Global Carbon Cycle: A Test of Our Knowledge of Earth as a System, Science, 2000, 290, 291–296 CrossRef CAS PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Irreversible climate change due to carbon dioxide emissions, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- R. S. Haszeldine, Carbon Capture and Storage: How Green Can Black Be?, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- D. W. Keith, Why Capture CO2 from the Atmosphere?, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge?, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Life cycle assessment of CO2 capture and utilization: a tutorial review, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Using carbon dioxide as a building block in organic synthesis, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, Reactive Capture of CO2: Opportunities and Challenges, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Carbon Dioxide Capture: Prospects for New Materials, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Carbon Dioxide Capture in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Direct Capture of CO2 from Ambient Air, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Metal-Organic Frameworks for Carbon Dioxide Capture and Methane Storage, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- A. C. Forse and P. J. Milner, New chemistry for enhanced carbon capture: beyond ammonium carbamates, Chem. Sci., 2021, 12, 508–516 RSC.
- J. Hack, N. Maeda and D. M. Meier, Review on CO2 Capture Using Amine-Functionalized Materials, ACS Omega, 2022, 7, 39520–39530 CrossRef CAS PubMed.
- R. Ayyappan, I. Abdalghani, R. C. Da Costa and G. R. Owen, Recent developments on the transformation of CO2 utilising ligand cooperation and related strategies, Dalton Trans., 2022, 51, 11582–11611 RSC.
- (a) U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Effective and Reversible Carbon Dioxide Insertion into Cerium Pyrazolates, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef CAS PubMed; (b) U. Bayer and R. Anwander, Carbonyl group and carbon dioxide activation by rare-earth-metal complexes, Dalton Trans., 2020, 49, 17472–17493 RSC.
- F. Kracht, P. Rolser, P. Preisenberger, C. Maichle-Mössmer and R. Anwander, Organomagnesia: Reversibly High Carbon Dioxide Uptake by Magnesium Pyrazolates, Adv. Sci., 2024, 11, 2403295 CrossRef CAS PubMed.
- K. Ziegler, F. Krupp, K. Weyer and W. Larbig, Metallorganische Verbindungen, XLI Reaktionen der Aluminiumtrialkyle mit Kohlendioxyd und Schwefeldioxyd, Justus Liebigs Ann. Chem., 1960, 629, 251–256 CrossRef CAS.
- J. Weidlein, CO2-Einschiebungsreaktionen bei Aluminium-, Gallium- und Indiumalkylverbindungen, Z. Anorg. Allg. Chem., 1970, 378, 245–262 CrossRef CAS.
- (a) D. B. Dell'Amico, F. Calderazzo, M. Dell'Innocenti, B. Guldenpfennig, S. Ilanelli, G. Pelizzi and P. Robino, N,N-Dialkylcarbamates of Silicon and Aluminium, Gazz. Chim. Ital., 1993, 123, 283–288 Search PubMed; (b) D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Converting Carbon Dioxide into Carbamato Derivatives, Chem. Rev., 2003, 103, 3857–3897 CrossRef PubMed.
- C.-C. Chang, B. Srinivas, W. Mung-Liang, C. Wen-Ho, M. Y. Chiang and H. Chung-Sheng, Fixation of CO2 by a Series of Ethynyl-Bridged Polynuclear Aluminum-Magnesium Complexes. Synthesis, Characterization, and Crystal Structures of [Me2Al(μ-i-Pr2N)2Mg(μ-C≡CR)]2 (R = C6H5, C6H4-p-CH3, t-Bu, SiMe3), [Me2Al(μ-Et2N)2Mg(μ-C≡CC6H5)]2, {(Me2Al)2[μ-OO-C(i-Pr2N)]2}, and {(Me2Al)2[(μ-OOC(i-Pr2N))2]2Mg}, Organometallics, 1995, 14, 5150–5159 CrossRef CAS.
- T. W. Yokley, H. Tupkar, N. D. Schley, N. J. DeYonker and T. P. Brewster, CO2 Capture by 2-(Methylamino)pyridine Ligated Aluminum Alkyl Complexes, Eur. J. Inorg. Chem., 2020, 2020, 2958–2967 CrossRef CAS PubMed.
- For more reccent examples of CO2 insertion into Al-pnictogenyl moieties, see: (a) W. Haider, M. D. Calvin-Brown, I.-A. Bischoff, V. Huch, B. Morgenstern, C. Müller, T. Sergeieva, D. M. Andrada and A. Schäfer, Diarylpnictogenyldialkylalanes – Synthesis, Structures, Bonding Analysis, and CO2 Capture, Inorg. Chem., 2022, 61, 1672–1684 CrossRef CAS PubMed; (b) E. Schumann, E. Brendler, U. Böhme and F. Mertens, Synthesis of Aluminum N,N-Dialkylcarbamates by Insertion of CO2 into Al−N Bonds, Eur. J. Inorg. Chem., 2023, 26, e202200568 CrossRef CAS.
- P. Federmann, R. Müller, F. Beckmann, C. Lau, B. Cula, M. Kaupp and C. Limberg, Synthesis of Intramolecular P/Al-Based Frustrated Lewis Pairs via Aluminum-Tin-Exchange and their Reactivity toward CO2, Chem. – Eur. J., 2022, 28, e202200404 CrossRef CAS PubMed.
- N. Takeda and S. Inoue, Activation of Carbon Dioxide by Tetraphenylporphinatoaluminium Methoxide. Reaction with Epoxide, Bull. Chem. Soc. Jpn., 1978, 51, 3564–3567 CrossRef CAS.
- T. Aida and S. Inoue, Activation of carbon dioxide with aluminum porphyrin and reaction with epoxide. Studies on (tetraphenylporphinato)aluminum alkoxide having a long oxyalkylene chain as the alkoxide group, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- F. Kojima, T. Aida and S. Inoue, Fixation and activation of carbon dioxide on aluminum porphyrin. Catalytic formation of a carbamic ester from carbon dioxide, amine, and epoxide, J. Am. Chem. Soc., 1986, 108, 391–395 CrossRef CAS PubMed.
- D. J. Darensbourg and M. W. Holtcamp, Catalysts for the reactions of epoxides and carbon dioxide, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- M. Alvaro, C. Baleizao, E. Carbonell, M. El Ghoul, H. García and B. Gigante, Polymer-bound aluminium salen complex as reusable catalysts for CO2 insertion into epoxides, Tetrahedron, 2005, 61, 12131–12139 CrossRef CAS.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, Formation of Cyclic Carbonates from Carbon Dioxide and Epoxides Coupling Reactions Efficiently Catalyzed by Robust, Recyclable One-Component Aluminum-Salen Complexes, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, A Powerful Aluminum Catalyst for the Synthesis of Highly Functional Organic Carbonates, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Highly Active Aluminium Catalysts for the Formation of Organic Carbonates from CO2 and Oxiranes, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed.
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, An aluminum porphyrin complex with high activity and selectivity for cyclic carbonate synthesis, Green Chem., 2015, 17, 2853–2858 RSC.
- M. A. St. Clair and B. D. Santarsiero, Structure of a scandium–carboxylate complex: (η5-C5Me5)2Sc(O2C)C6H4CH3, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 850–852 CrossRef.
- H. Schumann, J. A. Meese-Marktscheffel, A. Dietrich and F. H. Görlitz, Metallorganische Verbindungen der Lanthanoide: LXX. Bis(cyclopentadienyl)seltenerdkomplexe: Synthese und Röntgen-strukturanalyse von dem intramolekular stabilisierten Lutetiumalkyl Cp2Lu(CH2)3NMe2 und dem monomeren basenfreien Yttriumcarboxylat Cp2Y[η2-O2C(CH2)3NMe2], J. Organomet. Chem., 1992, 430, 299–315 CrossRef CAS.
- W. J. Evans, C. A. Seibel, J. W. Ziller and R. J. Doedens, CO2 Insertion Chemistry as a Probe of Organosamarium Allyl Reactivity, Organometallics, 1998, 17, 2103–2112 CrossRef CAS.
- F. A. LeBlanc, A. Berkefeld, W. E. Piers and M. Parvez, Reactivity of Scandium β-Diketiminate Alkyl Complexes with Carbon Dioxide, Organometallics, 2012, 31, 810–818 CrossRef CAS.
- O. T. Summerscales, C. M. Moore, B. L. Scott, M. P. Wilkerson and A. D. Sutton, Cerium(III) Carbonate Formation from {CeCp*2H}2 and Carbon Dioxide: Structure and Mechanistic Insights, Organometallics, 2017, 36, 4682–4685 CrossRef CAS.
- D. W. Beh, W. E. Piers, I. del Rosal, L. Maron, B. S. Gelfand, C. Gendy and J.-B. Lin, Scandium alkyl and hydride complexes supported by a pentadentate diborate ligand: reactions with CO2 and N2O, Dalton Trans., 2018, 47, 13680–13688 RSC.
- D. Cui, M. Nishiura, O. Tardif and Z. Hou, Rare-Earth-Metal Mixed Hydride/Aryloxide Complexes Bearing Mono(cyclopentadienyl) Ligands. Synthesis, CO2 Fixation, and Catalysis on Copolymerization of CO2 with Cyclohexene Oxide, Organometallics, 2008, 27, 2428–2435 CrossRef CAS.
- L. A. M. Steele, T. J. Boyle, R. A. Kemp and C. Moore, The selective insertion of carbon dioxide into a lanthanide(III) 2,6-di-t-butyl-phenoxide bond, Polyhedron, 2012, 42, 258–264 CrossRef CAS.
- M. N. Bochkarev, E. A. Fedorova, Yu. F. Radkov, S. Ya. Khorshev, G. S. Kalinina and G. A. Razuvaev, Carbon dioxide fixaion by lathanide complexes, J. Organomet. Chem., 1983, 258, C29–C33 CrossRef CAS.
- Y. F. Rad'kov, E. A. Fedorova, S. Y. Khorshev, G. S. Kalinina, M. N. Bochkarev and G. A. Razuvaev, Reactions of carbon dioxide with bis(trimethylsilyl)amino derivatives of lanthanides, Zh. Obshch. Khim., 1986, 56, 386–389 Search PubMed.
- W. J. Evans, C. H. Fujimoto and J. W. Ziller, Organolanthanide-Based Coordination and Insertion Reactivity of the Anion Formed by Deprotonation of ε-Caprolactam, Organometallics, 2001, 20, 4529–4536 CrossRef CAS.
- H. Yin, P. J. Carroll and E. J. Schelter, Reactions of a cerium(III) amide with heteroallenes: insertion, silyl-migration and de-insertion, Chem. Commun., 2016, 52, 9813–9816 RSC.
- D. V. Vitanova, F. Hampel and K. C. Hultzsch, Rare earth metal complexes based on β-diketiminato and novel linked bis(β-diketiminato) ligands: Synthesis, structural characterization and catalytic application in epoxide/CO2-copolymerization, J. Organomet. Chem., 2005, 690, 5182–5197 CrossRef CAS.
- D. Cui, M. Nishiura and Z. Hou, Alternating Copolymerization of Cyclohexene Oxide and Carbon Dioxide Catalyzed by Organo Rare Earth Metal Complexes, Macromolecules, 2005, 38, 4089–4095 CrossRef CAS.
- A. Decortes, R. M. Haak, C. Martín, M. M. Belmonte, E. Martin, J. Benet-Buchholz and A. W. Kleij, Copolymerization of CO2 and Cyclohexene Oxide Mediated by Yb(salen)-Based Complexes, Macromolecules, 2015, 48, 8197–8207 CrossRef CAS.
- B. Xu, P. Wang, M. Lv, D. Yuan and Y. Yao, Transformation of Carbon Dioxide into Oxazolidinones and Cyclic Carbonates Catalyzed by Rare-Earth-Metal Phenolates, ChemCatChem, 2016, 8, 2466–2471 CrossRef CAS.
- H. Yasuda, L.-N. He and T. Sakakura, Cyclic Carbonate Synthesis from Supercritical Carbon Dioxide and Epoxide over Lanthanide Oxychloride, J. Catal., 2002, 209, 547–550 CrossRef CAS.
- A. Ion, V. Parvulescu, P. Jacobs and D. de Vos, Sc and Zn-catalyzed synthesis of cyclic carbonates from CO2 and epoxides, Appl. Catal., A, 2009, 363, 40–44 CrossRef CAS.
- J. Qin, P. Wang, Q. Li, Y. Zhang, D. Yuan and Y. Yao, Catalytic production of cyclic carbonates mediated by lanthanide phenolates under mild conditions, Chem. Commun., 2014, 50, 10952–10955 RSC.
- C. Wang, X. Liu, Z. Dai, Y. Sun, N. Tang and J. Wu, Yttrium complex supported by a sterically encumbering N-anchored tris-arylphenoxide ligand: Heteroselective ROP of rac-lactide and CO2/epoxide coupling, Inorg. Chem. Commun., 2015, 56, 69–72 CrossRef CAS.
- J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, An Efficient and Versatile Lanthanum Heteroscorpionate Catalyst for Carbon Dioxide Fixation into Cyclic Carbonates, ChemSusChem, 2017, 10, 2886–2890 CrossRef PubMed.
- Z. Zhao, J. Qin, C. Zhang, Y. Wang, D. Yuan and Y. Yao, Recyclable Single-Component Rare-Earth Metal Catalysts for Cycloaddition of CO2 and Epoxides at Atmospheric Pressure, Inorg. Chem., 2017, 56, 4568–4575 CrossRef PubMed.
- O. Sodpiban, S. D. Gobbo, S. Barman, V. Aomchad, P. Kidkhunthod, S. Ould-Chikh, A. Poater, V. D'Elia and J.-M. Basset, Synthesis of well-defined yttrium-based Lewis acids by capturing a reaction intermediate and catalytic application for cycloaddition of CO2 to epoxides under atmospheric pressure, Catal. Sci. Technol., 2019, 9, 6152–6165 RSC.
- D. Pfeiffer, B. J. Ximba, L. M. Liable-Sands, A. L. Rheingold, M. J. Heeg, D. M. Coleman, H. B. Schlegel, T. F. Kuech and C. H. Winter, Synthesis, Structure, and Molecular Orbital Studies of Yttrium, Erbium, and Lutetium Complexes Bearing η2-Pyrazolato Ligands: Development of a New Class of Precursors for Doping Semiconductors, Inorg. Chem., 1999, 38, 4539–4548 CrossRef CAS PubMed.
- G. B. Deacon, A. Gitlits, P. W. Roesky, M. R. Bürgstein, K. C. Lim, B. W. Skelton and A. H. White, Simple Syntheses, Structural Diversity, and Tishchenko Reaction Catalysis of Neutral Homoleptic Rare Earth(II or III) 3,5-Di-tert-butylpyrazolates—The Structures of [Sc(t,Bu2pz)3], [Ln2(tBu2pz)6] (Ln=La, Nd, Yb, Lu), and [Eu4(tBu2pz)8], Chem. – Eur. J., 2001, 7, 127–138 CrossRef CAS PubMed.
- X. Zhou, L. Zhang, R. Ruan, L. Zhang, R. Cai and L. Weng, Synthesis of [(C5H5Y(η2-PzMe2)(η-PzMe2)]2 and [Y(η2-PzMe2)2(η-PzMe2)(η-THF)]2 and the insertion of Me2SiO into the Y-N bond, Chin. Sci. Bull., 2001, 46, 723–726 CrossRef CAS.
- D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Pyrazolates advance cerium chemistry: a CeIII/CeIV redox equilibrium with benzoquinone, Dalton Trans., 2017, 46, 6265–6277 RSC.
- N. E. Rad, P. C. Junk, G. B. Deacon and J. Wang, New Homoleptic Rare Earth 3,5-Diphenylpyrazolates and 3,5-Di-tert-butylpyrazolates and a Noteworthy Structural Discontinuity, Z. Anorg. Allg. Chem., 2019, 645, 877–881 CrossRef CAS.
- G. B. Deacon, E. E. Delbridge, C. M. Forsyth, P. C. Junk, B. W. Skelton and A. H. White, Main Group Pyrazolates—the X-Ray Structures of [Al(η2-But2pz)3], [SnMe3(η1-Ph2pz)] and [GePh3(η1-But2pz)] (R2pz = 3,5-Disubstituted Pyrazolate), Aust. J. Chem., 1999, 52, 733–740 CrossRef CAS.
- J. Lewiński, J. Zachara, P. Goś, E. Grabska, T. Kopeć, I. Madura, W. Marciniak and I. Prowotorow, Reactivity of Various Four-Coordinate Aluminum Alkyls towards Dioxygen: Evidence for Spatial Requirements in the Insertion of an Oxygen Molecule into the Al−C Bond, Chem. – Eur. J., 2000, 6, 3215–3227 CrossRef.
- S. R. Kosuru, T.-H. Sun, L.-F. Wang, J. K. Vandavasi, W.-Y. Lu, Y.-C. Lai, S. C. N. Hsu, M. Y. Chiang and H.-Y. Chen, Enhanced Catalytic Activity of Aluminum Complexes for the Ring-Opening Polymerization of ε-Caprolactone, Inorg. Chem., 2017, 56, 7998–8006 CrossRef CAS PubMed.
- C.-C. Chang, T.-Y. Her, F.-Y. Hsieh, C.-Y. Yang, M. Y. Chiang, G.-H. Lee, Y. Wang and S.-M. Peng, Chemical Reactivities of [Me2AI(μ-NiPr2)2MgMe]4 and [Me2AI(μ-NiPr2)2MgCI]2: Crystal Structures of [Me2AI(μ-Pz)2AIMe2], [Me2AI(μ-Pz)2Mg(μ-Pz)2AIMe2], and [Me2Al(μ-NiPr2)(μ-Me)Mg(μ-NiPr2)(μ-Me)AIMe2], J. Chin. Chem. Soc., 1994, 41, 783–789 CrossRef CAS.
- W. Zheng, H. Hohmeister, N. C. Mösch-Zanetti, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Syntheses and Characterization of μ,η1,η1-3,5-Di-tert-butylpyrazolato Derivatives of Aluminum, Inorg. Chem., 2001, 40, 2363–2367 CrossRef CAS PubMed.
- N. E. Rad, P. C. Junk, G. B. Deacon, I. V. Taidakov, J. Wang, N. E. Rad, P. C. Junk, G. B. Deacon, I. V. Taidakov and J. Wang, Synthesis and Structural Characterisation of Lithium, Zinc, and Aluminium Pyrazolate Complexes*, Aust. J. Chem., 2020, 73, 520–528 CrossRef CAS.
- S. A. Cortes-Llamas and M.-Á. Muñoz-Hernández, Sodium Polypyrazolylaluminates: Synthesis, Characterization, and Isolation of a Reaction Intermediate of a Trispyrazolylaluminate, Organometallics, 2007, 26, 6844–6851 CrossRef CAS.
- C. J. Snyder, M. J. Heeg and C. H. Winter, Poly(pyrazolyl)aluminate Complexes Containing Aluminum–Hydrogen Bonds, Inorg. Chem., 2011, 50, 9210–9212 CrossRef CAS PubMed.
- D. Sambade and G. Parkin, Synthesis and structural characterization of tris(pyrazolyl)hydroaluminate and tris(pyrazolyl)hydrogallate lithium compounds, Polyhedron, 2017, 125, 219–229 CrossRef CAS.
- O. J. Garcia, L. Vendier, M. Etienne, S. Gwaltney, A. Ressler and M.-Á. Muñoz-Hernández, Cyclooctadiene Rh(I) Bis- and Tris(pyrazolyl)aluminate Complexes and Their Catalytic Activity on the Polymerization of Phenylacetylene, Inorg. Chem., 2021, 60, 10757–10763 CrossRef CAS PubMed.
- S. R. Kosuru, F.-J. Lai, Y.-L. Chang, C.-Y. Li, Y.-C. Lai, S. Ding, K.-H. Wu, H.-Y. Chen and Y.-H. Lo, Collaboration between Trinuclear Aluminum Complexes Bearing Bipyrazoles in the Ring-Opening Polymerization of ε-Caprolactone, Inorg. Chem., 2021, 60, 10535–10549 CrossRef CAS PubMed.
- B. M. Louie, S. J. Rettig, A. Storr and J. Trotter, Synthesis and crystal structure of tri(μ-carbonyl)bis[methyltris(1-pyrazolyl)gallato]dirhodium – benzene (1:1), Can. J. Chem., 1984, 62, 633–643 CrossRef CAS.
- G. A. Banta, B. M. Louie, E. Onyiriuka, S. J. Rettig and A. Storr, Synthesis and characterization of LMo(CO)3M(PPh3)x (where L = tridentate pyrazolyl-gallate or -borate ligand; M = Rh, x = 2; M = Cu, x = 1). X-ray crystal structures of [MeGapz3]Mo(CO)3Rh(PPh3)2 and [MeGapz3]Mo(CO)3Cu(PPh3) (where pz = pyrazolyl, N2C3H3), Can. J. Chem., 1986, 64, 373–386 CrossRef CAS.
- E. C. Onyiriuka, S. J. Rettig and A. Storr, Synthesis and characterization of molybdenum–tin complexes derived from the molybdenum tricarbonyl anion, [MeGapz3]Mo(CO)3−, and organotin chlorides. X-ray crystal structure of [MeGapz3]Mo(CO)3SnPh3 (where pz = pyrazolyl, N2C3H3), Can. J. Chem., 1986, 64, 321–327 CrossRef CAS.
- A. Frazer, B. Piggott, M. Harman, M. Mazid and M. B. Hursthouse, Syntheses and X-ray crystal structures of [bis{(3,5-dimethylpyrazolyl)3hydridoborato}In]I and [bis{(pyrazolyl)3methylgallato}In][InI4], Polyhedron, 1992, 11, 3013–3017 CrossRef CAS.
- S. N. König, G. Gerstberger, C. Schädle, C. Maichle-Mössmer, E. Herdtweck and R. Anwander, Unusual reaction pathways of gallium(III) silylamide complexes, Main Group Met. Chem., 2013, 36, 169–180 Search PubMed.
- (a) M. J. Heeg, Z. Yu and C. H. Winter, CCDC 781781: Experimental Crystal Structure Determination, 2011; (b) C. T. Sirimanne, W. Zheng, Z. Yu, M. J. Heeg and C. H. Winter, Synthesis, Structure, and Bridge-Terminal Exchange Kinetics of Pyrazolate-Bridged Digallium and Diindium Complexes Containing Bridging Phenyl Groups, Organometallics, 2005, 24, 6184–6193 CrossRef CAS.
- For Ln3Na2 pyrazolate clusters with interstitial oxy ions, see: H. Schumann, P. R. Lee and J. Loebel, Angew. Chem., Int. Ed. Engl., 1989, 28, 1033–1035 CrossRef.
- U. Bayer, A. Jenner, J. Riedmaier, C. Maichle-Mössmer and R. Anwander, Effect of Substituents of Cerium Pyrazolates and Pyrrolates on Carbon Dioxide Activation, Molecules, 2021, 26, 1957 CrossRef CAS PubMed.
- (a) K. P. Kepp, A Quantitative Scale of Oxophilicity and Thiophilicity, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed; (b) K. A. Moltved and K. P. Kepp, The Chemical Bond between Transition Metals and Oxygen: Electronegativity, d-Orbital Effects, and Oxophilicity as Descriptors of Metal–Oxygen Interactions, J. Phys. Chem. C, 2019, 123, 18432–18444 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond. IV. The Energy of Single Bonds and the relative Electronegativity of Atoms, J. Am. Chem. Soc., 1932, 54, 3570–3582 CrossRef CAS.
- G. O. Kayode and M. M. Montemore, Factors controlling oxophilicity and carbophilicity of transition metals and main group metals, J. Mater. Chem. A, 2021, 9, 22325–22333 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2358935–2358943. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01656d |
| This journal is © the Partner Organisations 2024 |