
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional design of stimuli-responsive poly(phthalaldehyde)-based adhesives: depolymerization kinetics and mechanical strength management through plasticizer addition†‡
Patrick
Damacet
 ,
Hana J.
Yarbrough
,
Nicholas D.
Blelloch
,
Hyuk-Jun
Noh
and
Katherine A.
Mirica
*
,
Hana J.
Yarbrough
,
Nicholas D.
Blelloch
,
Hyuk-Jun
Noh
and
Katherine A.
Mirica
*
Department of Chemistry, Dartmouth College, 41 College Street, Hanover, NH 03755, USA. E-mail: Katherine.A.Mirica@dartmouth.edu
First published on 12th February 2024
Abstract
Through a strategic polymer engineering design, this paper shows that self-immolative poly(phthalaldehyde) (PPA) can be employed as a responsive debonding-on-demand adhesive for the detection of fluorides and acids in liquid and solid states. The engineered PPA with dimethyl phthalate plasticizer blends exhibits mechanical shear strength up to 1100 kPa when bonding smooth glass surfaces, which represents a significant enhancement compared to the o-phthalaldehyde monomer (12 kPa) and the pristine PPA polymer (up to 400 kPa). Exposure of acetyl-endcapped PPA to 8.0 eq. of trifluoroacetic acid (TFA) under ambient conditions resulted in a complete degradation of the polymer within 25 minutes via a backbone cleavage, whereas exposure of fluoride responsive silyl-ether endcapped PPA to 1.0 eq. of tetrabutylammonium fluoride (TBAF) resulted in a fast and simultaneous decomposition of the polymer within 2 minutes via end-cap cleavage. This study also illustrates the capability of PPA to undergo solid-state depolymerization, promoting on-demand release from adhesion. Overall, this study offers a unique design strategy for creating on-demand depolymerizable coatings that may find future potential applications in stimuli-responsive personal protective equipment (PPE) for the chemical detection and on-demand protection against chemical warfare agents (CWAs) in hostile environments.
Introduction
Self-immolative polymers (SIPs) represent a class of scissionable degradable materials that depolymerize end-to-end through a domino-like fragmentation upon exposure to an external stimulus.1–3 Originally designed by Katzenellenbogen and coworkers as a prodrug platform for drug delivery,4 modern applications of SIPs encompass chemical sensors, drug delivery, and lithographic patterning systems.5,6 The recent rising interest in SIPs lies in the opportunity to customize the selectivity of the material towards a particular stimulus through the molecular design of the end caps,7–10 cross-linkers,11–13 or polymer backbone.14–16 Depolymerizable SIPs respond to stimuli by undergoing either a reversible or an irreversible transformation.2 Irreversible SIPs degrade into products that differ from the monomers they were prepared from, rendering their repolymerization challenging to realize. On the other hand, reversible SIPs, characterized by a low ceiling temperature (TC), depolymerize to their initial starting monomers, making their repolymerization reaction possible.17 This ability to switch between monomeric and polymeric states through a reversible polymerization reaction gives rise to the rapid detection of the targeted molecular recognition event through an autonomous change in property (chemical structure, color, crystallinity, mechanical adhesion, and tensile strengths).18,19 While end-capping or cyclizing the polymer prevents it from self-depolymerizing, any bond cleavage, whether through the end-caps or the backbone, triggers a decomposition of the polymeric molecular structure due to the low TC of un-capped polymers.17,20 Despite their unique properties and ease of synthesis, the transient nature of SIPs poses several challenges that limit their usage and applicability in a variety of applications. In particular, these materials are known to have a broad polydispersity index (PDI) accompanied by a limited degree of polymerization, leading often to the formation of oligomers during polymerization.21 In addition, these materials are known to be hard and brittle, which limits their mechanical properties and flexibility.22Self-immolative poly(phthalaldehyde) (PPA) is unique in that its mechanical and transient properties can be readily tuned through the addition of a liquid-based plasticizer that turns the polymer into a robust, temporary adhesive able of debonding through a specific molecular recognition event.23,24 The addition of such plasticizer results in a dramatic improvement in the structural flexibility and thermal stability of PPA via the depression of its glass transition temperature (Tg), changing its physical state from hard and glassy to soft and leathery.23 In general, PPA exists in two different structures (linear and cyclic) based on the polymerization technique (living anionic or cationic, respectively) employed during its synthesis.10,25 While both linear and cyclic PPAs are intrinsically sensitive to acid, heat, and mechanical forces due to the vulnerable bonds in the hemiacetal units of the polymer backbone,25–27 the reactivity of linear PPAs can be altered by installing an end group that can be selectively cleaved by a pre-determined stimulus.26–28 Plenty of studies that demonstrate site-specific depolymerization of linear end-capped PPA exist in the literature, with a library of investigated stimuli including palladium,29 conjugate acids,30 UV light,27 fluorides,31 and ultrasound.32 Given the proven capability to functionally customize its end groups, in addition to its ability to depolymerize in the solid-state,29,33 linear PPA stands out as a strong candidate for adhesive modulation by chemical triggers.
Herein, we report the first use of linear end-capped PPA as a stimuli-responsive temporary adhesive with on-demand debonding through a self-immolative depolymerization mechanism triggered by chemical exposure. The molecular design of the end cap strategically controls the polymer selectivity towards an incoming chemical species, such that debonding only occurs upon exposure to the specific trigger. We reasoned that changes in mechanical properties of the polymer adhesive upon the degradation of the polymer will disrupt adhesion to the point of debonding and mechanical failure. Beyond improving the internal strength of PPA, the addition of a liquid-based dimethyl phthalate (DMP) plasticizing agent allowed the polymer to be melt-bonded without risking depolymerization by disrupting its intermolecular packing and suppressing its Tg. With the addition of DMP, we observed that linear PPA is capable of bonding smooth glass with shear stresses up to 1100 ± 282 kPa. We also showed that PPA depolymerizes in response to acid and fluoride in both liquid and solid phases resulting in complete delamination of smooth glass and textile substrates. This work expands the capabilities of controlling adhesion by reporting a temporary adhesive material that can offer a response to chemical stimuli, in particular the ones sharing similarities with chemical warfare and corrosive agents that release fluoride and acids, respectively. We believe this material can act as a versatile detection platform for such agents and may have potential applicability in the design of stimuli-responsive PPE for chemical detection and on-demand protection in hostile environments.
Results and discussion
Molecular design
The depolymerization kinetics of SIPs can be strategically mediated by the nature and concentration of a stimulus.34 Linear PPA has two structural locations from which depolymerization can be initiated: (i) the reactive end caps, and (ii) the hemiacetal linkages along the backbone. The reactivity of the end caps and incoming stimulus may encourage PPA to proceed through a site-specific depolymerization pathway, which promotes selective control of material properties at a molecular level. The site-specific depolymerization pathway enables programmable responses of PPA to chemical stimuli, which represents an exciting opportunity for realizing multifunctional temporary adhesives. Other types of stimuli, including extended sonication times, heat, or acids, may trigger depolymerization by cleaving the hemiacetal linkages of the polymer backbone in a non-specific pathway, resulting in a slow and unorganized degradation process.32,35,36 A significant drawback of the non-specific pathway is that cleavage along the backbone can cause random fragmentation, which sparks off random side reactions, rendering it difficult to recover o-phthalaldehyde for repolymerization. Another limitation of the abovementioned pathway is the lack of chemical selectivity toward stimuli, which the end-cap triggered polymerization is able to overcome.37 Our design of a chemically responsive temporary adhesive leverages the reactivity of cleavable end caps of the self-immolative polymer, PPA, for selective debonding (Fig. 1). Once the polymeric material strongly joins two surfaces, exposure to a stimulus is designed to trigger depolymerization through a site-specific pathway, as is in the case of fluoride, or site non-specific, as in the case of trifluoroacetic acid (TFA). The instability of the linear polymer without the end cap predisposes this material towards rapid depolymerization, regardless of the pathway.38 Strategic removal of the end cap by a pre-determined stimulus initiates depolymerization, which leads to release from adhesion due to dramatic difference in the mechanical properties between the polymer and newly formed monomer.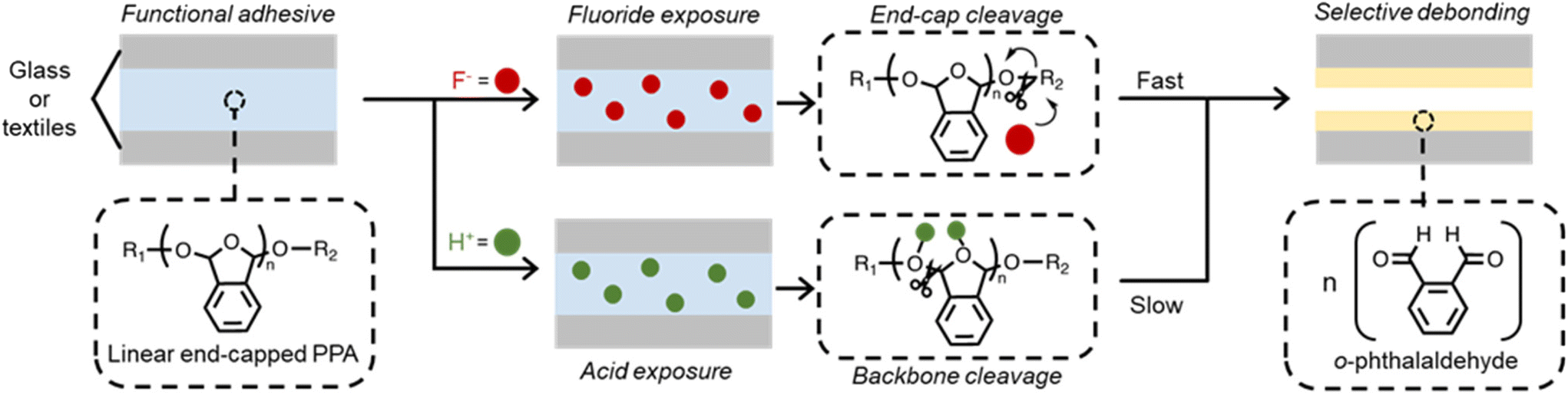 | ||
| Fig. 1 On-demand depolymerization of self-immolative PPA and the resulting delamination of surfaces. Two substrates, either glass slides or textiles, are joined with linear PPA, which consists of strategically selected end caps. Upon exposure to a specific chemical stimulus, self-immolative depolymerization rapidly ensues via either site-specific or unspecific pathways, thereby releasing the substrates due to structural changes of the material. | ||
Synthetic procedures & properties of PPA
We began our investigations by generating two linear PPA polymers using a living anionic polymerization method under air- and moisture-free conditions at −78 °C, employing an alcohol as initiator and an acyl chloride as terminator for the polymerization reaction (Fig. 2). A detailed synthetic route can be found in Section 1 of the ESI.‡ Two distinct sets of functional groups operating as end caps were chosen to probe adhesion and triggered debonding of PPA self-immolative systems. The first polymer consisted of an unreactive isopropyl ether (IPA) endcap along with a reactive acetyl ester (Ac) end cap and was denoted as IPA-PPA-Ac (Fig. S1‡). The second polymer comprised two reactive tert-butyldimethylsilyl (TBS) end caps and was denoted as TBS-PPA-TBS (Fig. S2‡). PPA, in general, whether cyclic or linear, possesses ether oxygen atoms in its central moiety. Hence, we reasoned that endcapping linear PPA with an oxygen-containing functional group, like acetyl ester would ameliorate the acid-degradation rate of the polymer as oxygen atoms tend to get protonated easily in the presence of an acid. On the other hand, the oxygen–silicon bond within the TBS end group can undergo a fluoride-mediated cleavage in the presence of a fluoride source, forming a much stronger fluoride–silicone bond.39 This transformation is well established in the literature, especially in protecting alcohol groups during reactions, in addition to probing degradation kinetics of self-immolative polymer systems.40,41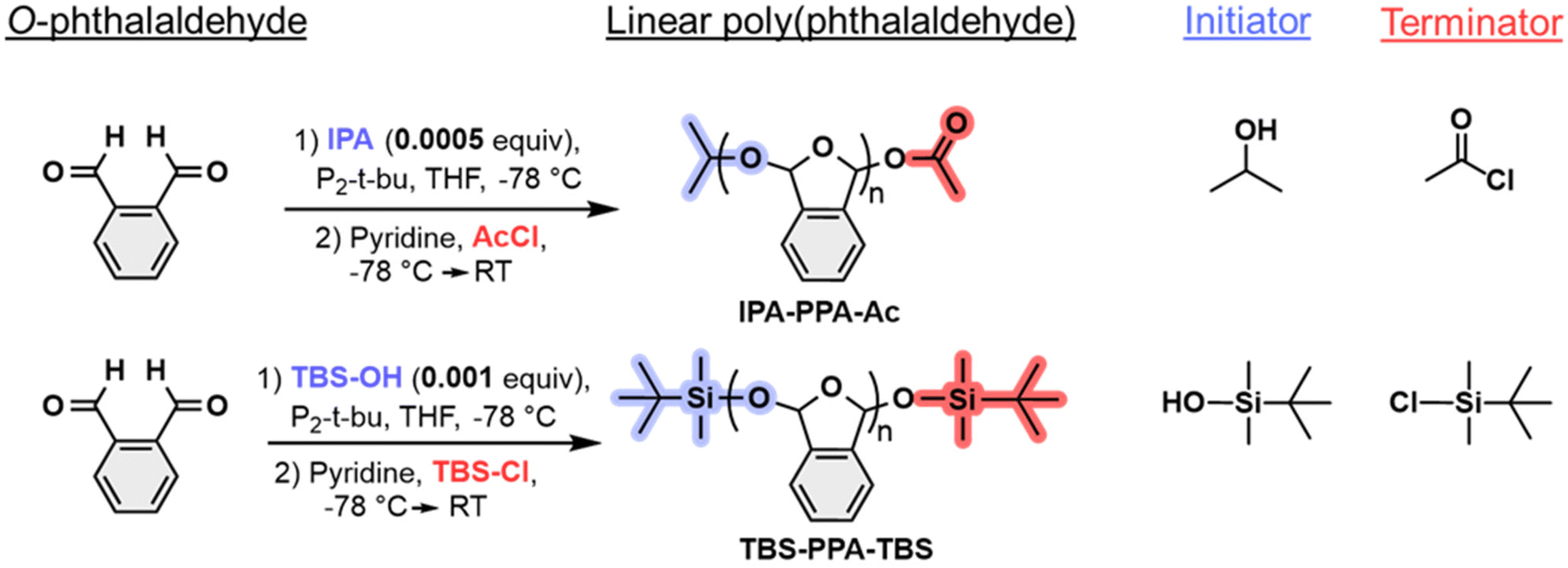 | ||
| Fig. 2 Synthetic route for the preparation of linear PPA polymers with different functional end groups. | ||
Following the synthesis of the polymeric materials, our focus shifted towards purifying the resulting products as the presence of any undesirable impurities carried over from polymerization, including unreacted monomer, excess solvent, or side products, may negatively impact the adhesion performance of PPA. We followed the purification procedures reported by Lutz and coworkers, which involved redissolving the crude polymer material in tetrahydrofuran followed by its slow precipitation into methanol, filtration, and drying under high vacuum.42 This purification technique was efficient and resulted in reaction yields of around 75%, as detailed in Section 1.3 of the ESI.‡
We characterized the chemical and thermal properties of the resulting polymers using a range of techniques. 1H-NMR confirmed the purity of PPA polymers and enabled calculations of their number average molecular weights (Fig. S3 and S4‡). By comparing the integrations of the peaks corresponding to both the polymer and the monomer (eqn (S1)‡), we demonstrated the high purity of the synthesized PPA relative to the starting monomer precursor (Table S1‡). We determined the degree of polymerization of each polymer, and subsequently the molar mass, through the end-group analysis method.43 By comparing the integral of a given end-group and the integral of the repeating chain unit (1,3-dioxyphthalan) using eqn (S2),‡ the molar mass was calculated by multiplying the number of repeating units by the molecular weight of 1,3-dioxyphthalan (eqn (S3)‡). Both polymers exhibited moderate molecular weights (14.7 kDa for IPA-PPA-Ac and 12.9 kDa for TBS-PPA-TBS) and a comparable number of repeating units (109 and 97, respectively). Gel permeation chromatography (GPC) was utilized as a secondary method to confirm the purity of synthesized PPA polymers where no signal corresponding to the monomer appeared in the elution curves of PPA, affirming the purity of the materials (Fig. S5‡). Moreover, the polymers showed relatively low PDI values of 1.46 and 1.54 for IPA-PPA-Ac and TBS-PPA-TBS, respectively, which are comparable to each other, and in most cases, lower than what has been reported in the literature for PPA.19,29,44
X-ray photoelectron spectroscopy (XPS) was used to analyze the chemical environment and surface composition of both polymers. Fig. S6a‡ shows survey spectra of both IPA-PPA-Ac and TBS-PPA-TBS, giving signals related to carbon (C 1s), oxygen (O 1s) and silicon (Si 2p). While the binding energies for the first two species are well matched for both polymers, an additional peak appears at around 101 eV in the TBS-PPA-TBS spectrum, which was attributed to the Si–C (100.5 eV) and Si–O (101.9 eV) binding energies, confirmed the successful functionalization of the polymer with the TBS endcap (inset of Fig. S6a‡).45 In addition, high resolution XPS spectra were applied to the C 1s region in both polymers to explore the interactions between the various carbon containing groups (Fig. S6b‡). Based on the deconvoluted spectra, peaks with binding energies of 284.4 eV and 287.3 eV were attributed to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O, respectively.46 Furthermore, an additional peak appearing at 289.9 eV in the IPA-PPA-Ac spectrum, which can be attributed to the carbonyl CO bond, confirmed the functionalization of the polymer with the carbonyl-based endgroup.47 The thermal stability of PPA was examined via differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The measurements showed that PPA is highly stable at temperatures up to 160 °C, where no endothermic peak appears before this temperature based on the DSC measurements (Fig. S7 and S8‡). Interestingly, both IPA-PPA-Ac and TBS-PPA-TBS depolymerize at a similar temperature, which suggests that the nature of the end groups utilized had a minimal effect on the melting point of PPA. This observation contrasts with that of the uncapped linear PPA, which depolymerizes readily at room temperature due to its low TC of −40 °C.28 Neither of the two synthesized polymers exhibited a Tg prior to depolymerization, which is consistent with the precedent literature.24
C and C–O, respectively.46 Furthermore, an additional peak appearing at 289.9 eV in the IPA-PPA-Ac spectrum, which can be attributed to the carbonyl CO bond, confirmed the functionalization of the polymer with the carbonyl-based endgroup.47 The thermal stability of PPA was examined via differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The measurements showed that PPA is highly stable at temperatures up to 160 °C, where no endothermic peak appears before this temperature based on the DSC measurements (Fig. S7 and S8‡). Interestingly, both IPA-PPA-Ac and TBS-PPA-TBS depolymerize at a similar temperature, which suggests that the nature of the end groups utilized had a minimal effect on the melting point of PPA. This observation contrasts with that of the uncapped linear PPA, which depolymerizes readily at room temperature due to its low TC of −40 °C.28 Neither of the two synthesized polymers exhibited a Tg prior to depolymerization, which is consistent with the precedent literature.24
Strength of adhesion of PPA
Annealing conditions significantly influence the adhesive strength of bonded samples.48,49 Given the sensitivity of linear PPA to sonication and elevated temperatures,50 we chose to employ a solvent-assisted bonding method to retain the polymer composition, while also providing the polymer chains with freedom of motion to form favorable intermolecular interactions with a substrate. We sought to develop a straightforward procedure for bonding substrates with linear PPA that (i) preserved the flexibility and properties of the polymer, (ii) maximized adhesive strength, and (iii) provided evidence regarding the structure–property relationships that contribute to adhesion. While most rigid substrates are compatible with lap shear testing, glass substrates were selected because glass microscope slides are readily available, do not require further preparation, and their transparency allows visualization of the bonded sample and the calculation of the adhesive area prior to testing.51–55 Moreover, polymer chains tend to be more rigid in a solvent that poorly dissolves them (e.g., methanol or acetone) compared to a good dissolving solvent (e.g., dichloromethane (DCM)). Thus, the polymer may not be as free to favorably interact with the neighboring functionalities of the substrate in the case of the former. Our results show that DCM readily solvates IPA-PPA-Ac (>200 mg mL−1), while acetone has a much lower solubilizing capacity (≈3.7 mg mL−1). These results are consistent with those of White et al. who showed that cyclic PPA films casted from DCM had a much higher tensile strength and elastic modulus compared to the same films casted from dioxane and chloroform.44 For these reasons, we adopted DCM in the solvent-assisted bonding method.To demonstrate adhesion, we bonded smooth glass slides with both IPA-PPA-Ac and TBS-PPA-TBS polymers using a solvent-assisted bonding method. In brief, a well dispersed suspension of the purified polymer in methanol was drop-casted onto a small area at the edge of a clean smooth glass microscope slide to achieve an even distribution of material across the intended bonded area. The wet polymeric suspension exhibited no adhesive character before or immediately after drop-casting. A small amount of DCM (<0.1 mL) was then added to saturate the polymer layer, followed by a second microscope slide. A calibration weight was placed on top to create the bonded assembly and set the thickness (Fig. S9‡). To remove any residual solvent that may disrupt cohesion within the polymer adhesive, the bonded assemblies were cured for 24 h under reduced pressure (≤200 mTorr) at room temperature. A detailed procedure for the preparation of these assemblies can be found in Section 4.1 of the ESI.‡
The maximum force that the bonded assembly can withstand prior to the separation of two glass substrates, known as the strength of adhesion, was quantified with lap shear testing under the ASTM D-1002-7 standard and carried out on a single column tensile tester (Fig. 3a, S10 and 11‡). Lap shear testing revealed comparative adhesive performances for both PPA polymers bearing distinct end caps, namely IPA-PPA-Ac (411 ± 135 kPa) and TBS-PPA-TBS (280 ± 96 kPa) (Fig. 3b, S12 and 13‡). As a control, monomer assemblies were prepared using the same solvent-bonding method where upon lap shear testing, an average shear strength value of 12.0 ± 5.5 kPa was attained. This result indicated that the monomer precursor lacks proper structural robustness needed for adhesion compared to PPA and has a low load-bearing capacity, rendering its usage impractical in bonding applications. To gain insight into the relative mechanical strength of the intermolecular interactions at the interface of these assemblies versus those within the bulk material, we quantitatively analyzed the adhesive joint after bond failure between PPA and the glass substrates. As can be seen in Fig. 3C and S14,‡ a mixed mechanical mode of failure was observed, portrayed by a combination of both adhesive and cohesive failures within these polymeric assemblies. Adhesive failure is an interfacial bond failure between the polymer, acting as an adhesive and the adherent substrate, which, in this case is a microscope glass slide. On the other hand, cohesive failure is the loss of mechanical strength in the bulk adhesive, which can be explained by the breakdown of intermolecular bonding forces within the polymeric chain.56 Quantitative image analysis of the lap joints after failure demonstrated that the cohesive failure prevailed over the adhesive failure with 77 ± 8% for the former compared to 23 ± 8% for the latter (Fig. 3c). These results are consistent with a qualitative assessment, where discrete and uneven amounts of PPA adhesives were visually observed remaining on both substrates after failure (inset of Fig. 3c). The results suggested that the forces holding both the polymer and glass substrate together, mostly hydrogen bonding and electrostatic interactions, are stronger than intermolecular forces holding the polymeric chains together. 1H-NMR measurements for both polymeric materials, before and after shearing, confirmed that the shear tests did not trigger depolymerization, nor resulted in any change in the chemical structure of PPA, demonstrating the ability of these materials to withstand large shearing forces without undergoing any decomposition (Fig. S15 and Table S2‡).
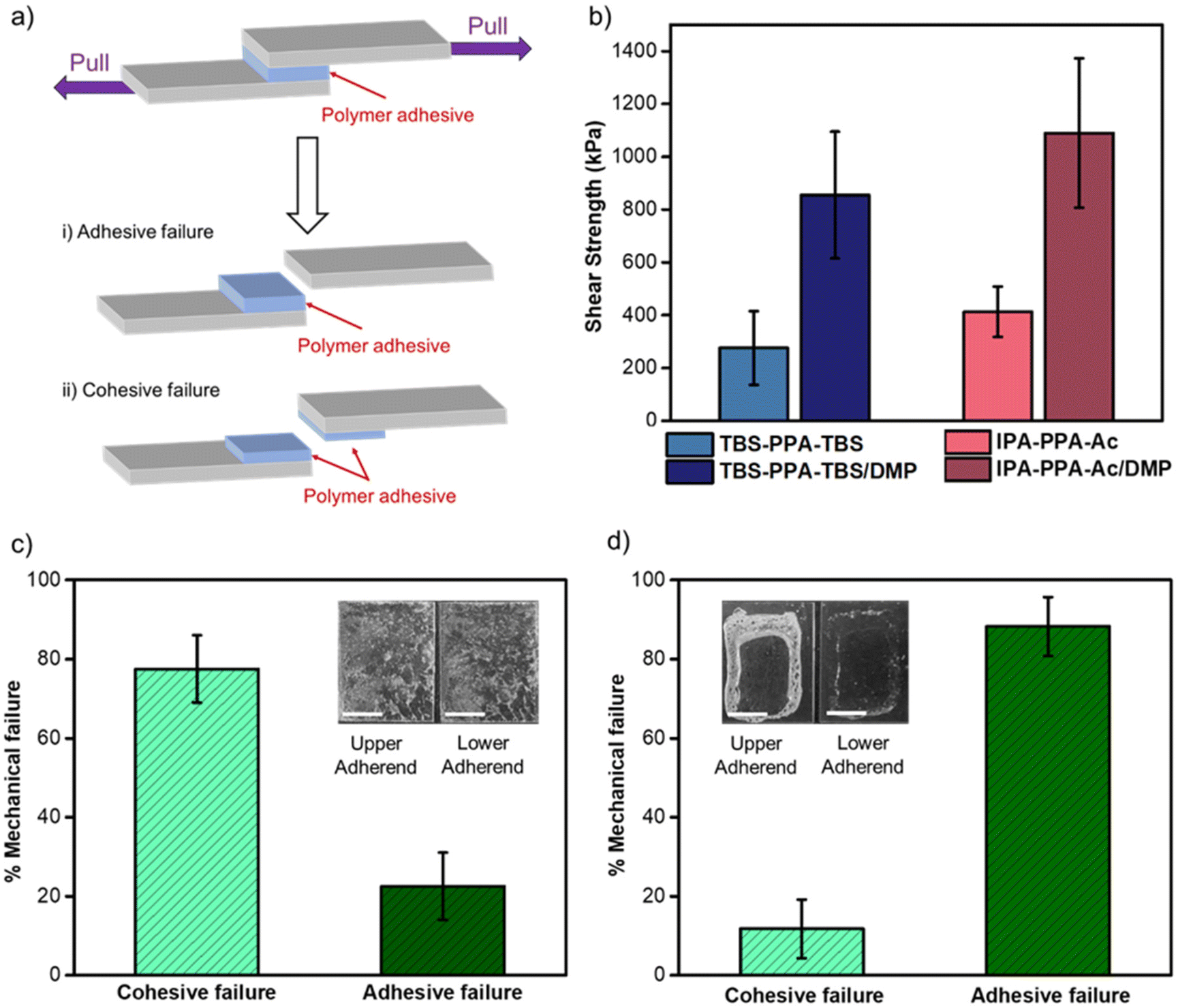 | ||
| Fig. 3 (a) Schematic illustration of a typical adhesive joint specimen for single-lap shear test along with the two possible types of mechanical failures (i) adhesive and (ii) cohesive failures. (b) Lap shear strengths of TBS-PPA-TBS and IPA-PPA-Ac assemblies prepared via solvent-assisted and melt-bonding methods. Error bars represent standard deviation from the mean value of 7–13 independent experiments. (c) Average values of adhesion vs. cohesion % failure in solvent-bonded IPA-PPA-Ac and TBS-PPA-TBS polymers (n = 16). Inset: a photograph of a lap shear test assembly after shear test. (d) Average values of adhesion vs. cohesion % failure in melt-bonded IPA-PPA-Ac/DMP and TBS-PPA-TBS/DMP polymers (n = 24). Inset: a photograph of a lap shear test assembly after shear test (scale bar: 1 cm). | ||
To improve the adhesive performance of PPA and alter the toughness, flexibility, and processability of the polymeric chains, we investigated the effect of adding a plasticizer on the mechanical properties and shear strength of PPA. Inspired by the study of Moore and coworkers,57 who showed that incorporating diethyl phthalate (DEP) plasticizer into cyclic PPA allowed the material to be processed with hot press molding, we hypothesized that a plasticizer would increase the adhesive strength of PPA by depressing its Tg, generally known to lie well above its thermal degradation temperature. This will result in increasing both the mobility of the chains and the free volume between the polymeric chains, rendering the polymer more flexible.24 Hence, we reasoned that a plasticizer would both increase the internal strength of PPA and enable melt-bonding through a glass transition without risking depolymerization.
Addition of DMP as a plasticizing agent was shown to depress Tg of both polymers. While several plasticizers have been utilized to tune the mechanical properties of cPPA,23,24,57 we settled on DMP due to its compatibility with PPA. It is soluble in most organic solvents and effectively disrupts the intermolecular packing of PPA. Thus, a small amount was sufficient to depress Tg well below the decomposition temperature of the polymers. To accurately determine Tg, we performed thermal analysis of DCM-casted films of both PPA polymers with 20% (w/w) DMP. While the pristine materials showed no glass transition between 0 °C and the onset of decomposition at 167 °C, the plasticized materials exhibited a glass transition at around 60 °C (Fig. S16‡). In addition, DSC thermal cycling experiments on both PPA/DMP blends cycled five times each from −40 to 80 °C and back to −40 °C at 10 °C min−1 confirmed the stability and robustness of the plasticized materials where Tg remained stable, and the properties of the materials were retained (Fig. S17‡). Based on these observations, we rationalized that glass substrates may be bonded with films of the plasticized PPA by heating the assemblies to a temperature ranging between the glass transition and degradation temperatures with the possibility of re-bonding after mechanical failure.
Glass bonded assemblies were prepared using the melt-bonding method described in Section 4.3 of the ESI.‡ Upon shear testing, assemblies bonded with IPA-PPA-Ac/DMP and TBS-PPA-TBS/DMP withstood substantial loads before failure, despite a low average adhesive area (1.40 ± 0.19 cm2 and 1.12 ± 0.44 cm2, respectively) (Fig. S18 and 19‡). Moreover, the lap shear strengths of both plasticized polymers with values of 1091 ± 282 kPa for IPA-PPA-Ac/DMP and 856 ± 239 kPa for TBS-PPA-TBS/DMP were ∼2.5-fold higher than the corresponding pristine polymers, demonstrating the effect of DMP on the adhesive strengths and bonding performance of PPA (Fig. 3b). Magnified optical images of the glass substrates after shear testing revealed a change in the mechanical failure mode from cohesive obtained for pristine PPA to mostly adhesive (88% ± 7%) for the polymer-plasticizer blends (Fig. 3d and S20‡). This result suggested that DMP helped increase the internal strength of the material by transforming the polymer from a brittle material that is easy to break, into a flexible and extensible material able to withstand substantial shear forces without breaking. Similar to the case of pristine PPA polymers, the structural integrity of the polymer-plasticizer blends was confirmed via1H-NMR measurements, where both polymer materials before and after shear tests exhibited indistinguishable chemical structures (Fig. S21‡).
Depolymerization of PPA via backbone cleavage
As part of this work, we aimed to probe the kinetics of the decomposition of PPA in the presence of several stimuli in ambient conditions. Starting with a proton-catalyzed degradation pathway, we found a lack of studies on the effect of the end cap of PPA on its degradation kinetics, as most previous research efforts have focused on the decomposition mechanism of cyclic uncapped PPA.16,58TFA, an organofluorine monocarboxylic acid with a pKa of 0.23, was used as an acidic model trigger. In brief, a 0.5 mM solution of IPA-PPA-Ac in deuterated DCM was exposed to different doses of TFA at room temperature. The depolymerization reactions were monitored in situ via1H-NMR spectroscopy wherein the entire reaction was carried out in an NMR sample tube within the spectrometer. In situ NMR analysis revealed a decrease in the integration ratios of the polymer peaks accompanied by a substantial increase of the integration ratios of the monomer peaks at chemical shifts 7.8, 7.9, and 10.55 ppm with time following TFA addition (Fig. S22‡). Varying the concentration of TFA had a significant impact on the depolymerization rate of IPA-PPA-Ac. While a complete, fast degradation was achieved within 35 min of exposure to 8.0 eq. of TFA, less than 9% of the polymer depolymerized within the same timeframe when exposed to 2.0 eq. of TFA relative to PPA (Fig. 4a). This result indicated the dominant dependency of the depolymerization reaction on the concentration of the specific applied signal. Closely examining the initial rate of depolymerization (inset of Fig. 4a), we observed a slow degradation of PPA, especially when small amounts of TFA were added as in the case of 2.0 eq. of TFA where less than 0.04% of the polymer degraded in the first 25 min compared to 80% when 8.0 eq. of TFA was added.
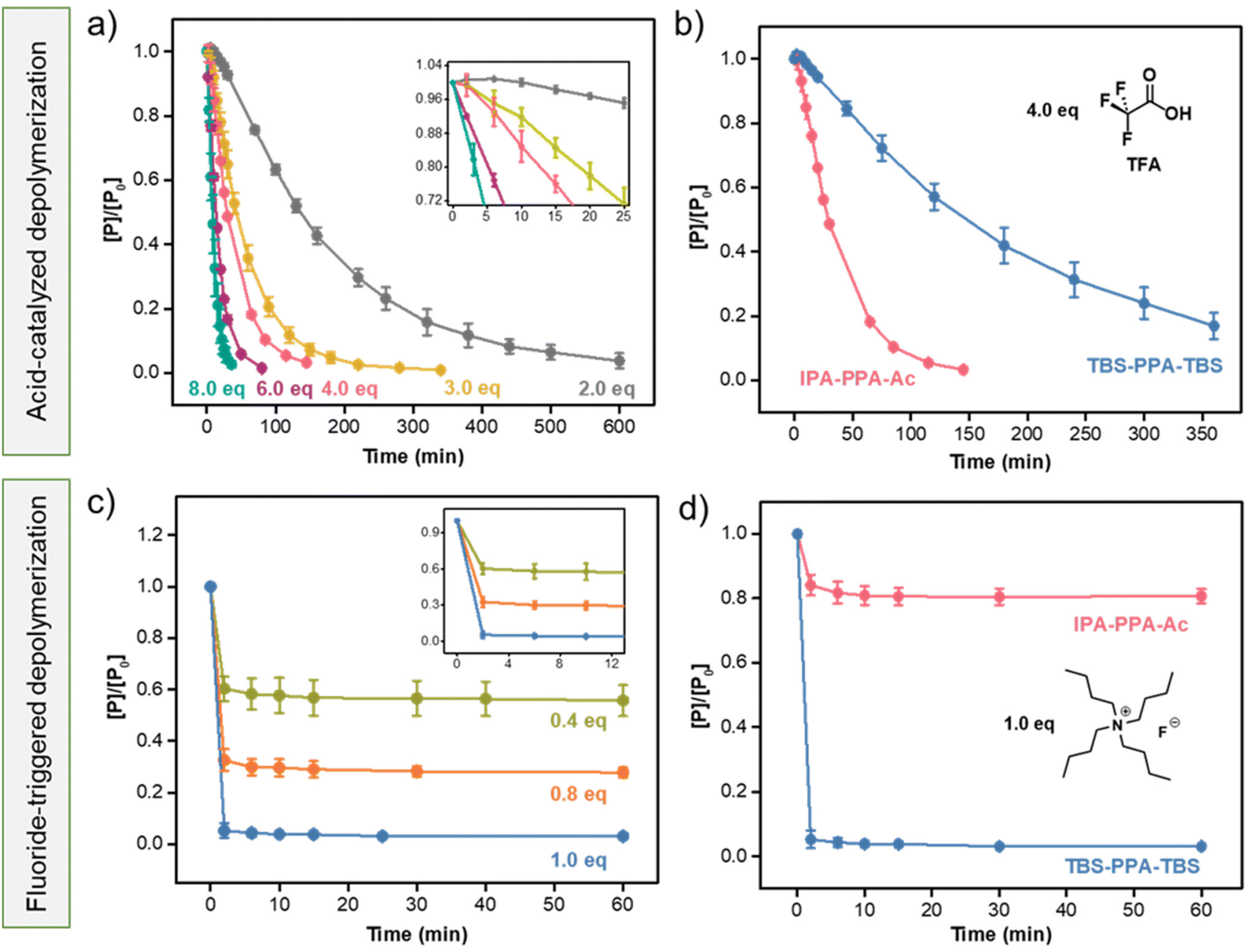 | ||
| Fig. 4 Acid-catalyzed degradation kinetics of (a) IPA-PPA-Ac at different equivalences of TFA in DCM-d2, (b) IPA-PPA-Ac and TBS-PPA-TBS (0.5 mM each) at 4.0 equivalences of TFA (2.0 mM) in DCM-d2, (c) fluoride-catalyzed degradation kinetics of TBS-PPA-TBS at different equivalences of TBAF in DMSO-d6, and (d) IPA-PPA-Ac and TBS-PPA-TBS at 1.0 equivalence of TBAF (0.25 mM for both PPA and TBAF) in DMSO-d6, displaying the importance of TBS endcaps in initiating depolymerization. All concentrations are calculated based on 1H-NMR integrations. Error bars represent standard deviation from the mean. | ||
To gain insight into the mechanism of degradation of IPA-PPA-Ac in the presence of TFA, the experimental data acquired from NMR for both, polymer degradation and monomer formation, were fitted into the three common integrated rate laws, pseudo zero-order, first-order, and second-order kinetics, also known as kinetic models (Fig. S23–27‡). As shown, the experimental data did not fit any of the abovementioned models. While the coefficient of determination for both zero and second orders was poor, indicating that these model-fitting methods were a poor fit, residual plots were used to assess whether the observed error in the kinetic models is consistent with the stochastic error, especially for the first-order polymer degradation models, which displayed correlation values of around 0.99. Interestingly, pseudo-first order residual plots for the polymer degradation indicated a cyclical trend as opposed to the null residual plot obtained in ideal models. Hence, none of the three linear models were adequate in describing the mechanism of acid-triggered depolymerization of PPA.
Based on the reported literature,35,57,59–61 the proposed mechanism of the backbone cleavage in PPA polymers in response to H+ starts with oxygen protonation, followed by bond dissociations through a cascade of intramolecular reactions. We aimed to determine whether the carbonyl oxygen in the end cap of IPA-PPA-Ac is also undergoing protonation and hence affecting the depolymerization rate of the polymer. For this reason, acid-catalyzed depolymerizations of two additional poly(phthalaldehyde)-based polymers, the previously synthesized TBS-endcapped linear polymer, having comparable molar mass and structure to IPA-PPA-Ac, but with distinct endcaps, in addition to a cyclic PPA (cPPA) generated via a cationic polymerization mechanism, and fully characterized via GPC, 1H-NMR, TGA, and DSC (Section 5.4 of the ESI, Fig. S28–31, and Table S3‡) were carried out under indistinguishable reaction conditions. Exposure of both polymers to 4.0 equivalences of TFA resulted in a notable decrease in the depolymerization rate compared to IPA-PPA-Ac. While IPA-PPA-Ac underwent a complete degradation in 150 min, more than 350 min was needed for both TBS-PPA-TBS and cPPA to decompose (Fig. 4b, S32 and 33‡). This result indicates that there exist at least three different starting routes for the decomposition of IPA-PPA-Ac, that is, two in the backbone (similar to TBS-PPA-TBS and cPPA), and one in the endcap (Fig. S34‡). In fact, Tsuda et al., studied the acid-catalyzed decomposition mechanism of linear PPA using a semi-empirical self-consistent-field (SCF) molecular orbital method.61 Based on their findings, degradation of PPA is highly dependent on the surrounding medium. For instance, while the protonation of the carbonyl oxygen is more favorable when PPA is in solution, both oxygen types in the central part of the polymer chain can be easily protonated in a rigid matrix, where it is hard for the protons to reach the end caps. In our case, depolymerization is taking place in solution, and hence the degradation rate can be predominantly attributed to the protonation of the carbonyl oxygen, which explains the considerable difference between the degradation rates of IPA-PPA-Ac on one side and TBS-PPA-TBS and cPPA on the other side, upon exposure to the same amount of TFA.
Depolymerization of PPA via end-cap cleavage
After studying the proton-catalyzed degradation pathway of linear PPA, we shifted our focus to another designed stimulus, mainly fluoride ions, known to depolymerize TBS-terminated SIPs via an end-to-end backbone cleavage mechanism.62 TBS derivatives are widely used in the literature for the protection of hydroxyl groups.63–66 Once the target chemical transformation reaction is carried out, deprotection of TBS can be achieved using any source of fluoride ions capable of reacting with TBS to form tert-butyl dimethyl silyl fluoride (TBS-F). Hence, we reasoned that end-capping PPA with two silyl-ether endcaps may accelerate the degradation kinetics, as fluoride ions will be cleaving the endcaps on both ends of the polymer resulting in a rapid domino-like fragmentation reaction.Similar depolymerization experiments to the ones we performed using TFA were carried out with fluoride ions (F−). However, we opted to use dimethyl sulfoxide (DMSO) as a solvent in this case due to the poor solubility of tetrabutylammonium fluoride (TBAF), the source of fluoride ions, in DCM. In addition, several control studies were carried out to affirm the stability of the polymers in both DCM and DMSO. As shown in Fig. S35 and S36,‡ no significant depolymerization was realized in the 1H-NMR spectra after dissolving PPA in both solvents for 7 days, excluding the possibility of solvent-induced depolymerization. Following exposure of TBS-PPA-TBS to 1.0 equivalent of TBAF, a near-complete decomposition of the polymer was noted within the first 2 minutes, as revealed by 1H-NMR (Fig. S37‡). This rapid cleavage was attributed to the sequential fragmentation of PPA into monomers following the cleavage of both reactive endcaps, which resulted in fast and orderly unzipping reactions from both sides of the polymer. To the best of our knowledge, there has not been any self-immolative polymer reported to date that completely depolymerizes upon exposure to 1.0 eq. of F− at the reported fast response rate (Table S4‡). Furthermore, lowering TBAF equivalents from 1.0 to 0.8 and 0.4 resulted in a decrease in the depolymerization extent from around 95% to 72% and 51%, respectively, as illustrated in Fig. 4c. A common observation realized in these tests is that PPA only decomposed during the first couple of minutes after exposure to fluoride ions, with no change in the concentration of the polymer thereafter (Fig. 4c). We attributed this observation to the lack of a sufficient amount of F− available to cleave TBS endcaps in all polymeric PPA chains, i.e., once the fluoride ions are consumed, the remaining polymeric chains in solution are left intact, resulting in a final mixture of PPA, o-phthalaldehyde, TBS-F, and additional side-products.
As a result of the fast degradation in these cases, we found it challenging to investigate the depolymerization rates of PPA using any of the three rate laws (Fig. S38–40‡). Therefore, we modified the NMR measurement procedure to be able to collect 1H-NMR spectra every 10 seconds during the first three minutes of the depolymerization reaction. Information regarding the experimental procedure can be found in Section 6.4 of the ESI.‡ Our results suggested that 60% of the polymer degraded within the first 10 seconds of the addition of 1.0 eq. of TBAF to the polymeric solution, suggesting a remarkably fast depolymerization rate (Fig. S41 and 42‡). While the degradation kinetics during the first three minutes did not follow any of the integrated rate laws, as depicted in Fig. S43,‡ we reasoned that PPA might be exhibiting mixed zero-first order degradation kinetics wherein the depolymerization undergoes an initial zero-order phase followed by a transition towards a first-order regime over the course of the decomposition. Such behavior implies that the concentration of the polymeric chains will not be altered until a given chain is fully degraded into its monomeric components according to Scheme 1.67 Inspired by the studies of Gillies and coworkers,21,68 which suggested mixed-mode degradation kinetics unique to linear self-immolative polymers, we proceeded by fitting our kinetic data to the modified Avrami equation shown in the ESI (eqn (S4)‡), which ended up providing a good fit and a sound evidence for the proposed mixed zero- and first-order depolymerization mechanism (Fig. S44‡).
 | ||
| Scheme 1 Degradation profile of TBS-PPA-TBS via a first-order intramolecular cyclization reactions. | ||
In order to verify the role of TBS endcaps in the depolymerization process of PPA, IPA-PPA-Ac was tested for fluoride triggered depolymerization under similar reaction conditions. As shown in Fig. 4d, the polymer did not respond to fluoride ions and thereby showed no noticeable degradation behavior when exposed to 2.0 eq. of F−. We attributed the sudden 16% degradation within the first 2 minutes to be the consequence of the rapid change in pH in the system from 13.36 to 9.46 upon the addition of TBAF. Nonetheless, the concentration of PPA remained mostly constant following the two minutes mark.
To assess the selectivity of TBS-PPA-TBS towards fluoride ions, we conducted a similar set of experiments with different tetra-n-butyl ammonium salts. Tetrabutylammonium chloride (TBACl), tetrabutylammonium bromide (TBABr), and tetrabutylammonium iodide (TBAI) were employed as sources of Cl−, Br−, and I−, respectively. Upon exposing TBS-PPA-TBS to 2.0 equivalences of the aforementioned ions, no tangible decomposition was observed (Fig. S45 and 46‡). Overall, these experiments provided evidence for (i) the successful incorporation of TBS endcaps in linear PPA polymer, (ii) the selectivity of TBS towards fluoride ions, and (iii) the selective endcap cleavage mechanism, which resulted in a complete depolymerization within a few minutes. Regarding the proposed depolymerization mechanism, it begins by a fluoride-mediated cleavage of the aliphatic TBS protecting groups on both ends of the polymer. End-cap cleavage results in the generation of alkoxide groups, which render the polymer thermodynamically unstable due to its low TC. The degradation mechanism proceeds by a cascade of irreversible intramolecular fragmentation reactions that yield different monomeric products including o-phthalaldehyde (Fig. S47‡).27
Solid-state depolymerization (SSD) of TBS-PPA-TBS
To further highlight the ability of TBS-PPA-TBS to selectively depolymerize in response to fluoride ions under different conditions, we elucidated the capability of the polymer to decompose in the solid-state form, in the presence of a minimal amount of fluoride ions. The first step was to prepare the TBS-PPA-TBS disks, which was carried out in silicon molds according to a procedure described in Section 7 of the ESI.‡ Once the PPA disk was suspended in acetone, one drop of a 2 mM solution of fluoride was added, leading to a fast and complete depolymerization within 140 seconds, which was visually confirmed by the change in the color of the solution to pale yellow (Fig. S48‡). Analysis by GPC revealed complete degradation of the polymer and the formation of o-phthalaldehyde and other unidentified by-products (Fig. S49‡). We noticed the retention peak corresponding to the product of the SSD experiment to be broader and slightly shifted to a higher value compared to the pure o-phthalaldehyde which supported the previously mentioned observation of the (i) complete degradation of PPA, (ii) formation of o-phthalaldehyde, and (iii) formation of additional by-products with various compositions and molar weights. We also noticed that the color of the solution kept getting darker even after complete degradation of PPA (Fig. S50‡). Inspecting this solution via1H-NMR revealed the transformation of the o-phthalaldehyde monomer into unrecognizable products over time (Fig. S51‡).Qualitative assessment of the adhesive strength of NYCO-bonded textiles with PPA/DMP
In addition to the electrostatic interactions at the adhesive-adherend structure interface, as in the case of the polymer-glass adhesion, polymeric materials can form adhesive interactions through chain entanglement to surfaces with the requisite porosity and mobility conditions.69 This adhesive diffusion phenomenon is not possible with glass substrates due to the insurmountable hardness of silicate glass at room temperature in addition to its low fatigue resistance and high sensitivity to abrasion during handling.70 Alternatively, textiles with a high availability of hydrogen bond donors, such as cotton or nylon-cotton blend (NYCO), offer a significant opportunity for diffusion and intermolecular interactions with linear PPA. For this reason, we aimed to qualitatively investigate the adhesive performance of PPA/DMP blend with NYCO-bonded textiles and assess their reusability after shearing.The preparation method of NYCO-bonded textiles with PPA/DMP blend along with a schematic illustration of the adhesive joint can be found in Section 8 and Fig. S52 of the ESI.‡ The adhesion performance was tested by suspending a 0.5 kg stainless steel weight from a bonded lap joint. No failure was observed within a 24-hour period suggesting a strong adhesion between the polymer and the NYCO textile (Fig. S53a and b‡). After 24 h, the 0.5 kg calibration weight was replaced by a 1.5 kg weight. This increase in load weakened the adhesion between the polymer and the textile resulting in the drop of the weight after a few seconds. The joint adhesive area after the adherents were pulled apart was analyzed, as seen in Fig. S53c.‡ The residual PPA/DMP on both textile surfaces was observed with some areas having no remaining residues, indicating a mixed adhesive/cohesive failure. The results revealed strong adhesion forces and interactions between NYCO textiles and the plasticized polymeric material suggesting a favorable interaction between these materials.
We then focused on assessing the reversibility of the adhesive by re-bonding the same NYCO substrates with the remaining PPA/DMP blend left on them. The textiles were joined together again following the previously described melt-bonding method by redissolving the PPA films left on both textiles with DCM and heating the assembly on a hot plate at 85 °C for a few seconds. After cooling down the material below its Tg, the same 0.5 kg weight was suspended from the bonded lap joint (Fig. S53d–f‡). No failure was observed over at least 48 hours demonstrating not only a robust adhesion performance, but also a sturdy and long-lasting adhesion between the polymer/plasticizer blend and NYCO textile.
Debonding-on-demand of NYCO-bonded textiles
To further investigate the controlled on-demand debonding process of PPA/DMP bonded textiles, several adhesion tests were carried out under different conditions. First, we bonded textiles with DMP/PPA blends using the melt bonding method described in Section 4.3 of the ESI.‡ For the first test, a 0.5 kg stainless steel weight was suspended from the bonding lap joint followed by the addition of a few drops of water to the adhesive area. After waiting a few minutes, no debonding was observed. We then added 2 mL of acetone, which similarly showed no debonding, demonstrating the stability and strength of the adhesive area. Finally, upon exposure to a 2 mM solution of fluoride anions, the polymer underwent a self-immolative disassembly in less than 40 seconds where the weight dropped and the adhesive layer experienced mechanical failure (Fig. 5 and Movie S1‡). Two additional control experiments were carried out to confirm the robustness of the bonded textiles in addition to their ability to endure harsh conditions. In the second control test, and after the 0.5 kg weight was mounted onto the sample, the textile assembly was continuously wetted for 24 hours with more than 30 mL of water, ethanol, methanol, and acetone each. No separation or displacement of the adhering area was observed, affirming the robustness of the bonded assembly (Movie S2‡). As for the third control experiment, the solvent wetting step was omitted and the textile assembly holding the calibration weight was immediately subjected to a few drops of a 2 mM solution of TBAF. This process caused an immediate mechanical failure within the polymer adhesive where the weight dropped in approximately two minutes (Movie S3‡). | ||
| Fig. 5 Snapshots showing textiles bonded PPA/DMP before and after exposure to different stimuli. The 0.5 kg stainless steel weight fell after 2 min of exposure to a diluted solution of TBAF. | ||
Conclusion
This work expands the molecular-scale control over adhesion by strategically engineering a modular self-immolative polymer that is capable of bonding a variety of surfaces and releasing them through a triggered debonding-on-demand process. The use of DMP plasticizer enabled the tuning of the transient and mechanical properties of linear PPA by depressing its Tg and enhancing its adhesive properties. Upon shear testing of smooth glass surfaces, we found that this polymer-plasticizer blend exhibited remarkable adhesion (up to 1100 kPa) compared to the monomer (12 kPa) and pristine polymer (up to 400 kPa). Also, we demonstrated, for the first time, that engineered PPA adhesive can successfully bond NYCO textile substrates, while withstanding a weight of 0.5 kg, which is a critical metric for using this adhesive in chemical protective garments. Exposure of acetyl ester endcapped IPA-PPA-Ac to 8.0 eq. of TFA at ambient conditions resulted in a complete polymer degradation within 25 minutes via a backbone cleavage. Exposure of the fluoride-responsive TBS-PPA-TBS to 1.0 eq. of TBAF resulted in a fast and simultaneous decomposition within 2 minutes via end-cap cleavage, which is among the fastest degradation rates reported to date. Integrated rate laws suggested a mixed zero-first order degradation kinetics of TBS-PPA-TBS characterized by an initial zero-order regime followed by a first order regime with little to no change in depolymerization rate. We have additionally expanded the known PPA thermal processing technique and shown that PPA retains its adhesive properties and self-immolation capabilities once processed with a plasticizer. Additional work focusing on optimization of the polymer–plasticizer blends preparation and annealing method may further enhance adhesive and debonding-on-demand performance. By joining surfaces with a self-immolative polymer, such as PPA, there is immense potential to develop highly customizable adhesive materials that exhibit strong adhesion when desirable, yet immediate debonding when exposed to a specific chemical trigger.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the United States Army Research Office Grant No. W911NF1920089 and the Camille Dreyfus Teacher-Scholar Award. The authors would like to thank Prof. Chenfeng Ke for providing access to the Instron and GPC instruments. P. D. would like to thank Mr Paul Defino for assistance in setting up the NMR kinetic screening experiment.References
- O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- S. H. Jung, G. Choi, S. Jeong, J. Park, H. Yoon, J.-J. Park and H. Kim, ACS Sustainable Chem. Eng., 2022, 10, 13816–13824 CrossRef CAS.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS PubMed.
- A. G. Gavriel, M. R. Sambrook, A. T. Russell and W. Hayes, Polym. Chem., 2022, 13, 3188–3269 RSC.
- Y. Xiao, X. Tan, Z. Li and K. Zhang, J. Mater. Chem. B, 2020, 8, 6697–6709 RSC.
- J. Deng, S. Bailey, R. Ai, A. Delmonico, G. Denbeaux, S. Jiang and C. K. Ober, ACS Macro Lett., 2022, 11, 1049–1054 CrossRef CAS PubMed.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- P. G. Maschmeyer, X. Liang, A. Hung, O. Ahmadzai, A. L. Kenny, Y. C. Luong, T. N. Forder, H. Zeng, E. R. Gillies and D. A. Roberts, Polym. Chem., 2021, 12, 6824–6831 RSC.
- J. A. Kaitz, C. E. Diesendruck and J. S. Moore, J. Am. Chem. Soc., 2013, 135, 12755–12761 CrossRef CAS PubMed.
- T. S. Babra, M. Wood, J. S. Godleman, S. Salimi, C. Warriner, N. Bazin, C. R. Siviour, I. W. Hamley, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 119, 260–271 CrossRef CAS.
- H. Kim, H. Mohapatra and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 13063–13067 CrossRef CAS PubMed.
- Y. Oh, J. Park, J.-J. Park, S. Jeong and H. Kim, Chem. Mater., 2020, 32, 6384–6391 CrossRef CAS.
- C.-H. Whang, K. S. Kim, J. Bae, J. Chen, H.-W. Jun and S. Jo, Macromol. Rapid Commun., 2017, 38, 1700395 CrossRef PubMed.
- E. C. Feinberg, O. Davydovich, E. M. Lloyd, D. G. Ivanoff, B. Shiang, N. R. Sottos and J. S. Moore, ACS Cent. Sci., 2020, 6, 266–273 CrossRef CAS PubMed.
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1198–1204 CrossRef CAS.
- Q. E. A. Sirianni and E. R. Gillies, Polymer, 2020, 202, 122638 CrossRef CAS.
- S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2017, 139, 10002–10008 CrossRef CAS PubMed.
- A. M. DiLauro, G. G. Lewis and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 6200–6205 CrossRef CAS PubMed.
- M. Hansen-Felby, A. Sommerfeldt, M. L. Henriksen, S. U. Pedersen and K. Daasbjerg, Polym. Chem., 2022, 13, 85–90 RSC.
- E. K. Y. Chen, R. A. McBride and E. R. Gillies, Macromolecules, 2012, 45, 7364–7374 CrossRef CAS.
- J. A. Kaitz and J. S. Moore, Macromolecules, 2014, 47, 5509–5513 CrossRef CAS.
- J. Jiang, M. Warner, O. Phillips, A. Engler and P. A. Kohl, Polymer, 2019, 176, 206–212 CrossRef CAS.
- M. Warner, A. Engler and P. A. Kohl, Polymer, 2020, 202, 122588 CrossRef CAS.
- A. Engler and P. A. Kohl, Macromolecules, 2020, 53, 1543–1549 CrossRef CAS.
- J. A. Kaitz and J. S. Moore, Macromolecules, 2013, 46, 608–612 CrossRef CAS.
- A. M. DiLauro, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 2963–2968 CrossRef CAS.
- S. Soars, J. Kamps, B. Fairbanks and C. Bowman, Macromol. Chem. Phys., 2021, 222, 2100111 CrossRef CAS.
- A. M. DiLauro and S. T. Phillips, Polym. Chem., 2015, 6, 3252–3258 RSC.
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1656–1662 CrossRef CAS.
- W. Seo and S. T. Phillips, J. Am. Chem. Soc., 2010, 132, 9234–9235 CrossRef CAS PubMed.
- G. I. Peterson and A. J. Boydston, Macromol. Rapid Commun., 2014, 35, 1611–1614 CrossRef CAS PubMed.
- H. Kim, A. D. Brooks, A. M. DiLauro and S. T. Phillips, J. Am. Chem. Soc., 2020, 142, 9447–9452 CrossRef CAS PubMed.
- P. S. Addy, M. Shivrayan, M. Cencer, J. Zhuang, J. S. Moore and S. Thayumanavan, ACS Macro Lett., 2020, 9, 855–859 CrossRef CAS PubMed.
- H. Ito, E. Flores and A. F. Renaldo, J. Electrochem. Soc., 1988, 135, 2328 CrossRef CAS.
- L. Pessoni, J. De Winter, M. Surin, N. Hergué, N. Delbosc, R. Lazzaroni, P. Dubois, P. Gerbaux and O. Coulembier, Macromolecules, 2016, 49, 3001–3008 CrossRef CAS.
- A. M. DiLauro and S. T. Phillips, Polym. Chem., 2015, 6, 3252–3258 RSC.
- J. M. Schwartz, A. Engler, O. Phillips, J. Lee and P. A. Kohl, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 221–228 CrossRef CAS.
- A. M. DiLauro, W. Seo and S. T. Phillips, J. Org. Chem., 2011, 76, 7352–7358 CrossRef CAS PubMed.
- T. D. Nelson and R. D. Crouch, Synthesis, 1996, 1031–1069 CrossRef CAS.
- X.-W. Kan, L.-J. Zhang, Z.-Y. Li, F.-S. Du and Z.-C. Li, Macromol. Rapid Commun., 2021, 42, 2100169 CrossRef CAS PubMed.
- J. P. Lutz, O. Davydovich, M. D. Hannigan, J. S. Moore, P. M. Zimmerman and A. J. McNeil, J. Am. Chem. Soc., 2019, 141, 14544–14548 CrossRef CAS PubMed.
- T. F. Page and W. E. Bresler, Anal. Chem., 1964, 36, 1981–1985 CrossRef CAS.
- H. L. Hernandez, S. K. Takekuma, E. B. Mejia, C. L. Plantz, N. R. Sottos, J. S. Moore and S. R. White, Polymer, 2019, 162, 29–34 CrossRef.
- A. Kaur, P. Chahal and T. Hogan, IEEE Electron Device Lett., 2016, 37, 142–145 CAS.
- L. Shi, L. Jin, Z. Meng, Y. Sun, C. Li and Y. Shen, RSC Adv., 2018, 8, 39937–39947 RSC.
- S. P. Ponnappa, Q. Liu, M. Umer, J. MacLeod, J. Jickson, G. Ayoko, M. J. A. Shiddiky, A. P. O'Mullane and P. Sonar, Polym. Chem., 2019, 10, 3722–3739 RSC.
- Z. Dong, J. Wu, X. Shen, Z. Hua and G. Liu, Chem. Sci., 2023, 14, 3938–3948 RSC.
- R. Jamaati and M. R. Toroghinejad, Mater. Des., 2010, 31, 4508–4513 CrossRef CAS.
- G. I. Peterson and A. J. Boydston, Macromol. Rapid Commun., 2014, 35, 1611–1614 CrossRef CAS PubMed.
- X. Ou, X. Zou, Q. Liu, L. Li, S. Li, Y. Cui, Y. Zhou and F. Yan, Chem. Mater., 2023, 35, 1218–1228 CrossRef CAS.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H.-J. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS PubMed.
- D. K. Hohl and C. Weder, Adv. Opt. Mater., 2019, 7, 1900230 CrossRef.
- L. Liu, Z. Liu, Y. Ren, X. Zou, W. Peng, W. Li, Y. Wu, S. Zheng, X. Wang and F. Yan, Angew. Chem., Int. Ed., 2021, 60, 8948–8959 CrossRef CAS PubMed.
- M. Inada, T. Horii, T. Fujie, T. Nakanishi, T. Asahi and K. Saito, Mater. Adv., 2023, 4, 1289–1296 RSC.
- Y. Yuan, X. Zhu and L. Chen, Mater. Des., 2020, 185, 108272 CrossRef.
- E. C. Feinberg, H. L. Hernandez, C. L. Plantz, E. B. Mejia, N. R. Sottos, S. R. White and J. S. Moore, ACS Macro Lett., 2018, 7, 47–52 CrossRef CAS PubMed.
- H. L. Hernandez, O. P. Lee, C. M. Possanza Casey, J. A. Kaitz, C. W. Park, C. L. Plantz, J. S. Moore and S. R. White, Macromol. Rapid Commun., 2018, 39, 1800046 CrossRef PubMed.
- C. Wu, J. Jiang, H. Guo, X. Pu, L. Liu, W. Ding, P. A. Kohl and Z. L. Wang, Adv. Electron. Mater., 2019, 5, 1900725 CrossRef CAS.
- H. L. Hernandez, S.-K. Kang, O. P. Lee, S.-W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef CAS PubMed.
- M. Tsuda, M. Hata, R. Nishida and S. Oikawa, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 77–89 CrossRef CAS.
- C. Ergene and E. F. Palermo, J. Mater. Chem. B, 2018, 6, 7217–7229 RSC.
- A. H. Jadhav, A. Chinnappan, R. H. Patil, W.-J. Chung and H. Kim, Chem. Eng. J., 2014, 236, 300–305 CrossRef CAS.
- S.-i. Kawahara, T. Wada and M. Sekine, J. Am. Chem. Soc., 1996, 118, 9461–9468 CrossRef CAS.
- A. Iida, H. Okazaki, T. Misaki, M. Sunagawa, A. Sasaki and Y. Tanabe, J. Org. Chem., 2006, 71, 5380–5383 CrossRef CAS PubMed.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190–6191 CrossRef CAS.
- R. A. McBride, Degradation Kinetics and Functional Design of Linear Self-Immolative Polymers, The University of Western Ontario, Canada, 2013 Search PubMed.
- R. A. McBride and E. R. Gillies, Macromolecules, 2013, 46, 5157–5166 CrossRef CAS.
- E. M. Petrie, in Joining Textiles, ed. I. Jones and G. K. Stylios, Woodhead Publishing, 2013, pp. 225–274, DOI:10.1533/9780857093967.2.225.
- G. Raos and B. Zappone, Macromolecules, 2021, 54, 10617–10644 CrossRef CAS.
Footnotes |
| † This work is dedicated to Professor George M. Whitesides on his 85 birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01154b |
| This journal is © The Royal Society of Chemistry 2024 |