
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the optoelectronic properties of emerging solar absorbers through cation disorder engineering
Yi-Teng
Huang
 and
Robert L. Z.
Hoye
*
and
Robert L. Z.
Hoye
*
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, UK. E-mail: robert.hoye@chem.ox.ac.uk
First published on 2nd May 2024
Abstract
Chalcogenide solar absorbers, such as AgBiS2 and kesterites, have gained a resurgence of interest recently, owing to their high stability compared to metal–halide compounds, as well as their rising efficiencies in photovoltaic devices. Although their optical and electronic properties are conventionally tuned through the composition and structure, cation disorder has increased in prominence as another important parameter that influences these properties. In this minireview, we define cation disorder as the occupation of a cation crystallographic site with different species, and the homogeneity of this cation disorder as how regular the alternation of species in this site is. We show that cation disorder is not necessarily detrimental, and can lead to increases in absorption coefficient and reductions in bandgap, enabling the development of ultrathin solar absorbers for lightweight photovoltaics. Focusing on kesterites and ABZ2 materials (where A = monovalent cation, B = divalent cation, and Z is a chalcogenide anion), we discuss how the degree and homogeneity of cation disorder influences the optical properties, charge-carrier transport and photovoltaic performance of these materials, as well as how cation disorder could be tuned and quantified. We finish with our perspectives on the important questions moving forward in making use of cation disorder engineering as a route to achieve more efficient solar absorbers.
 Yi-Teng Huang | Yi-Teng Huang is currently a postdoctoral researcher at Department of Chemistry of University of Oxford, under the supervision of Prof. Robert Hoye. He completed his PhD in Physics at University of Cambridge (2019–2023) before joining Prof. Robert Hoye's group at Oxford. His research interests include understanding the mechanism behind defect tolerance, cation disorder, charge-carrier phonon coupling, and developing optoelectronic devices based on novel semiconductors. |
 Robert L. Z. Hoye | Robert L. Z. Hoye is an Associate Professor at the University of Oxford. He completed his PhD at the University of Cambridge (2012–2014), followed by a postdoc at MIT (2015–2016), and two College Research Fellowships at the University of Cambridge (2016–2020). He began his independent academic career at the University of Cambridge with a Royal Academy of Engineering (RAEng) Research Fellowship (2018–2019), followed by a Lectureship (2020–2022) and Senior Lectureship (2022) at Imperial College London, before moving to the University of Oxford as Associate Professor (2022−present). He leads a group working on developing optoelectronic materials for energy harvesting and conversion, light emission and radiation detection. |
1. Introduction
Discovering solar absorbers that are simultaneously efficient, cost-effective, stable, scalable, and comprised of nontoxic, earth-abundant elements, has long been the overarching goal of the thin film photovoltaics (PVs) community.1–3 These emerging solar absorbers are important not only for single-junction outdoor PVs, but also as the bottom- or top-cell for multi-junction devices,4,5 devices to harvest energy from indoor lighting to more sustainably power Internet of Things electronics,6,7 as well as building-integrated PVs and agrivoltaics.8 As such, these materials have significant potential to fulfil energy harvesting requirements that are complementary to the capabilities of crystalline silicon PVs, and many of the compounds investigated are inorganic materials, including kesterites,9–11 chalcopyrites,12–14 chalcogenides,15–17 chalcohalides18–20 and metal–halide perovskites.21–23 Historically, efforts at discovering these materials have focussed on the optical properties of the solar absorbers, namely bandgap and absorption coefficient.3 More recently, charge-carrier transport has taken on a much more prominent role in materials discovery than before, especially to account for the effects of defect states and carrier localization, which have limited the potential of many emerging materials.24–27A key property that influences these optoelectronic properties is cation disorder. Disorder has long been a prominent and common feature28 in inorganic compounds. Although the links between the transport of charge-carriers and phonons with cation disorder has been widely studied in the thermoelectrics field,29,30 such effects have either not been widely considered in emerging inorganic solar absorbers, or have typically been thought to be detrimental. Recent work on AgBiS2 and related materials have shown that this is not necessarily the case, and processing the samples to achieve a homogeneous arrangement of cations occupying the same crystallographic lattice site can lead to improvements in the absorption coefficient,31 as well as reduce the degree of carrier localization, which enhances mobilities.32
In this minireview, we discuss the effects of cation disorder on the optoelectronic properties and PV performance of inorganic solar absorbers. We define cation disorder as the occupation of a cation lattice site by a different cation species, and that homogeneous cation disorder is when there is perfect alternation between the cation species on the same lattice site, as displayed in Fig. 1(a) and (b). That is, we would define full cation order as when the cation species occupy different lattice sites (Fig. 1(a)). The degree of cation disorder can be further quantified by the “order parameter S”,33,34 which is expressed as
| S = 2(PAA − 0.5) | (1) |
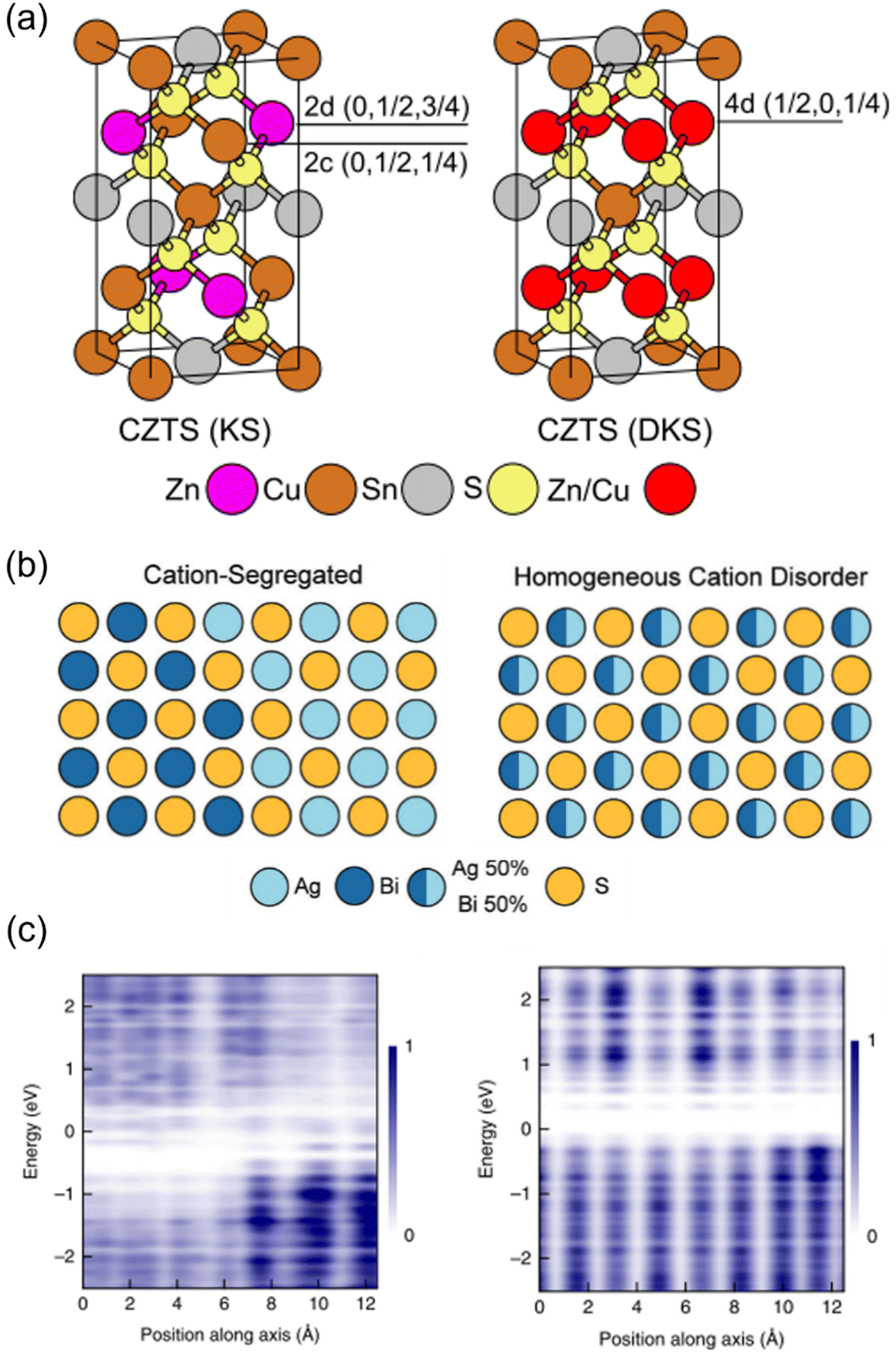 | ||
Fig. 1 Cation disorder in different materials. (a) Unit cells of ordered (left) and disordered (right) Cu2ZnSnS4. During the order–disorder transition, the Wyckoff positions of Cu2ZnSnS4 will be changed from 2c and 2d to position 4d. The space group will also change from I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) to I2m. Reprinted with permission from ref. 39. Copyright 2017 Elsevier. (b) Schematic of AgBiS2 crystal structures with different degrees of cation disorder. In the homogeneous cation disorder configuration, Ag+ and Bi3+ cations can randomly occupy the same lattice sites. Reprinted with permission under the CC-BY-4.0 license from ref. 32. (c) The change on the planar-averaged local electronic density of states (DOS) for AgBiS2 when changing from inhomogeneously (left) to homogeneously cation disordered. The color bar refers to the normalized to I2m. Reprinted with permission from ref. 39. Copyright 2017 Elsevier. (b) Schematic of AgBiS2 crystal structures with different degrees of cation disorder. In the homogeneous cation disorder configuration, Ag+ and Bi3+ cations can randomly occupy the same lattice sites. Reprinted with permission under the CC-BY-4.0 license from ref. 32. (c) The change on the planar-averaged local electronic density of states (DOS) for AgBiS2 when changing from inhomogeneously (left) to homogeneously cation disordered. The color bar refers to the normalized  . Reprinted with permission from ref. 31. Copyright 2022, Springer Nature. . Reprinted with permission from ref. 31. Copyright 2022, Springer Nature. | ||
In the following sections, we will discuss how the degree and homogeneity of cation disorder impacts several optoelectronic properties, and how it could be controlled or experimentally quantified. The materials focussed on in this minireview are kesterites and ABZ2 materials, which have been the most studied solar absorbers from the perspective of cation disorder. It should be noted that although the concept of cation disorder discussed here mainly involves the interchange between two types of cations, it can be generalized to three or more cations if the cations involved have similar ionic radii and chemical properties, as has been reported in some mixed-cation ternary chalcogenides,35 perovskites,36 and high-entropy materials.37,38 The discussion and points made in this minireview can hence be applied across the wider set of emerging inorganic solar absorbers, as well as provide insights into how cation disorder can be more consciously tuned as a design parameter to improve the optoelectronic properties of photovoltaic materials.
2. Impact of cation disorder
2.1 Optical properties
It has been experimentally and theoretically verified that cation disorder can change the bandgaps of several kesterites and AgBiS2. Owing to the broadening of the density of states (DOS) at band edges, the bandgaps of these materials tend to decrease with increases in the degree of cation disorder. For example, in AgBiS2, the bandgap was predicted (based on the hybrid functional HSE06) to decrease from over 1.5 eV in the fully ordered state (matildite phase) to 0 eV in the fully homogeneous Ag+–Bi3+ disordered state (cubic phase).40 In other words, AgBiS2 could be transformed from a semiconductor into a metal when cation ordering is reduced.Interestingly, such a bandgap change is reversible in kesterites, and is highly associated with the order–disorder transition in these materials. Rey et al. have shown that the degree of cation disorder in Cu2ZnSnSe4 could be controlled by different post-synthesis annealing treatments.33 As shown in Fig. 2(a), the bandgaps of Cu2ZnSnSe4 films determined by their Tauc plots could not only be reduced at higher annealing temperatures, but also be enlarged again at lower annealing temperatures, with a very minor hysteresis effect. This bandgap-temperature curve strongly indicates a second-order phase transition, where a continuous rather than an abrupt change with temperature was present. It could be also seen that the bandgap was more or less the same at temperatures above 200 °C, which could be understood if complete disorder on Cu+–Zn2+ has been reached, and 200 °C is therefore considered to be the critical temperature TC for the order–disorder transition in Cu2ZnSnSe4.
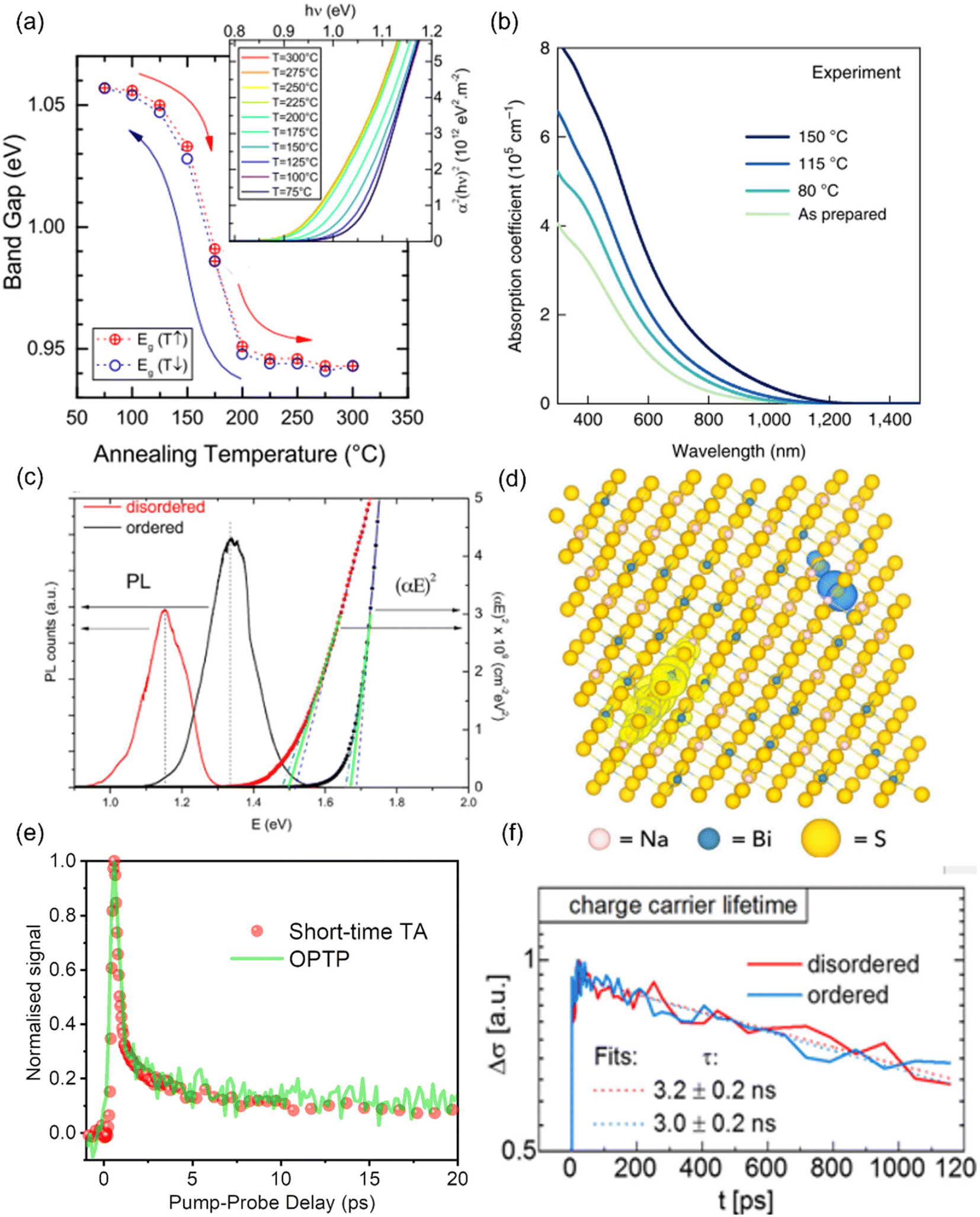 | ||
| Fig. 2 Impact of cation disorder on optoelectronic properties. (a) The change in the bandgap of Cu2ZnSnS4 with different degrees of cation disorder (controlled by different annealing temperatures). The inset shows the Tauc plots of Cu2ZnSnSe4 annealed at different temperatures. Reprinted with permission from ref. 33. Copyright 2014, AIP Publishing. (b) Absorption coefficient spectra of AgBiS2 nanocrystals (NCs) annealed at different temperatures. Reprinted with permission from ref. 31. Copyright 2022, Springer Nature. (c) The Tauc plots and photoluminescence (PL) spectra of Cu2ZnSnS4 with different degrees of cation disorder. Reprinted with permission from ref. 41. Copyright 2017, John Wiley and Sons. (d) Charge density isosurfaces of a relaxed electron–hole pair in a cation-disordered NaBiS2 supercell. Na, Bi, and S atoms are colored pink, blue and yellow, respectively. The translucent yellow and blue isosurfaces represent electron and hole densities, respectively. Reprinted under the CC-BY-4.0 license from ref. 45. (e) The normalized kinetics of NaBiS2 NCs acquired from the short-time transient absorption (TA) and optical-pump-terahertz-probe (OPTP) measurements. Reprinted under the CC-BY-4.0 license from ref. 45. (f) Photoconductivity transients (Δσ) of Cu2ZnSnSe4 in different cation ordering configurations. Reprinted with permission from ref. 48. Copyright 2018, Elsevier. | ||
Since the bandgap reduction in these cation-disordered materials has been attributed to band-edge broadening, it is intuitive to assume an enlarged Urbach energy EU in a more disordered phase. Although some works did report a larger EU in a more cation-disordered Cu2ZnSnS4 film,41 other works have also reported the opposite trend.42 Additionally, AgBiS2 has exhibited a significant reduction in EU from 173 meV to 26 meV when cation disorder became more homogeneous (i.e., closer to an arrangement where completely random Ag+ and Bi3+ cations are distributed in the octahedral holes in the rocksalt structure). On the one hand, a more disordered structure could cause bandgap or band edge fluctuations, raising the possibility of band tailing. But on the other side, the spread of the band-edge DOS might be decreased when the structure approaches the homogeneously cation-disordered phase, as displayed in Fig. 1(c). We hence note that the correlation between EU values and cation disorder is not as straightforward as expected.
If the upper valence band (VB) and lower conduction band (CB) of a cation-disordered material are mainly composed of the orbitals from a specific species, then the band edge DOS of such a material will be significantly influenced by the degree of cation disorder. As an example, the band edge DOS of disordered rocksalt AgBiS2 is shown in Fig. 1(c), where we can see that both the Ag-dominated VB maximum (VBM) and Bi-dominated CB minimum (CBM) are homogenized following an increase in the uniformity of the Ag+–Bi3+ distribution. The more homogeneous band-edge DOS then leads to enhanced transition dipole moments, and therefore higher absorption coefficients, as shown in Fig. 2(b), where a more homogeneous cation distribution in AgBiS2 NCs could be reached with higher annealing temperatures. A similar enhancement in the absorption coefficients with increased cation homogeneity is also theoretically predicted in some ternary nitrides such as MgGeN2,43 although this is yet to be experimentally proven.
Finally, since cation disorder is basically a type of antisite defect (but can occur with much higher concentrations than typical point defects in the dilute limit), the non-radiative recombination rate might be also increased in materials with high degree of cation disorder, which would lead to reduced photoluminescence (PL) intensities or quantum yields. This detrimental effect of cation disorder on PL has been commonly observed in both Cu2ZnSnS4 and Cu2ZnSnSe4 films,41,44 as displayed in Fig. 2(c). It should be noted that although the mechanism behind this effect is still unclear, the difference between the PL peak energy and the bandgap was almost constant, independent of the bandgap changes. Furthermore, similar to the bandgap, all the aforementioned PL properties showed high reversibility when kesterites experienced repeated order–disorder transitions.42 These results clearly indicate the great impact of cation disorder on PL properties, potentially via changing the corresponding defect densities.
2.2 Charge-carrier transport
It has been shown that cation disorder can significantly impact charge-carrier transport in some ABZ2 materials, such as NaBiS2 and AgBiS2, which is also highly associated with the significant impact of cation disorder on the band-edge DOS of these materials. For example, Na+ cations do not contribute any orbitals to the band extrema in NaBiS2, while Bi s orbitals make weak contributions to the upper VB, thus leading to a very flat VBM and hence large hole effective mass.45 Moreover, S2− anions coordinated by 5 or 6 Na+ cations (i.e., in Na+-rich clusters) tend to exhibit higher energy levels relative to the VBM due to the reduced Coulomb interaction from Na+ compared to Bi3+ cations. As a result, S p orbitals from these sulfur sites with high Na+ coordination form flat bands just above the VBM, which could tightly capture holes to form small polarons, with hole wavefunctions constrained to within approximately one unit cell. For electrons, a similar localization process takes place at Bi3+-rich clusters, albeit the electrons are less tightly bound, with their wavefunctions spreading over a slightly larger region compared to holes. Therefore, electrons and holes are spatially separated in cation-disordered NaBiS2, and there is an ultrafast decay in the sum mobility after photo-excitation owing to the strong carrier localization, as shown in Fig. 2(d).On the other hand, the VB of AgBiS2 is composed of a large proportion of Ag d orbitals, leading to a more disperse VBM and hence a smaller hole effective mass. Nevertheless, since the VB and CB of AgBiS2 are dominantly contributed by Ag d–S p orbitals and Bi p–S p orbitals, respectively, cation disorder can still lead to localized DOS in Ag+- and Bi3+-rich regions,31,32 as shown in Fig. 1(c). It has been claimed that such a localized DOS will lead to a reduced dimensionality in the electronic structure of AgBiS2 despite its 3D crystal structure, which can potentially limit the charge-carrier transport. In addition, according to Holstein-Anderson theory, even minimal disorder can also lead to the formation of small polarons.46,47 As a result, significant charge-carrier localization could be still present in cation-disordered AgBiS2.
Interestingly, for kesterite materials, such as Cu2ZnSnSe4, the degree of cation disorder barely affects the sum mobilities or carrier kinetics,48 as shown in Fig. 2(e). A possible explanation can be seen from the electronic structure of Cu2ZnSnSe4, for which the upper VB and lower CB primarily consist of Cu d orbitals and Sn s–S p orbitals, respectively. Since calculations have predicted the Cu–Zn antisites to be the most favourable defects in Cu2ZnSnSe4, cation disorder in this material is expected to primarily affect the VB states and hole transport. Nevertheless, due to the fluctuating VB,49 the hole mobility of Cu2ZnSnSe4 is lower than its electron mobility by over an order of magnitude, and therefore, the cation disorder will not significantly limit the charge-carrier transport, which is dominated by the much more mobile electrons, of this material. In terms of Cu2ZnSnS4, although direct experimental evidence is currently lacking, calculations have shown no signs of either small electron or hole polaron formation in its disordered phase.50
2.3 PV performance
Given the significant impact of cation disorder on the optoelectronic properties of materials, it is not surprising to see different PV performance in solar cells fabricated based on materials with different degrees of cation disorder. It has been shown that AgBiS2 nanocrystals (NCs) with more homogeneous cation disorder can exhibit stronger absorption and hence a higher spectroscopic limited maximum efficiency (SLME), as displayed in Fig. 3(a). Therefore, improving the homogeneity of cation disorder was critical for AgBiS2 solar cells to reach its current certified record power conversion efficiency (PCE) of 8.85%, which is one of the highest values among all bismuth-based solar absorbers.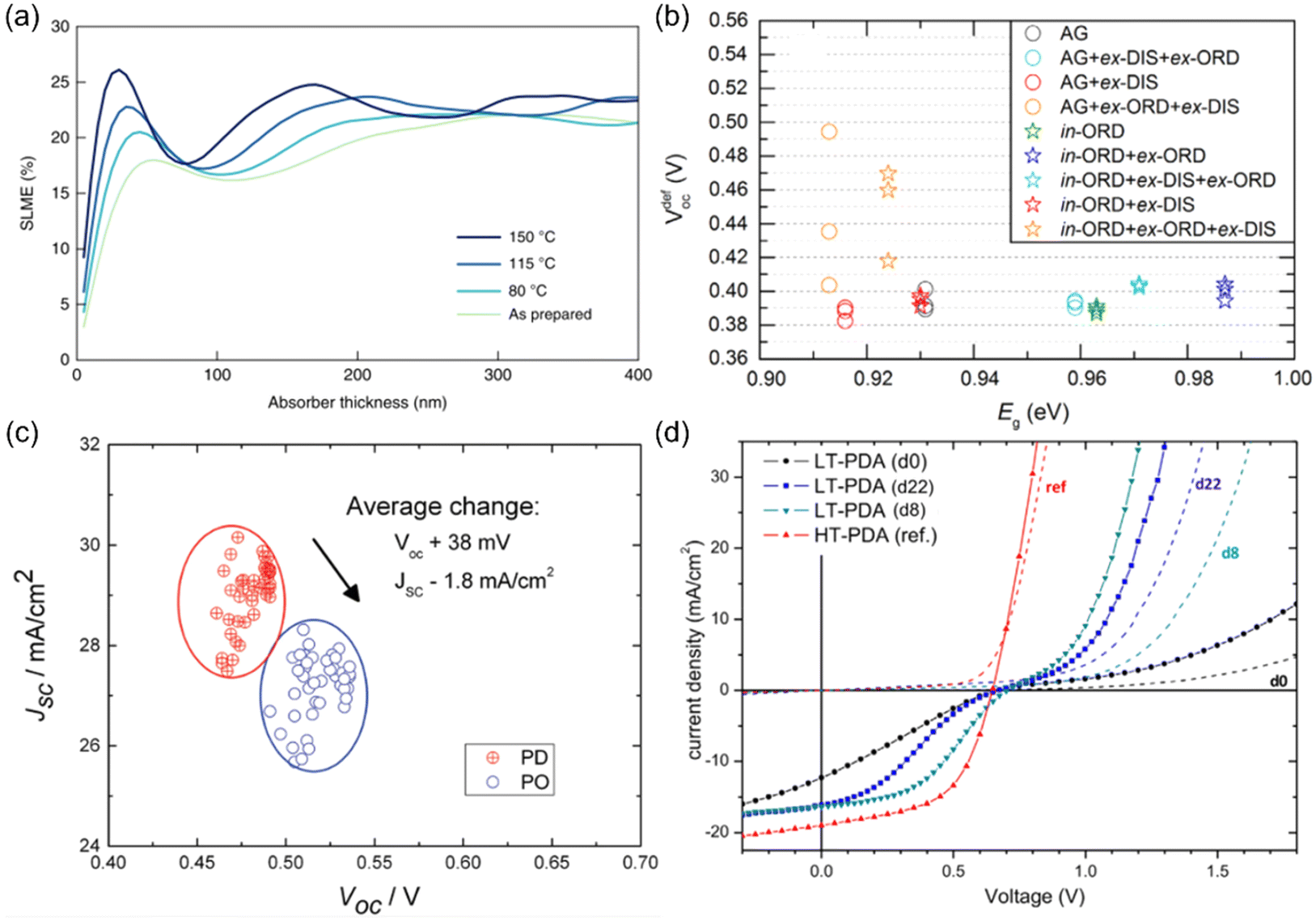 | ||
| Fig. 3 Impact of cation disorder on photovoltaic performance. (a) The spectroscopic limited maximum efficiencies (SLMEs) of AgBiS2 NC films as a function of thickness and annealing temperature, which changes the degree of cation disorder. Reprinted with permission from ref. 31. Copyright 2022, Springer Nature. (b) Open-circuit voltage deficit (VdefOC) of solar cells fabricated based on Cu2ZnSnSe4 with different degree of cation disorder and hence different bandgaps Eg. Reprinted with permission from ref. 53. Copyright 2016, Elsevier. (c) A set of short-circuit current density (JSC)-open-circuit voltage (VOC) data points acquired from the same batch of Cu2ZnSnSe4 solar cells. PD and PO refer to highly cation disordered and relatively cation ordered Cu2ZnSnSe4, respectively. Reprinted with permission from ref. 42. Copyright 2016, John Wiley and Sons. (d) Current density–voltage curves of Cu2ZnSnS4 devices with different degrees of cation disorder. LT-PDA and HT-PDA refer to the post deposition annealing at low (160 °C) and high temperatures (300 °C), respectively. LT-PDA could lead to a higher degree of cation order, while HT-PDA tended to lower the degree of cation order. “dx” in the brackets refers to the xth day after device fabrication. Reprinted with permission from ref. 41. Copyright 2017, John Wiley and Sons. | ||
More studies on the role of cation disorder in kesterite solar cells have been reported. Earlier work used to regard cation disorder as the main cause of the large open-circuit voltage (VOC) deficit42,51–53 (i.e., difference between the experimental VOC values and theoretical VOC values, usually >380 meV), which was believed to be the culprit limiting the PCEs of kesterite solar cells. However, more recent studies have indicated that the VOC deficit of kesterite solar cells was in fact not directly related to the degree of cation disorder in kesterites.53 As shown in Fig. 3(b), the minimum VOC deficit that could be achieved in Cu2ZnSnSe4 solar cells with different degrees of cation disorder was almost unchanged. Such a lack of a dependence of the VOC on cation disorder has been rationalized by the shallow levels of CuZn/ZnCu antisite defects, as well as the limited effect of these defects on the band edges.50,54 However, it has been computationally predicted that the formation energies for deep-level SnZn antisite defects and their defect complexes can be lowered when cation disorder is present, even at a relatively low degrees.50 Since a fully ordered configuration can never be achieved at room temperature, the VOC deficit in kesterite solar cells is most likely caused by the inevitable non-radiative recombination resulting from deep-level defects. Based on the Shockley-Read-Hall recombination model with these disorder-facilitated defects taken into account, the maximum PCE for Cu2ZnSnS4 solar cells has been estimated to drop from 25% to 14%,50 which is close to the present record value.
At the same time, we can expect the VOC values to increase in solar cells fabricated with more ordered kesterites, not necessarily because of reduced VOC deficits, but rather the wider bandgaps compared to their disordered counterparts, which can lead to larger quasi-Fermi level splitting. However, this VOC increase is usually accompanied by a decrease in short-circuit current density (JSC) (Fig. 3(c)) because of the reduced absorption range. As a result, improved and reduced PCEs/external quantum efficiencies (EQEs) in solar cells with a more ordered configuration have been reported simultaneously in literature.41,55 Therefore, compared to AgBiS2 solar cells, it is still too early to conclude if cation disorder has a similarly positive impact on kesterite solar cells.
Apart from the main characteristics discussed above, some additional considerations that could affect the performance of kesterite solar cells were also reported recently. For the conventional Cu2ZnSnS4 solar cell architecture (glass/MoS2/Cu2ZnSnS4/CdS/ZnO/ITO), a parasitic diode at the MoS2/Cu2ZnSnS4 interface may block hole transport and lead to “S-shaped” J–V curves with very low fill factors (FFs), as displayed in Fig. 2(d). It has been shown that with more disordered Cu2ZnSnS4, such an S-shaped feature could be removed, leading to smoother J–V curves with improved FFs. The proposed explanation is that this phenomenon is caused by the shallower VBM in more disordered Cu2ZnSnS4, which could make a better energy level alignment between MoS2/Cu2ZnSnS4 interface and Cu2ZnSnS4. Consequently, both the hole transport and FF were improved.41 In addition, from capacitance–voltage (C–V) measurements, a smaller charge-carrier density was observed in more ordered Cu2ZnSnSxSe1−x solar cells, which could result in a wider space charge region and enhance charge-carrier collection efficiency.42 However, such a large space charge region might also lead to a more voltage-dependent photocurrent and reduce the FF. More analyses on devices will be hence needed to elucidate the actual impacts of a reduced charge-carrier density in kesterite solar cells.
3. Strategies to tune cation disorder
Since cation disorder originates from the similar ionic radii of the two cations, introducing a third type of cation with a much different size is an apparent strategy to mitigate cation disorder. For kesterites, substituting Ag+ for Cu+ or Ba2+ for Zn2+ have been found to be effective on tuning their degree of cation disorder. In both cases, the ionic radius of Ag+ (1.14 Å) and Ba2+ (1.56 Å) is significantly larger than that of Cu+ and Zn2+, which are both approximately 0.74 Å. Calculations have shown that the formation energies of AgCu and CuBa antisites and even their related defect complexes, such as AgZn + ZnAg, are higher than the common antisite defects in conventional kesterites.56,57 Particularly, the large charge difference between Ba2+ and Sn4+ also increases the formation energies of Sn-related defects,57 which tend to have deep levels and are more detrimental to PV performance. As a result, both the degree of cation disorder and also the total defect concentrations of kesterites are predicted to decrease after Ag+- or Ba2+- substitution. It has been verified that (Cu,Ag)2ZnSnSe4 with different Ag compositions could show different bandgaps and low-temperature (10 K) PL peaks,58 as shown in Fig. 4(a). These results provide further evidence that the degree of cation disorder varies with different Ag/(Ag + Cu) ratios. However, it should be noted that not all of the cations with a large size mismatch with Cu+ or Zn2+ could have similar effects on suppressing cation disorder. For example, Cd2+ was believed to be favorable for substituting Zn2+ because of its larger ionic radius (0.92 Å), while calculations have shown that the formation energy of CuCd is still the lowest among all defects in Cu2ZnSnS4 and only slightly higher than that of CuZn, indicating that the degree of cation disorder cannot be changed effectively via Cd2+ substitution.56 Such an unexpected result originates from the narrow chemical potential region for stabilizing Cu2CdSnS4, which can only be formed when the chemical potential of Cd is below −0.9 eV. At such low chemical potentials, the formation energy for CuCd is also low, and cation disorder due to the prevalence of these antisite defects will still occur.59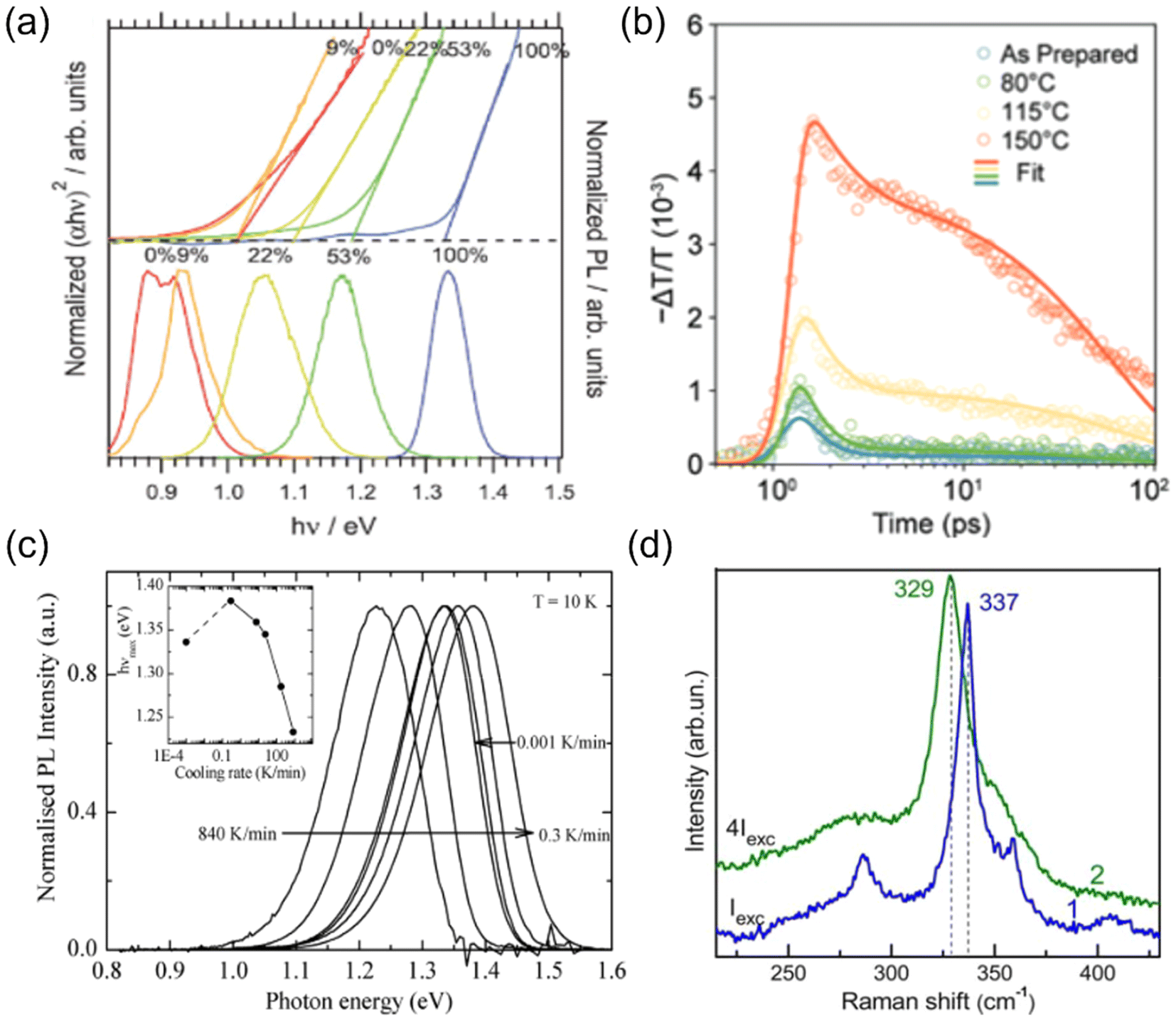 | ||
| Fig. 4 Strategies to tune cation disorder. (a) Normalized Tauc plots (top) and normalized PL spectra (bottom) of (Cu,Ag)2ZnSnSe4. The labelled ratios correspond to Ag/(Ag + Cu) values. α and hν represent the absorption coefficient and photon energy, respectively. Reprinted with permission from ref. 58. Copyright 2016, John Wiley and Sons. (b) Photoconductivity (proportional to −ΔT/T) kinetics of AgBiS2 NC films annealed (post-synthesis) at different temperatures for 10 min. Higher post-synthesis annealing temperatures can lead to higher degrees of cation disorder in AgBiS2. Reprinted under the CC-BY-4.0 license from ref. 32. (c) Low-temperature (10 K) PL spectra of Cu2ZnSnS4 powders quenched from 573 K to 473 K at different cooling rates. The inset shows the PL peak positions (hνmax) for the samples quenched at different cooling rates. Reprinted with permission from ref. 61. Copyright 2014, Elsevier. (d) Raman spectra of a Cu-rich Cu2ZnSnS4 sample excited by a 514.5 nm wavelength laser at a low (blue line) and high (green line) power. Reprinted with permission ref. 64. Copyright 2013, John Wiley and Sons. | ||
Additionally, another commonly used strategy to tune cation disorder is through annealing. Such a treatment can be conducted during or after synthesis. For the annealing treatment during synthesis, it has been shown that kesterite powders annealed at different temperatures followed by quenching could have different degrees of cation disorder, which was verified from the gradual change in bandgaps and lattice parameters.60 This result suggests that the cation distribution configuration formed during synthesis could be fixed by swift quenching. Interestingly, the cooling rates were also found to lead to different degrees of cation disorder. As shown in Fig. 4(c), using lower cooling rates seemed to further mitigate the cation disorder in Cu2ZnSnS4 powders, leading to blue shifts of the bandgaps and corresponding PL peak positions.61
On the other hand, post-synthesis annealing treatment has been widely adopted in kesterites and ABZ2 materials since it is the most convenient strategy to tune cation disorder in thin film samples, which could be potentially applied in PV devices. As indicated in Section 2.1 and Fig. 2(a), post-synthesis annealing could lead to a continuous transition of Cu2ZnSnS4 from a more ordered phase to a completely disordered phase when passing through the critical temperature TC.33 It should be noted that although most post-synthesis annealing treatments only required around 1 h to achieve the final disordered phases, a longer time (>20 h) was usually needed for the transformation into (relatively) ordered phases at temperatures below 150 °C. Such a long treatment time can then allow the slow diffusion process of cations to fully complete, and also enable the structure to reach thermal equilibrium. Similar phase transitions through a critical temperature TC were also observed in AgBiS2 and AgBiSe2,62,63 which exhibited a sudden drop in conductivity and thermopower (i.e., thermoelectric voltage divided by the temperature difference across the material) when a homogeneous disordered configuration was reached in these materials. Since the TC for these ABZ2 materials was usually above 300 °C, we might expect post-synthesis annealing at temperatures below 300 °C should mitigate the cation disorder of these materials, as observed in kesterites. However, the latest experimental results showed that for AgBiS2 NC films, even post-synthesis annealing at temperatures below 150 °C seemed to be sufficient to increase the degree of cation disorder.31,32 This clear difference between AgBiS2 and kesterites is not yet explained until now.
Finally, apart from thermal energy discussed above, light energy or an applied electric field might also change the cation disorder of materials. Valakh et al. have shown that when a Cu2ZnSnS4 particle was excited by a high-power 514.5 nm wavelength laser, its Raman peaks shifted and broadened, indicating an increased degree of cation disorder.64 In terms of the impact of an applied electric field on cation disorder, although a direct investigation has not yet been reported, significant ion migration has been observed in AgBiS2 and NaBiS2 devices under bias,65,66 suggesting that the cations may be mobile. Nevertheless, more quantitative investigations will be needed to verify the effectiveness and reproducibility of these light- or electric field-induced changes in cation disorder.
4. Quantifying and comparing cation disorder
The most common technique for characterizing cation disorder is Raman spectroscopy.67–69 As shown in Fig. 5(a), disordered Cu2ZnSnS4 powders (VF) showed clearly broadened Raman peaks compared to relatively ordered ones (VS) owing to a distribution of vibrational modes caused by different cation configurations.67 The difference between the ordered and disordered samples can be further enhanced if resonance Raman spectroscopy is used67,68 (lower part of Fig. 5(a), which used a 785 nm wavelength laser excitation). Scragg et al. also proposed that the order parameter S of a specific sample can be quantified by its “Q value”, which is defined as the ratio of the characteristic Raman m2A peak to m3A peak intensity.67 The former peak corresponds to the main A-symmetry mode, while the latter is believed to highly associate with the Cu–Zn distribution. Both peaks have been found to be highly sensitive to cation disorder. Fig. 5(b) presents the Q values and full-width-at-half-maximum (FWHM) values of the main m1A peaks for a series of Cu2ZnSnS4 films annealed at different temperatures, where we can deduce a critical temperature TC for the order–disorder transition.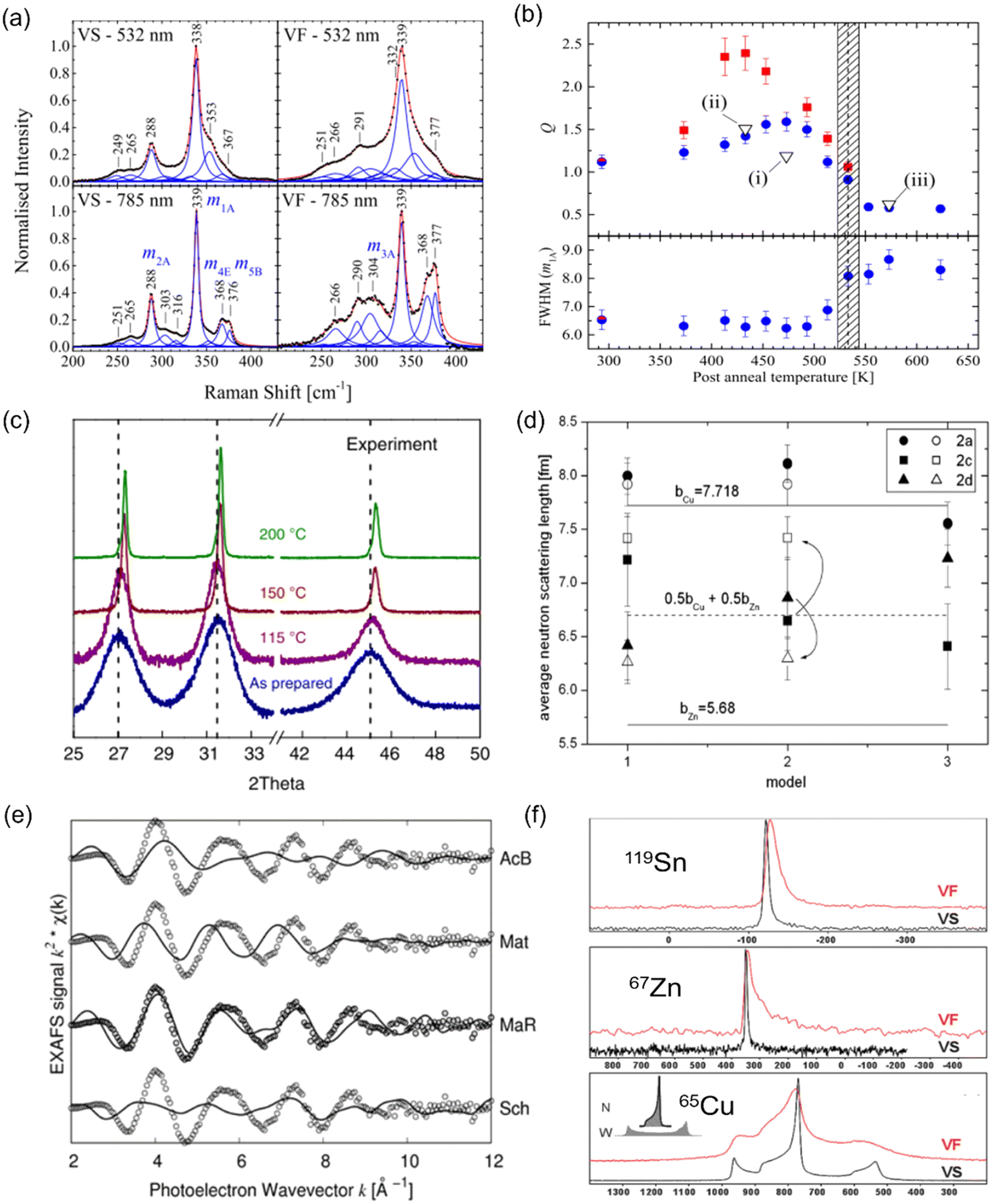 | ||
| Fig. 5 Characterization techniques for understanding and quantifying cation disorder. (a) Normalized Raman spectra with peak fitting for disordered (VF) and relatively ordered (VS) Cu2ZnSnS4 powders. The spectra taken under 532 nm (non-resonance) and 785 nm (resonance) laser excitations were recorded for comparison. Reprinted with under the terms of the CC-BY-4.0 license from ref. 67. (b) Variation in the Q value, which is defined as the ratios of the Raman peak intensity on m2A to m3A modes (part Fig. (a)), for Cu2ZnSnS4 films annealed at different temperatures for 1 h (blue circles) and 24 h (red squares). The full-width-at-half-maximum (FWHM) values of the Raman peak on the m1A mode for these samples are also displayed. The shaded area indicates the probable region of the critical temperature Tc for order–disorder transition. Reprinted under the terms of the CC-BY-4.0 license from ref. 67. (c) X-ray diffraction (XRD) patterns of pristine and AgBiS2 NC films annealed at different temperatures. Reprinted with permission from ref. 31. Copyright 2022, Springer Nature. (d) Fitted average neutron scattering lengths in the Cu2ZnSnS4 powders using different structural models. Closed and open symbols refer to the disordered and relatively ordered samples, respectively. The two solid lines indicate the neutron scattering length for Cu+ and Zn2+ cations, and the dotted line refers to the average neutron scattering length of Cu+ and Zn2+ cations. Reprinted with permission from ref. 72. Copyright 2011, Elsevier. (e) Extended X-ray absorption fine structure (EXAFS) spectra of a AgBiS2 NC film (open circles) along with the simulated curves (solid lines) based on 4 structures: acanthite (AcB), matildite (Mat), matildite with random cation occupation (MaR), and schapbachite (Sch). Reprinted under the terms of the CC-BY-4.0 license from ref. 73. (f) Solid-state nuclear magnetic resonance (NMR) spectra of 119Sn, 67Zn, and 65Cu for Cu2ZnSnS4 powders cooled down at different rates. The VF and VS samples are as indicated in Fig. (a). The unit of the horizontal axis (chemical shift) is in ppm. Reprinted with permission from ref. 74. Copyright 1999, Royal Society of Chemistry. | ||
Additionally, different cation configurations can also slightly change the bond lengths and hence the corresponding X-ray diffraction (XRD) patterns. An example can be seen in AgBiS2 NC films annealed at higher temperatures (Fig. 5(c)), which tended to have shorter Ag–S bonds when cation disorder became more homogeneous, causing a shift of the XRD peaks towards larger angles.31 Furthermore, if single-crystalline materials can be synthesized, single-crystal XRD can serve as another powerful tool for characterizing the degree of cation disorder since it can provide information on lattice parameters and cation occupancies at specific lattice sites.70,71 Unfortunately, this technique cannot apply to the kesterites discussed in this review, due to the identical electron densities of Cu+ and Zn2+ cations. In this respect, neutron scattering measurements can be more suitable for studying cation disorder in kesterites because the neutron scattering length of Cu+ and Zn2+ cations is very different. By fitting the average neutron scattering lengths determined from neutron diffraction patterns, the cation occupancies at each lattice site can be precisely extracted. An example for Cu2ZnSnS4 is displayed in Fig. 5(d), where we can see that site 2a is almost exclusively occupied by Cu+ independent of the models used or degree of disorder, while site 2c and 2d can be occupied by both Cu+ and Zn2+ cations,72 leading to the fitted neutron scattering length closer to the average value of Cu+ and Zn2+.
Recently, X-ray absorption spectroscopy (XAS) and nuclear magnetic resonance (NMR) spectroscopy were also used to probe cation disorder in both ABZ2 materials and kesterites. The extended X-ray absorption fine structure (EXAFS) spectra of materials include information on the bond lengths and coordination numbers of the target atoms, which can help to predict the level of cation segregation, as well as the crystal structure,73 as displayed in Fig. 5(e). On the other hand, NMR spectroscopy is particularly sensitive to the local chemical environments of the target nuclei, and similar to Raman spectroscopy, its characteristic peak widths and shifts will be affected by changes in the cation configurations,68,74 as seen from the example shown in Fig. 5(f) for Cu2ZnSnS4.
It can be seen that techniques commonly used to characterize cation disorder now mainly rely on the comparison between ordered and disordered samples, while a more direct way to probe cation arrangements is still lacking. Techniques such as high-resolution transmission electron microscopy (HRTEM) or atom probe tomography (APT),75 which can visualize different atoms with sub-nanometer resolution, can be useful for more directly characterizing cation disorder. However, the critical challenge for these techniques will be to characterize the materials without altering them during sample preparation or during the measurement itself.
5. Conclusion and outlook
In conclusion, cation disorder, which is widely found in inorganic materials, has proven to not necessarily be detrimental to the performance of these materials as solar absorbers, but rather can be an important tuning parameter to optimize their efficiency in PV devices. The effect of the degree of cation disorder and its homogeneity on the properties of solar absorbers has mostly been studied thus far in kesterites (Cu2ZnSnS4 and Cu2ZnSnSe4) and AgBiS2. Among these materials, it has been shown that cation disorder affects the bandgap, steepness of the absorption onset, absorption coefficient, and charge-carrier mobility, which all determine the ultimate efficiencies of these materials in solar cells. However, there is still much to be learned regarding how cation disorder affects these properties, and how cation disorder can be controlled and quantified. For example, whilst it has been shown that improving the homogeneity of cation disorder in AgBiS2 flattens the energy landscape,31 which leads to reduced Urbach energies, Cu2ZnSnS4 has shown the opposite trend.42 Furthermore, it was previously believed that an increased degree of cation disorder in Cu2ZnSnS4 led to increased VOC deficits, but more recently there has been debate about whether this is the case, and whether cation disorder influences VOC through non-radiative recombination or simply through changes in the bandgap (and therefore the achievable quasi-Fermi level splitting).41,42As the field moves forward, it will be important to actively look for cation disorder in novel solar absorber materials and to establish its link with its optoelectronic properties. What makes this challenging at present is that there is no straightforward method to quantify the degree and homogeneity of cation disorder in materials. Current approaches are all indirect and rely firstly on establishing correlations between cation disorder and the signals measured. For example, EXAFS can probe the local environment around the target cations, but requires simulations in order to interpret the measured spectra. The same can be said for neutron scattering measurements or NMR. More direct methods of probing cation disorder would be beneficial, such as using HRTEM techniques. But at the same time, it will be important to ensure that the measurement conditions or sample preparation methods do not alter cation disorder in the materials. For example, materials that are ionic conductors may readily change the arrangement of cations when energy is input from the probing beam during imaging, or from the mechanical/thermal energy input when samples are crushed to finer powder to measure.
Being able to quantify cation disorder more accurately will be critical for developing more reliable methods to control the degree and homogeneity of cation disorder. Currently, cation disorder is mostly tuned by the ionic radii of the species present and post-annealing. However, it has been found that the ionic radius is only a proxy for how likely it is for two cations to occupy the same crystallographic lattice site, and it is important to evaluate this by directly calculating the energy for antisite defect formation.56,57 Furthermore, whilst post-synthesis annealing at 115 °C has been effective for tuning the homogeneity of cation disorder in AgBiS2,31 low-temperature annealing has not been found to have similar effects in NaBiS2.45 Efforts should therefore be made to uncover alternative strategies for tuning these parameters, for example through the application of light, electric field, or the use of ligands and dopants. Realizing these routes can potentially open up more opportunities to achieve efficient photovoltaics from emerging solar absorbers. Additionally, it is important to monitor the changes in cation disorder when solar cells are in operation. That is, either under bias or over the temperature range from −40 °C to +85 °C in standard thermal cycling stability tests.76 Given the absence of such studies, we encourage more investigations into such in-operando measurements in the future, possibly through the use of in situ Raman spectroscopy or time-resolved XRD.
Author contributions
R. L. Z. H. conceived of the topic for this minireview. Y.-T. H. and R. L. Z. H. wrote the review and edited it together.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like financial support from the Engineering and Physical Sciences Research Council (no. EP/V014498/2).References
- L. Meng, J. You and Y. Yang, Nat. Commun., 2018, 9, 5265 CrossRef CAS PubMed.
- A. M. Ganose, D. O. Scanlon, A. Walsh and R. L. Z. Hoye, Nat. Commun., 2022, 13, 4715 CrossRef CAS PubMed.
- V. Steinmann, R. E. Brandt and T. Buonassisi, Nat. Photonics, 2015, 9, 355–357 CrossRef CAS.
- G. E. Eperon, M. T. Hörantner and H. J. Snaith, Nat. Rev. Chem., 2017, 1, 0095 CrossRef CAS.
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS.
- H. Michaels, M. Rinderle, I. Benesperi, R. Freitag, A. Gagliardi and M. Freitag, Chem. Sci., 2023, 14, 5350–5360 RSC.
- V. Pecunia, L. G. Occhipinti and R. L. Z. Hoye, Adv. Energy Mater., 2021, 11, 2100698 CrossRef CAS.
- J. C. Blakesley, R. S. Bonilla, M. Freitag, A. M. Ganose, N. Gasparini, P. Kaienburg, G. Koutsourakis, J. D. Major, J. Nelson, N. K. Noel, B. Roose, J. S. Yun, S. Aliwell, P. P. Altermatt, T. Ameri, V. Andrei, A. Armin, D. Bagnis, J. Baker, H. Beath, M. Bellanger, P. Berrouard, J. Blumberger, S. A. Boden, H. Bronstein, M. J. Carnie, C. Case, F. A. Castro, Y.-M. Chang, E. Chao, T. M. Clarke, G. Cooke, P. Docampo, K. Durose, J. R. Durrant, M. R. Filip, R. H. Friend, J. M. Frost, E. A. Gibson, A. J. Gillett, P. Goddard, S. N. Habisreutinger, M. Heeney, A. D. Hendsbee, L. C. Hirst, M. S. Islam, K. D. G. I. Jayawardena, M. B. Johnston, M. Kauer, J. Kettle, J.-S. Kim, D. Lamb, D. Lidzey, J. Lim, R. MacKenzie, N. Mason, I. McCulloch, K. P. McKenna, S. B. Meier, P. Meredith, G. Morse, J. D. Murphy, C. Nicklin, P. Ortega-Arriaga, T. Osterberg, J. B. Patel, A. Peaker, M. Riede, M. Rush, J. W. Ryan, D. O. Scanlon, P. J. Skabara, F. So, H. J. Snaith, L. Steier, J. Thiesbrummel, A. Troisi, C. Underwood, K. Walzer, T. Watson, J. M. Walls, A. Walsh, L. D. Whalley, B. Winchester, S. D. Stranks and R. L. Z. Hoye, arXiv, 2023, preprint, arXiv:2310.19430, DOI:10.48550/arXiv.2310.19430.
- S. Chen, A. Walsh, X. G. Gong and S. H. Wei, Adv. Mater., 2013, 25, 1522–1539 CrossRef CAS PubMed.
- D. B. Mitzi, O. Gunawan, T. K. Todorov, K. Wang and S. Guha, Sol. Energy Mater. Sol. Cells, 2011, 95, 1421–1436 CrossRef CAS.
- T. K. Todorov, K. B. Reuter and D. B. Mitzi, Adv. Mater., 2010, 22, 156–159 CrossRef PubMed.
- A. Chirilǎ, P. Reinhard, F. Pianezzi, P. Bloesch, A. R. Uhl, C. Fella, L. Kranz, D. Keller, C. Gretener, H. Hagendorfer, D. Jaeger, R. Erni, S. Nishiwaki, S. Buecheler and A. N. Tiwari, Nat. Mater., 2013, 12, 1107–1111 CrossRef PubMed.
- U. Rau and H. W. Schock, Appl. Phys. A: Mater. Sci. Process., 1999, 69, 131–147 CrossRef CAS.
- D. A. R. Barkhouse, O. Gunawan, T. Gokmen, T. K. Todorov and D. B. Mitzi, Prog. Photovolt.: Res. Appl., 2015, 20, 6–11 CrossRef.
- C. Shen, T. Li, Y. Zhang, R. Xie, T. Long, N. M. Fortunato, F. Liang, M. Dai, J. Shen, C. M. Wolverton and H. Zhang, J. Am. Chem. Soc., 2023, 145, 21925–21936 CrossRef CAS PubMed.
- S. A. Mcdonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. D. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138–142 CrossRef CAS PubMed.
- J. Tang, K. W. Kemp, S. Hoogland, K. S. Jeong, H. Liu, L. Levina, M. Furukawa, X. Wang, R. Debnath, D. Cha, K. W. Chou, A. Fischer, A. Amassian, J. B. Asbury and E. H. Sargent, Nat. Mater., 2011, 10, 765–771 CrossRef CAS PubMed.
- H. Shi, W. Ming and M.-H. Du, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 104108 CrossRef.
- K. T. Butler, S. McKechnie, P. Azarhoosh, M. VanSchilfgaarde, D. O. Scanlon and A. Walsh, Appl. Phys. Lett., 2016, 108, 112103 CrossRef.
- U. V. Ghorpade, M. P. Suryawanshi, M. A. Green, T. Wu, X. Hao and K. M. Ryan, Chem. Rev., 2023, 123, 327–378 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- G. E. Eperon, G. M. Paternò, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 RSC.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- S. R. Rondiya, R. A. Jagt, J. L. Macmanus-Driscoll, A. Walsh and R. L. Z. Hoye, Appl. Phys. Lett., 2021, 119, 220501 CrossRef CAS.
- R. A. Jagt, I. Bravić, L. Eyre, K. Gałkowski, J. Borowiec, K. R. Dudipala, M. Baranowski, M. D. Yksik, T. W. J. van deGoor, T. Kreouzis, M. Xiao, A. Bevan, P. Płochocka, S. D. Stranks, F. Deschler, B. Monserrat, J. L. MacManus-Driscoll and R. L. Z. Hoye, Nat. Commun., 2023, 14, 2452 CrossRef CAS PubMed.
- L. R. V. Buizza and L. M. Herz, Adv. Mater., 2021, 33, 202007057 CrossRef PubMed.
- A. Walsh and A. Zunger, Nat. Mater., 2017, 16, 964–967 CrossRef CAS PubMed.
- J. Leeman, Y. Liu, J. Stiles, S. B. Lee, P. Bhatt, L. M. Schoop and R. G. Palgrave, PRX Energy, 2024, 3, 011002 CrossRef.
- V. Vijay, S. Harish, J. Archana and M. Navaneethan, CrystEngComm, 2021, 23, 5522–5530 RSC.
- S. Roychowdhury, T. Ghosh, R. Arora, M. Samanta, L. Xie, N. K. Singh, A. Soni, J. He, U. V. Waghmare and K. Biswas, Science, 2021, 371, 722–727 CrossRef CAS PubMed.
- Y. Wang, S. R. Kavanagh, I. Burgués-Ceballos, A. Walsh, D. Scanlon and G. Konstantatos, Nat. Photonics, 2022, 16, 235–241 CrossRef CAS.
- M. Righetto, Y. Wang, K. A. Elmestekawy, C. Q. Xia, M. B. Johnston, G. Konstantatos and L. M. Herz, Adv. Mater., 2023, 35, 2305009 CrossRef CAS PubMed.
- G. Rey, A. Redinger, J. Sendler, T. P. Weiss, M. Thevenin, M. Guennou, B. ElAdib and S. Siebentritt, Appl. Phys. Lett., 2014, 105, 112106 CrossRef.
- G. H. Vineyard, Phys. Rev., 1956, 102, 981–992 CrossRef CAS.
- A. Baqais, N. Tymińska, T. LeBahers and K. Takanabe, Chem. Mater., 2019, 31, 3211–3220 CrossRef CAS.
- S. Zeiske, O. J. Sandberg, N. Zarrabi, C. M. Wolff, M. Raoufi, F. Peña-Camargo, E. Gutierrez-Partida, P. Meredith, M. Stolterfoht and A. Armin, J. Phys. Chem. Lett., 2022, 13, 7280–7285 CrossRef CAS PubMed.
- M. C. Folgueras, Y. Jiang, J. Jin and P. Yang, Nature, 2023, 621, 282–288 CrossRef CAS PubMed.
- C. Y. He, Y. Li, Z. H. Zhou, B. H. Liu and X. H. Gao, Adv. Mater., 2024, 2400920, 1–36 Search PubMed.
- M. Quennet, A. Ritscher, M. Lerch and B. Paulus, J. Solid State Chem., 2017, 250, 140–144 CrossRef CAS.
- F. Viñes, G. Konstantatos and F. Illas, Phys. Chem. Chem. Phys., 2017, 19, 27940–27944 RSC.
- C. Malerba, M. Valentini and A. Mittiga, Sol. RRL, 2017, 1, 1700101 CrossRef.
- S. Bourdais, C. Choné, B. Delatouche, A. Jacob, G. Larramona, C. Moisan, A. Lafond, F. Donatini, G. Rey, S. Siebentritt, A. Walsh and G. Dennler, Adv. Energy Mater., 2016, 6, 1502276 CrossRef.
- Q. Feng, Ceram. Int., 2023, 49, 19533–19536 CrossRef CAS.
- D. M. Bishop, B. McCandless, T. Gershon, M. A. Lloyd, R. Haight and R. Birkmire, J. Appl. Phys., 2017, 121, 065704–065701 CrossRef.
- Y.-T. Huang, S. R. Kavanagh, M. Righetto, M. Rusu, I. Levine, T. Unold, S. J. Zelewski, A. J. Sneyd, K. Zhang, L. Dai, A. J. Britton, J. Ye, J. Julin, M. Napari, Z. Zhang, J. Xiao, M. Laitinen, L. Torrente-Murciano, S. D. Stranks, A. Rao, L. M. Herz, D. O. Scanlon, A. Walsh and R. L. Z. Hoye, Nat. Commun., 2022, 13, 4960 CrossRef CAS PubMed.
- F. X. Bronold, F. X. Bronold, H. Fehske and H. Fehske, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 731021–731024 Search PubMed.
- F. X. Bronold, A. Alvermann and H. Fehske, Philos. Mag., 2004, 84, 673–704 CrossRef CAS.
- H. Hempel, R. Eichberger, I. Repins and T. Unold, Thin Solid Films, 2018, 666, 40–43 CrossRef CAS.
- H. Hempel, C. J. Hages, R. Eichberger, I. Repins and T. Unold, Sci. Rep., 2018, 8, 14476 CrossRef PubMed.
- W. Chen, D. Dahliah, G. M. Rignanese and G. Hautier, Energy Environ. Sci., 2021, 14, 3567–3578 RSC.
- T. Gokmen, O. Gunawan, T. K. Todorov and D. B. Mitzi, Appl. Phys. Lett., 2013, 103, 103506 CrossRef.
- J. J. S. Scragg, J. K. Larsen, M. Kumar, C. Persson, J. Sendler, S. Siebentritt and C. Platzer Björkman, Phys. Status Solidi B, 2016, 253, 247–254 CrossRef CAS.
- G. Rey, T. P. Weiss, J. Sendler, A. Finger, C. Spindler, F. Werner, M. Melchiorre, M. Hála, M. Guennou and S. Siebentritt, Sol. Energy Mater. Sol. Cells, 2016, 151, 131–138 CrossRef CAS.
- U. Rau and J. H. Werner, Appl. Phys. Lett., 2004, 84, 3735–3737 CrossRef CAS.
- M. Kauk-Kuusik, K. Timmo, M. Pilvet, K. Muska, M. Danilson, J. Krustok, R. Josepson, V. Mikli and M. Grossberg-Kuusk, J. Mater. Chem. A, 2023, 11, 23640–23652 RSC.
- Z.-K. K. Yuan, S. Chen, H. Xiang, X.-G. G. Gong, A. Walsh, J.-S. S. Park, I. Repins and S.-H. H. Wei, Adv. Funct. Mater., 2015, 25, 6733–6743 CrossRef CAS.
- D. Shin, B. Saparov and D. B. Mitzi, Adv. Energy Mater., 2017, 7, 1602366 CrossRef.
- T. Gershon, Y. S. Lee, P. Antunez, R. Mankad, S. Singh, D. Bishop, O. Gunawan, M. Hopstaken and R. Haight, Adv. Energy Mater., 2016, 6, 4–10 Search PubMed.
- Z. K. Yuan, S. Chen, H. Xiang, X. G. Gong, A. Walsh, J. S. Park, I. Repins and S. H. Wei, Adv. Funct. Mater., 2015, 25, 6733–6743 CrossRef CAS.
- A. Ritscher, M. Hoelzel and M. Lerch, J. Solid State Chem., 2016, 238, 68–73 CrossRef CAS.
- M. Grossberg, J. Krustok, T. Raadik, M. Kauk-Kuusik and J. Raudoja, Curr. Appl. Phys., 2014, 14, 1424–1427 CrossRef.
- H. Jang, S. Abbey, W. H. Nam, B. Frimpong, C. V. Nguyen, S. J. Joo, H. S. Shin, J. Y. Song, E. N. Cho, M. Kim, Y. S. Jung and M. W. Oh, J. Mater. Chem. A, 2021, 9, 4648–4657 RSC.
- S. N. Guin, S. Banerjee, D. Sanyal, S. K. Pati and K. Biswas, Inorg. Chem., 2016, 55, 6323–6331 CrossRef CAS PubMed.
- M. Y. Valakh, O. F. Kolomys, S. S. Ponomaryov, V. O. Yukhymchuk, I. S. Babichuk, V. Izquierdo-Roca, E. Saucedo, A. Perez-Rodriguez, J. R. Morante, S. Schorr and I. V. Bodnar, Phys. Status Solidi RRL, 2013, 7, 258–261 CrossRef CAS.
- Y. T. Huang, D. Nodari, F. Furlan, Y. Zhang, M. Rusu, L. Dai, Z. Andaji-Garmaroudi, D. Darvill, X. Guo, M. Rimmele, T. Unold, M. Heeney, S. D. Stranks, H. Sirringhaus, A. Rao, N. Gasparini and R. L. Z. Hoye, Small, 2023, 2310199 Search PubMed.
- Y. T. Huang, M. Schleuning, H. Hempel, Y. Zhang, M. Rusu, T. Unold, A. Musiienko, O. Karalis, N. Jung, S. J. Zelewski, A. J. Britton, N. Ngoh, W. Song, L. C. Hirst, H. Sirringhaus, S. D. Stranks, A. Rao, I. Levine and R. L. Z. Hoye, Adv. Funct. Mater., 2024, 2310283 CrossRef.
- J. J. S. Scragg, L. Choubrac, A. Lafond, T. Ericson and C. Platzer-Björkman, Appl. Phys. Lett., 2014, 104, 041911 CrossRef.
- M. Paris, L. Choubrac, A. Lafond, C. Guillot-Deudon and S. Jobic, Inorg. Chem., 2014, 53, 8646–8653 CrossRef CAS PubMed.
- X. Fontané, V. Izquierdo-Roca, E. Saucedo, S. Schorr, V. O. Yukhymchuk, M. Y. Valakh, A. Pérez-Rodríguez and J. R. Morante, J. Alloys Compd., 2012, 539, 190–194 CrossRef.
- A. Zucchini, P. Comodi, A. Katerinopoulou, T. Balic-Zunic, C. McCammon and F. Frondini, Phys. Chem. Miner., 2012, 39, 319–328 CrossRef CAS.
- A. Nicolson, J. Breternitz, S. R. Kavanagh, Y. Tomm, K. Morita, A. G. Squires, M. Tovar, A. Walsh, S. Schorr and D. O. Scanlon, J. Am. Chem. Soc., 2023, 145, 12509–12517 CrossRef CAS PubMed.
- S. Schorr, Sol. Energy Mater. Sol. Cells, 2011, 95, 1482–1488 CrossRef CAS.
- J. K. Kesavan, F. D′acapito, P. Scardi, A. Stavrinadis, M. Z. Akgul, I. Burgués-Ceballos, G. Konstantatos and F. Boscherini, Nanomaterials, 2020, 10, 316 CrossRef CAS PubMed.
- L. Choubrac, M. Paris, A. Lafond, C. Guillot-Deudon, X. Rocquefelte and S. Jobic, Phys. Chem. Chem. Phys., 2013, 15, 10722–10725 RSC.
- A. Sharma, A. H. Pandey, M. K. Jangid, V. Srihari, H. K. Poswal and A. Mukhopadhyay, ACS Appl. Mater. Interfaces, 2023, 15, 782–794 CrossRef CAS PubMed.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. DiCarlo, R. Cheacharoen, Y. B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. DiGirolamo, M. Grätzel, A. Hagfeldt, E. vonHauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. (Frank)Liu, Y. L. Loo, J. M. Luther, C. Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N. G. Park, M. O. Reese, F. DeRossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |