
In situ carbonization metamorphoses porous silica particles into biodegradable therapeutic carriers of lesser consequence on TGF-β1 mediated fibrosis†
Akanksha
Dohare‡
a,
Niranjan
Chatterjee‡
a and
Santosh K.
Misra
 *ab
*ab
aDepartment of Biological Sciences & Bioengineering, Indian Institute of Technology Kanpur, Kanpur, UP 208016, India. E-mail: skmisra@iitk.ac.in; Tel: +91-512-259-4013
bThe Mehta Family Centre for Engineering in Medicine, Indian Institute of Technology Kanpur, Kanpur, UP 208016, India
First published on 11th April 2024
Abstract
Extensive modifications have been made to the synthesis protocol for porous silica particles to improve the shape, size and yield percentage, but problems associated with improvement in biodegradability and decrease in chances to induce side effects still remain a concern. To circumvent these limitations, a facile modification strategy has been employed through in situ carbonization of porous silica particles. Herein, carbon particles were integrated within porous silica core–shell particles (Si-P-CNPs) during the synthesis process and found to preserve the ordered structural morphology. Curcumin was used as a model drug for loading in prepared Si-P-CNPs whereas lung cancer cells were used as a model system to study the in vitro fate. These Si-P-CNPs showed improved drug loading, drug effectivity, biodegradability and avoidance of interaction with transforming growth factor β1 (TGF-β1) indicating the possibility of reducing the chances of lung fibrosis and thereby enhancing the safety profile over conventional porous silica particles.
Porous silica materials receive significant attention due to their large surface area, high pore diameter, facile methods of synthesis, possibility for easy surface modification, tunable biocompatibility, and economical production.1 Such qualities make these materials suited for different research and application domains such as drug delivery, impurity extraction, catalysis, sensing etc.1–5 The production of silica particles in various forms, sizes, and volumes has been achieved by using different synthetic methodologies.6 Among the many available methods, the mono-dispersity, spherical form, and diameter of the particle can be controlled using the Stöber method.6,7 On the other hand, mesoporous nanomaterials with a high surface area, defined channels, and pore structures are generally generated either by a liquid templating method or by using inorganic precursors as well as surfactants in specific formulations.8–11 Here, a modified synthesis protocol has been used to prepare carbonized porous silica particles (Si-P-CNPs), using a combination of nanocarbon with other precursors in a one pot synthesis method. It was found that the prepared Si-P-CNPs could show complete biodegradation after completion of their role delivering the loaded drug. Further, these carbonized particles were also found to avoid interaction with transforming growth factor β1 (TGF-β1) to a significant extent in comparison with non carbonized porous particles and therefore would be capable of reducing the chances of lung fibrosis. The possibility of inducing fibrosis is otherwise a major drawback of silica based particulate systems. Furthermore, the efficiencies of newly synthesized Si-P-CNPs were verified and compared with other control systems, namely carbonized non-porous silica particles (Si-NP-CNPs), non-porous silica particles (Si-NPs) and porous silica (Si-Ps) core–shell particles.
In order to make spherical Si-NP and Si-P core–shell particles, a synthesis protocol from the report of Dong et al. was taken into consideration.12 Cetyltrimethylammonium bromide (CTAB), n-tridecane, and non-porous particles (d = ∼800 nm) previously synthesized by a Stöber procedure were treated with the addition of ammonium hydroxide, containing ammonium fluoride, at 90 °C while being stirred at 240–280 rpm.13 For the synthesis of Si-P-CNPs, a suspension of carbon nanoparticles (CNPs), which was already prepared using a previously established protocol,14,15 was added during the synthesis of silica nonporous structures. Similar reaction conditions were used to those generally used for synthesis of a nonporous silica core covered with a silica shell, with MCM-41 (Mobil Composition of Matter-41) type mesopores on the surface.9,16,17 By using these reaction conditions, considering that variation in reaction time can be utilized to modify the relative core–shell sizes, multiple variations were included to optimize the synthesis conditions. Additionally, Si-NP-CNP particles were also synthesized for better comparison of various physiochemical properties. All of the synthesized materials were characterized for shape, size, morphology, porosity, optical properties, cell interaction, safety and biodegradability status. The process by which nonporous spherical particles transform into spherical silica core shell particles has been well discussed elsewhere.12 In brief, the negatively charged surface of the silica particles adsorbs micelles containing the cationic surfactant CTAB. A negatively charged silica species is dissolved from the particle surface in a templated dissolution process. The particle is typically wrapped in a shell with wormhole-like pores as shown in the results. It is significant that the use of NH4F as an etching agent caused many of the Si-OH groups on the the silica core surface to be replaced by SiF. As a result, the shell of the spherical dense core is coated with SiF features after the dissolution process, which diminishes the surface's negative charge. The measurement of hydrodynamic diameter through the dynamic light scattering technique showed that the Si-P particles were of ∼1056 ± 88 nm in size with a zeta potential of −36 ± 1 mV whereas Si-P-CNPs were of size 750 ± 86 nm with an average zeta potential of −20 ± 1 mV. The incorporated CNPs were of ∼270 ± 21 nm in diameter and zeta potential of −5 ± 1 mV. The hydrodynamic diameters of the Si-NPs and Si-NP-CNPs were around 756 ± 221 nm and 870 ± 239 nm, respectively, with zeta potentials of around −52 ± 1 mV and −51 ± 1 mV, respectively (Fig. S1A and S1B, ESI†). The decreased zeta potential values for Si-Ps and Si-P-CNPs in comparison with Si-NPs and Si-NP-CNPs are presumably due to the use of the cationic surfactant CTAB as a pore forming agent. The X-ray diffraction (XRD) pattern showed a wide peak at 2θ ∼22.3° with a d spacing of ∼3.99 Å in the case of Si-Ps, which indicates the amorphous nature of the synthesized particles.18,19 A similar type of XRD pattern was also observed in the case of Si-P-CNPs without any significant alteration of the position of the diffraction peak at 2θ ∼22.75° with a d spacing of ∼3.90 Å (Fig. 1A). Si-NP and Si-NP-CNP particles also showed broad diffraction peaks centered at 2θ ≈ 23.1° and 22.3° with d spacings of 3.85 Å and 3.98 Å, respectively (Fig. S2A, ESI†). At the same time, the similar small angle XRD patterns of Si-Ps and Si-P-CNPs with a small hump at 2θ ≈ 1.6° signify the equivalent levels of orderedness of the pores (Fig. S2B, ESI†).12 This study revealed the preservation of the native ordered structures of Si-Ps even after incorporation of CNPs. Fourier transform infrared spectroscopy (FTIR) was performed to evaluate the impact of incorporation of nanosized carbon particles on the chemical bonds responsible for making the framework of the silica nanostructures. All characteristic spectral features indicating Si–O bending vibration, Si–O–Si stretching vibration and Si–OH stretching vibration were observed in both Si-P and Si-P-CNP particles (Fig. 1B). Intriguingly, we noticed the transmittance peak at 807 cm−1 and 1108 cm−1 implying Si–O bending vibration and Si–O–Si stretching vibration in Si-Ps20,21 were shifted to 801 cm−1 and 1088 cm−1 in the case of Si-P-CNPs. This shifting of vibrational bands to lower wavenumber in Si-P-CNPs probably indicates the weakening of two important chemical bonds Si–O and Si–O–Si in the silica nanostructures which might suggest some biological benefits including better degradability. No shift of the transmittance peak was observed for the Si-H2O flexion at 1637 cm−1; however, the broad transmittance peak at 3445 cm−1 attributed to Si-OH stretching vibration in Si-P was shifted to higher wavelength in Si-P-CNP.
 | ||
| Fig. 1 Characterization of silica particles for orderedness, surface functionality, porosity and drug loading efficiency. (A) XRD spectra of Si-Ps and Si-P-CNPs; (B) FTIR spectra of Si-Ps and Si-P-CNPs where blue arrows are indicating some of the characteristic absorption peaks at different wavenumbers; (C) N2 adsorption/desorption isotherms of Si-NP, Si-P and Si-P-CNP particles; (D) FTIR spectra of cur and Si-P-CNP-Cur with the allocation of some characteristic absorption peaks at different wavenumbers. | ||
Furthermore, the quantitative measurement of the porous shells of Si-Ps and Si-P-CNPs was carried out by measuring nitrogen adsorption–desorption isotherms. The adsorption–desorption behavior could also be associated with the efficiency of these particles to act as therapeutic carriers. These Si-P and Si-P-CNP particles could demonstrate nitrogen adsorption and desorption isotherms in a reversible trend that shows a classic type IV isotherm hysteresis (Fig. 1C).22,23 As shown by the Si-Ps, the well-arranged MCM-41 type of hexagonal mesopores might be present as indicated by the distinctive H1 loop, whereas Si-NPs don't show any hysteresis or loop due to their nonporous nature. The BJH method shows that a higher average pore size (Table 1) that varied from 0.2 to 3 nm exists, while the specific surface area of Si-Ps was around 228 m2 g−1. Interestingly, Si-P-CNPs show a specific surface area of ∼1864 m2 g−1 which is much higher than that of Si-P particles.
| Sample name | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| Si-NPs | 13.986 | 0.01 | 0.2 |
| Si-Ps | 228.358 | 0.3 | 3 |
| Si-P-CNPs | 1864.930 | 0.4 | 1.8 |
The therapeutic drug carrying capacities of Si-P and Si-P-CNP particles were investigated by measuring their loading efficiency of curcumin as a model drug. Curcumin is a naturally derived polyphenol that shows potential anticancer activity by interfering in nuclear factor kappa B (NF-κB) signaling, and eventually induction of apoptosis etc.24,25 But, its inadequate aqueous solubility reduces its bioavailability to be used as a potent drug for cancer treatment.26 It was observed that Si-Ps and Si-P-CNPs could load around 21.6 ± 4.8 and 63.2 ± 14.4% of curcumin, respectively. A higher loading percentage of drug by carbon enhanced porous silica particles, Si-P-CNPs, was observed compared to Si-Ps, despite the pore size being smaller in the case of Si-P-CNPs i.e., ∼1.8 nm compared to 3.0 nm for Si-Ps. This indicates that drugs are probably loaded in the pores of the Si-P-CNPs by some additional mode of loading. The existence of an additional mode could be because of the presence of more low volume pores present on the Si-P-CNP surface which translates into more pores to load drug molecules and added to this, 29Si NMR spectroscopic investigations revealed an additional level of interaction of curcumin in the Si-P-CNP pores in comparison to the Si-P ones. Spectroscopic study on curcumin loaded Si-P-CNPs (Si-P-CNP-Cur) and curcumin loaded Si-Ps (Si-P-Cur) showed the presence of Q3 (Si(Osi)3OH) and Q4 (Si(Osi)4) species27 (Fig. S3, ESI†). The peak position of Q4 species in Si-P-Cur at ∼−112.5 ppm was shifted to ∼−115.5 ppm in Si-P-CNP-Cur, while the Q3 species was found to be in the same position (∼−101.89) in both Si-P-Cur and Si-P-CNP-Cur. The broad spectral appearance of Q3 and Q4 species was also noticeable in Si-P-CNP-Cur compared to Si-P-Cur. This suggested an additional level of interaction of curcumin in Si-P-CNP pores in comparison to that in Si-P ones. On performing drug release studies, it was found that Si-P-CNPs could release a higher amount of drug at the 72 h time point whereas Si-Ps were releasing the drug at the earlier time point of 24 h (Fig. S4, ESI†). This points to sustained release of drug from Si-P-CNPs compared to Si-Ps which also supports the difference in the mode of drug loading in Si-P-CNPs compared to Si-Ps. Probably, the presence of carbon in Si-P-CNPs provides better interaction with curcumin in the pores which requires a longer time to allow the release of the drug.
Despite the excellent therapeutic benefits of curcumin, its proper clinical translation is limited because of its low bioavailability, short half-life, low aqueous solubility etc. To circumvent these issues various types of engineered nano-formulations including mesoporous silica particles have been developed for the improvement of its cellular internalization and suspendability in physiological buffer28,29etc. It has been previously reported that folic acid conjugated mesoporous silica particles increased curcumin levels in plasma as well as in tumor tissue.29 Based on these previously established reports we believe that curcumin loaded Si-P-CNP particles will also be helpful to improve the problems associated with bioavailability by improving the aqueous suspendability. Further the sustained release of curcumin from Si-P-CNP particles will help in slowly reaching the therapeutic concentration inside cells while preventing its unnecessary and premature loss.30 The sustained release of curcumin from mesoporous silica particles under physiological conditions has also previously been reported.31 As the half-life of curcumin drug is around 6–7 h, it becomes more important to release a smaller amount at different time points rather than releasing it at once, wherein most of it would get biotransformed into degraded metabolites in turn reducing the therapeutic efficiency of the formulation. But it would be possible to present a better assessment of bioavailability, half-life and biotransformation of curcumin released from Si-P-CNPs after extensive in vivo study.32 Analysis of the FTIR spectrum of curcumin (Cur) showed the presence of transmittance peaks at 1429 cm−1 and 1457 cm−1 attributed to the stretching vibration of the aromatic benzene ring of curcumin33 which also appeared in Si-P-CNP-Cur at wavenumbers of 1406 cm−1 and 1455 cm−1. Further, the transmittance peak at 3512 cm−1, generally corresponding to the –OH stretching vibration of curcumin, reduces significantly in Si-P-CNP-Cur, probably due to successful entrapment of curcumin in the Si-P-CNP particles (Fig. 1D).34
The anhydrous morphology of the synthesized Si-P, Si-NP and Si-P-CNP particles was visualized by transmission electron microscopy (TEM). Analysis of TEM images showed that Si-P particles were of 1.673 ± 1 μm in diameter with 161.2 ± 3 nm porous shell thickness. Representative TEM images of Si-Ps (Fig. 2A and B) confirmed the presence of highly organized porous shells which could be a result of the use of CTAB as a pore-forming agent during the reaction.35 Although Si-P-CNP particles also showed similarly organized porous shells (Fig. 2C), a significant reduction in anhydrous diameter was noticed in comparison to Si-P particles. Si-P-CNPs were of 687 ± 28 nm in size with 84.5 ± 2 nm porous shell thickness. As expected, no shell structure was observed in Si-NP particles which were of 760.6 ± 17 nm in diameter (Fig. 2D). These surface pores are a distinguishing property of these particles. According to the MCM-41 materials pattern, the new particles Si-Ps and Si-P-CNPs have hexagonally arranged shells with tunnels that have layer thicknesses of 161.2 ± 3 nm and 84.5 ± 2 nm, respectively, around the dense particles. The thickness of the shell could be influenced by reaction time, much like in the case of Si-Ps and Si-P-CNPs.
 | ||
| Fig. 2 Transmission electron microscopy images of (A) Si-Ps, (B) magnified portion of the porous shell of an Si-P, (C) an Si-P-CNP and (D) an Si-NP. | ||
The incorporation of CNPs inside the core of Si-P particles was verified by confocal microscopy-based investigation. The autofluorescent nature of CNPs36,37 makes Si-P-CNP particles traceable under a confocal microscope. To accurately visualize the location of CNPs, negatively charged Si-P-CNP particles were loaded with another cationic dye rhodamine 6G.38 The appearance of bright red fluorescence on the shell of the Si-P particles indicates the successful loading of rhodamine 6G (Fig. 3A), which was not visible on the non-porous silica surface (Fig. 3B). CNPs are known to emit in the green region upon excitation with a 488 nm wavelength laser39,40 as evident in Fig. 3C. The presence of an orange color on Si-P-CNP particles (Fig. 3D) denotes the co-localization of red and green color fluorescence which were contributed by rhodamine 6G and incorporated CNPs, respectively (Fig. S5, ESI†). This study confirms the successful incorporation of CNPs inside the Si-P-CNP particles. Cellular internalization of Si-P-CNP-Cur particles was evaluated by flow cytometry based investigations. Vero kidney epithelial cells were taken as a healthy control whereas A549 lung epithelial cells were used as cancer cells for this investigation. It was evident that Si-P-CNP-Cur treated A549 cells showed a significant shift in fluorescence intensity in comparison with Si-P-CNP-Cur treated Vero cells. This investigation revealed more internalization of Si-P-CNP-Cur particles in cancer cells with respect to the healthy control (Fig. 4A). Probably, a highly receptive nature of cancer cells toward silica particles makes the particles get delivered into high percentage of cancer cells (area under the curve) with high numbers of silica particles per cancer cell compare to normal Vero cells (fluorescence intensity).41 A cell viability assay using thiazolyl blue tetrazolium bromide (MTT) was performed to investigate the functional efficacy of the curcumin loaded Si-P-Cur and Si-P-CNP-Cur.
 | ||
| Fig. 3 Confocal microscopy images of (A) rhodamine loaded Si-P particles, (B) Si-NP particles, (C) CNPs and (D) rhodamine loaded Si-P-CNP particles. Scale bar (A–D) = 20 μm. | ||
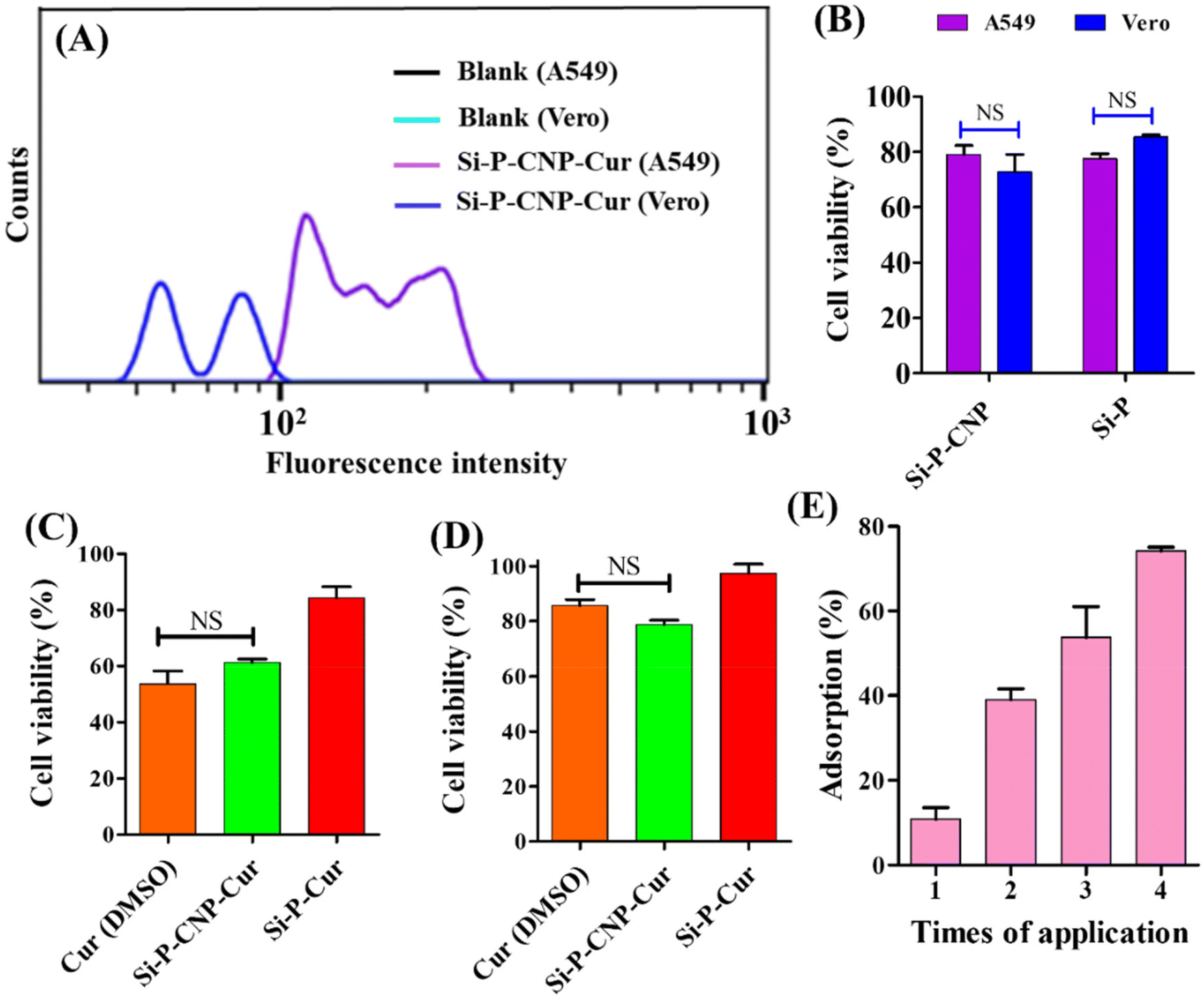 | ||
| Fig. 4 Cellular interaction and biological fate of silica particles. (A) Flow cytometry analysis of the green subset from A549 and Vero cells treated with Si-P-CNP-Cur with respect to their untreated control (y-axis 0–40, a.u.). (B) Cell viability assay of Si-P-CNP and Si-P treated A549 and Vero cells. Cell viability assay of Cur, Si-P-CNP-Cur and Si-P-Cur treated (C) A549 cells and (D) Vero cells. (E) Mucus adsorption assay using Si-P-CNP particles. | ||
It was noticeable that the respective amount of vehicle, e.g., Si-P (5.4 μg ml−1) and Si-P-CNP (20 μg ml−1), which was finally used for cell viability studies with loaded curcumin, didn't show any significant cell death in either of the cell lines (Fig. 4B). A 25 μM concentration of curcumin loaded in Si-P-CNP-Cur particles could inhibit the cell viability by ∼40% for A549 cancer cells within 48 h of application which was very similar to treatment with curcumin dissolved in DMSO (Fig. 4C). No significant cell death was observed in Vero cells treated with either the same amount of curcumin loaded Si-P-CNP-Cur particles or curcumin dissolved in DMSO (Fig. 4D). Probably, this distinguishability might have occurred due to the intrinsic properties of curcumin along with differential delivery of curcumin into cancer cells over normal cells.41–43
The interacting ability of Si-P-CNP particles with mucus of the respiratory tract was evaluated in a simulated nasal mucus (SNM) which was prepared using a previously reported composition.44 The SNM was further mixed with 20 μM rhodamine 6G and incubated with 20 μg Si-P-CNP particles. It was observed that the extent of SNM adsorption increased gradually with the increment of number of times of application of Si-P-CNP particles (Fig. 4E) and reached up to ∼74% after 4 cycles of application. This study revealed the possible capability of these particles to entrap excessively produced and accumulated airway mucus which otherwise confers a detrimental effect on airway obstruction and lung function in several disease conditions.45–47 It is believed that this mucus entrapment ability of Si-P-CNP particles will further be able to reduce the use of drugs for inhibiting hypersecretion of mucus like mucolytics in different disease conditions including chronic obstructive pulmonary diseases (COPD), cystic fibrosis, asthma etc.,46,48 although, just based on this experiment, it is hard to comment on the permeation behavior of the synthesized particles through the mucus layer of the respiratory tract before performing an in vivo experiment in an animal model.
The safety profile of the Si-P-CNP particles for use in the treatment of lung complications was investigated through multiple experiments including an interaction study with macrophages (RAW 264.7 cells), a biodegradation study in simulated body fluid (SBF), and silicosis induction assay. Previously multiple research groups have reported the ability of silica nanoparticles to interact with macrophage cells.49,50 A scanning electron microscopy (SEM) based study was performed to find the interaction ability of Si-P-CNPs with RAW 264.7 cells.
Intriguingly Si-P-CNP particles were found to be attached to the surface of macrophages (Fig. 5A and Fig. S6, ESI†). The measurement of cell viability dictated the cytocompatibility of both Si-P-CNP-Cur and Si-P-CNP particles (Fig. 5B and C). This study revealed the possibility of phagocytosis of Si-P-CNP particles by macrophages without creating any toxicity. One of the most reported side-effects of silica-based materials during biological applications is the induction of silicosis. In this situation, there is the formation of nodules in the lung, massive inflammation and lung fibrosis,51,52 but, various fabrication and porosity induction possibilities make silica-based materials desirable for biological applications. Thus, a silica-based biomaterial with other beneficial properties but without significant onset of silicosis would be a great achievement. It was hypothesized that the produced carbonaceous silica porous material might show such a property. To assess this, a silicosis induction assay was performed in A549 cells by the application of Si-P-CNP and Si-P particles incubated with TGF-β1, named Si-P-CNP-TGF-β1 and Si-P-TGF-β1, respectively. TGF-β1 is considered an important contributary factor for epithelial–mesenchymal transition which is one of the major steps in lung fibrosis.51,53–55 Flow cytometry-based analysis using an antibody against p-Smad2/3, a downstream mediator of the TGF-β signaling pathway,56–58 demonstrated that Si-P-CNP-TGF-β1 and Si-P-CNP treated cells showed no shift in fluorescence intensity with respect to the untreated control. Cells treated with Si-P-TGF-β1 and Si-Ps gave a shift in fluorescence intensity to some extent. This indicates the possibility of induction of lung fibrosis which didn't exist in the case of Si-P-CNP-TGF-β1 treatment (Fig. 5D and E). Cells treated with TGF-β1 alone also showed a shift in fluorescence intensity indicating the activation of the TGF-β1 signaling pathway (Fig. S7, ESI†). The biological safety of produced biomaterials generally depends on the properties of bioavailability and biodegradation.
 | ||
| Fig. 5 Immune cell interaction and biological fate of silica particles. (A) SEM images Si-P-CNP-Cur particles interacting with RAW 264.7 cells. Scale bar = 10 μM. Cell viability assay of RAW 264.7 cells treated with (B) Si-P-CNP-Cur and (C) Si-P-CNP particles. Flow cytometry analysis of the red subset from A549 cells treated with (D) Si-P-CNP-TGF-β1 and Si-P-TGF-β1, and (E) Si-P-CNP and Si-P, with respect to untreated controls. | ||
The biodegradability of Si-P-CNP particles was monitored in SBF by measuring the hydrodynamic diameter at an interval of 24 h for a total time span of 168 h and SEM based investigations at 0 h and 168 h. It was observed that Si-P-CNP particles were of ∼710 nm in diameter just after incubation in SBF which was similar to the hydrodynamic diameter measured in aqueous medium. This revealed the stability of the Si-P-CNP particles in SBF. Further the reduction of hydrodynamic diameter to ∼110 nm after 48 h of incubation was observed in SBF, which indicated the degradation of the Si-P-CNP particles. Thereafter a further increase in diameter from 72 h to 168 h was noticeable, which was probably because of aggregation of degraded Si-P-CNP particles (Fig. 6D). This type of aggregation of degraded silica particles was also previously reported.59 Further, to confirm the degradation process, a SEM based investigation was performed. It was found that Si-P-CNPs showed complete degradation after 168 h of incubation in SBF in comparison with the image obtained at 0 h (Fig. 6E and F). On the other hand, Si-P particles showed a substantial increment in hydrodynamic diameter to ∼2083 nm in SBF from ∼989 nm when studied in aqueous medium. This demonstrated the loss of stability of Si-P particles in SBF. The maximum reduction of the diameter (∼1045 nm) was noticeable after 72 h of incubation in SBF, which was increased to ∼1293 nm after 168 h of incubation (Fig. 6A). This is probably because of partial degradation and aggregation of Si-P particles as observed in Si-P-CNP particles. SEM based investigations demonstrated a lesser extent of reduction of Si-P particle diameter after 168 h of incubation in SBF (Fig. 6B and C). This observation confirms the superior biodegradability of Si-P-CNP particles over Si-P particles.
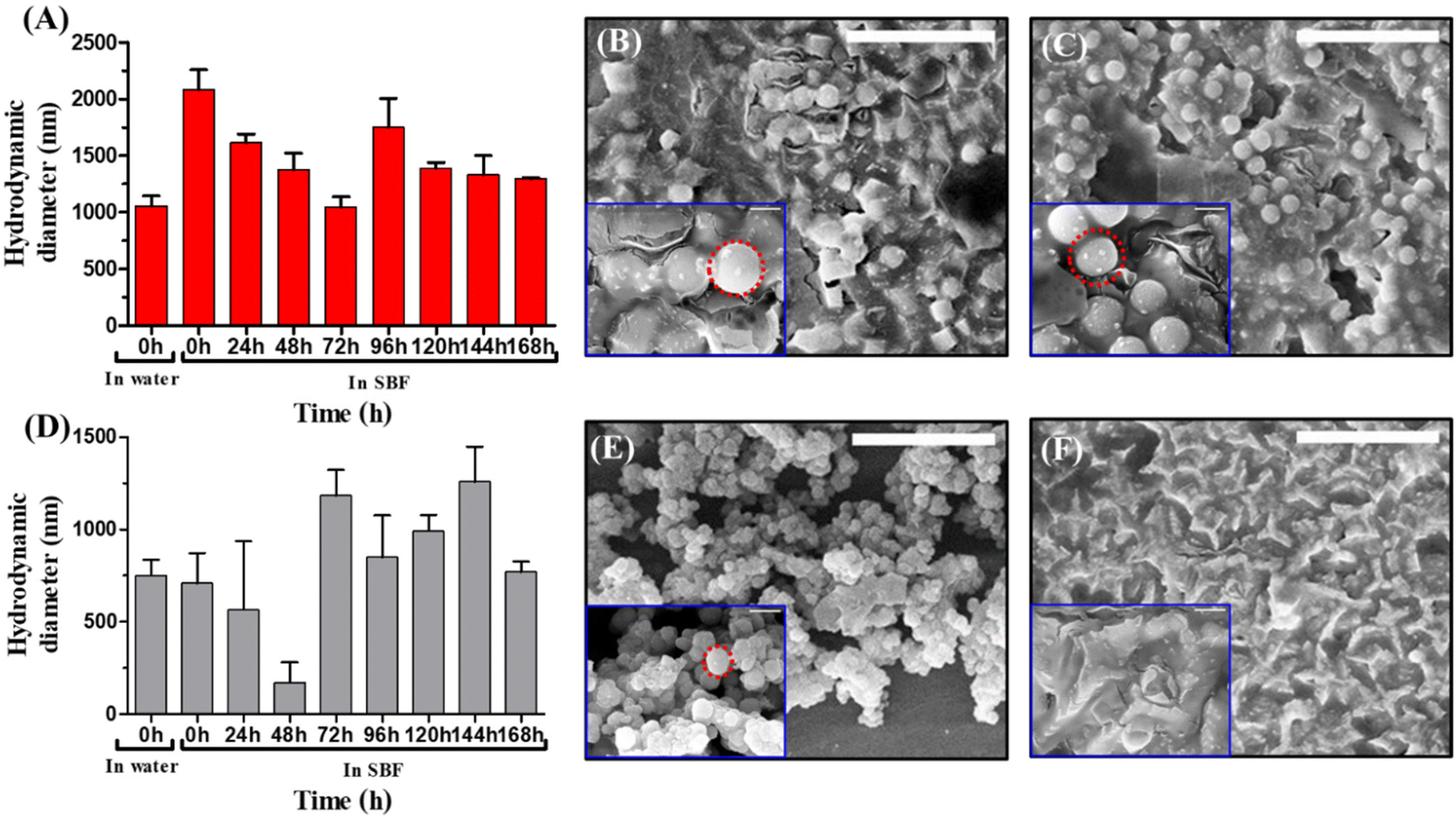 | ||
| Fig. 6 Biodegradability of silica particles in simulated body fluid. (A) Comparison of the hydrodynamic diameters of Si-P particles in water and SBF. SEM images of Si-P particles after incubation in SBF at (B) 0 h and (C) 168 h. (D) Comparison of hydrodynamic diameters of Si-P-CNP particles in water and SBF. SEM images of Si-P-CNP particles after incubation in SBF at (E) 0 h and (F) 168 h. Red circles indicate the sizes of the particles. Scale bar = 10 μm in the main figures and scale bar in = 1 μm in insets. | ||
Conclusions
In summary, this study describes the state of the art of preparing carbonized porous silica particles to improve their biological applications. Though porous silica particles have been utilized for many years in multiple applications, issues with their biodegradability and triggering cellular signaling pathways through forming protein coronas were a matter of concern for a long time. The carbonized porous silica particles were found to have multiple beneficial qualities like enhanced biodegradability after releasing the payloads and non-interacting ability with fibrosis inducing factors like TGF-β1 over unmodified porous silica particles. The developed nondestructive methodology of integrating CNPs was effective enough to maintain an ordered structural arrangement of the silica particles even after carbonization. We believe this facile modification strategy of silica particles will pave the way for a greater number of other applications in physiological systems. Finally, this study also indicates the potential of nanosized carbon particles to bring a combination of multiple functional attributes to silica based porous particles. Investigating the mechanism of CNPs interaction with different silica species during synthesis will be of potential interest to gain more insight for further development of the field.Author contributions
The project was conceptualized by SKM; synthesis of silica particles, DLS, drug loading studies, and FTIR spectroscopy, BET and TEM characterization were performed by AD; zeta potential, XRD, cell viability and confocal studies, mucous entrapment assay, macrophage interaction assay, silicosis induction assay and biodegradation studies were performed by NC whereas confocal studies for synthesized silica particles were performed by both AD and NC. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Overall, NC and AD have contributed equally to this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the help of Ms Shraddha Singh of BSBE, IIT Kanpur for help in obtaining SEM images and Mr Jay of Advanced Imaging Center, IIT Kanpur for help in performing TEM. This study was funded by IITK. The TOC image was created using BioRender.com.References
- Q. He and J. Shi, J. Mater. Chem., 2011, 21, 5845–5855 RSC.
- G. Bapat, C. Labade, A. Chaudhari and S. Zinjarde, Adv. Colloid Interface Sci., 2016, 237, 1–14 CrossRef CAS PubMed.
- X. Yu and C. T. Williams, Catal. Sci. Technol., 2022, 12, 5765–5794 RSC.
- M. C. Prihatiningsih, T. Ariyanto, E. G. R. Putra, V. Y. Susilo, I. Mahendra and I. Prasetyo, ACS Omega, 2022, 7, 13494–13506 CrossRef CAS PubMed.
- Z. Li, D. Dang, X. Yang, J. Wu, W. Yang, Z. Tian, H. Zhang and W. Liang, ACS Appl. Nano Mater., 2023, 6, 4121–4131 CrossRef CAS.
- N. Plumeré, A. Ruff, B. Speiser, V. Feldmann and H. A. Mayer, J. Colloid Interface Sci., 2012, 368, 208–219 CrossRef PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- A. C. Kresge, M. E. Leonowicz, W. J. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. Kresge, K. Schmitt, C. Chu, D. H. Olson, E. Sheppard and S. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- J. Tu, A. L. Boyle, H. Friedrich, P. H. Bomans, J. Bussmann, N. A. Sommerdijk, W. Jiskoot and A. Kros, ACS Appl. Mater. Interfaces, 2016, 8, 32211–32219 CrossRef CAS PubMed.
- H. J. Dong and J. D. Brennan, J. Mater. Chem., 2012, 22, 13197–13203 RSC.
- J. Maisch, F. Jafarli, T. Chassé, F. Blendinger, A. Konrad, M. Metzger, A. J. Meixner, M. Brecht, L. Dähne and H. A. Mayer, Chem. Commun., 2016, 52, 14392–14395 RSC.
- N. Chatterjee and S. K. Misra, ACS Appl. Mater. Interfaces, 2023, 15, 27533–27547 CrossRef CAS PubMed.
- A. Naik, K. Kumar, N. Chatterjee and S. K. Misra, ACS Appl. Bio Mater., 2022, 6, 288–295 CrossRef PubMed.
- A. Steel, S. W. Carr and M. W. Anderson, J. Chem. Soc., Chem. Commun., 1994, 1571–1572 RSC.
- H. Ahmed, S. S. Gomte, A. Prabakaran, M. Agrawal and A. Alexander, J. Drug Delivery Sci. Technol., 2022, 103729 CrossRef.
- S. Rovani, J. J. Santos, P. Corio and D. A. Fungaro, ACS Omega, 2018, 3, 2618–2627 CrossRef CAS PubMed.
- F. Yan, J. Jiang, X. Chen, S. Tian and K. Li, Ind. Eng. Chem. Res., 2014, 53, 11884–11890 CrossRef CAS.
- R. Dubey, Y. Rajesh and M. More, Mater. Today: Proc., 2015, 2, 3575–3579 CAS.
- H. El Rassy and A. Pierre, J. Non-Cryst. Solids, 2005, 351, 1603–1610 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- A. Dohare, S. Sudhakar, B. R. Brodbeck, A. Mukherjee, M. Brecht, A. Kandelbauer, E. Schäffer and H. A. Mayer, Langmuir, 2021, 37, 13460–13470 CrossRef CAS PubMed.
- S. Banerjee and A. R. Chakravarty, Acc. Chem. Res., 2015, 48, 2075–2083 CrossRef CAS PubMed.
- A. Safavy, K. P. Raisch, S. Mantena, L. L. Sanford, S. W. Sham, N. R. Krishna and J. A. Bonner, J. Med. Chem., 2007, 50, 6284–6288 CrossRef CAS PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- M. Wu, Q. Meng, Y. Chen, L. Zhang, M. Li, X. Cai, Y. Li, P. Yu, L. Zhang and J. A. M. Shi, Adv. Mater., 2016, 28, 1963–1969 CrossRef CAS PubMed.
- Y. Song, L. Cai, Z. Tian, Y. Wu and J. Chen, ACS Omega, 2020, 5, 15083–15094 CrossRef CAS PubMed.
- J. Wang, Y. Wang, Q. Liu, L. Yang, R. Zhu, C. Yu and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 26511–26523 CrossRef CAS PubMed.
- N. S. Elbialy, S. F. Aboushoushah, B. F. Sofi and A. Noorwali, Microporous Mesoporous Mater., 2020, 291, 109540 CrossRef CAS.
- R. Kotcherlakota, A. K. Barui, S. Prashar, M. Fajardo, D. Briones, A. Rodríguez-Diéguez, C. R. Patra and S. Gómez-Ruiz, Biomater. Sci., 2016, 4, 448–459 RSC.
- N. M. Khalil, T. C. F. do Nascimento, D. M. Casa, L. F. Dalmolin, A. C. de Mattos, I. Hoss, M. A. Romano and R. M. Mainardes, Colloids Surf., B, 2013, 101, 353–360 CrossRef CAS PubMed.
- S. S. Hettiarachchi, S. P. Dunuweera, A. N. Dunuweera and R. G. Rajapakse, ACS Omega, 2021, 6, 8246–8252 CrossRef CAS PubMed.
- K. Pan, Q. Zhong and S. J. Baek, J. Agric. Food Chem., 2013, 61, 6036–6043 CrossRef CAS PubMed.
- N. Hao, K. W. Jayawardana, X. Chen and M. Yan, ACS Appl. Mater. Interfaces, 2015, 7, 1040–1045 CrossRef CAS PubMed.
- S. K. Misra, I. Srivastava, I. Tripathi, E. Daza, F. Ostadhossein and D. Pan, J. Am. Chem. Soc., 2017, 139, 1746–1749 CrossRef CAS PubMed.
- S. Chahal, J.-R. Macairan, N. Yousefi, N. Tufenkji and R. Naccache, RSC Adv., 2021, 11, 25354–25363 RSC.
- S. Rajoriya, S. Bargole and V. K. Saharan, Ultrason. Sonochem., 2017, 34, 183–194 CrossRef CAS PubMed.
- S. Kundu, N. Chatterjee, S. Chakraborty, A. Gupta, D. Goswami and S. K. Misra, Spectrochim. Acta, Part A, 2022, 270, 120778 CrossRef CAS PubMed.
- I. Tripathi, S. K. Misra, F. Ostadhossein, I. Srivastava and D. Pan, ACS Appl. Mater. Interfaces, 2018, 10, 37886–37897 CrossRef CAS PubMed.
- R. Kotcherlakota, A. K. Barui, S. Prashar, M. Fajardo, D. Briones, A. Rodríguez-Diéguez, C. R. Patra and S. Gómez-Ruiz, Biomater. Sci., 2016, 4, 448–459 RSC.
- R. K. Gangwar, G. B. Tomar, V. A. Dhumale, S. Zinjarde, R. B. Sharma and S. Datar, J. Agric. Food Chem., 2013, 61, 9632–9637 CAS.
- V. S. Bollu, A. K. Barui, S. K. Mondal, S. Prashar, M. Fajardo, D. Briones, A. Rodriguez-Dieguez, C. R. Patra and S. Gómez-Ruiz, Mater. Sci. Eng., C, 2016, 63, 393–410 CrossRef CAS PubMed.
- A. R. Clementino, G. Pellegrini, S. Banella, G. Colombo, L. Cantù, F. Sonvico and E. Del Favero, Mol. Pharm., 2021, 18, 3132–3146 CrossRef CAS PubMed.
- Z. Zhou-Suckow, J. Duerr, M. Hagner, R. Agrawal and M. A. Mall, Cell Tissue Res., 2017, 367, 537–550 CrossRef CAS PubMed.
- D. F. Rogers and P. J. Barnes, Ann. Med., 2006, 38, 116–125 CrossRef CAS PubMed.
- E. Prescott, P. Lange and J. Vestbo, Eur. Respir. J., 1995, 8, 1333–1338 CrossRef CAS PubMed.
- D. Zhao, D. Li, X. Cheng, Z. Zou, X. Chen and C. He, ACS Nano, 2022, 16, 11161–11173 CrossRef CAS PubMed.
- D. Kwon, B. G. Cha, Y. Cho, J. Min, E.-B. Park, S.-J. Kang and J. Kim, Nano Lett., 2017, 17, 2747–2756 CrossRef CAS PubMed.
- N. Encinas, M. Angulo, C. Astorga, M. Colilla, I. Izquierdo-Barba and M. Vallet-Regí, Acta Biomater., 2019, 84, 317–327 CrossRef CAS PubMed.
- C. C. Leung, I. T. S. Yu and W. Chen, Lancet, 2012, 379, 2008–2018 CrossRef CAS PubMed.
- S. Zhuravskii, G. Yukina, O. Kulikova, A. Panevin, V. Tomson, D. Korolev and M. Galagudza, Toxicol. Mech. Methods, 2016, 26, 260–269 CrossRef CAS PubMed.
- Z. Wang, C. Wang, S. Liu, W. He, L. Wang, J. Gan, Z. Huang, Z. Wang, H. Wei and J. Zhang, ACS Nano, 2017, 11, 1659–1672 CrossRef CAS PubMed.
- W. Yan, L. Xiaoli, A. Guoliang, Z. Zhonghui, L. Di, L. Ximeng, N. Piye, C. Li and T. Lin, Toxicol. Lett., 2016, 259, 28–34 CrossRef PubMed.
- J. Guo, Z. Yang, Q. Jia, C. Bo, H. Shao and Z. Zhang, Toxicol. Lett., 2019, 300, 59–66 CrossRef CAS PubMed.
- A. C. Tecalco-Cruz, D. G. Ríos-López, G. Vázquez-Victorio, R. E. Rosales-Alvarez and M. Macías-Silva, Signal Transduction Targeted Ther., 2018, 3, 15 CrossRef PubMed.
- L. Xu, W. H. Cui, W. C. Zhou, D. L. Li, L. C. Li, P. Zhao, X. T. Mo, Z. Zhang and J. Gao, J. Cell. Mol. Med., 2017, 21, 1545–1554 CrossRef CAS PubMed.
- Y. Zhang, W. Lu, X. Zhang, J. Lu, S. Xu, S. Chen, Z. Zhong, T. Zhou, Q. Wang and J. Chen, J. Pharmacol. Res., 2019, 147, 104307 CrossRef CAS PubMed.
- E. Choi, D.-K. Lim and S. Kim, J. Colloid Interface Sci., 2020, 560, 416–425 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00123k |
| ‡ These authors have contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |