
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent research progress on building C–N bonds via electrochemical NOx reduction
Shaktiswaran R.
Udayasurian
 and
Tengfei
Li
*
and
Tengfei
Li
*
School of Chemistry and Environment, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: t.li@mmu.ac.uk
First published on 10th January 2024
Abstract
The release of NOx species (such as nitrate, nitrite and nitric oxide) into water and the atmosphere due to human being's agricultural and industrial activities has caused a series of environmental problems, including accumulation of toxic pollutants that are dangerous to humans and animals, acid rain, the greenhouse effect and disturbance of the global nitrogen cycle balance. Electrosynthesis of organonitrogen compounds with NOx species as the nitrogen source offers a sustainable strategy to upgrade the waste NOx into value-added organic products under ambient conditions. The electrochemical reduction of NOx species can generate surface-adsorbed intermediates such as hydroxylamine, which are usually strong nucleophiles and can undergo nucleophilic attack to carbonyl groups to build C–N bonds and generate organonitrogen compounds such as amine, oxime, amide and amino acid. This mini-review summarizes the most recent progress in building C–N bonds via the in situ generation of nucleophilic intermediates from electrochemical NOx reduction, and highlights some important strategies in facilitating the reaction rates and selectivities towards the C–N coupling products. In particular, the preparation of high-performance electrocatalysts (e.g., nano-/atomic-scale catalysts, single-atom catalysts, alloy catalysts), selection of nucleophilic intermediates, novel design of reactors and understanding the surface adsorption process are highlighted. A few key challenges and knowledge gaps are discussed, and some promising research directions are also proposed for future advances in electrochemical C–N coupling.
 Shaktiswaran R. Udayasurian | Shaktiswaran is currently a PhD student at Manchester Metropolitan University under the supervision of Dr Tengfei Li, specializing in the design of electrocatalysts for the electrocatalytic conversion of nitrate, ammonia and CO2. He received his MEng in Materials Science and Engineering with Corrosion from University of Manchester in 2019. He received his MSc in Data Science and Artificial Intelligence from Goldsmiths, University of London in 2020. Previously, he was a research assistant at Agency for Science, Technology and Research Institute of High-Performance Computing and Nanyang Technological University, Singapore from 2020 to 2022. |
 Tengfei Li | Dr Tengfei Li is a Senior Lecturer in Chemistry at Manchester Metropolitan University in the UK. He obtained his PhD degree in Chemistry with Prof. Curtis Berlinguette at the University of British Columbia, Canada. He then moved to the University of Cambridge and joined Prof. Erwin Reisner's lab as a Canadian Banting Postdoc Fellow. His independent lab is currently focused on sustainable electrocatalysis and photocatalysis, including organic electrosynthesis, nitrate reduction, CO2 reduction and plastic degradation. |
1. Introduction
The global environmental and energy crisis due to the combustion of fossil fuels demands a transition in the synthetic industry from dependence on fossil fuels to sustainable energy, e.g., renewable electricity.1,2 Organic electrosynthesis has emerged as a promising strategy as it can enable ambient reaction conditions and drive redox reactions by clean electrons instead of toxic or expensive chemical reagents.3–5 Electrochemical upgrading of pollutants and greenhouse gases into value-added organic products is particularly attractive, because it can not only be driven by clean electricity, but also utilize the environmentally unfriendly wastes as the feedstocks for synthesis.6–8The emission of NOx species, such as nitrate (NO3−), nitrite (NO2−), nitric oxide (NO) and nitrogen dioxide (NO2), has become a serious environmental challenge. For example, the accumulation of NO3− in water due to excessive fertiliser usage and industrial sewage has disturbed the global nitrogen cycle balance.9–11 Although NO3− itself is not dangerous to human beings, NO3− can be easily reduced to NO2−, which is toxic as it can cause damages of red blood cells.9 Furthermore, the emission of gaseous nitrogen oxide compounds has also significantly contributed to the global warming effect: the greenhouse effect of N2O is 265 times that of CO2.12 On the other side, organonitrogen compounds, such as urea, amide, amino acid and oxime, are very important in agricultures and polymer and pharmaceutical industries. Therefore, it is desirable to upgrade waste NOx species into value-added organonitrogen compounds, which can enable the removal of environmental pollutants and the synthesis of high-value products simultaneously.
Electrochemical NOx reduction reaction (NOxRR) offers a sustainable method to convert the NOx species into ammonia (NH3).13–30 Compared to the electrochemical reduction of N2 into NH3, NOxRR can utilize the activated N species in environment and avoid the requirement of breaking the stable N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, and thus can make NH3 at higher faradaic efficiency (FE) and current density. The state-of-the-arts NOxRR systems can produce NH3 with ∼90% FE at ∼1 A cm−2.31–34 Although NH3 is a very important chemical building block, it is still desirable to make nitrogen products via electrochemical NOxRR that bear higher economic values than NH3, such as organonitrogen compounds.
N bond, and thus can make NH3 at higher faradaic efficiency (FE) and current density. The state-of-the-arts NOxRR systems can produce NH3 with ∼90% FE at ∼1 A cm−2.31–34 Although NH3 is a very important chemical building block, it is still desirable to make nitrogen products via electrochemical NOxRR that bear higher economic values than NH3, such as organonitrogen compounds.
Recently, a series of surface-adsorbed reaction intermediates have been identified in the electrochemical NOxRR process, such as *NH2OH and *NH2. These intermediates are typically stronger nucleophiles compared to the final product of NOxRR (i.e., NH3), and therefore can be coupled with chemical reactions and undergo nucleophilic attack to carbonyl species (e.g. ketone, aldehyde, CO2, CO) to build C–N bonds in a wide range of organic products (Fig. 1). These C–N coupling reactions are usually more difficult to occur between NH3 and the carbonyl species (i.e. typically require high temperatures/pressures).35 Furthermore, these highly active N-containing intermediates are unstable over long period of time. For example, the transportation and storage of concentrated NH2OH are challenged by the risk of explosion.36 Therefore, the electrochemical NOxRR also enables an in situ generation of nucleophilic N species on demand for the C–N coupling reaction. Moreover, compared to the traditional chemical methods, the electrosynthesis of organonitrogen compounds via NOxRR offers a more sustainable synthesis strategy, as it can be performed under ambient conditions (e.g. room temperature and pressure, aqueous media, without expensive or toxic catalyst) and can be coupled with clean electrons to replace redox reagents.
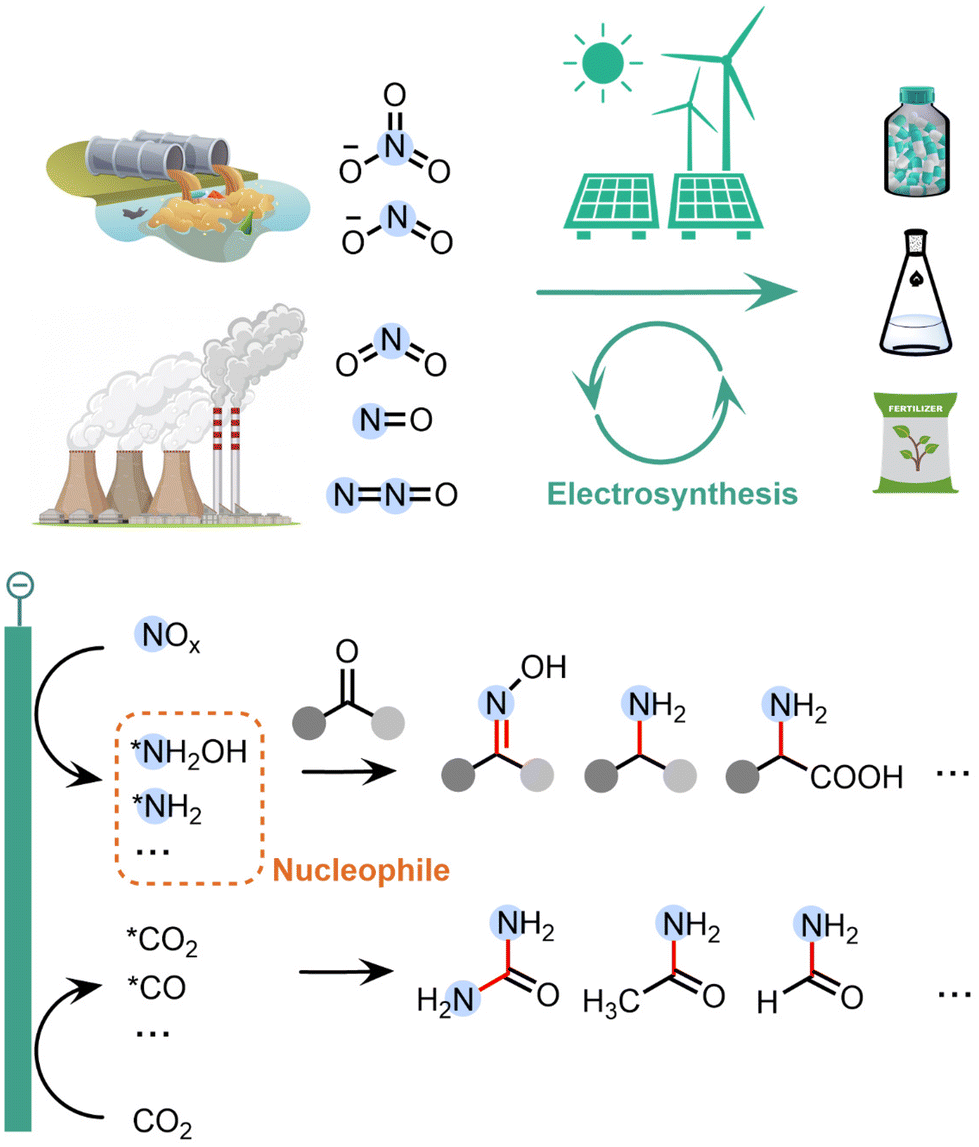 | ||
| Fig. 1 Electrosynthesis of value-added organonitrogen products from gas- and liquid-phase NOx compounds. The electroreduction of NOx can generate a reaction intermediate that can undergo nucleophilic attack to a carbonyl group, including organic ketone/aldehyde and CO2 reduction intermediates. | ||
However, building organic C–N bonds by electrochemical NOxRR is challenged by the generation of a series of by-products, such as N2, NH3 and H2, as well as the hydrogenation products of the organic species, which all decrease the selectivity towards the desired C–N coupling products. In this minireview, we will highlight some emerging strategies to facilitate the electrochemical C–N bond formation via NOxRR, such as choosing a suitable nucleophile for C–N coupling, tuning the composition and morphology of the electrocatalysts, making catalysts with nano-/atomic-scale and single-atom structures, designing novel reactors, and controlling surface adsorption by understanding the reaction mechanism, etc. As this area is developing quite rapidly, this minireview does not aim to provide a complete compilation of publications (readers may refer to other reviews to get a comprehensive understanding of this area).37,38 Instead, we will first give a brief summary of building C–N bonds by electrochemical NOxRR, and then mainly focus the most recent reports in this area in the last two years, and summarize the main challenges and emerging opportunities to develop more efficient NOxRR systems to drive electrosynthesis of organonitrogen compounds.
2. Electrochemical C–N bond formation systems
2.1. Initial explorations on electrochemical C–N coupling: which N species should be used to undergo nucleophilic attack to carbonyl species?
The first electrocatalytic C–N coupling process was demonstrated in 2019 by Jiao's group.39 During the electrochemical reduction of CO on a commercial-available Cu nanoparticle electrocatalyst, NH3 was added into the electrolyte, which can undergo a C–N coupling reaction to produce acetamide with a FE of ∼40% at 300 mA cm−2 and an operating potential at −0.68 V vs. reversible hydrogen electrode (VRHE). The mechanistic study showed that the C–N coupling process occurs through the nucleophilic attack of NH3 to the *C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO intermediate generated in CO electroreduction to make acetamide, which is in competition with the nucleophilic attack of OH− to the same intermediate to generate acetate (Fig. 2a). This strategy can be extended to produce other amide compounds by co-electrolysis of CO with different amines, and an increase for the selectivity towards amide was observed when the nucleophilicity of the N species increased, i.e., NH3 < CH3NH2 < CH3CH2NH2 < (CH3)2NH (Fig. 2b). It is also notable that adding NH3 into the electrolysis of CO2 (instead of CO) did not generate any amide. Later Kornienko et al. showed that the co-electrolysis of NH3 and CO2 can generate a small amount of formamide and acetamide (FE = 0.4% and 10%, respectively).40 These results proved the importance of choosing a suitable nucleophile (i.e., N species) and electrophile (i.e., carbonyl species) with high reactivities for the C–N coupling reaction.
CO intermediate generated in CO electroreduction to make acetamide, which is in competition with the nucleophilic attack of OH− to the same intermediate to generate acetate (Fig. 2a). This strategy can be extended to produce other amide compounds by co-electrolysis of CO with different amines, and an increase for the selectivity towards amide was observed when the nucleophilicity of the N species increased, i.e., NH3 < CH3NH2 < CH3CH2NH2 < (CH3)2NH (Fig. 2b). It is also notable that adding NH3 into the electrolysis of CO2 (instead of CO) did not generate any amide. Later Kornienko et al. showed that the co-electrolysis of NH3 and CO2 can generate a small amount of formamide and acetamide (FE = 0.4% and 10%, respectively).40 These results proved the importance of choosing a suitable nucleophile (i.e., N species) and electrophile (i.e., carbonyl species) with high reactivities for the C–N coupling reaction.
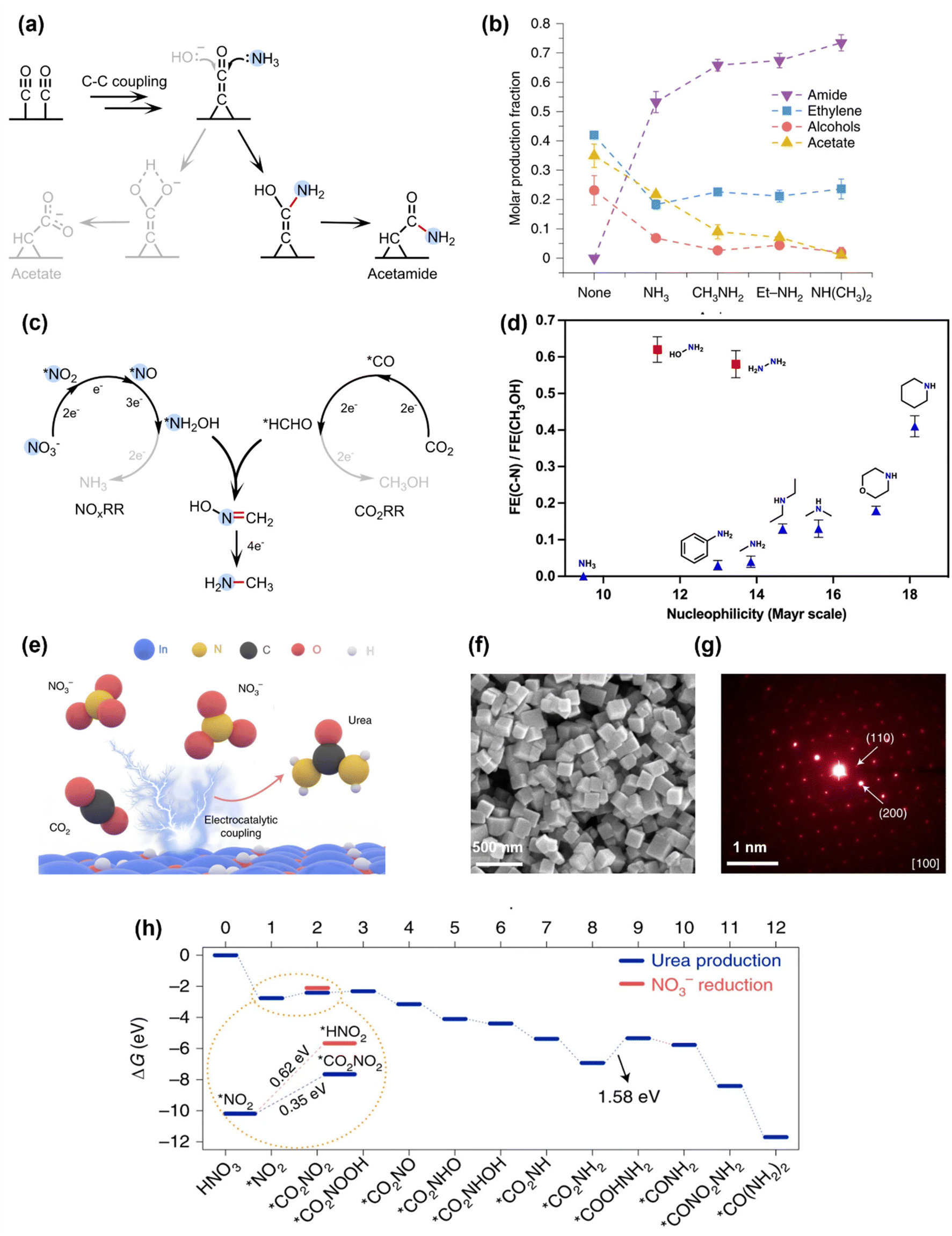 | ||
| Fig. 2 Initial studies on electrochemical C–N coupling. (a) and (b) Formation of amides by adding NH3 or amines into the electrochemical CO reduction systems.39 Copyright 2019, Springer Nature. (c) Electrochemical co-reduction of NO3− and CO2 to form methylamine via the coupling of *NH2OH and *HCHO intermediates.41 Copyright 2021, Springer Nature. (d) Electrochemical C–N bond formation by coupling CO2RR with different N nucleophiles.42 Copyright 2021, American Chemical Society. (e) Electrochemical co-reduction of NO3− and CO2 to produce urea on an In(OH)3 catalyst. (f) and (g) Structural characterizations of the In(OH)3 catalyst by SEM and SAED. (h) Theoretical calculations for the free energy changes during the reaction, where the key C–N coupling step is thermodynamically more favorable than NO3− reduction.43 Copyright 2021, Springer Nature. | ||
In order to increase the nucleophilicity of the N species, different N sources have been investigated for electrochemical C–N bond formation, including NH3, N2, and NOx species. The intermediates generated in electrochemical NOxRR, such as *NH2OH and *NH2, are particularly attractive as they are more nucleophilic than NH3. One of the first successful C–N coupling reactions via electrochemical NOxRR was reported in 2021 by Wang and coworkers,41 where the co-reduction of CO2 and NO3− on a cobalt molecule catalyst (CoPc–NH2, previously reported to drive CO2RR to methanol by the same group44) can generate methylamine (CH3NH2) with a FE of 13% at ∼25 mA cm−2 and an operating potential of −0.92VRHE. The key C–N bond formation step was found to be the nucleophilic addition–elimination reaction between the NH2OH intermediate generated by NOxRR and the formaldehyde (HCHO) intermediate generated by CO2 reduction reaction (CO2RR) to form formaldoxime (H2CNOH), which is then further reduced to CH3NH2 (Fig. 2c). Later, Wang's group also investigated coupling CO2RR with different N nucleophiles (e.g., NH2OH, NH2NH2, primary and secondary amines) and demonstrated that the C–N coupling efficiency was mainly determined by the nucleophilicity of the N species (Fig. 2d).42 These works highlighted that the in situ generated *NH2OH in NOxRR process is a promising nucleophile to build C–N bond, which can be used to make a wide range of organonitrogen compounds (e.g., amine, oxime, amino acid) in following works as described in section 2.2.
Another important organonitrogen compound that can be produced by electrochemical NOxRR is urea, which accounts for two thirds of the global fertilizer market. Urea is mainly produced via the Bosch–Meiser process from CO2 and NH3, which requires harsh reaction conditions and is energy intensive (150–200 °C, 150–250 bar, 7.1 GJ per ton).35 Electrochemical co-reduction of CO2 and NOx can provide an alternative method to produce urea under ambient conditions. In 2021, Yu et al. reported that CO2 and NO3− can be reduced by a single-crystal-facet In(OH)3 electrocatalyst to make urea with a FE of 53% at 0.6 mA cm−2 and −0.6VRHE (Fig. 2e).43 Despite of the relatively low current density, the nitrogen and carbon selectivities towards urea are high (83% and 100%, respectively), which are quite remarkable considering that NOxRR and CO2RR can usually generate a series of by-products such as NH3, N2, CO and formate. The high selectivities were attributed to the low energy barrier for the C–N coupling step on the (100) facets of the In(OH)3 catalyst. The single-crystal-facet In(OH)3 electrocatalyst with well-defined (100) facets via a reported solvothermal approach45 (the structural characterizations of the catalyst are shown in Fig. 2f and g). Computational studies (Fig. 2h) revealed that C–N bond was formed between the surface adsorbed *CO2 and *NO2 species and the energy barrier for forming *CO2NO2 (0.35 eV) was lower than the protonation of *NO2 to *HNO2 (0.62 eV), thus favoring the C–N coupling reaction over further NOxRR. Although *NO2 is not as nucleophilic as the more reduced form of N species (e.g., *NH2OH, *NH2), the formation of C–N bond at an early stage of *NO2 can prevent the by-product generation of NOxRR and CO2RR, and therefore enhance the selectivities towards urea, highlighting that nucleophilicity is not the only consideration when designing an in situ generated NOxRR intermediate for C–N coupling.
A summary of key works in building C–N bond by electrochemical NOxRR is shown in Table 1 as below, which highlights the electrocatalyst, C and N sources, C–N coupling product, reaction conditions and electrochemical performance. Reports that utilize N2 as the nitrogen source46–49 are not listed in the table, as those reactions do not involve NOxRR processes. Next, we will focus on the most recent research that couple NOxRR with organic compounds (section 2.2) and CO2RR (or CO2-derived molecules such as CO and formate; section 2.3).
| Catalyst | C source | N source | Additional electrolyte | Product | Yield | FE | Potential | Partial current density | Cell configuration | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Cu | CO | NH3 | 1 M KOH | Acetamide | — | 38% | −0.68VRHE | 114 mA cm−2 | Flow cell (3-electrode) | 39 |
| CoPc–NH2 | CO2 | KNO3 (0.5 M) | 0.1 M KHCO3 | Methylamine | — | 13% | −0.92VRHE | 3.3 mA cm−2 | H-cell (3-electrode) | 41 |
| In(OH)3 | CO2 | KNO3 (0.1 M) | 0.1 M KNO3 | Urea | — | 53% | −0.6VRHE | 0.3 mA cm−2 | H-cell (3-electrode) | 43 |
| Cu–S | Cyclohexanone (0.01 M) | NaNO2 (0.1 M) | 0.5 M phosphate buffer (pH 5.8) | Cyclohexanone oxime | 92% | 26% | −0.35VRHE | 26 mA cm−2 | H-cell (3-electrode) | 50 |
| Fe | Cyclohexanone (0.1 M) | KNO3 (1 M) | 0.5 M K2CO3 | Cyclohexanone oxime | 100% | 20% | −0.3VRHE | — | H-cell (3-electrode) | 51 |
| 100% | 20% | 2.34 V (cell) | 100 mA cm−2 | Flow cell (2-electrode) | ||||||
| Zn–Cu | Cyclohexanone (0.025 M) | KNO3 (0.1 M) | 0.5 M phosphate buffer (pH 7) | Cyclohexanone oxime | 97% | 27% | −0.82VRHE | 27 mA cm−2 | H-cell (3-electrode) | 52 |
| Al-based MOFs | Pyridine aldehyde (5 mM) | NO | 0.1 M KOH | Pyridine oxime | 92% | 50% | ∼−0.1VRHE | 1.6 mA cm−2 | H-cell (3-electrode) | 53 |
| Pd–Cu | Pyruvic acid (0.05 M) | KNO3 (1 M) | 0.2 M KH2PO4 + 1 M KOH (pH 5) | Alanine | 55% | ∼50% | −0.3VRHE | ∼10 mA cm−2 | H-cell (3-electrode) | 54 |
| — | ∼30% | 1.7 V (cell) | ∼100 mA cm−2 | Flow cell (2-electrode) | ||||||
| Ag | Pyruvic acid (unspecified concentration) | NO | 0.1 M NaOH + 0.9 M NaClO4 (with H2SO4 for reactor 2) | Alanine | 90% | 70% | ∼3 V (cell) | 100 mA cm−2 | Two separate flow cells | 55 |
| Co–Fe | α-Ketoisocaproic acid (0.04 M) | NO | 0.1 M HCl | Leucine | 57% | 32% | −0.7VRHE | 5.4 mA cm−2 | H-cell (3-electrode) | 56 |
| Cu | CO2 | KNO3 (0.1 M) | 1 M KOH | Urea | — | 52% | −0.41VRHE | 115 mA cm−2 | Flow cell (3-electrode) | 57 |
| Zn–Cu | CO2 | KNO3 (1000 ppm) | 0.1 M KHCO3 | Urea | — | 75% | −0.8VRHE | ∼7 mA cm−2 | Flow cell (3-electrode) | 58 |
| Pd4Cu1 | CO2 | KNO3 (0.1 M) | 0.1 M KHCO3 | Urea | — | 66% | −0.6VRHE | ∼5 mA cm−2 | Flow cell (3-electrode) | 59 |
| Cu–Hg | Oxalic acid (0.25 M) | KNO3 (0.25 M) | 15 wt% H2SO4 | Glycine | 46% | 43% | −1.2VRHE | 39 mA cm−2 | H-cell (3-electrode) | 60 |
| Cu | Formic acid (0.2 M) | NaNO2 (0.02 M) | 0.5 M NaOH | Formamide | 90% | 30% | −0.6VRHE | ∼3 mA cm−2 | H-cell (3-electrode) | 61 |
| Ru–Cu | CO | KNO2 (1 M) | 1 M KOH | Formamide | — | 46% | −0.5VRHE | ∼5 mA cm−2 | H-cell (3-electrode) | 62 |
| Te–Pd | CO2 | KNO2 (0.01 M) | 0.1 M KHCO3 | Urea | — | 12% | −1.1VRHE | 0.8 mA cm−2 | H-cell (3-electrode) | 63 |
| ZnO | CO2 | NaNO2 (0.1 M) | 0.2 M NaHCO3 | Urea | — | 23% | −0.79VRHE | 5.3 mA cm−2 | H-cell (3-electrode) | 64 |
| TiO2–Nafion | CO2 | KNO3 (0.1 M) | — | Urea | — | 40% | −0.5VRHE | 0.8 mA cm−2 | H-cell (3-electrode) | 65 |
| Cu–TiO2 | CO2 | KNO2 (0.02 M) | 0.2 M KHCO3 | Urea | — | 43% | −0.4VRHE | 3.2 mA cm−2 | H-cell (3-electrode) | 66 |
| Cu | CO2 | KNO3 (0.1 M) | — | Ethylamine | — | 0.3% | −1.0VRHE | 0.3 mA cm−2 | H-cell (3-electrode) | 67 |
2.2. Couple electrochemical NOxRR with organic compounds: utilizing *NH2OH as the nucleophile
The strong nucleophilicity of the *NH2OH intermediate generated in NOxRR offers an ideal platform to undergo the C–N coupling process with a ketone/aldehyde to make an oxime. One of the most important oxime compounds is cyclohexanone oxime, as it can undergo Beckmann rearrangement to make caprolactam, the monomer for Nylon-6.68 The NH2OH required for cyclohexanone oxime production is typically generated via chemical hydrogenation of NOx on palladium catalysts69 or oxidation of ammonia (NH3) by O2/H2O2.68,70,71 However, these chemical processes generally require harsh reaction conditions (e.g., high temperatures and/or pressures, strong acidic/alkaline solutions) and are also challenged by the safety concerns for the downstream transportation and storage of NH2OH.36 In contrast, electrochemical NOxRR provides an attractive alternative strategy to generate NH2OH in situ as needed to produce cyclohexanone oxime (Fig. 3a).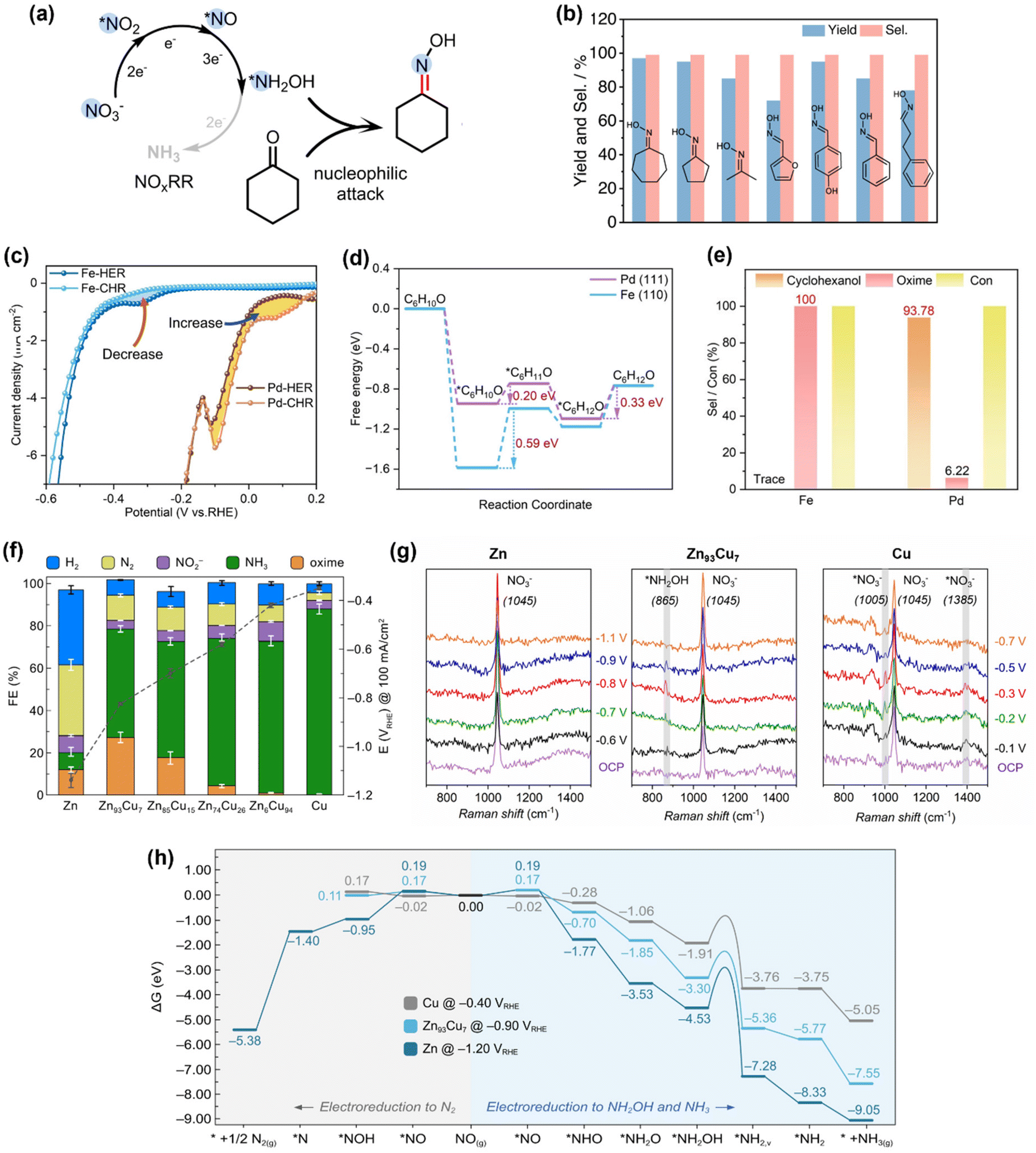 | ||
| Fig. 3 Electrosynthesis of cyclohexanone oxime via the *NH2OH intermediate generated in NOxRR. (a) Schematic illustration for the electrosynthesis process. (b) Reaction scope for the synthesis of oximes compounds by electrochemical NOxRR.50 Copyright 2023, Springer Nature. (c)–(e) Avoiding the production of cyclohexanol in electrosynthesis of cyclohexanone oxime, including the cyclic voltammetry (c) for cyclohexanone hydrogenation reaction (CHR), energy diagram (d) for CHR process, and selectivities towards cyclohexanol and oxime (e) on Fe and Pd electrocatalysts.51 Copyright 2023, Wiley-VCH GmbH. (f)–(h) Synthesis of cyclohexanone oxime via electrochemical NOxRR on a series of Zn–Cu alloy catalysts, including the electrochemical performance of different Zn/Cu catalysts (f), in situ Raman for Zn, Zn93Cu7 and Cu (g), and theoretical calculations (h) to explain the product selectivities on different Zn/Cu catalysts.52 Copyright 2023. | ||
Recently, Zhang and coworkers demonstrated the electrosynthesis of cyclohexanone oxime by reduction of aqueous NO2− on a Cu–S electrocatalyst.50 The *NH2OH intermediate made in electrochemical NOxRR can undergo a subsequent chemical reaction with cyclohexanone to produce oxime with a FE of 26% at 100 mA cm−2 and >90% yield (carbon selectivity). The Cu–S electrocatalyst was prepared by a sulfuration–electroreduction method that was previously developed by the same group.72 A techno-economic analysis (TEA) was also performed and confirmed that the electrosynthesis strategy is profitable. Reaction intermediates were confirmed by in situ attenuated total reflection infrared (ATR-IR) spectroscopy, including *NO, *NH2OH, *NH2, and *CN, demonstrating the key roles of the surface-adsorbed intermediates. Moreover, this electrosynthesis strategy can also be extended to a variety of ketones and aldehydes and produce the corresponding oximes with high yields and carbon selectivities (Fig. 3c). It is notable that the aromatic substrates that are usually quite active for electrochemical hydrogenation into alcohols (e.g. furfural, benzaldehyde) can still bear >90% selectivities towards oximes, which can potentially be explained by the low loading of the organic substrate (0.01 M ketone/aldehyde). This work demonstrates a promising strategy to make oximes with NO2− as the N source. Li's group has also reported a similar method to synthesize a wide range of pyridine oximes via the *NH2OH intermediate generated in NOxRR.73
In the meantime, the groups of Zou and Wang have also developed an electrosynthesis method for cyclohexanone oxime by utilizing the in situ generated *NH2OH in NOxRR.51 Compared to Zhang's work, they used a 10-times higher concentration of cyclohexanone (0.1 M) and therefore the side reaction of cyclohexanone reduction into cyclohexanol must be considered. By comparing two catalyst materials (Fe and Pd, which were prepared via an electrical explosion method and an ethylene glycol reduction approach, respectively), they demonstrated that a good electrocatalyst for this conversion should have: (1) a strong adsorption of cyclohexanone, so that the surface-enriched cyclohexanone and *NH2OH intermediate can form C–N bonds on the catalyst surface; and (2) a poor reduction activity for cyclohexanone to avoid the formation of the cyclohexanol byproduct. The cyclic voltammetry (CV, Fig. 3c) showed that after adding cyclohexanone into the electrolyte (without NOx species), the Pd electrode exhibited a higher current which indicates a strong electrochemical activity for cyclohexanone reduction reaction (CHR), while the Fe electrode showed a lower current which suggests suppressed CHR activity. Theoretical calculations for the CHR processes on Fe and Pd (Fig. 3d) revealed that the adsorptions of cyclohexanone on Fe surface and Pd surface are both thermodynamically favorable, while the first hydrogenation step is endergonic (and potentially the rate determining step) for both metals. The Gibbs energy change for this hydrogenation step on Fe (+0.59 eV) is much higher than Pd (+0.20 eV), indicating that Fe has a weaker activity for cyclohexanone reduction compared to Pd. The computation results are also consistent the CV data, as Fe has a strong surface adsorption of cyclohexanone but the subsequent hydrogenation of cyclohexanone is difficult, and therefore the surface-adsorbed cyclohexanone would ‘poison’ the Fe catalyst for hydrogen evolution reaction (HER) and decrease the electrochemical current. Meanwhile, the surface-enriched cyclohexanone on Fe can facilitate the nucleophilic reaction with the *NH2OH generated in NOxRR, and therefore Fe exhibited a ∼100% carbon selectivity towards cyclohexanone oxime, whereas Pd mainly form cyclohexanol, the hydrogenation product (Fig. 3e).
Zou and Wang's work highlighted the importance of controlling the surface coverage, particularly, the adsorption of the organic substrate, in electrochemical C–N bond formation. Recently, our group also reported an electrochemical method to produce cyclohexanone oxime via NOxRR and investigated the surface adsorption of NOxRR intermediates.52 We prepared a series of Zn/Cu alloys via electrodeposition and conducted a systematic study on the product selectivities on different Zn/Cu catalysts (Fig. 3f). As the Cu content increased, the NO3RR could be driven at lower potentials and the FE for NH3 was significantly enhanced, but the FEs for cyclohexanone oxime was quite low (<1%). The highest FE for the oxime product was achieved on Zn93Cu7 (FE = 27% at 100 mA cm−2 and −0.82VRHE). These observations can be explained by the trend that the weak surface adsorption of NOxRR intermediates on pure Zn can lead to high potentials required for electroreduction, whereas strong adsorption on a Cu-rich catalyst18 can promote this process at lower potentials but results in further reduction of *NH2OH to NH3 instead of forming oxime. The surface-adsorbed species was analyzed by in situ Raman spectroscopy (Fig. 3g), where pure Zn only showed a Raman peak at 1045 cm−1 for the NO3− in solution,19,74 while Zn93Cu7 exhibited a small peak at 865 cm−1 that can be attributed to the surface-adsorbed *NH2OH intermediate,19,75 providing direct experimental evidence for this surface-adsorbed species. The pure Cu electrocatalyst exhibited two peaks at 1005 cm−1 and 1385 cm−1 corresponding to adsorbed *NO3− species,19,74 indicating that Cu provides a stronger binding for NO3− compared to Zn, which is beneficial for electrochemical NO3RR. Theoretical calculations (Fig. 3h) revealed that pure Cu has a lower kinetic barrier for the electroreduction of *NH2OH to *NH2 compared to pure Zn and Zn93Cu7, and thus favours the further electroreduction of *NH2OH to NH3 (rather than the nucleophilic reaction with ketone to make oxime). Computational results also showed that pure Zn is disadvantaged by the competing NO3RR pathway towards N2 (rather than NH2OH or NH3), which is consistent with the experimental observation that a substantial amount of N2 was produced on Zn (FE ∼ 33%). These works collectively demonstrated that the surface adsorption of both organic substrate and NOxRR intermediates plays a central role in the C–N coupling process.
Following the findings that the *NH2OH generated in NOxRR can be used to produce oxime and the Pd electrocatalyst favors the organic hydrogenation (instead of C–N coupling), Zou and Wang groups next utilized this technique to drive the electrosynthesis of alanine (Ala), an important amino acid, from pyruvic acid (PA) and NO3−.54 The *NH2OH generated in NOxRR can undergo nucleophilic attack to the ketone group in PA to form pyruvate oxime (PO), which needs to be electrochemically hydrogenated to produce Ala (Fig. 4a). In order to suppress the competing reactions, including further reduction of *NH2OH to NH3 and hydrogenation of PA to lactic acid (LA), they developed a Cu–Pd alloy catalyst and drive the one-pot synthesis of Ala (noted as strategy 1). Pure Cu (metal A) can drive the NOxRR and C–N bond formation process to produce PO, but the hydrogenation of PO into Ala is sluggish on pure Cu (Fig. 4b). On the other side, pure Pd (metal B) is more active for hydrogenation reactions and therefore mainly produces LA from PA. To combine the advantages of the two metals, they prepared a Pd–Cu nanobead wires alloy catalyst via a solvothermal method (SEM and TEM images shown in Fig. 4c and d), where a maximum yield at 55% for Ala can be achieved on the Pd1Cu1 alloy catalyst (Fig. 4b). In a similar way, Li and coworkers also demonstrated the electrosynthesis of 13 different amino acids through NOxRR on a Co–Fe alloy electrocatalyst.56
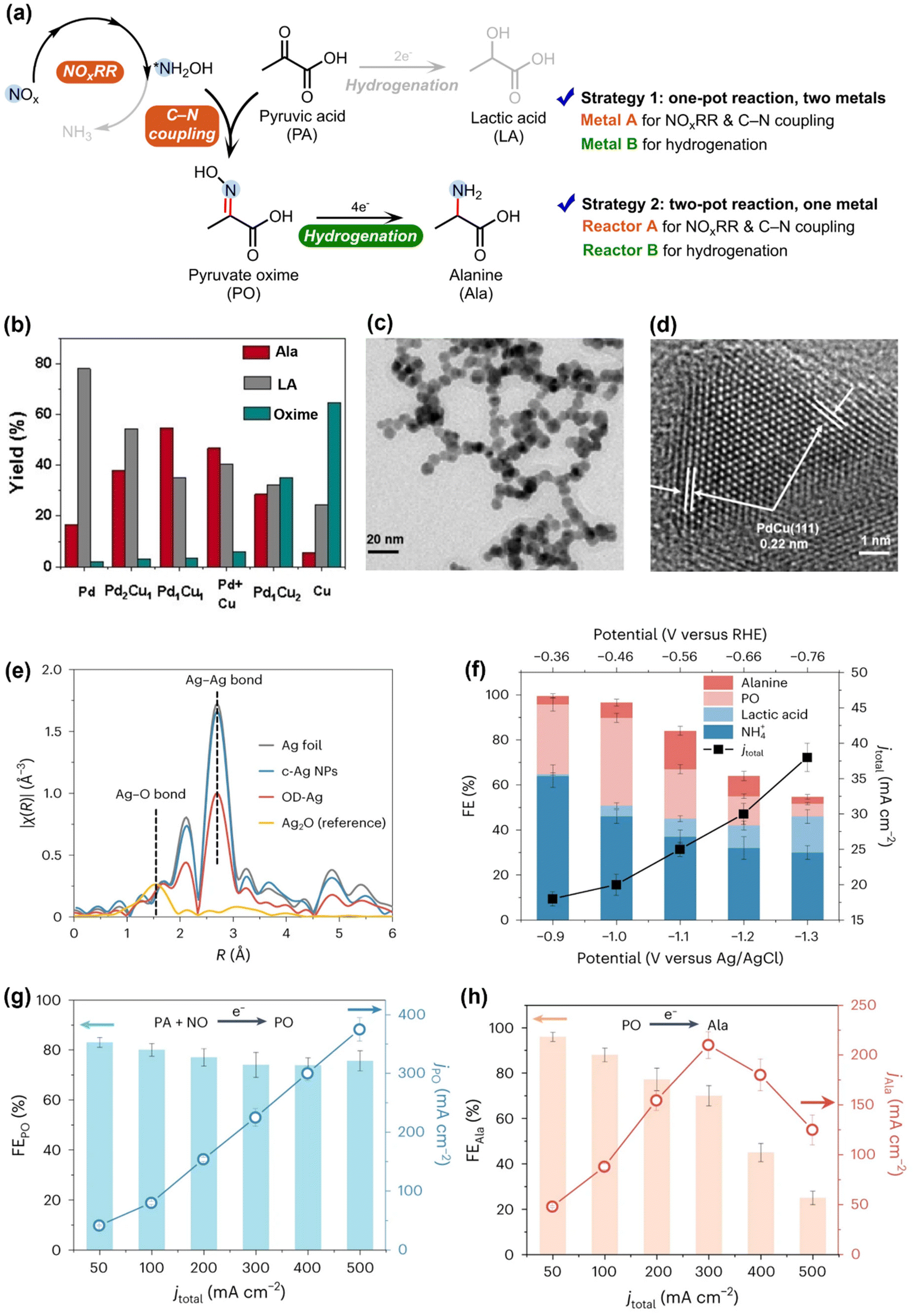 | ||
| Fig. 4 Electrosynthesis of alanine via NOxRR. (a) Reaction pathway for making analine and possible byproducts. Two strategies can be used to improve the selectivity towards analine. (b) Strategy 1, where two metals (Cu and Pd) are used to the two steps of the reaction in a one-pot system. (c) and (d) SEM and TEM images for the Pd1Cu1 alloy catalyst.54 Copyright 2023, Wiley-VCH GmbH. (e)–(h) Strategy 2, where two separate reactors are used to drive the two steps of the reaction with the same OD–Ag catalyst. (e) EXAFS spectra of the OD–Ag catalyst and other Ag materials. (f) Electrochemical performance when the OD–Ag catalyst was used to drive the reaction in one-pot. (g) and (h) Electrochemical performance when the OD–Ag catalyst was used to drive the reaction in two separate reactors.55 Copyright 2023, Springer Nature. | ||
Another strategy to overcome the challenges in making Ala is to spatially decouple the synthesis of PO and the subsequent hydrogenation of PO to Ala, and drive the reaction in a two-pot process. Zhang et al. recently developed an electrosynthesis method to produce Ala from PA and NO with two separate reactors (denoted as strategy 2, Fig. 4e–g), where NOxRR and C–N coupling occur in reactor A, and the PO produced in reactor A is further converted to Ala in reactor B.55 They prepared an oxide-derived Ag (OD–Ag) catalyst via in situ electroreduction to drive both steps. As the shown in the EXAFS spectra (Fig. 4e), the Ag–O bond has been removed in OD–Ag, indicating the full conversion from Ag2O to OD–Ag catalyst, and the smaller Ag–Ag bond in OD–Ag compared to Ag foil also revealed the low-coordination sites, which are also important in the electrocatalysis process. When the OD–Ag catalyst was used to drive the one-pot synthesis of Ala (Fig. 4f), the FE for Ala was quite low (<20%) and a series of byproducts (e.g., NH4+, LA) were formed due the rapid PO formation but the slow PO hydrogenation steps. In contrast, when the OD–Ag catalyst was used in two separate reactors (Fig. 4g and h), both the PO formation (PA + NO → PO, reactor A) and the PO hydrogenation (PO → Ala, reactor B) can be driven with high FEs. The aforementioned works collectively demonstrated the importance of designing complementary bi-metal (strategy 1) or bi-reactor (strategy 2) systems to match the different requirements in PO formation and PO hydrogenation.
2.3. Couple electrochemical NOxRR and CO2RR
Based on the similarities between electrochemical CO2RR and NOxRR, such as surface adsorption, electron transfer and catalyst design, a series of bi-functional catalysts have been developed to enable the co-reduction of CO2 and NOx into small organonitrogen molecules. For example, urea is the most widely studied co-reduction product due to its large market demands as a fertilizer. Following the study by Yu's lab in 2021, where urea is produced with a FE as high as 53% but a relatively low current density at 0.6 mA cm−2 (as described in 2.1),43 researchers have recently significantly improved the FE and current density for the co-reduction of CO2 and NO3− into urea. In 2023, Kwon and coworkers prepared a Cu electrocatalyst with atomic scale spacings (ds) between two facets of Cu, which can facilitate the production of urea (Fig. 5a).57 The atomic-scale spacings were created by an electrochemical lithiation process on the Cu nanoparticles and the size of spacings was controlled by degree of lithiation, as revealed by TEM and STEM in Fig. 5b. The Cu with a ds of 6 Å can achieves a FE for urea of 52% at 220 mA cm−2 and −0.41VRHE, which are significantly higher than the bare Cu without lithiation treatment (Fig. 5c). Computational studies revealed that atomic-scale spacings can efficiently lower the kinetic barriers for C–N coupling (Fig. 5d): the kinetic barriers for CO2 coupling with a variety of possible NOxRR intermediates (*NO, *NOH, *N, *NH, *NH2) are all decreased in the Cu with 6 Å spacing. Recently, Ye's lab has also reported that an atomically-dispersed Cu in Pd catalyst (Pd4Cu1) can convert CO2 and NO3− into urea with a FE of 66% and 436.9 mmol gcat.−1 h−1 at −0.6VRHE, as well as a long cycling stability of 1000 h.59 These works highlighted the importance of controlling the morphology of the electrocatalyst at atomic scale in facilitating the electrochemical urea production.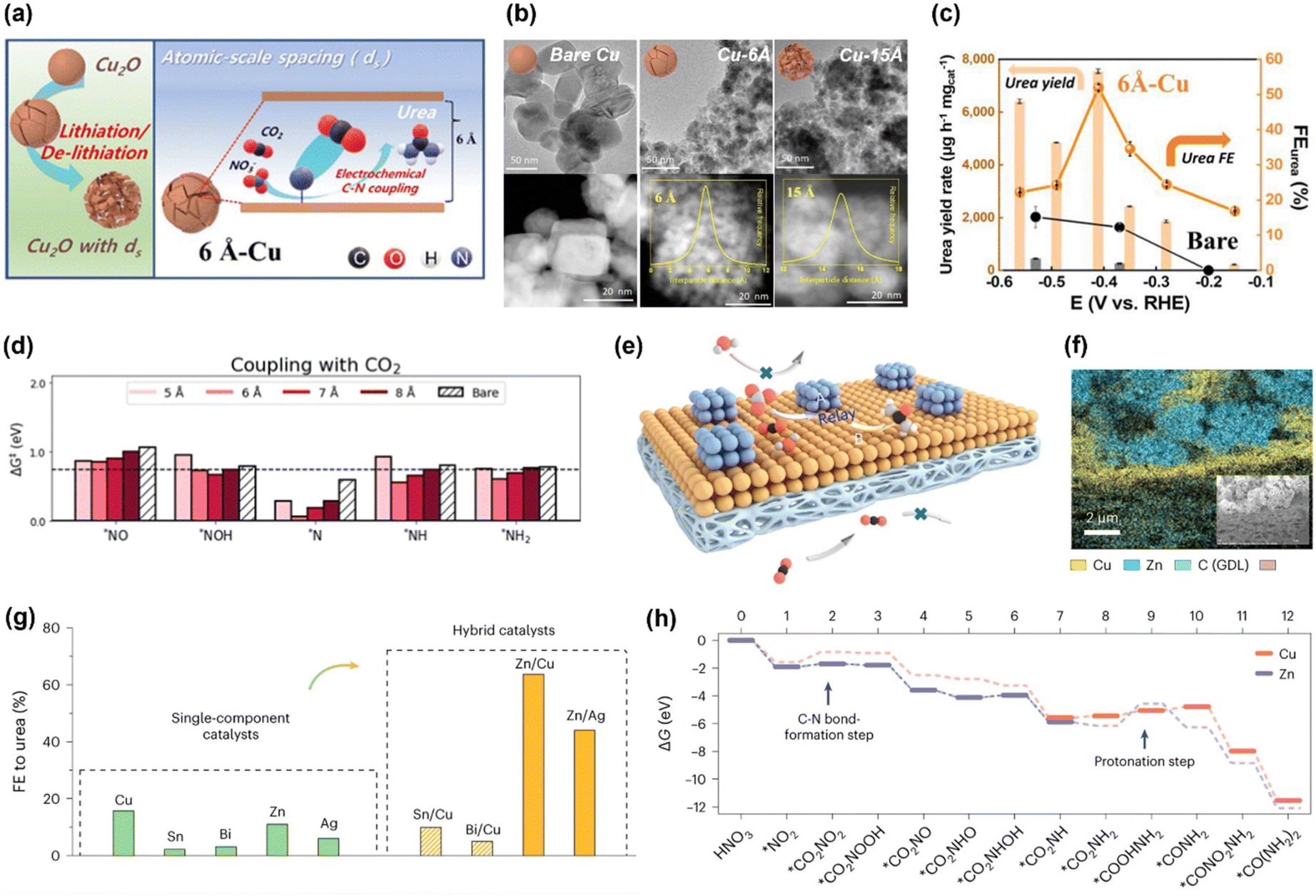 | ||
| Fig. 5 Electrochemical co-reduction of CO2 and NO3− into urea. (a) Schematic illustration of the Cu electrocatalyst with atomic scale spacings. (b) TEM and STEM images of the bare Cu, Cu with 6 Å spacing and Cu with 15 Å spacing. (c) Electrochemical performance for urea production on the 6 Å-Cu and bare Cu. (d) Kinetic barriers for the C–N coupling for CO2 with possible NOxRR intermediates.57 Copyright 2023, Royal Society of Chemistry. (e) Schematic illustration of the Zn–Cu alloy catalyst for urea production. (f) Elemental mapping of Zn and Cu in the alloy catalyst. (g) FE for urea on different metals and alloys. (h) Theoretical calculations for the free energy changes during the reaction.58 Copyright 2023, Springer Nature. | ||
Similar to the electrosynthesis of Ala, another strategy to enhance the electrochemical co-reduction of CO2 and NO3− into urea is making a bimetallic alloy catalyst, where one metal can facilitate the key C–N coupling step, and the other metal can facilitate the electron and proton transfer steps (co-reduction of CO2 and NO3− into urea is a 16-electron process). Recently, Sargent's lab developed a Zn–Cu hybrid catalyst for electrosynthesis of urea, where Zn can facilitate the C–N coupling step and Cu can reduce the energy required for a key protonation step in making urea (Fig. 5e and f).58 The Zn–Cu hybrid catalyst was prepared by spray-coating an incomplete layer of Zn on top of the Cu layer (Zn and Cu were both commercially available nanoparticles). Compared to single metal catalysts, bimetallic alloy catalysts such as Zn–Cu and Zn–Ag can significantly improve the FE for urea (Fig. 5g). Theoretical calculations indicated that the C–N bond formation (CO2 + *NO2 → *CO2NO2) is more favorable on the surface of Zn compared to Cu, whereas the endergonic step for the protonation of *CO2NH2 to *COOHNH2 (potentially the rate determining step) requires less energy on Cu compared to Zn (Fig. 5h). With this relay catalyst design, CO2 gas and simulated wastewater containing 1000 ppm NO3− can be converted to urea with a FE of 75% and a total current density of 10 mA cm−2 at −0.8VRHE, which can last for 32 hours.
In addition to urea, other small organonitrogen molecules can also be produced via the coupling CO2RR and NOxRR, such as methylamine41 as described in 2.1. Researchers have also proved that NOxRR intermediates can undergo nucleophilic attack to CO2-derived molecules. For example, Nam and coworkers reported in 2021 that oxalic acid, a CO2RR product, can be converted to glycine via electrochemical NO3− reduction.60 Recently, Zhang's lab demonstrated that the electrochemical co-reduction of NO2− and the CO2-derived formic acid can yield formamide on a Cu catalyst (Fig. 6a), which was prepared by electroreduction of Cu2O nanocubes.61 At −0.6VRHE, NO2− and formic acid can be converted to formamide with a FE ∼ 30% (Fig. 6b). Mechanistic studies suggested that the C–N coupling step occurs between the *NH2 and *CHO intermediates (ΔG = −1.12 eV), which is thermodynamically favored compared to thermoneutral step of further reduction of *CHO (ΔG = −0.08 eV), whereas the further reduction of *NH2 is also thermodynamically feasible (ΔG = −0.48 eV). These calculations results are consistent with the experimental observations that NH3 (i.e., over-reduction of NO2−) was the main by-product, while the generation of CH3OH (i.e., over-reduction of CO2) is minimal.
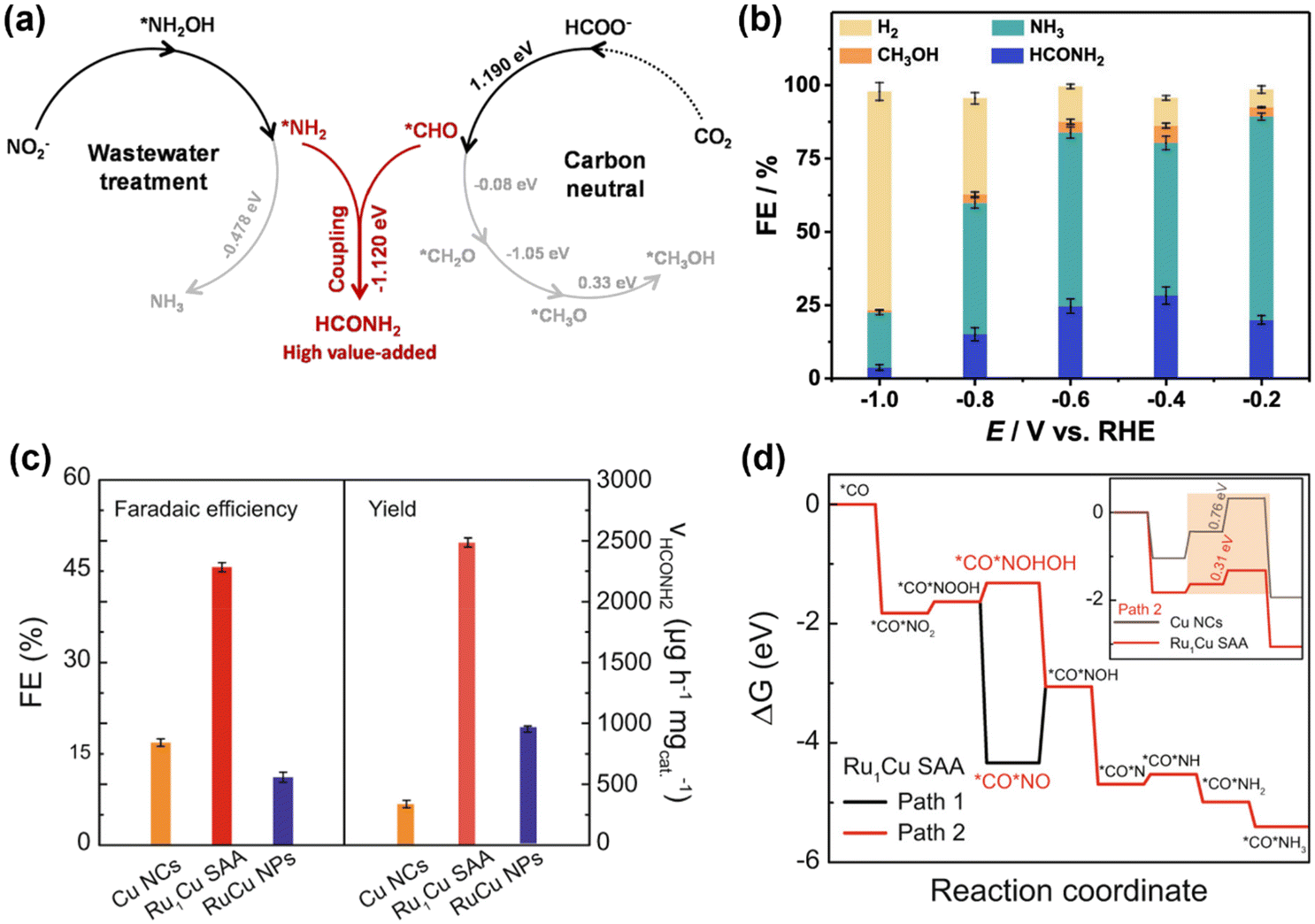 | ||
| Fig. 6 Electrosynthesis of formamide from NO2− and CO2-derived molecules. (a) and (b) Electrochemical co-reduction of NO2− and formic acid into formamide.61 Copyright 2023, American Chemical Society. (c) and (d) Electrochemical co-reduction of NO2− and CO into formamide.62 Copyright 2023, Springer Nature. | ||
Tan and Coworkers also reported an electrochemical method to convert NO2− and CO into formamide on a single-atom alloy (SAA) electrocatalyst with atomically dispersed Ru atoms on Cu nanoclusters.62 The Ru–Cu SAA catalysts were prepared by thermal reduction of Cu2+ to make Cu nanoclusters and then introducing Ru single atom onto the surface of Cu nanoclusters through galvanic replacement reaction. Formamide can be produced with a FE of 46% at −0.5VRHE, which is significantly higher than pure Cu or Ru–Cu nanoparticles (Fig. 6c), thus highlighting the importance of bimetallic alloy as well as single-atom catalyst. Computational studies (Fig. 6d) showed that Ru can lower the energy barriers for the hydrogenation steps of the *CO*NO2 intermediate (*CO*NO2 → *CO*NOOH → *CO*NOHOH), and thus can facilitate the formamide production compared to pure Cu.
3. Summary and outlook
The aforementioned works provide attractive methods to build C–N bonds via electrochemical NOxRR. In particular, some key strategies and considerations are highlighted to enhance the selectivities towards the desired C–N coupling products, as summarized as below:(1) Choosing a suitable nucleophile for C–N coupling. The NOxRR intermediates, such as *NH2OH and *NH2, are usually more nucleophilic than both the reactants (e.g., NO3−, NO2−) and the final product of NOxRR (NH3) and therefore can facilitate the C–N coupling step. On the other side, utilizing a NOxRR intermediate at the early stage (e.g., *NO2) as the nucleophile for C–N coupling can prevent the formation of by-products (e.g., N2, NH3).
(2) Controlling the surface adsorptions of both nucleophile (i.e., NOxRR intermediates) and electrophile (i.e., CO2RR intermediates or organic substrate). The electrocatalyst should have sufficient adsorptions of the reactants and intermediates so that the electroreduction can proceed at low potentials, but the adsorptions should not be too strong so that the product can desorb from the catalyst and avoid over-reduction.
(3) Designing bimetallic alloy catalysts or bi-reactor system to drive the C–N coupling step and hydrogenation step separately. The C–N coupling step and electroreduction step can have quite different requirement for reaction conditions, such as catalyst, potential, pH, etc., and therefore it would be helpful to decouple the two steps by designing an alloy catalyst containing two metals or a tandem system with two separate reactors.
(4) Preparing electrocatalysts with nano-/atomic-scale surface structures, single-atom structures, single crystal facets, or alloy compositions. The catalysts with nano-/atomic-scale morphologies or single-atom structures can usually enhance the reaction rate by lowering the energy barriers and a specific single crystal facet can potentially improve the selectivity towards a certain product. Making alloy electrocatalysts can also tune the surface adsorptions of intermediates, meet different requirements for C–N coupling step and electroreduction step, and allow synergetic effects to improve the FE.
Despite these distinctive methods, there are still some challenges and problems in this area as listed below:
(1) The formation of the desired C–N coupling product is in competition of other reactions. For example, the co-reduction of NOx and CO2 can obviously generate a series of byproducts without C–N coupling, such as NH3, N2, CO, formate, etc. The HER can also be a significant competing reaction. When organic compounds (e.g. cyclohexanone) are present in the electrolyte, the organic hydrogenation reaction can also lower the FE for C–N coupling. Furthermore, the C–N coupling products could also be further reduced and the desired chemical structures (e.g. oxime) may be lost.
(2) The reaction pathway and key intermediates for C–N coupling are still not very clear, particularly for the formation of urea. The NH2OH-mediated C–N coupling pathways (e.g. in the formations of amine, oxime, amino acid) are relatively better understood and researchers can confirm that the surface-adsorbed *NH2OH is the key intermediate to build the C–N bond. However, it is still unclear that the C–N bond in urea is formed at which stage of electrochemical NOxRR. Several possible intermediates have been proposed as key species for C–N coupling, such as *NO2, *NO, *N, and *NH2, but the direct experimental evidence is still rarely reported. The intermediates at the early stage of NOxRR (e.g. *NO2, *NO) can potentially avoid the generation of byproducts in NOxRR and CO2RR, whereas the intermediates at the later stage (e.g. *N, *NH2) are more nucleophilic and therefore more energy-favorable. More in-depth mechanistic studies, ideally in situ spectroscopies, are needed to better understand the reaction pathway for making urea.
(3) The NOxRR to build C–N bond is also challenged by similar factors in the pure electrochemical NOxRR (i.e. without C–N coupling). For example, the electrochemical reduction of NO3− is usually limited by the large overpotential caused by the sluggish rate-determining step of NO3− reduction to NO2−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 (the theoretical potential is +0.69VRHE whereas the operation potential is usually from −1.0VRHE to 0VRHE). The C–N coupling reaction is also challenged by other common problems in pure electrochemical NOxRR, such as catalyst poisoning caused by the excessive adsorption of *NO2,18 degradation of metallic catalysts in concentrated NH3 solution, difficulties in utilizing diluted NOx in real-world wastewater, etc.
32 (the theoretical potential is +0.69VRHE whereas the operation potential is usually from −1.0VRHE to 0VRHE). The C–N coupling reaction is also challenged by other common problems in pure electrochemical NOxRR, such as catalyst poisoning caused by the excessive adsorption of *NO2,18 degradation of metallic catalysts in concentrated NH3 solution, difficulties in utilizing diluted NOx in real-world wastewater, etc.
We also look into a few possible future directions that can be pursued to improve the performance, profitability and impact of this research area:
(1) The nitrogen selectivity/utilization efficiency should be improved. Current electrochemical C–N coupling systems typically contain NOx species with a concentration that is about one order of magnitude higher than the organic substrate to allow the full conversion of organic substrate. However, the high loading of NOx species also decreases the FE and N utilization efficiency for the desired product. The main N-containing product is usually NH3 (and N2 in some cases). Considering many studies utilize the *NH2OH intermediate as the nucleophile, it is necessary to tune the surface adsorption of *NH2OH to prevent its over-reduction into NH3 and reduce the loading NOx required for full conversion of organic substrate. For example, Zhang's lab recently proved that the electroreduction of NO favored the formation of NH2OH (FE up to 80%) on Co single-atom catalysts while the NO reduction on Co nanoparticles mainly generated NH3.76 The selectivity between NH2OH and NH3 was tuned by regulating the adsorption configuration of NO: the linear adsorption of NO on the single-atom catalysts can maintain the N–O bond and allow the NO reduction to stop at the stage of NH2OH, while the bridge adsorption of NO on the nanoparticles would weaken the N–O bond to produce NH3. It is also notable that the NOxRR may not proceed through the *NH2OH-mediated pathway (i.e., NOx → *NH2OH → *NH2 → NH3); instead, it could also undergo the *N-mediated pathway: NOx → *N, and then the surface-adsorbed *N can either make N2 (2 *N → N2), or produce NH3 (*N → *NH → *NH2 → NH3).9,37 In either case, *NH2OH will not be produced. Therefore, the reaction mechanism that involves surface-adsorbed intermediates should be further understood (as discussed below) to prevent the *N-mediated pathway and enhance formation of *NH2OH. In addition, pulsed electrolysis methods, which are known to tune the local concentration of NOx species near the electrode and optimize the surface adsorption configuration of intermediates in electrochemical NOxRR to NH3,77–79 could also be used to improve the N selectivity in C–N coupling.
(2) Mechanistic study by detecting the reaction intermediates with in situ technologies. The surface-adsorbed reaction intermediates play a central role in electrochemical NOxRR and C–N coupling, such as *NO, *NOH, *N, *NH2OH and *NH2. Those intermediates can be studied by in situ spectroscopies, such as ATR-IR and Raman.43,50 The hydrogen radicals at the electrode surface, which determine the electrochemical hydrogenation of NOxRR intermediates, can be investigated by electron paramagnetic resonance (EPR).14,32 The catalytic sites of electrocatalyst can be monitored by in situ X-ray diffraction (XRD), X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) technologies.31,32 Moreover, the gaseous products and intermediates can also be analyzed by on-line differential electrochemical mass spectrometry (DEMS) during the electrochemical reaction.14,32 More in situ technologies should be adapted together with computational studies to systematically understand the reaction pathways towards different N-containing products (N2, NH3, C–N coupling products, etc.), and how the selectivities are controlled by catalyst structure and reaction conditions (potential, concentrations, pH, etc.).
(3) Electrochemical co-reduction of CO2 and NOx into C2+ organonitrogen molecules. Co-reduction of CO2 and NOx to form C–N bonds have provided an attractive route to produce organonitrogen compounds. Despite these distinctive methods, current studies mainly focus on producing C1 organonitrogen compounds. A co-reduction system that can build the C–N bonds in C2+ organonitrogen molecules, which tend to be more valuable, has barely been reported. In CO2RR studies, a microfluidic flow reactor has been developed:80–84 an ion exchange membrane and an aqueous alkaline liquid layer, which is known to facilitate CO2RR towards C2+ products,81,82 are sandwiched between the anode and cathode. In a similar way, this flow reactor design could also enhance the formation of C2+ organonitrogen molecules in CO2–NOx co-reduction. The reaction can occur at the gas–liquid–solid interface, where the electrocatalyst is in contact with aqueous electrolyte (OH− and NO3−/NO2−) and gaseous CO2. A key challenge here that making C2+ organonitrogen products will require the formation of both C–C and C–N bonds, where the strategies of making bimetallic alloy catalysts could be employed to facilitate the C–C and C–N bonds formation separately.
(4) Utilize industrial/agricultural wastewater containing NO3−/NO2− to produce organonitrogen compounds. Compared to the NO3−/NO2− solutions in lab, the real-world wastewater usually has lower concentrations of NO3−/NO2−, unsuitable pH values, and complicated components such as organic residues, metal cations that can poison the catalyst through electrodeposition on the electrode,85,86 anions that may change the product selectivity (e.g., Cl− is known to facilitate NOxRR towards N2),87–89etc., these factors will affect the C–N coupling reaction. Therefore, some pre-treatment of wastewater may be needed and a more robust electrocatalyst will be required. Furthermore, the scalability, energy efficiency, economic cost, separation of the organonitrogen products, and long-term stability will also be important considerations to develop a profitable NOxRR electrolyzer for industrial implementation.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We appreciate the financial support by the RSC Research Fund (R21-3641011632) and Manchester Metropolitan University Research Accelerator Grant.References
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 350 CrossRef PubMed.
- H. Xu, Y. Ma, J. Chen, W.-X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- W. Chen, X. Yang, Z. Chen, Z. Ou, J. Hu, Y. Xu, Y. Li, X. Ren, S. Ye, J. Qiu, J. Liu and Q. Zhang, Adv. Funct. Mater., 2023, 2300512, DOI:10.1002/adfm.202300512.
- R. Cassia, M. Nocioni, N. Correa-Aragunde and L. Lamattina, Front. Plant Sci., 2018, 9, 273 CrossRef PubMed.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, L. Han, S. Wang, T. Cui, Y. Yan, M. Xu, H. Duan, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202213711 CrossRef CAS PubMed.
- R. Zhao, Q. Yan, L. Yu, T. Yan, X. Zhu, Z. Zhao, L. Liu and J. Xi, Adv. Mater., 2023, 35, 2306633 CrossRef CAS PubMed.
- L. Wu, J. Feng, L. Zhang, S. Jia, X. Song, Q. Zhu, X. Kang, X. Xing, X. Sun and B. Han, Angew. Chem., Int. Ed., 2023, e202307952, DOI:10.1002/anie.202307952.
- W. Gao, K. Xie, J. Xie, X. Wang, H. Zhang, S. Chen, H. Wang, Z. Li and C. Li, Adv. Mater., 2023, 35, 2202952 CrossRef CAS PubMed.
- M. Xie, S. Tang, Z. Li, M. Wang, Z. Jin, P. Li, X. Zhan, H. Zhou and G. Yu, J. Am. Chem. Soc., 2023, 145, 13957–13967 CrossRef CAS PubMed.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J.-F. Li, Z.-Q. Tian, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- X. He, Z. Li, J. Yao, K. Dong, X. Li, L. Hu, S. Sun, Z. Cai, D. Zheng, Y. Luo, B. Ying, M. S. Hamdy, L. Xie, Q. Liu and X. Sun, iScience, 2023, 26, 107100 CrossRef CAS PubMed.
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- L. Yue, W. Song, L. Zhang, Y. Luo, Y. Wang, T. Li, B. Ying, S. Sun, D. Zheng, Q. Liu, A. Farouk, M. S. Hamdy, S. Alfaifi and X. Sun, Small Struct., 2023, 4, 2300168 CrossRef CAS.
- X. He, X. Li, X. Fan, J. Li, D. Zhao, L. Zhang, S. Sun, Y. Luo, D. Zheng, L. Xie, A. M. Asiri, Q. Liu and X. Sun, ACS Appl. Nano Mater., 2022, 5, 14246–14250 CrossRef CAS.
- H. Wang, F. Zhang, M. Jin, D. Zhao, X. Fan, Z. Li, Y. Luo, D. Zheng, T. Li, Y. Wang, B. Ying, S. Sun, Q. Liu, X. Liu and X. Sun, Mater. Today Phys., 2023, 30, 100944 CrossRef CAS.
- X. Fan, C. Liu, Z. Li, Z. Cai, L. Ouyang, Z. Li, X. He, Y. Luo, D. Zheng, S. Sun, Y. Wang, B. Ying, Q. Liu, A. Farouk, M. S. Hamdy, F. Gong, X. Sun and Y. Zheng, Small, 2023, 19, 2303424 CrossRef CAS PubMed.
- Z. Li, Q. Zhou, J. Liang, L. Zhang, X. Fan, D. Zhao, Z. Cai, J. Li, D. Zheng, X. He, Y. Luo, Y. Wang, B. Ying, H. Yan, S. Sun, J. Zhang, A. A. Alshehri, F. Gong, Y. Zheng and X. Sun, Small, 2023, 19, 2300291 CrossRef CAS PubMed.
- F. Y. Chen, Z. Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- D.-X. Liu, Z. Meng, Y.-F. Zhu, X.-F. Sun, X. Deng, M.-M. Shi, Q. Hao, X. Kang, T.-Y. Dai, H.-X. Zhong, J. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2023, e202315238, DOI:10.1002/anie.202315238.
- J. Shao, H. Jing, P. Wei, X. Fu, L. Pang, Y. Song, K. Ye, M. Li, L. Jiang, J. Ma, R. Li, R. Si, Z. Peng, G. Wang and J. Xiao, Nat. Energy, 2023, 8, 1273–1283 CrossRef CAS.
- L. Shi, L. Liu, B. Yang, G. Sheng and T. Xu, Sustainability, 2020, 12, 3793 CrossRef CAS.
- L. O. Cisneros, W. J. Rogers and M. Sam Mannan, Thermochim. Acta, 2004, 414, 177–183 CrossRef CAS.
- P. Liao, J. Kang, R. Xiang, S. Wang and G. Li, Angew. Chem., Int. Ed., 2023, e202311752, DOI:10.1002/anie.202311752.
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed.
- M. Jouny, J.-J. Lv, T. Cheng, B. H. Ko, J.-J. Zhu, W. A. Goddard and F. Jiao, Nat. Chem., 2019, 11, 846–851 CrossRef CAS PubMed.
- J. Li and N. Kornienko, Chem. Sci., 2022, 13, 3957–3964 RSC.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustainability, 2021, 4, 725–730 CrossRef.
- C. L. Rooney, Y. Wu, Z. Tao and H. Wang, J. Am. Chem. Soc., 2021, 143, 19983–19991 CrossRef CAS PubMed.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustainability, 2021, 4, 868–876 CrossRef.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- J. Liu, G. Chen, Y. Yu, Y. Wu, M. Zhou, W. Zhang, H. Qin, C. Lv and W. Fu, New J. Chem., 2015, 39, 1930–1937 RSC.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- Y. Wu, J. Zhao, C. Wang, T. Li, B.-H. Zhao, Z. Song, C. Liu and B. Zhang, Nat. Commun., 2023, 14, 3057 CrossRef CAS PubMed.
- Y. Wu, W. Chen, Y. Jiang, Y. Xu, B. Zhou, L. Xu, C. Xie, M. Yang, M. Qiu, D. Wang, Q. Liu, Q. Liu, S. Wang and Y. Zou, Angew. Chem., Int. Ed., 2023, e202305491, DOI:10.1002/anie.202305491.
- J. Sharp, A. Ciotti, H. Andrews, S. Udayasurian, M. García-Melchor and T. Li, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-q265h.
- R. Xiang, S. Wang, P. Liao, F. Xie, J. Kang, S. Li, J. Xian, L. Guo and G. Li, Angew. Chem., Int. Ed., 2023, e202312239, DOI:10.1002/anie.202312239.
- J. Wu, L. Xu, Z. Kong, K. Gu, Y. Lu, X. Wu, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2023, e202311196, DOI:10.1002/anie.202311196.
- M. Li, Y. Wu, B.-H. Zhao, C. Cheng, J. Zhao, C. Liu and B. Zhang, Nat. Catal., 2023, 6, 906–915 CrossRef CAS.
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, R. Xiang, Y. Zhang, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202306726 CrossRef CAS PubMed.
- S. Shin, S. Sultan, Z.-X. Chen, H. Lee, H. Choi, T.-U. Wi, C. Park, T. Kim, C. Lee, J. Jeong, H. Shin, T.-H. Kim, H. Ju, H. C. Yoon, H.-K. Song, H.-W. Lee, M.-J. Cheng and Y. Kwon, Energy Environ. Sci., 2023, 16, 2003–2013 RSC.
- Y. Luo, K. Xie, P. Ou, C. Lavallais, T. Peng, Z. Chen, Z. Zhang, N. Wang, X.-Y. Li, I. Grigioni, B. Liu, D. Sinton, J. B. Dunn and E. H. Sargent, Nat. Catal., 2023, 6, 939–948 CrossRef CAS.
- M. Xu, F. Wu, Y. Zhang, Y. Yao, G. Zhu, X. Li, L. Chen, G. Jia, X. Wu, Y. Huang, P. Gao and W. Ye, Nat. Commun., 2023, 14, 6994 CrossRef CAS PubMed.
- J. E. Kim, J. H. Jang, K. M. Lee, M. Balamurugan, Y. I. Jo, M. Y. Lee, S. Choi, S. W. Im and K. T. Nam, Angew. Chem., Int. Ed., 2021, 60, 21943–21951 CrossRef CAS PubMed.
- C. Guo, W. Zhou, X. Lan, Y. Wang, T. Li, S. Han, Y. Yu and B. Zhang, J. Am. Chem. Soc., 2022, 144, 16006–16011 CrossRef CAS PubMed.
- J. Lan, Z. Wei, Y.-R. Lu, D. Chen, S. Zhao, T.-S. Chan and Y. Tan, Nat. Commun., 2023, 14, 2870 CrossRef CAS PubMed.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- D. Saravanakumar, J. Song, S. Lee, N. H. Hur and W. Shin, ChemSusChem, 2017, 10, 3999–4003 CrossRef CAS PubMed.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- Z. Tao, Y. Wu, Z. Wu, B. Shang, C. Rooney and H. Wang, J. Energy Chem., 2022, 65, 367–370 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- G. R. Tauszik and P. Crocetta, Appl. Catal., 1985, 17, 1–21 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Qin, T. E. Davies, D. J. Morgan, A. Stenner, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, L. Chen, Y. Yamamoto and G. J. Hutchings, ACS Catal., 2023, 13, 1934–1945 CrossRef CAS.
- A. Thangaraj, S. Sivasanker and P. Ratnasamy, J. Catal., 1991, 131, 394–400 CrossRef CAS.
- Y. Wu, C. Liu, C. Wang, Y. Yu, Y. Shi and B. Zhang, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed.
- R. Xiang, S. Wang, P. Liao, F. Xie, J. Kang, S. Li, J. Xian, L. Guo and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202312239 CrossRef CAS PubMed.
- D. P. Butcher and A. A. Gewirth, Nano Energy, 2016, 29, 457–465 CrossRef CAS.
- T. J. Hager and L. R. Sadergaski, Measuring Hydroxylammonium, Nitrate, and Nitrite Concentration with Raman Spectroscopy for 238Pu Supply Program, 2021 Search PubMed.
- J. Zhou, S. Han, R. Yang, T. Li, W. Li, Y. Wang, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202305184 CrossRef CAS PubMed.
- Y. Huang, C. He, C. Cheng, S. Han, M. He, Y. Wang, N. Meng, B. Zhang, Q. Lu and Y. Yu, Nat. Commun., 2023, 14, 7368 CrossRef CAS PubMed.
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed.
- M. He, Y. Wu, R. Li, Y. Wang, C. Liu and B. Zhang, Nat. Commun., 2023, 14, 5088 CrossRef CAS PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- T. Li, E. W. Lees, Z. S. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- J. He, A. Huang, N. J. J. Johnson, K. E. Dettelbach, D. M. Weekes, Y. Cao and C. P. Berlinguette, Inorg. Chem., 2018, 57, 14624–14631 CrossRef CAS PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- G. Pérez, R. Ibáñez, A. M. Urtiaga and I. Ortiz, Chem. Eng. J., 2012, 197, 475–482 CrossRef.
- X. Ma, M. Li, C. Feng and Z. He, J. Hazard. Mater., 2020, 388, 122085 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhao, Z. Chen, L. Wang, P. Wu and F. Wang, Electrochim. Acta, 2018, 291, 151–160 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |