
Recent progress of heterogeneous catalysts for transfer hydrogenation under the background of carbon neutrality
Guangyu
Chen†
a,
Jun
Ma†
ab,
Wanbing
Gong
 *a,
Jiayi
Li
a,
Zheyue
Li
a,
Ran
Long
*a and
Yujie
Xiong
*ab
*a,
Jiayi
Li
a,
Zheyue
Li
a,
Ran
Long
*a and
Yujie
Xiong
*ab
aNational Synchrotron Radiation Laboratory, School of Nuclear Science and Technology, Hefei National Research Center for Physical Sciences at the Microscale, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: yjxiong@ustc.edu.cn; longran@ustc.edu.cn; wbgong2021@ustc.edu.cn
bSuzhou Institute for Advanced Research, University of Science and Technology of China, Suzhou, Jiangsu 215123, P. R. China
First published on 21st December 2023
Abstract
Under the background of carbon neutrality, the direct conversion of greenhouse CO2 to high value added fuels and chemicals is becoming an important and promising technology. Among them, the generation of liquid C1 products (formic acid and methanol) has made great progress; nevertheless, it encounters the problem of how to use it efficiently to solve the overcapacity issue. In this review, we suggest that the catalytic transfer hydrogenation using formic acid and methanol as the hydrogen sources is a critical and potential route for the substitution for the fossil fuel-derived H2 to generate essential bulk and fine chemicals. We mainly focus on summarizing the recent progress of heterogeneous catalysts in such reactions, including thermal- and photo-catalytic processes. Finally, we also propose some challenges and opportunities for this development.
 Guangyu Chen | Guangyu Chen received his bachelor's degree in energy chemical engineering from the Hefei University of Technology in 2022. Presently, he is a master's student in Prof. Yujie Xiong's group at the University of Science and Technology of China. His current research interests focus on developing new catalytic materials for photo- and thermo-driven hydrogen production and hydrogenation reactions. |
 Jun Ma | Jun Ma received his B.S. degree in material chemistry in 2016 and Ph.D. in inorganic chemistry in 2021 under the supervision of Professor Yujie Xiong, both from the University of Science and Technology of China. His research interest is focused on the controlled synthesis of oxide materials and photocatalytic C–H bond activation. |
 Wanbing Gong | Wanbing Gong received his PhD degree from the University of Science and Technology of China in 2018 under the supervision of Professor Huijun Zhao. Then, he joined the Hefei Institutes of Physical Science, Chinese Academy of Sciences as a postdoc. He is currently a research associate professor at the University of Science and Technology of China. His current research interests focus on searching for new nonprecious catalytic materials and novel catalytic systems to meet the need of industries. |
 Ran Long | Ran Long received her BSc degree in chemistry in 2009 and PhD in inorganic chemistry under the tutelage of Professor Yujie Xiong in 2014, both from the University of Science and Technology of China. After postdoctoral training with Professors Yujie Xiong and Li Song, she is currently a professor working at the USTC. Her research interests focus on the controlled synthesis and catalytic applications of metal nanocrystals. |
 Yujie Xiong | Yujie Xiong received his B.S. degree in chemical physics in 2000 and Ph.D. in inorganic chemistry in 2004, both from the University of Science and Technology of China. During 2004–2009, he worked as a postdoc at the University of Washington in Seattle and as a research associate at the University of Illinois at Urbana-Champaign, respectively. He was a principal scientist of the National Nanotechnology Infrastructure Network (NSF-NNIN) site at Washington University in St Louis in 2009–2011. He joined the USTC in 2011, and currently is the chair professor of chemistry. His research centers on artificial cycles of elements and energy toward ecosystem reconstruction. |
1. Introduction
The massive increase in the world population inevitably leads to energy shortage and environmental crisis, which is one of the greatest challenges of this century on Earth. The overuse of nonrenewable fossil resources such as coal, oil and natural gas greatly increases CO2 emission and the accompanying greenhouse effect, and thus it is necessary to develop renewable resources and CO2 capture and utilization technologies.1–7 For this, many chemists and materials scientists focus on the development of new renewable resources (wind, light, electricity, etc.) and new materials such as nanomaterials. In particular, in the context of carbon neutrality, the highly efficient conversion of CO2 to formic acid and methanol is regarded as one of the most promising CO2 direct conversion technologies for industrialization, which is undergoing rapid development.5,8–14 For example, Yang et al. recently synthesized a layered {001}-oriented Bi oxyhalide (i.e. BiOX, X = Cl, Br or I) electrocatalyst to produce formic acid from CO2 in a flow cell. They found that the bromide activated bismuth catalyst can achieve a high current density (>100 mA cm−2) and formic acid selectivity (>90%), thus paving the way for the rational design of excellent electrocatalysts for renewable chemical and fuel production.9 In addition, Su et al. employed monodisperse cobalt phthalocyanine (CoPc) on single-walled CNTs (CoPc/SWCNTs) to catalyze the formation of methanol in a tandem-flow electrolyser, which achieves a methanol partial current density of >90 mA cm−2 with >60% selectivity.11 These great advances are accelerating the industrialization of direct CO2 utilization technologies, and thus the overall pace of carbon neutrality. However, formic acid and methanol can also be obtained in quantity from traditional chemical routes such as petroleum and biomass,15–17 which leads to a temporary oversupply of these two chemicals (Scheme 1). Therefore, how to use them via an efficient and green route will be a dilemma for the future society. | ||
| Scheme 1 Main synthetic paths of formic acid and methanol. | ||
In this review, we therefore propose that formic acid and methanol can be the ideal hydrogen sources to replace high pressure H2 in hydrogenation reactions. Hydrogenation is one of the most critical and fundamental processes in the modern chemical industry, and can produce important chemical and pharmaceutical intermediates.18–23 However, the use of high pressure hydrogen inevitably leads to some disadvantages such as safety, storage and transportation, and the selectivity of the desired product is difficult to control.24–27 In this respect, the catalytic transfer hydrogenation using organic hydrogen donors is therefore considered as a good alternative to H2 hydrogenation, which not only reduces the requirements of special equipment but also improves the desired product selectivity due to the diversity of organic hydrogen donors (Scheme 2). In recent years, some progress has been made in catalytic transfer hydrogenation.21,23,24,28–33 For example, Gilkey and Xu have reviewed the recent progress in catalytic transfer hydrogenation for the conversion of biomass-derived feedstocks to fuels and chemicals with a focus on mechanistic interpretation, and they also discussed future challenges and opportunities.34
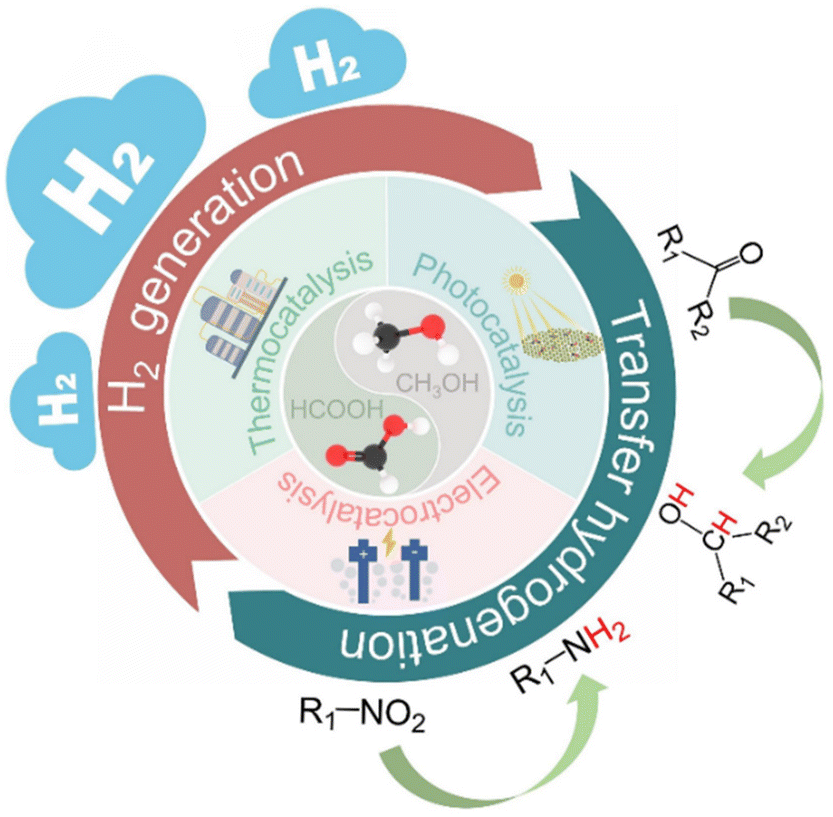 | ||
| Scheme 2 Proposed transfer hydrogenation process using formic acid and methanol as hydrogen sources. | ||
Currently, the common hydrogen donors mainly include low molecular mass alcohols (e.g., methanol, ethanol, and isopropanol), and formic acid is also an ideal hydrogen storage medium with a high hydrogen content of 4.4 wt% and stable chemical properties.35–40 Recently, Zhai et al. reviewed the developments in CO2 hydrogenation to formic acid/formate and reverse formic acid/formate dehydrogenation as a liquid organic hydrogen donor under mild conditions.41 Such a CO2–formic acid hydrogenation cycle not only promotes CO2 capture and utilization, but also provides great guidance for the subsequent hydrogen-related energy and chemical industry. Considering that the CO2 hydrogenation to formic acid has been discussed by many researchers,8,9 here we focus only on the progress in transfer hydrogenation using formic acid as the hydrogen source, especially in terms of catalysts. In the past few decades, researchers have devoted much effort to developing highly efficient homogeneous and heterogeneous catalysts for the dissociation of formic acid into H2 and subsequent hydrogenation.15,38,42–48 Homogeneous catalysts are mainly transition metal–organic complexes, including ruthenium, iridium, nickel, etc., which usually have high activity and good selectivity.49–51 Compared with homogeneous catalysts, heterogeneous catalysts are easy to separate from the reaction and can be recycled.52 At present, heterogeneous catalysts used for formic acid transfer hydrogenation are mainly divided into supported precious metal (e.g., Pd, Pt, Au, and Ag) catalysts53–55 and carbon coated transition metal (e.g., Co, Ni, and Fe) catalysts.25,56–58 Precious metals usually have high catalytic activity and stability in corrosive formic acid solution, but their scarcity and high price limit their further development.25,57–59 Fortunately, the low activity and poor stability of transition metals are being gradually overcome via carbon coating, which can efficiently inhibit the direct contact between nonprecious nanoparticles (NPs) and formic acid.57,58,60–62 Moreover, the electron transfer between the coated carbon layer and the metal NPs endows it with excellent catalytic activity to accelerate formic acid adsorption and subsequent dissociation. Compared with direct H2 hydrogenation, formic acid-driven transfer hydrogenation can not only hydrogenate specific functional groups, but also trigger a series of reactions such as the methylation of aromatic amines using the abundant reaction sites of formic acid itself.33,63–66
As another liquid hydrogen source, methanol is also an ideal substitute for high pressure hydrogen.67–71 Currently, traditional thermocatalysis and electrocatalysis systems have exhibited great potential for the production of methanol on a larger scale, but efficient use of excessive methanol remains a headache in the future chemical industry. Industrially, methanol can be used for producing basic building blocks such as ethylene or propylene, and can also be used as a gasoline additive.72–77 More importantly, methanol has been regarded as an ideal liquid hydrogen source due to its high hydrogen capacity of 12.5 wt%.37,39,71 However, it is rarely used for transfer hydrogenation due to high energy consumption for in situ hydrogen production and utilization. There are four primary routes for hydrogen production from the traditional thermal catalytic process: methanol steam reforming (MSR),78–80 methanol decomposition (MD),81,82 partial oxidation of methanol (POM),83,84 and a combination of oxidative methanol steam reforming (OMSR, MSR, and POM), also known as auto-thermal reforming of methanol (ATRM).85–87 The main approach for producing hydrogen at present is methanol steam reforming (CH3OH + H2O → CO2 + 3H2). However, this process is energy-intensive, demanding specific temperature and pressure conditions, thus leading to substantial fossil energy consumption and the generation of carbon dioxide as a by-product. This is not conducive to environmentally friendly sustainable development. While Lin et al. have reported that platinum (Pt) atoms dispersed on α-molybdenum carbide (α-MoC) can achieve an outstanding low-temperature MSR performance, significantly increasing the H2 generation rate and minimizing fossil energy consumption in MSR reactions,69 the reliance on precious metal catalysts like Pt, Pd, and Ru remains unavoidable.88 This poses challenges for practical implementation in industrial production.
Compared to thermal catalysis, electro-reforming of methanol, also known as methanol–water co-electrolysis, presents a compelling alternative to traditional reforming methods.89 This is because it employs an indirect methanol oxidation reaction (MOR) to replace the sluggish oxygen evolution reaction (OER), thereby effectively accelerating the hydrogen production rate of the cathodic hydrogen evolution reaction (HER).90–92 Additionally, this process transforms methanol into some high-value byproducts instead of CO2 or CO. Moreover, the energy input for electrocatalytic methanol reforming is significantly lower than that required for complete decomposition of water and thermal catalytic methanol reforming. This aspect holds great significance for reducing the overall energy consumption.93,94 For instance, Guo et al. developed a bi-functional co-doped Rh electrocatalyst, where an enhanced HER on the cathode was coupled with an accelerated MOR on the anode. This design achieved an efficient rate for complete hydrogen decomposition and provided an impressive mass activity of up to 889 mA mg−1 for the MOR.95
Simultaneously, given solar energy's status as a high-quality sustainable resource, attention has shifted towards driving methanol steam reforming (MSR) through light and the exploration of nonprecious metal catalysts.96–99 Li et al. successfully prepared a novel L–Cu catalyst for photo-driven MSR by reducing CuAl layered double hydroxide (CuAl-LDH) nanosheets, demonstrating an exceptional hydrogen production activity of 160.5 μmol gcat−1 s−1.100 Despite substantial efforts to improve the H2 production rate in MSR reactions, there has been limited focus on investigating carbon-containing by-products. The predominant emphasis in most studies has been on reducing CO production. This emphasis is driven by the imperative to prevent CO poisoning of precious metal catalysts,101 a factor detrimental to the application of proton exchange membrane fuel cells (PEMFC), which mandate CO concentrations below 10 ppm at the anode.102 Unfortunately, the potential utilization of carbon-containing products derived from semi-oxidation reactions is frequently overlooked, leading to significant resource wastage. Therefore, achieving efficient hydrogen production and highly selective generation of valuable carbon-containing products through light-driven methanol under mild conditions holds substantial practical significance. Tang's group recently developed an environmentally friendly methanol photocatalytic hydrogen production system that harnesses the synergy between Cu single atoms and Pt nanodots on a PtCu–TiO2 catalyst. The results showcase a remarkable H2 formation rate of 2383.9 μmol h−1, an apparent quantum efficiency reaching 99.2%, and the high-value carbon-containing product, formaldehyde, with a selectivity of up to 98.6%.103 Additionally, Wang's group engineered single-atom rhodium-doped metal sulfide nanorods, featuring alternately stacked wurtzite/zinc blende segments. The design efficiently activated the methanol C–H bond to generate hydrogen under light, yielding ethylene glycol at a rate of 34 mmol gcat−1 h−1, maintaining a consistent selectivity of approximately 85%.104 These examples illustrate significant advancements in methanol hydrogen production, underscoring the substantial application potential of clean and efficient photocatalysis and electrocatalysis in this domain. Such progress can effectively propel the seamless integration of subsequent hydrogen production and hydrogenation processes. It also encourages the advancement of transfer hydrogenation reactions using methanol as a hydrogen source, paving the way for crucial breakthroughs in the future.
In the past few years, much research has been done on the hydrogen production and hydrogenation systems of formic acid and methanol; however, few reviews have focused on the transfer hydrogenation reactions using CO2-derived methanol and formic acid as hydrogen sources, let alone catalytic material design. In this review, we first summarize the developments of catalysts in the transfer hydrogenation process (e.g., thermal catalysis and photocatalysis) using formic acid as a hydrogen source, mainly including Pd-based precious metal catalysts and Co-based nonprecious metal catalysts. Similarly, we also analyze the catalyst design strategies for methanol-driven transfer hydrogenation reactions, and Pd-based and Cu-based materials are mainly discussed. Finally, in view of the current developments of catalysts in this field, we propose some key challenges and corresponding opportunities for transfer hydrogenation.
2. Transfer hydrogenation using formic acid as a hydrogen source
2.1. Catalyst design for the thermocatalytic process
Thermocatalytic hydrogenation using formic acid as a hydrogen source is one of the fundamental processes in modern chemical industries.45,56 In theory, two kinetic pathways may occur when formic acid is used as a hydrogen source: (i) dehydrogenation of formic acid to form H2 and CO2 (dehydrogenation pathway) and (ii) dehydration of formic acid to form H2O and CO (dehydration pathway). The latter is undesired because the dehydration pathway will not only result in a decrease in the concentration of hydrogen, but also produce by-product CO, leading to the poisoning of heterogeneous catalysts (Scheme 3).15,38,47,62,105,106 Currently, thermo-catalytic hydrogenation using formic acid as a hydrogen source usually requires harsh reaction conditions (high temperature or long time) and highly efficient catalysts.56 For example, the strong corrosive properties of formic acid hamper the long-term stability of catalysts.61,62 Therefore, it is necessary to summarize the recent progress in catalyst development for thermocatalytic hydrogenation using formic acid as a hydrogen source.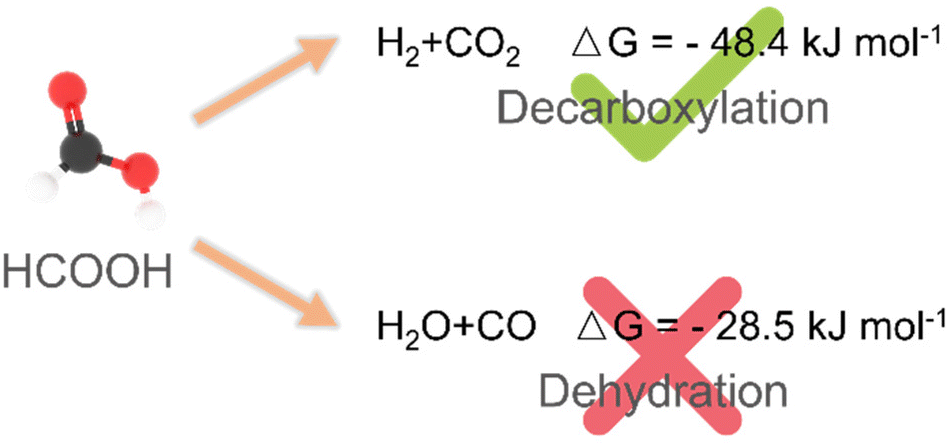 | ||
| Scheme 3 The possible routes for formic acid decomposition. | ||
Palladium-based NP catalysts. Among all the precious metal catalysts, Pd based catalysts are the most effective and thus widely studied candidates because of their special electronic structures and adsorption patterns.107–112 As we all know, the catalytic performance of heterogeneous catalysts greatly depends on their support;113–118 we therefore categorize and summarize the recent developments of Pd catalysts for the hydrogenation reactions using formic acid as a hydrogen source according to the type of support.
First of all, carbon materials have been regarded as a typical catalyst support for formic acid transfer hydrogenation because of their large surface area, high porosity, easy modification, strong chemical stability, etc.119–123 In particular, heteroatoms such as N or S doped carbon supports have obvious electronic effects, which leads to excellent formic acid dehydrogenation and subsequent hydrogenation performance.23,58,124–128
López Granados et al. investigated and found that Pd/C was the most effective and hydrogenation of hydromaleic acid identified specific active sites to succinic acid using formic acid as a hydrogen source for different combinations of precious metals (Pd, Au, Ru, Pt, and Rh) and supports (γ-Al2O3, TiO2, CeO2, ZrO2, WO3, CeZrO4, C, Nicanite, SiO2 and TS-1).55 Zhang et al. also found that the hydrogenation activity followed the trend of Pd/activated carbon (AC) > Pd/TiO2-AC > Pd/MIL-101 > Pd/TiO2 > Pd/Al2O3 for the catalytic transfer hydrogenation of phenol to cyclohexanone, and the optimum Pd/AC catalyst can be recycled at least 6 times. The effect of formic acid on the hydrogenation activity of supported Pd catalysts was also studied. For the MIL-101 and oxide (TiO2 and Al2O3) supports, they revealed that the low activity may be due to the competitive adsorption of formic acid with phenol at a single Pd site (Fig. 1a).129 Afterwards, heteroatoms such as N- or S-doped carbon supported Pd catalysts were further studied. Hu et al. showed that nitrogen functionalized carbon nanotube supported Pd NPs (Pd/NCNT) were especially suitable for the transfer hydrogenation of phenol using formic acid as a hydrogen donor, compared to oxygen functionalized Pd/OCNT (Fig. 1b).130 For the transfer hydrogenation of Lindane using formic acid as a hydrogen source, Yang et al. found that the pyridine N doped porous carbon supported Pd catalyst exhibited good catalytic activity due to the promotional effect of pyridine N, which enhanced the Pd NP dispersion and thus formic acid decomposition. Compared with the commercial bare carbon-supported Pd catalyst, this catalyst can obtain a 99.7% conversion and 100% dechlorination selectivity at 25 °C under atmospheric pressure.135 For this, Zhang et al. prepared ultrafine Pd NPs anchored on N-doped carbon (Pd/N-XC72R), where N-XC72R was prepared by annealing commercial Vulcan XC72R with urea in advance. They showed that N doping on the carbon surface can significantly enhance the affinity between metal NPs and the support, and avoid the aggregation and over-growth of NPs. Compared with the bare Vulcan XC72R supported Pd catalyst, the prepared Pd/N-XC72R catalyst exhibited better catalytic activity for the transfer hydrogenation of nitroaromatic hydrocarbons at room temperature (Fig. 1c).131 In addition, graphitized carbon nitride (g-C3N4) containing nitrogen is a potential nitrogen-doped carbon material, which has been gradually applied to perform transfer hydrogenation. For example, Xu et al. found that the 5 wt% Pd/g-C3N4 catalyst can completely convert nitrobenzene to aniline using formic acid as the hydrogen donor and water as the solvent at room temperature. Meanwhile, the Pd/g-C3N4 catalyst also had good activity in the one-pot reductive amination of carbonyl compounds and nitro compounds, and the corresponding secondary amines can be obtained with excellent selectivity (>90%) (Fig. 1d).132 Cheng et al. also prepared a Pd@g-C3N4 nanosheet catalyst by a liquid-phase exfoliation method for the transfer hydrogenation of nitroaromatic hydrocarbons. The superior activity and stability can be attributed to the amphiphilic g-C3N4 support, the uniform dispersion of Pd NPs and the Mott–Schottky effect between g-C3N4 and Pd NPs (Fig. 1e).133 Chou et al. prepared the bulk- and nanoflake-g-C3N4 supported Pd NP catalysts (Pd/b-CN and Pd/ns-CN) for the catalytic transfer hydrogenation of furfural alcohol to tetrahydrofurfuryl alcohol. The characterization analysis showed that Pd/ns-CN had a higher specific surface area, more surface bases and higher Pd dispersion than Pd/b-CN, which are the key factors for the activation of formic acid.136 Li et al. further synthesized porous g-C3N4 nanosheets/reduced graphene oxide composites by hydrothermal co-assembly of graphene oxide and g-C3N4 nanosheets. Owing to the synergistic support effects, the obtained Pd catalyst showed significant catalytic activity for olefin hydrogenation with formic acid and formates as hydrogen sources under atmospheric pressure (Fig. 1f).134
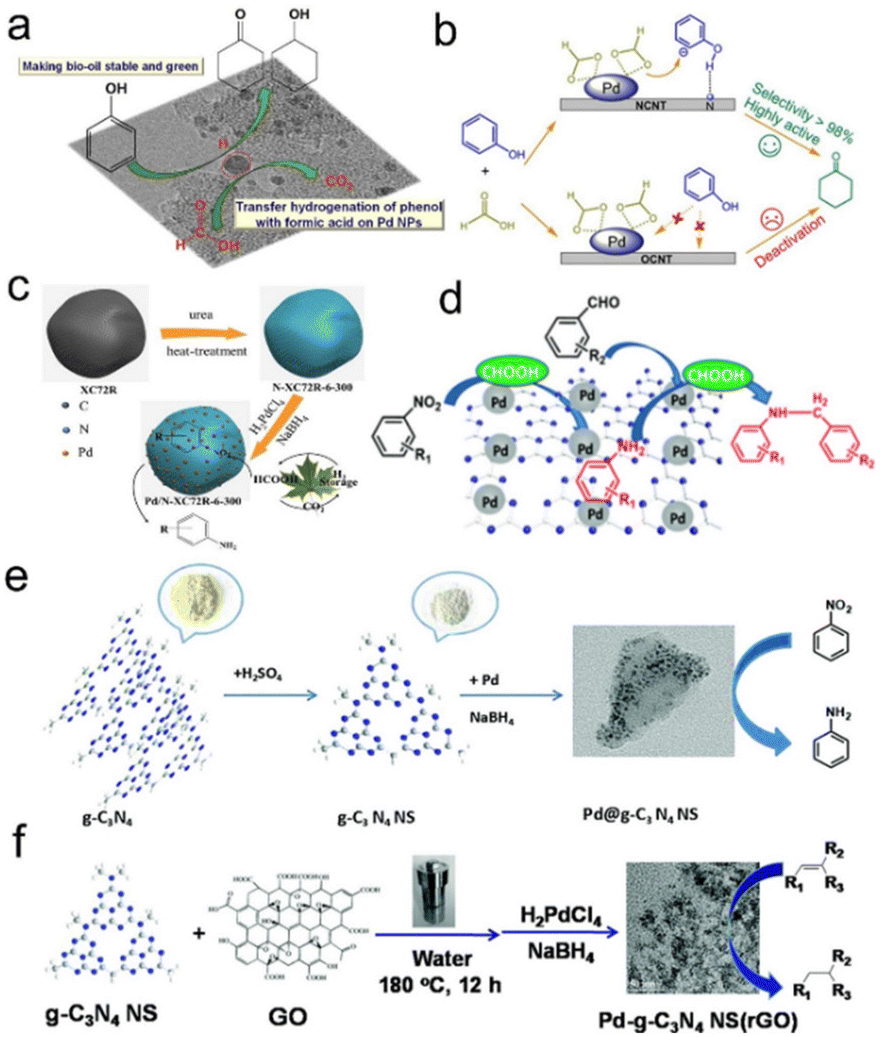 | ||
| Fig. 1 (a) Schematic illustration of the transfer hydrogenation of phenol on supported Pd catalysts. Reproduced with permission from ref. 129. Copyright 2014 Elsevier. (b) Schematic illustration of different reaction paths on Pd/OCNT and Pd/NCNT. Reproduced with permission from ref. 130. Copyright 2021 Wiley-VCH. (c) Schematic illustration of the preparation progress of Pd/N-XC72R. Reproduced with permission from ref. 131. Copyright 2018 American Chemical Society. (d) Schematic illustration for the preparation of primary and secondary amines over g-C3N4 supported Pd NPs. Reproduced with permission from ref. 132. Copyright 2018 Royal Society of Chemistry. (e) Schematic illustration of the preparation process of the Pd@g-C3N4 NS catalyst. Reproduced with permission from ref. 133. Copyright 2018 Royal Society of Chemistry. (f) Schematic illustration of the preparation process of the Pd-g-C3N4 NS/rGO catalyst. Reproduced with permission from ref. 134. Copyright 2018 Royal Society of Chemistry. | ||
In addition to carbon materials, other supports were also studied. For instance, Zhang et al. synthesized a layered Ti3C2 MXene supported Pd NP catalyst, which could achieve efficient and selective hydrogenation of nitrobenzene in the presence of formic acid. They confirmed that the electron-deficient Pd NPs regulated the reaction pathway and promoted the dissociation of formic acid, thus improving the production of active H* to hydrogenate nitrobenzene.137 Neeli et al. synthesized Pd NPs with an average diameter of 2.2 nm by anion exchange and chemical reduction on NH2-UiO-66. The as-prepared Pd/NH2-UiO-66 catalyst can catalyze the transfer hydrogenation of nitrobenzene to aniline in mild water with formic acid as a hydrogen source, and the excellent catalytic activity was mainly attributed to the synergistic effect of Pd NPs and NH2-UiO-66 (Fig. 2a).138 For the ![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) nsupported Pd catalysts, our group identified specific active sites (i.e., surfaces, corners and edges) for formic acid decomposition and olefin/nitrobenzene hydrogenation. The results showed that the decomposition of formic acid mainly occurs at the edge of cubic nanocrystals and the plane of octahedral/tetrahedral nanocrystals, and hydrogenation mainly occurs at the edge of cubic and octahedral/tetrahedral nanocrystals.140
nsupported Pd catalysts, our group identified specific active sites (i.e., surfaces, corners and edges) for formic acid decomposition and olefin/nitrobenzene hydrogenation. The results showed that the decomposition of formic acid mainly occurs at the edge of cubic nanocrystals and the plane of octahedral/tetrahedral nanocrystals, and hydrogenation mainly occurs at the edge of cubic and octahedral/tetrahedral nanocrystals.140
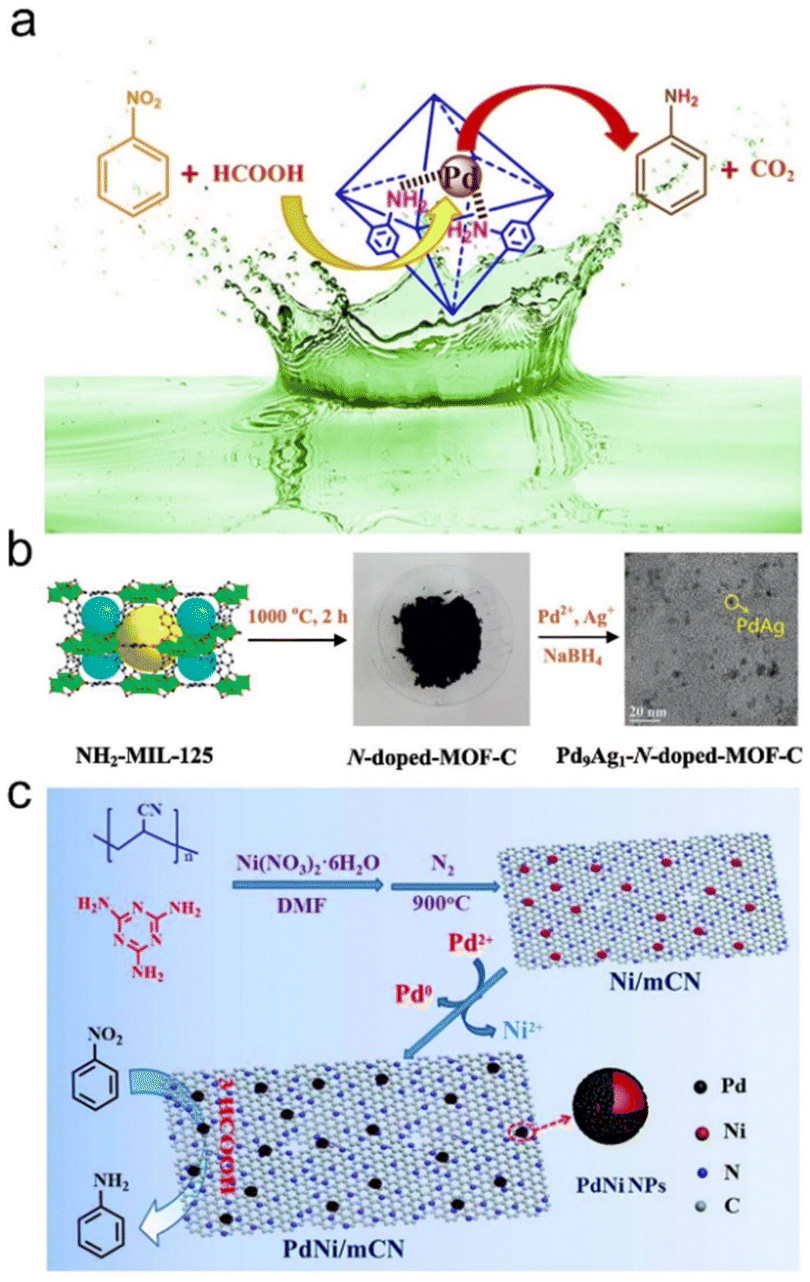 | ||
| Fig. 2 (a) Schematic illustration of the transfer hydrogenation of nitrobenzene on Pd nanoparticles supported on NH2-UiO-66. Reproduced and modified with permission from ref. 138. Copyright 2018 Elsevier. (b) Schematic illustration of the preparation progress of Pd9Ag1-N-doped-MOF-C. Reproduced with permission from ref. 139. Copyright 2017 Elsevier. (c) The preparation procedure and application of the PdNi/mCN nanocatalyst. Reproduced with permission from ref. 54. Copyright 2018 Royal Society of Chemistry. | ||
In addition to single metal sites, Pd-based bimetallic catalysts usually exhibit strong catalytic properties due to the electronic and geometric synergistic effects of bimetallic sites. Liu et al. prepared Pd–Ag bimetallic NPs on N-doped porous carbon by using one-step doped NH2-MIL-125 as the precursor. Due to the synergistic interaction between the support and Pd-Ag bimetallic NPs, the as-synthesized Pd9Ag1-N-doped-MOF-C catalyst exhibited excellent catalytic activity and selectivity for the catalytic transfer hydrogenation of nitroaromatics using formic acid as a renewable hydrogen donor (Fig. 2b).139 Cui et al. first prepared mesoporous N-doped carbon supported Ni NPs (Ni/mCN) through pyrolysis of a mixture of polyacrylonitrile, melamine and Ni(NO3)2·6H2O. Then, Ni/mCN was treated with Pd(AcO)2 to form a PdNi bimetallic NP catalyst (PdNi/mCN). The as-prepared PdNi/mCN catalysts showed higher catalytic activity at room temperature for the transfer hydrogenation of nitroarenes using formic acid as a reducing agent, which may be due to the high dispersion of PdNi bimetallic NPs, the maximum utilization rate of Pd atoms and the special structure of mesoporous N-doped carbon (Fig. 2c).54
Other precious metal-based NP catalysts. In addition to Pd-based catalysts, other precious metal materials have also been studied and they exhibited excellent catalytic performance. Li et al. developed a simple and reliable process for the selective transfer hydrogenation of semi-reduced alkynes using formic acid as a hydrogen source and supported Au as a catalyst, which exhibited high activity, selectivity and recyclability under mild reaction conditions (Fig. 3a).141 Furthermore, Zhang et al. developed a TiO2-supported Au catalyst coated with an N-doped porous carbon layer using a polydopamine-coating-carbonization strategy. For the conversion of 5-hydroxymethylfurfural to 5-methylfurfural, the obtained Au/TiO2@NPC showed high catalytic performance with a >95% 5-methylfurfural yield. The experimental results showed that the catalytic performance was related to the suitable combination of highly dispersed Au NPs in the core–shell nanostructure and good interfacial interaction (Fig. 3b).142
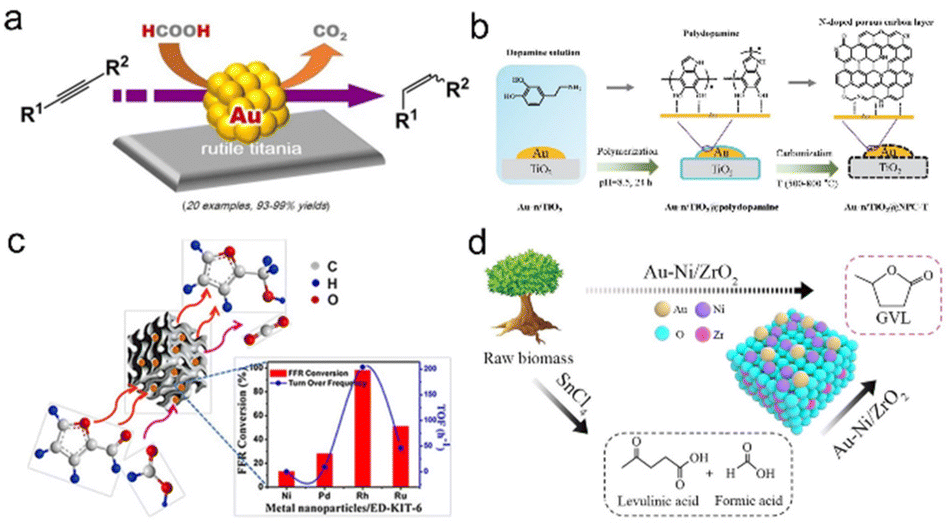 | ||
| Fig. 3 (a) Heterogeneous gold-catalyzed selective semireduction of alkynes. Reproduced with permission from ref. 141. Copyright 2016 Wiley-VCH. (b) Schematic illustration of the synthesis procedure of Au-n/TiO2@NPC-T. Reproduced with permission from ref. 142. Copyright 2022 Elsevier. (c) Ultrasmall Rh nanoparticles embedded on diamine-functionalized KIT-6. Reproduced with permission from ref. 143. Copyright 2017 Wiley-VCH. (d) γ-Valerolactone production from furfural via an integrated strategy on Au–Ni/ZrO2. Reproduced with permission from ref. 144. Copyright 2020 American Chemical Society. | ||
Besides, Neeli et al. synthesized the Rh/ED-KIT-6 catalyst with an average particle size of 1.2 nm by chemical reduction of the Rh3+ precursor on KIT-6. The synergistic effect of the nitrogen function and ultrafine Rh NPs endowed it with excellent catalytic activity for transfer hydrogenation of furfural to furfuryl alcohol with formic acid as a hydrogen source without other additives (Fig. 3c).143 Zhou et al. used formic acid as the only hydrogen resource to selectively synthesize γ-valerolactone from raw lignocellulosic biomass. The first step used SnCl4 as the catalyst to prepare levulinic acid and formic acid, and the second step used the developed Au-Ni/ZrO2 bimetallic catalyst to produce γ-valerolactone from levulinic acid. Compared to the Au/ZrO2 catalyst, an appropriate Ni loading amount can promote the reduction of Auδ+ to Au0 and the dispersion of Au0 without aggregation, which can increase the hydrogenation reaction and the subsequent dehydration reaction (Fig. 3d).144
Cobalt-based NP catalysts. The high price and scarcity of precious metal catalysts limit their industrial application, which urgently requires efficient design and precise preparation of nonprecious metals. However, nonprecious metal catalysts still face some challenges such as low activity and poor stability, and harsh reaction conditions such as high temperature inevitably increase overall energy consumption.40,128,145–148 Moreover, strong corrosion of formic acid also put forward a big challenge to improve the stability of nonprecious metal catalysts for hydrogenation reactions.
The carbon supported or coated Co catalysts were therefore used as highly efficient transfer hydrogenation samples when using formic acid as a hydrogen source. Beller's group pioneerly prepared cobalt oxide based catalyst (Co3O4-NGr@C) by pyrolysis of the cobalt–phenanthroline complex on commercial Vulcan XC72R. It achieved excellent performance for the transfer hydrogenation of nitroaromatics, olefins, aldehydes, ketones, esters, amides and nitriles. They also prepared a carbon supported nitrogen-modified cobalt catalyst by pyrolyzing a mixture of Co(AcO)2 in melamine or waste melamine resin solutions. The selective transfer hydrogenation of N-isoaromatic hydrocarbons can be achieved with formic acid as a hydrogen source and ethanol as the solvent. At the same time, this nonprecious Co catalyst also showed certain activity and selectivity of formic acid dehydrogenation (Fig. 4a).57,60 As far as carbon sources are concerned, melamine is a cheap and readily available carbon source and contains nitrogen species. Yuan et al. prepared the N-doped carbon coated Co NP catalyst (Co@NC) by using melamine and Co(AcO)2 through hydrothermal and carbonization treatment, this structure effectively avoided the corrosion and leaching of Co NPs. As a result, Co@NC greatly promoted the catalytic transfer hydrogenation of various functionalized nitroaromatic hydrocarbons with formic acid as a hydrogen donor in water/ethanol solution without alkali (Fig. 4b).61 Kar et al. prepared a porous N-doped carbon coated cobalt catalyst (Co@C-N800) using o-phenylenediamine (OPD) as C and N sources and melamine as additional N sources. Using formic acid as a hydrogen source, this catalyst achieved 92.1% quinoline conversion and 87% selectivity for 1,2,3,4-tetrahydroquinoline (Fig. 4c).62 Additionally, Zhou et al. prepared a multilayer N-doped graphene catalyst with highly dispersed cobalt NPs (Co@NG) using carboxymethyl cellulose as the raw material. Using urea as a non-corrosive activator can introduce porous strip nanostructures and abundant nitrogen. With formic acid as a hydrogen source, alkaline transfer hydrodeoxygenation can be realized with a 99% 2-methoxy-p-cresol yield on optimized Co@NG-6 with the highest proportion of pyridine-N. The superior catalytic performance can be attributed to the synergistic effect of the strong interaction between doped N atoms and coated Co NPs. Deuterium kinetic isotope experiments showed that protic N–H+ and hydrogenated Co–H− were two active intermediates. Finally, the graphene shell prevented the corrosion and aggregation of Co NPs under real reaction conditions (Fig. 4d).149 Cobalt-containing metal–organic frameworks (MOFs) such as ZIF-67 are often used as cobalt and carbon sources. For example, Jiang et al. used the ZIF-67/GO composite by depositing ZIF-67 on the GO layer to prepare the N-doped carbon-based cobalt catalyst. The transfer hydrogenation reaction can be performed on this catalyst using formic acid as a hydrogen donor and tetrahydrofuran as the solvent. The structurally diverse secondary amines were produced with excellent yields without reducing other functional groups (Fig. 4e).150
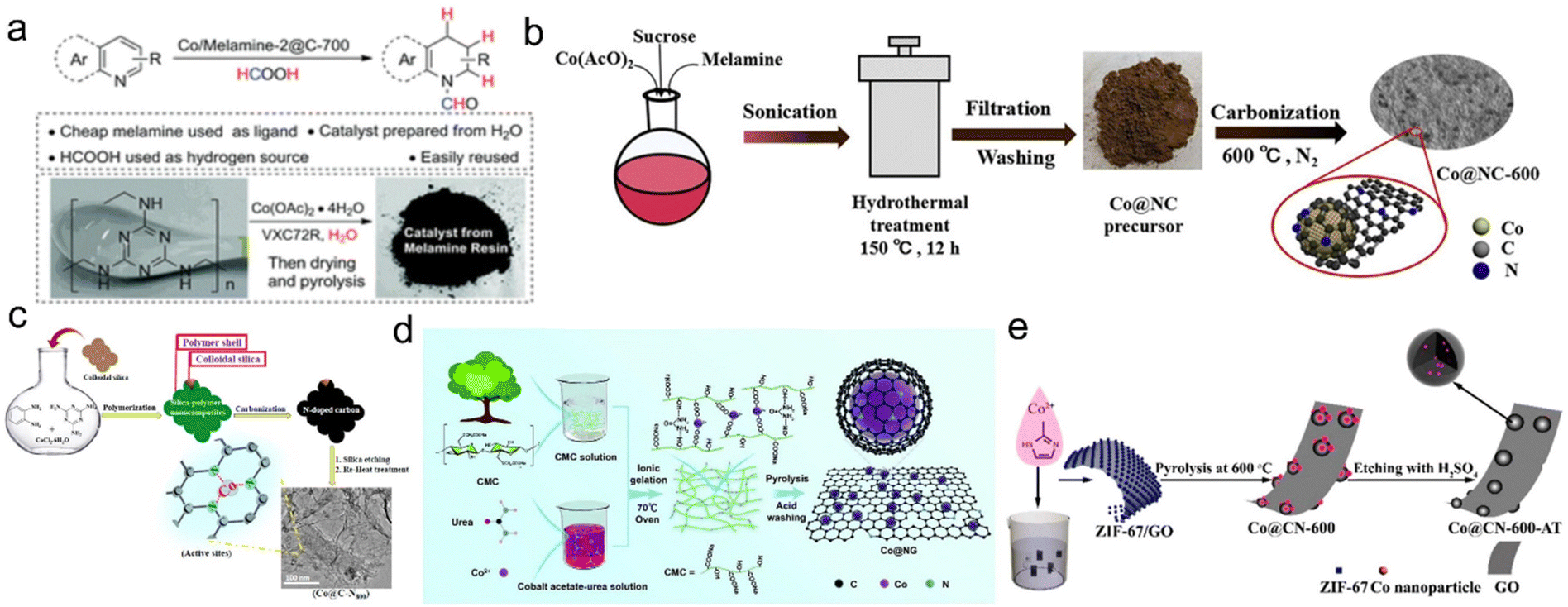 | ||
| Fig. 4 (a) Preparation of cobalt nanoparticles from melamine in water. Reproduced with permission from ref. 60. Copyright 2017 Royal Society of Chemistry. (b) Preparation of Co@NC catalysts. Reproduced with permission from ref. 61. Copyright 2018 Wiley-VCH. (c) Stepwise synthesis protocol adopted for the preparation of Co@C–Nx materials. Reproduced with permission from ref. 62. Copyright 2019 American Chemical Society. (d) The fabrication process of Co@NG. Reproduced with permission from ref. 149. Copyright 2019 Royal Society of Chemistry. (e) Schematic illustration of the synthesis of the Co@CN-600-AT catalyst. Reproduced with permission from ref. 150. Copyright 2017 American Chemical Society. | ||
The morphology and structure of carbon materials are also very important for catalytic hydrogenation performance. Xu et al. prepared Co NPs coated with the N-doped carbon nanotube catalyst (Co@NCNTs-800) through multiple pyrolyses of dicyandiamide and cobalt(II) acetylacetone. The outer nitrogen-doped carbon nanotubes can prevent the agglomeration and leaching of the enclosed Co NPs, and the defects formed by N doping were beneficial for embedding active molecules on the surface of Co NPs (Fig. 5a).151 Besides, some researchers have done much investigations about the N and S co-doped carbon supported Co materials. Zhu et al. prepared Co NPs coated with N and S co-doped carbon nanotubes by one-pot pyrolysis of melamine, thiocyanic acid and cobalt nitrate hexahydrate. The corrosion, leaching and agglomeration of Co NPs could be effectively avoided by double-heteroatomic co-doped porous carbon materials. They found that the synergistic interaction between Co NPs and N,S atoms can promote the ability of transfer hydrogenation and formylation of nitroaromatic hydrocarbons, especially the incorporation of S atom greatly improved the selectivity of the N-formylation reaction (Fig. 5b).152 In addition, Chen's group first synthesized an organic precursor (POPs) through the solvothermal condensation of melamine and terephthalic acid in dimethyl sulfoxide. Then pyrolysis of POPs impregnated with cobalt nitrate was used to successfully embed Co in a porous carbon catalyst with N,S co-doping. The catalyst can be used for the hydrogenation of nitrobenzene to aniline with formic acid as a hydrogen source without alkali. In their experiments, Co NPs were confirmed as the active sites of the reaction, and their electron transfer to the carbon shell was crucial to the hydrogenation performance. The incorporation of the S heteroatom in the N-doped carbon layer can improve the electron transfer process and the positive charge of Co, thus improving acid resistance. To further improve the hydrogenation performance of this catalyst, they further prepared N,S co-doped carbon embedded with Co NPs (1.14 wt%) by pyrolyzing the complex of the Schiff-base network (SNW-1) and Co(Ac)2 coated with glucose. It can be seen that the glucose coating prevented highly dispersed Co NPs from directly bonding with S species. Consequently, the catalyst showed excellent catalytic activity and stability for the catalytic transfer hydrogenation using formic acid as a hydrogen source in the alkali-free system, and thus generated multiple secondary amines with excellent yields through hydrogenation and reducing amination domino aggregate reaction (Fig. 5c and d).127,153 In addition to N,S co-doped carbon, Duan et al. developed N,P co-doped carbon coated Co NPs (Co@NPC-800) using cheap and easily available PPh3 as the P source via a simple and green synthesis method. When N and P were uniformly added to the carbon lattice, they showed that the functionalized nitroaromatics can be efficiently and selectively hydrogenated to the corresponding aniline under the condition of formic acid or ammonium formate as the hydrogen donor and tetrahydrofuran/water as the solvent (Fig. 5e).58 In addition to monometallic sites, bimetallic sites have also received some attention and developments. Duan et al. synthesized a novel Ni–Co–P/HAP amorphous alloy catalyst by the impregnation–reduction method, and achieved a high yield of 2-methoxy-4-methylphenol by hydrodeoxygenation of vanillin using formic acid as a hydrogen source and isopropyl alcohol as the solvent. They revealed that the Co site can increase the disorder of Ni-based amorphous alloy and reduce the agglomeration of the catalyst (Fig. 5f).154 Zhao et al. prepared a CoFeOx-modified CoFe bimetallic catalyst by high-temperature calcination with Co–Mg–Fe layered dihydroxide as the precursor. Under the condition of formic acid as the hydrogen source, the efficient transfer hydrogenation of 5-hydromethylfurfural to 2,5-dimethylfuran was achieved due to the synergistic interaction between bimetallic CoFe NPs, CoFeOx species with abundant defects and surface base sites, effectively promote the dehydrogenation of formic acid. Owing to the high degree of alloying, and strong interaction between CoFe NPs and the support, the as-prepared CoFe catalyst also exhibited excellent acid resistance.155 Yang et al. achieved the catalytic transfer hydrogenation of 5-hydroxymethylfurfural to the 2,5-dimethylfuran with a >90% yield on a carbon-supported nickel-cobalt catalyst (Ni–Co/C) using formic acid as a hydrogen donor.156
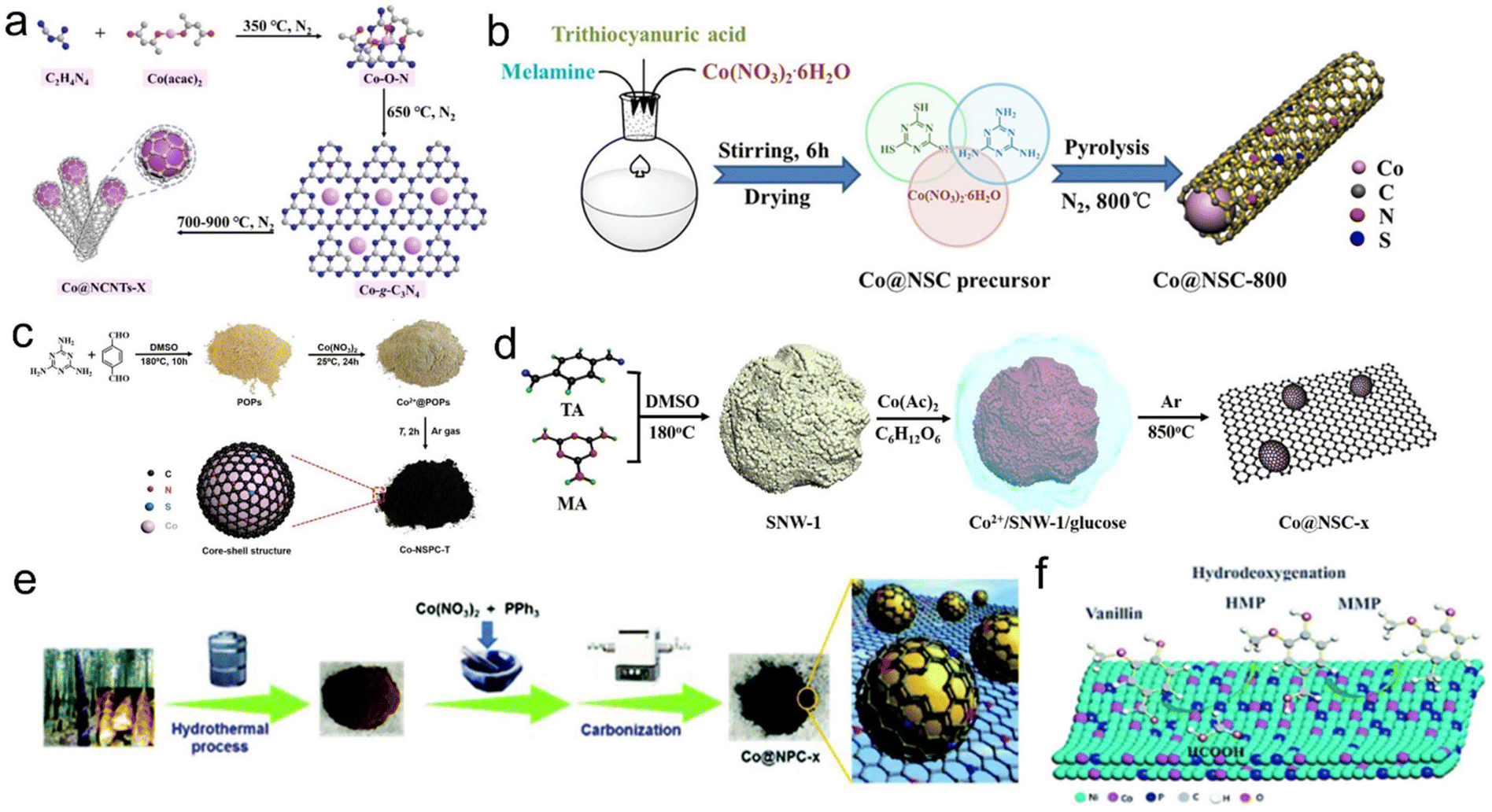 | ||
| Fig. 5 (a) Schematic of the synthesis process of Co@NCNTs-X. Reproduced with permission from ref. 151. Copyright 2019 Wiley-VCH. (b) Preparation process of the Co@NSC-800 catalyst. Reproduced with permission from ref. 152. Copyright 2020 American Chemical Society. (c) The fabrication of the Co-NSPC-T catalysts. Reproduced with permission from ref. 153. Copyright 2018 Wiley-VCH. (d) Diagram for the fabrication of Co@NSC-x. Reproduced and modified with permission from ref. 127. Copyright 2019 American Chemical Society. (e) Schematic illustration of the preparation of Co NPs coated with an N,P-codoped carbon shell. Reproduced with permission from ref. 58. Copyright 2018 Royal Society of Chemistry. (f) In situ hydrodeoxygenation of vanillin over Ni–Co–P/HAP. Reproduced with permission from ref. 154. Copyright 2021 Royal Society of Chemistry. | ||
Recently, with the rapid development of single-atomic materials, single-atomic catalysts have demonstrated excellent performance in transfer hydrogenation using formic acid as a hydrogen source. Lu's group used the Co(phen)2(OAc)2 complex as precursor and SBA-15 as a hard template to prepare an ordered mesoporous N-doped carbon-confined highly dispersed Co–Nx as the catalyst (Co–NC). Compared with direct H2 hydrogenation, formic acid inhibited the hydrogenation reaction during the formation of 2,5-furandimethyl alcohol. Subsequently, they further developed a cascade catalytic system (NiCl2/FA/Co-NC) to produce 2,5-furandimethanol from glucose and di/polysaccharide in a one-pot reaction (Fig. 6a).157,158 Han et al. successfully used template-assisted pyrolysis to stabilize isolated cobalt single-atomic sites on an ordered porous nitrogen-doped carbon (ISAS-Co/OPNC) using SBA-15 as a sacrificial template. When formic acid or exogenous hydrogen was used as a hydrogen source, both the release of H2 by N-heterocyclic dehydrogenation and the storage of H2 by the reverse transfer of N-heterocyclic hydrogenation (or hydrogenation) showed high catalytic activity (Fig. 6b).59 Huang et al. reported that a Co(acac)2 metal precursor was encapsulated in an aerotocan/sodium dodecyl sulfate hydrogel, and S,N co-doped carbon-supported Co single-atomic catalyst (Co/SNC) was synthesized by the microencapsulation pyrolysis method. The transfer hydrogenation of N-isoaromatic hydrocarbons was performed with formic acid as a hydrogen donor without acid leaching (Fig. 6c).159 Gong et al. prepared an N-doped carbon based Co catalyst (Co–Nx/C-800-AT) by pyrolyzing cobalt phthalocyanine-silica colloidal composite and removing silica templates and Co NPs by pickling, in which cobalt–nitrogen species (Co–Nx) was the active site of the reaction. N-substituted pyrrole compounds have been synthesized by the heterocyclic reaction of nitrocompounds with 2,5-hexadione using formic acid as a hydrogen source and acid catalyst using the Paal–Knorr shrinkage method (Fig. 6d).160 Zhang et al. proposed a template-assisted spatial restriction strategy for the preparation of ultra-high Co single-atomic sites (10.26 wt%) embedded N-doped graphene-like carbon by one-step pyrolysis. In the presence of formic acid, the optimum Co SSCs@NG-800-50 catalyst exhibited excellent catalytic performance for the transfer hydrogenation of nitrobenzene with a 99% conversion and 96.3% selectivity of aniline. DFT calculations combined with detailed experiments revealed that active H was first generated by the dissociation of the C–H bond in HCOOH rather than the known O–H bond, and then used for hydrogenation of nitroaromatics at the CoN3 site (Fig. 6e).161 Li et al. prepared cobalt single-atoms anchored on N-doped ultra-thin carbon nanosheets (CoSAs/NCNS) to achieve selective nitroaromatic hydrogenation with formic acid as a hydrogen donor and the turnover frequency (TOF) reached 110.6 h−1. Due to the ultra-thin mesoporous structure, the catalytic activity of CoSAs/NCNS was 20 times higher than that of transition metal Co NPs reported in the literature under similar reaction conditions (Fig. 6f).162
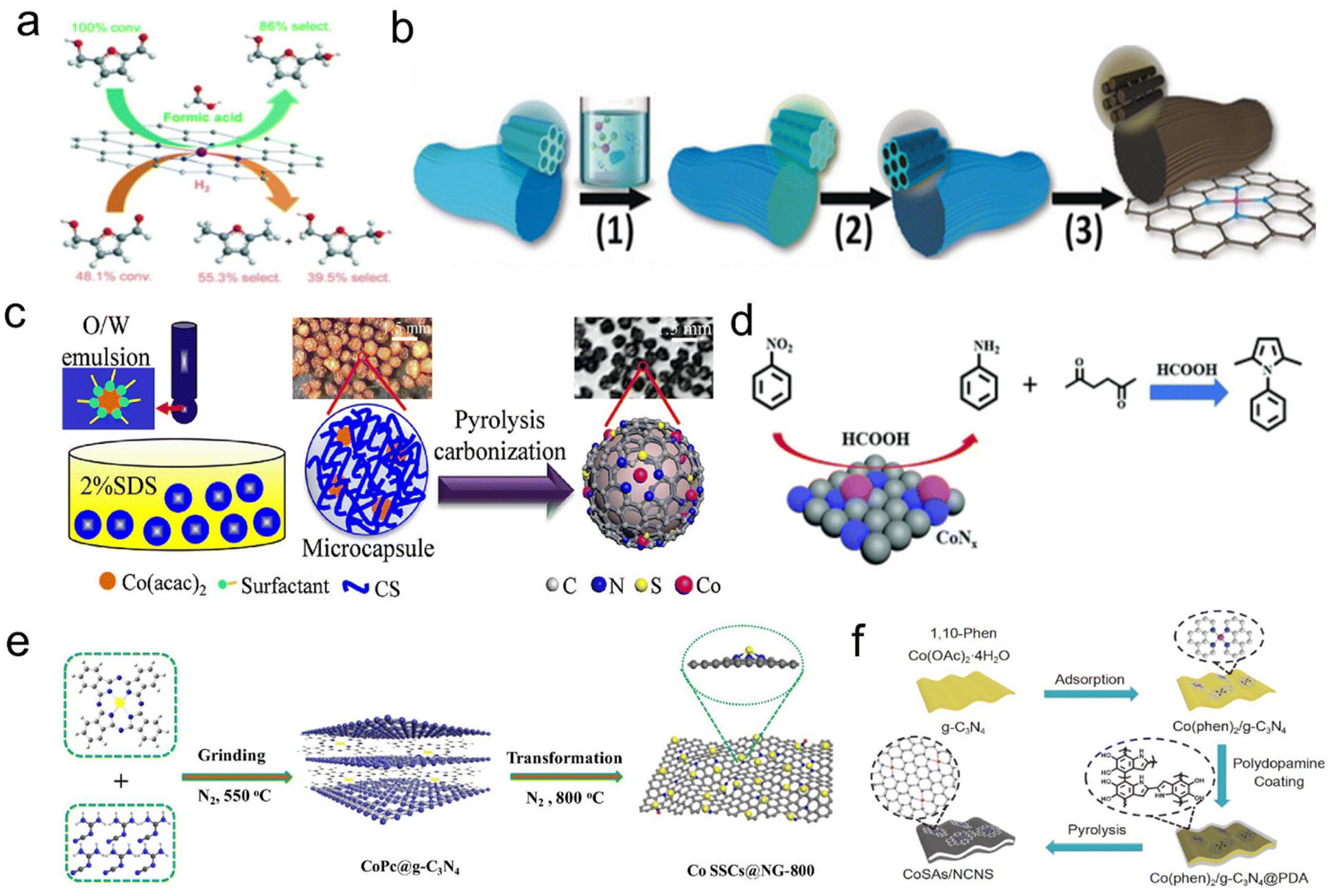 | ||
| Fig. 6 (a) Schematic illustration of the transfer hydrogenation on highly dispersed Co–Nx sites. Reproduced with permission from ref. 157. Copyright 2021 Royal Society of Chemistry. (b) Illustration of the synthesis of ISAS-Co/OPNC. Reproduced and modified with permission from ref. 59. Copyright 2018 Wiley-VCH. (c) Scheme of the quasi-continuous synthesis of Co/SNC. Reproduced and modified with permission from ref. 159. Copyright 2022 Elsevier. (d) Schematic illustration of the Co–Nx/C-800-AT catalyst. Reproduced with permission from ref. 160. Copyright 2017 Royal Society of Chemistry. (e) Schematic illustration for the preparation of Co SSCs@NC-800. Reproduced and modified with permission from ref. 161. Copyright 2021 Elsevier. (f) Schematic illustration of CoSAs/NCNS preparation. Reproduced with permission from ref. 162. Copyright 2019 Springer Nature. | ||
Other nonprecious metal-based NP catalysts. In addition to cobalt, other nonprecious metals such as Ni and Cu are also used in formic acid driven transfer hydrogenation reactions. Liu et al. prepared Ni/CeO2 catalysts by loading Ni oxides onto CeO2 nanorods, and the addition of Ni reduced the reduction temperature of the catalyst, forming the Ni–O–Ce interfacial site. In the hydrogenation of levulinic acid with formic acid as a hydrogen source, the Ni/CeO2 catalyst with 6.9 nm particle size and appropriate acidity distribution showed excellent catalytic performance, the conversion of levulinic acid was 73%, and the selectivity of γ-valerolactone was 90%.163 Wang et al. reported in situ encapsulation and stabilization of Cu/CuOx in a porous carbon matrix (Cu/CuOx@C). Under appropriate reaction conditions, the Cu/CuOx@C-450 catalyst showed a high furfural conversion of 99.1% with a 98% furfuryl alcohol selectivity due to the high dispersion of Cu/CuOx NPs and the suitable Cu0/Cun+ ratio.164 Lomate et al. found the Cu-SiO2 catalyst showed a higher levulinic acid conversion and valerolactone selectivity when using formic acid as a hydrogen source compared to that of only the SiO2 support. The key to the high activity and selectivity lies in the strong interaction of some copper oxide monomers with the support and the binding of more acid sites in Cu-SiO2.165 Bimetallic synergies also play a nonnegligible role in promoting nonprecious metal based systems. Lin's group investigated the catalytic conversion of 5-formyloxymethylfurfural to 2,5-dimethylfuran over the Ni–Cu bimetallic catalyst using formic acid as a hydrogen donor. They showed that copper was more likely to catalyze the hydrogenation of C–O ester bonds, while nickel species played a dual role in catalyzing the hydrogenation of C–O ester bonds and the hydrodehydration of hydroxyl groups in furan rings. They also developed a Cu–Cs bimetallic catalyst for the catalytic transfer hydrogenation of furfural to furfuryl alcohol. The Cu in CuCs-MCM had the dual function of catalyzing the decomposition of formic acid into hydrogen and hydrogenating furfural into furfuryl alcohol. Meanwhile, the doping of Cs decreased the size of Cu NPs and improved the dispersion of Cu active sites. As an accelerator, Ce played a good role in improving hydrogenation activity by regulating the surface acidity of Cu to an appropriate level (Fig. 7a and b).166,167
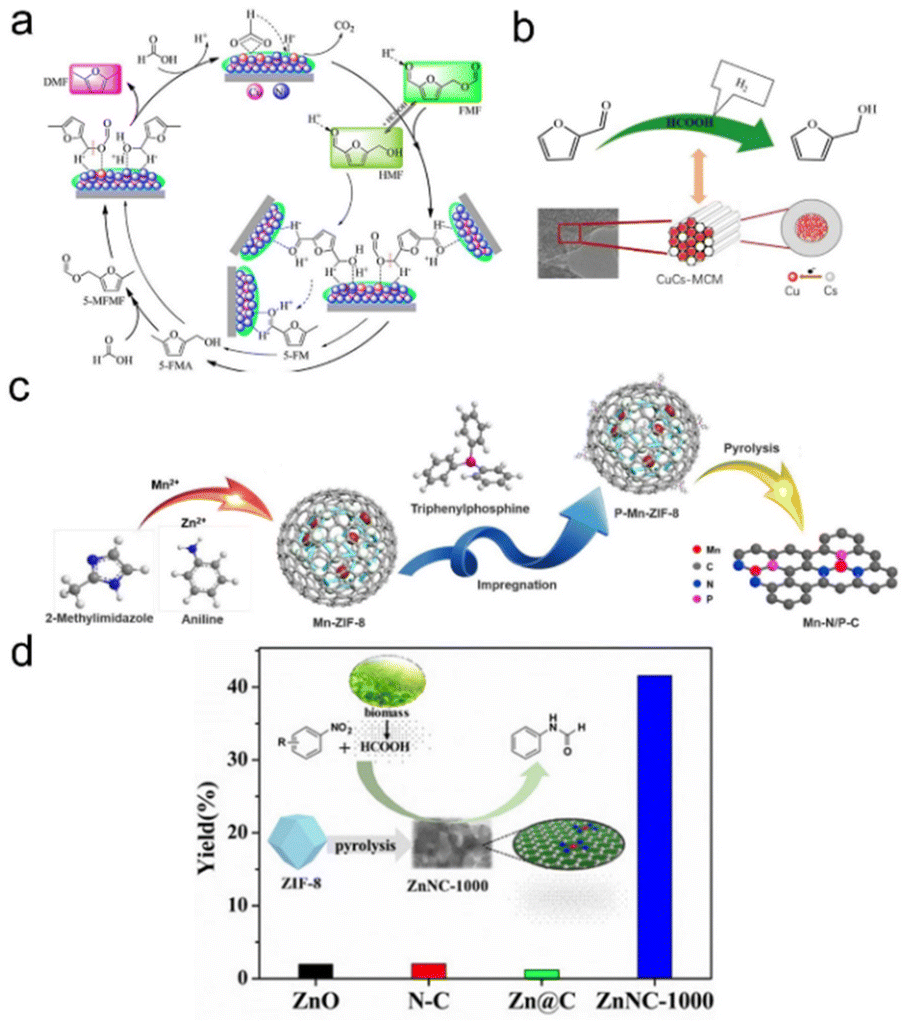 | ||
| Fig. 7 (a) Schematic illustration for the conversion of FMF to DMF over the Ni–Cu bimetallic catalyst. Reproduced with permission from ref. 166. Copyright 2019 American Chemical Society. (b) Catalytic transfer hydrogenation of furfural to furfuryl alcohol over the CuCs-MCM catalyst. Reproduced with permission from ref. 167. Copyright 2021 Elsevier. (c) Schematic illustration of the synthetic process for Mn–N/P–C. Reproduced and modified with permission from ref. 168. Copyright 2022 American Chemical Society. (d) Atomically dispersed Zn–Nx sites in N-doped carbon for reductive N-formylation of nitroarenes. Reproduced with permission from ref. 169. Copyright 2019 Wiley-VCH. | ||
The stability of nonprecious metal NPs in the formic acid system has always been a big problem, but the appearance of single-atomic catalysts can not only solve the stability problem but also improve the activity and selectivity. In this respect, Su et al. successfully prepared a porous carbon loaded N,P bi-coordination Mn single-atomic catalyst by phosphating a zeolite imidazole skeleton and subsequent pyrolysis. The Mn1–N/P–C catalysts with an Mn–N2–P atomic structure showed better catalytic activity than the related catalysts with an Mn–Nx structure for the transfer hydrogenation of nitroaromatics with formic acid as the donor. It was found that doping of the P source played an important role in affecting the morphology and electronic properties of the catalyst, thus improving the performance of the catalyst. This was the first example of a Mn heterogeneous catalyst for nitroaromatic reduction, and also revealed that the Mn–N2–P structure was a more promising alternative in heterogeneous catalysis (Fig. 7c).168 Li et al. prepared a highly active, inexpensive and stable single-atomic Zn/N-doped porous carbon catalyst using a zeolite imidazoline skeleton precursor. The optimized ZnNC-1000 had a higher Zn content (5.2 wt%) and higher nitrogen content (6.73 wt%), thus showing promising catalytic performance in N-formylation of nitroaromatics in one pot with formic acid as the hydrogen donor and formylation agent. H2–D2 exchange reactions and HCOOH chemisorption experiments showed that atomically dispersed Zn–Nx species were essential for the activation of hydrogen and HCOOH molecules, which ultimately resulted in the highest catalytic activity for reducing carbamylation (Fig. 7d).169
2.2. Photocatalytic hydrogenation catalysts
Efficient and cost-effective usage of solar energy represents an extremely promising technology to solve global issues of energy and environment, thus realizing carbon neutrality.170–176 Among them, photocatalytic hydrogenation has been regarded as an important strategy to convert solar energy into hydrogen energy and some key chemicals, involving three primary steps: light absorption, charge separation and transfer and subsequent catalytic hydrogenation.22,23,177–180 However, the current systems still suffer from low conversion efficiency. The key scientific problem lies in how to design and develop highly efficient photocatalysts to improve the efficiency of each step and the whole process.Lu et al. used SAPO-31 supported Pd single atoms and ultra-small cluster catalyst (PdSA+C/SAPO-31) to achieve hydrodeoxygenation of vanillin through a simple photochemical pathway. Under mild conditions (1 atm, 80 °C, 30 min), the selectivity of 2-methoxy-4-methylphenol and TOF were >99% and 3000 h−1 using formic acid as a hydrogen source, respectively. The high activity of the catalyst was due to the synergistic catalysis between the positively charged Pd single atoms and the fully exposed clusters and the strong metal–support interactions (Fig. 8a).181 Tsutsumi et al. prepared a transition metal-supported silicon NP catalyst that achieved a photoinduced reduction of nitrobenzene to aniline using formic acid as a hydrogen source. By comparing M/Si (M = Pt, Ru, and Pd), Pd/Si was the most suitable catalyst for the photocatalytic conversion of nitrobenzene to aniline. They revealed that formic acid acted both as a hydrogen source and as a sacrificial reagent to introduce electrons into the semiconductor to form holes, and avoided the use of high-pressure equipment (Fig. 8b).182 Considering that MOFs usually have high surface areas, high porosities and discrete active sites, they can therefore achieve efficient light absorption and charge transport by various modifications. Dong et al. prepared the Pd/MIL-101(Fe)-NH2 catalyst with highly dispersed and uniform Pd NPs (∼1.8 nm) by an in situ photodeposition method. The dual role of the amine group on the support was embodied in stabilizing Pd NPs and enhancing the electron density in the Pd center. Under the conditions of triethylamine as the electron donor and HCOOH as the proton source, visible light induced selective transfer hydrogenation of aromatic aldehydes to produce corresponding alcohols was successfully realized (Fig. 8c).183 Kar et al. prepared a visible-light photosensitive catalyst (Pd/Ti-MOF) by embedding Pd NPs in a titanium MOF, which can be used for photocatalytic reductive N-formylation of nitroarenes in water. The high photocatalytic performance of Pd/Ti-MOF can be attributed to the direct contact between Pd NPs and MOF, resulting in an abundance of catalytically active sites as well as superior electrical conductivity for a fast electron transfer process (Fig. 8d).184
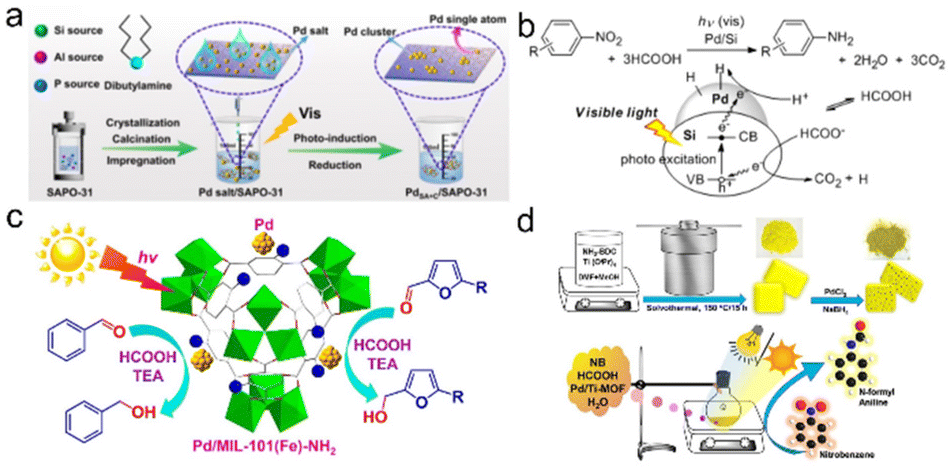 | ||
| Fig. 8 (a) Schematic illustration for the construction and characterization of PdSA + C/SAPO-31. Reproduced and modified with permission from ref. 181. Copyright 2021 Springer Nature. (c) Photoinduced reduction of nitroarenes using a transition-metal-loaded silicon semiconductor. Reproduced with permission from ref. 182. Copyright 2016 American Chemical Society. (b) Photoinduced transfer hydrogenation of aromatic aldehydes by using Pd/MIL-101(Fe)-NH2. Reproduced with permission from ref. 183. Copyright 2018 American Chemical Society. (d) Schematic Illustration of the synthesis of the Pd/Ti-MOF and the reaction setup used for the reductive N-formylation of NB. Reproduced with permission from ref. 184. Copyright 2022 American Chemical Society. | ||
3. Transfer hydrogenation using methanol as a hydrogen source
3.1. Catalyst design for thermocatalytic hydrogenation
In addition to formic acid, methanol produced from CO2 direct reduction and petroleum and biomass chemical industry is also considered as a good hydrogen donor since it is inexpensive and easy to handle, involving environmentally benign by-products, do not require elaborate experimental setups, such as high pressure reactors, and are more stable than molecular hydrogen.23,103,185 Compared with formic acid, methanol is less corrosive to experimental equipment and catalysts.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation maintained a good balance in the transfer hydrogenation reaction.188
O hydrogenation maintained a good balance in the transfer hydrogenation reaction.188
Copper-based NP catalysts. Due to the superior activity of copper in hydrogen production from methanol, copper based catalysts have received the most attention in methanol promoted transfer hydrogenation. Yfanti et al. investigated the role of different hydrogen donors in the liquid-phase hydrogenation of glycerol to deoxypropylene glycol over the Cu
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:Al catalyst. They found that methanol was the best H donor (74% yield), followed by 2-propyl alcohol (59.6% yield) in terms of hydrogen production and 1,2-propylene glycol production. Formic acid was also a potential H donor, since its activity in catalyzing hydrogen transfer was more favorable at low H2 donor/glycerol molar ratios (Fig. 9a).189 Tang et al. performed the hydrocyclization of methyl levulinate in methanol on a nano-Cu catalyst without external H2. The nanocrystalline Cu catalyst can be used as a bifunctional catalyst for hydrogen production by methanol reforming and methyl levulinate hydrogenation.190 Zhang et al. synthesized a series of Cu-based catalysts with different supports (Cu/Al2O3, Cu/ZnO, Cu/ZrO2 and Cu/CeO2) to study the in situ hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran using methanol as an economic hydrogen supplier. The results showed that different supports exhibited significant effects on the activity of methanol dehydrogenation and 5-hydroxymethylfurfural hydrogenation of Cu-based catalysts. The best catalytic activity of Cu/Al2O3 with the smallest grain size of Cu and the strongest acidity was attributed to the highest in situ hydrogen production activity of methanol.191 Awan et al. tested a copper–nickel mixed catalyst prepared by calcination of iron and aluminium hydrotalcites (layered double hydroxides, LDH) in the conversion of a lignin model dimer in subcritical methanol. The presence of copper was essential for efficient hydrogenation, either through the direct transfer of hydrogen from methanol to aldehyde groups or through reactivity of methanol reforming products. TPR experiments showed that the enhanced reducibility of Cu catalysts promoted the hydrogenation activity, which was related to the presence of other oxide components. They showed that copper was formed by the reduction of CuO by methanol, and the modification of the oxide catalyst in the reaction medium played a major role in the formation of the active site.192 Zhang et al. synthesized a ZrO2-supported nano-Cu catalyst (monooblique ZrO2: m-ZrO2; tetragonal ZrO2: t-ZrO2). In the methanol–water system, the catalytic performance of the Cu/t-ZrO2 catalyst for lauric acid hydrogenation was significantly better than that of the Cu/m-ZrO2 catalyst. The low efficiency of m-ZrO2 as a support was attributed to the weak adsorption of reactants and intermediate molecules and the absence of oxygen vacancy in the crystal phase. The DFT calculations further indicated that the metal–support interface played an important role in promoting the cleavage of C–O or H–H bonds in both possible reaction paths, thereby reducing the activation barrier of the whole reaction (Fig. 9b).26 Zhang et al. screened a series of cheap Cu-based catalysts from hydrotalcite precursors using methanol as the solvent and hydrogen donor to selectively transfer hydrogenate furfural to furfuryl alcohol and 2-methylfuran. The Cu-based catalyst had good selectivity for transfer hydrogenation of furfural to furfuryl alcohol, and the furfuryl alcohol yield in 473 K methanol was 94.0 mol%. The Cu catalyst used CuO and Cu2O as catalysts and methanol as a hydrogen source to conduct transfer hydrogenation of furfural. The characterization and experimental results showed that the conversion of furfural–furfuryl alcohol and furfural-2-methylfuran depended largely on the hydrogen production, and furfural-2-methylfuran was further treated with H2/N2 at 773 K and Cu was formed in the activated sample. Interestingly, the resulting activated copper catalyst was favorable for the conversion of furfural to 2-methylfuran with a yield of 94.1 mol% at 513 K.193 Zhang et al. determined the capacity of methanol reforming and the adsorption strength of furfural on CuOx. DFT calculations showed that the CuO (200) surface had low free energy and energy barrier for methanol reforming and furfural transfer hydrogenation (Fig. 9c).194 Besides, Li et al. prepared the Cu0/Cu2O·SiO2 catalyst, which can selectively generate 2-methylfuran with a yield of 90% from furfural at 220 °C within 2 h. Methanol was used in situ to produce high purity hydrogen (92% content), and the nearly CO free atmosphere avoided toxicity at Cu sites. The structure–activity relationship showed that Cu0 or Cu2O or the physical mixture of Cu0 and Cu2O had no activity, but the hydrogen-reduced layered copper silicate samples with a rich Cu0/Cu2O·SiO2 interface had high activity in both methanol decomposition and furfural hydrodeoxygenation reactions.195
:Zn:Al catalyst. They found that methanol was the best H donor (74% yield), followed by 2-propyl alcohol (59.6% yield) in terms of hydrogen production and 1,2-propylene glycol production. Formic acid was also a potential H donor, since its activity in catalyzing hydrogen transfer was more favorable at low H2 donor/glycerol molar ratios (Fig. 9a).189 Tang et al. performed the hydrocyclization of methyl levulinate in methanol on a nano-Cu catalyst without external H2. The nanocrystalline Cu catalyst can be used as a bifunctional catalyst for hydrogen production by methanol reforming and methyl levulinate hydrogenation.190 Zhang et al. synthesized a series of Cu-based catalysts with different supports (Cu/Al2O3, Cu/ZnO, Cu/ZrO2 and Cu/CeO2) to study the in situ hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran using methanol as an economic hydrogen supplier. The results showed that different supports exhibited significant effects on the activity of methanol dehydrogenation and 5-hydroxymethylfurfural hydrogenation of Cu-based catalysts. The best catalytic activity of Cu/Al2O3 with the smallest grain size of Cu and the strongest acidity was attributed to the highest in situ hydrogen production activity of methanol.191 Awan et al. tested a copper–nickel mixed catalyst prepared by calcination of iron and aluminium hydrotalcites (layered double hydroxides, LDH) in the conversion of a lignin model dimer in subcritical methanol. The presence of copper was essential for efficient hydrogenation, either through the direct transfer of hydrogen from methanol to aldehyde groups or through reactivity of methanol reforming products. TPR experiments showed that the enhanced reducibility of Cu catalysts promoted the hydrogenation activity, which was related to the presence of other oxide components. They showed that copper was formed by the reduction of CuO by methanol, and the modification of the oxide catalyst in the reaction medium played a major role in the formation of the active site.192 Zhang et al. synthesized a ZrO2-supported nano-Cu catalyst (monooblique ZrO2: m-ZrO2; tetragonal ZrO2: t-ZrO2). In the methanol–water system, the catalytic performance of the Cu/t-ZrO2 catalyst for lauric acid hydrogenation was significantly better than that of the Cu/m-ZrO2 catalyst. The low efficiency of m-ZrO2 as a support was attributed to the weak adsorption of reactants and intermediate molecules and the absence of oxygen vacancy in the crystal phase. The DFT calculations further indicated that the metal–support interface played an important role in promoting the cleavage of C–O or H–H bonds in both possible reaction paths, thereby reducing the activation barrier of the whole reaction (Fig. 9b).26 Zhang et al. screened a series of cheap Cu-based catalysts from hydrotalcite precursors using methanol as the solvent and hydrogen donor to selectively transfer hydrogenate furfural to furfuryl alcohol and 2-methylfuran. The Cu-based catalyst had good selectivity for transfer hydrogenation of furfural to furfuryl alcohol, and the furfuryl alcohol yield in 473 K methanol was 94.0 mol%. The Cu catalyst used CuO and Cu2O as catalysts and methanol as a hydrogen source to conduct transfer hydrogenation of furfural. The characterization and experimental results showed that the conversion of furfural–furfuryl alcohol and furfural-2-methylfuran depended largely on the hydrogen production, and furfural-2-methylfuran was further treated with H2/N2 at 773 K and Cu was formed in the activated sample. Interestingly, the resulting activated copper catalyst was favorable for the conversion of furfural to 2-methylfuran with a yield of 94.1 mol% at 513 K.193 Zhang et al. determined the capacity of methanol reforming and the adsorption strength of furfural on CuOx. DFT calculations showed that the CuO (200) surface had low free energy and energy barrier for methanol reforming and furfural transfer hydrogenation (Fig. 9c).194 Besides, Li et al. prepared the Cu0/Cu2O·SiO2 catalyst, which can selectively generate 2-methylfuran with a yield of 90% from furfural at 220 °C within 2 h. Methanol was used in situ to produce high purity hydrogen (92% content), and the nearly CO free atmosphere avoided toxicity at Cu sites. The structure–activity relationship showed that Cu0 or Cu2O or the physical mixture of Cu0 and Cu2O had no activity, but the hydrogen-reduced layered copper silicate samples with a rich Cu0/Cu2O·SiO2 interface had high activity in both methanol decomposition and furfural hydrodeoxygenation reactions.195
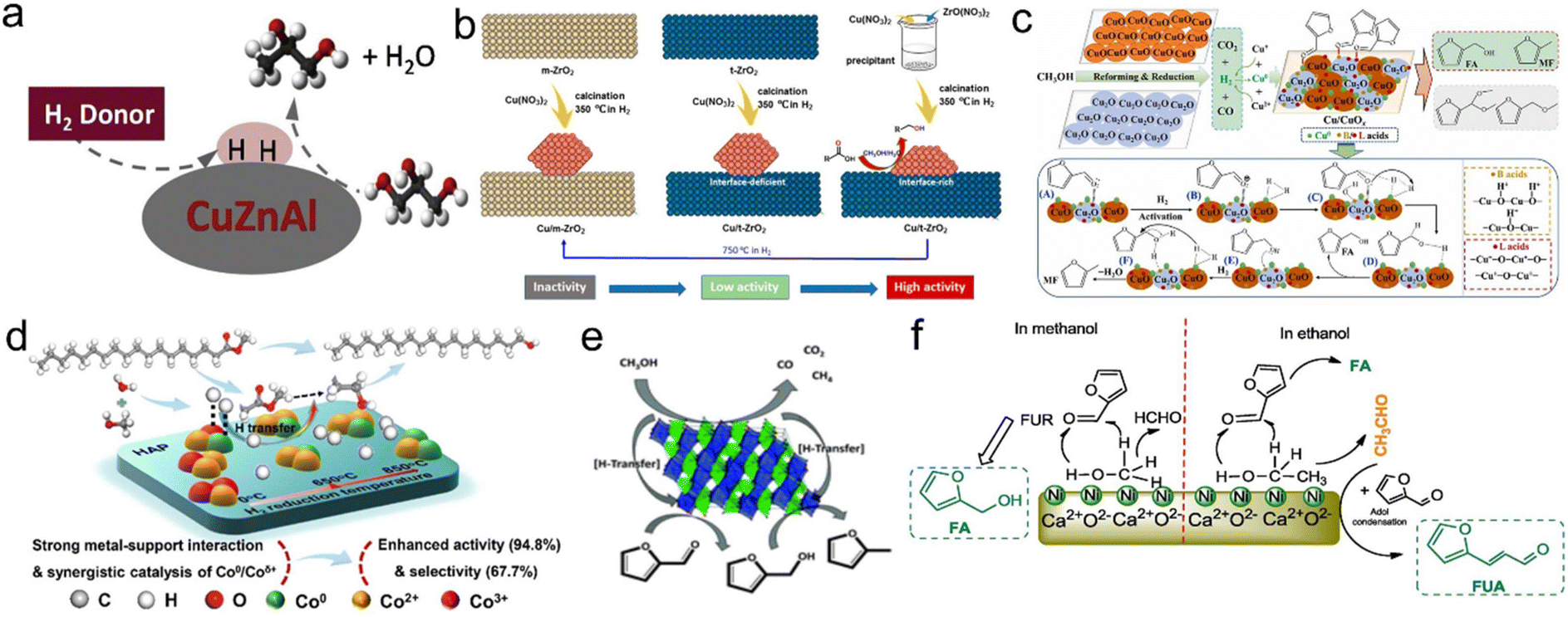 | ||
| Fig. 9 (a) Hydrodeoxygenation of glycerol to 1,2-propanediol over the Cu:Zn:Al catalyst. Reproduced and modified with permission from ref. 189. Copyright 2020 Elsevier. (b) Schematic diagram of the synthesis of Cu/ZrO2via different preparation methods and reduction temperatures. Reproduced with permission from ref. 26. Copyright 2020 American Chemical Society. (c) The catalytic mechanism for methanol-mediated H-transfer transformation of furfural over CuOx. Reproduced with permission from ref. 194. Copyright 2022 Elsevier. (d) Schematic illustration of the catalytic transfer hydrogenation over synergistic Co/HAP catalysts. Reproduced with permission from ref. 196. Copyright 2021 American Chemical Society. (e) The gas-phase production of 2-methylfurfural using a FeVO4 catalyst. Reproduced with permission from ref. 197. Copyright 2017 Royal Society of Chemistry. (f) The catalytic reaction mechanism for conversion of FUR in aliphatic alcohol. Reproduced with permission from ref. 198. Copyright 2019 Elsevier. | ||
In addition to the copper site, Yao et al. studied the catalytic transfer hydrogenation of methyl stearate to octadecyl alcohol with methanol and water as hydrogen donors over the Co/hydroxyapatite catalyst. Characterization and DFT calculations showed that the strong interaction between Co and hydroxyapatite support in the catalyst enhanced the electron transfer ability, and the combination of Co0/Coδ+ active sites can synergically promote H2 formation and hydrogenation of methyl stearate in the methanol/water system (Fig. 9d).196 Grazia et al. studied the process of producing 2-methylfuran in the gas phase of furfural using methanol as H transfer agent and FeVO4 as the catalyst. At 320 °C, the yield of 2-methylfuran can reach 80%, with small amounts of 2,5-dimethylfuran and 2-vinylfuran as byproducts. The characterization of the catalyst indicated that the reduction of FeVO4 occurred under real conditions, leading to the in situ development of the truly active phase (Fig. 9e).197 Zhang et al. prepared a novel Ni@R–Ca catalyst for catalytic transfer hydrogenation of furfural under mild reaction conditions. With methanol as the reaction medium, furfuryl alcohol can be obtained as the only product with a 99% yield. The synergistic effect of metal Ni and R–Ca lattice played a crucial role in the efficient transformation of furfural (Fig. 9f).198 Luo et al. investigated the synergistic effect of Fe and Ni over Ni–Fe/SBA-15 catalysts for the synthesis of γ-valerolactone from levulinic acid with methanol as the only hydrogen donor. The results of H2-TPD and XPS indicated that electron transfer occurred from Fe to Ni, which was favorable for the activation of the CO bond and dissociative activation of the H2 molecule.199
3.2 Photocatalytic hydrogenation catalysts
First, pure semiconductors such as TiO2 show excellent conversions and selectivities for this photocatalytic hydrogenation. Wu et al. found that selective conversion of the key lignocellulosic platform compounds such as furfural and vanillin could be achieved through photocatalysis by controlling the TiO2 exposure surface. The surface dependent density of oxygen vacancy determined the charge distribution and adsorption strength of surface species, thus controlling the selectivity of products (Fig. 10a).200 Besides, Zhao et al. designed a photocatalytic transfer hydrogenation system using solar energy as energy input and methanol as the hydrogen source. In this respect, the Pd–Pt/TiO2 bimetallic alloy catalyst showed significantly better photocatalytic performance than Pd/TiO2 or Pt/TiO2 alone. They revealed holes with strong oxidation ability spread to the surface of TiO2, triggering the oxidation of adsorbed methanol into formaldehyde or other deep oxidation products, accompanied by the release of hydrogen ions. Photoinduced electrons on the surface of the precious metal reduced protons to hydrogen atoms, thus forming surface hydrides that can react with the unsaturated CO or CC groups of the substrate molecules adsorbed on the surface (Fig. 10b).201 Jian et al. developed a new strategy to control the activity/selectivity of biomass reduction upgrading to biofuels at room temperature by engineering oxygen vacancies and manipulating the exposed crystal surfaces of TiO2-based catalysts. The (101) crystal plane was more likely to generate more oxygen vacancies in situ than the (001) and (110) crystal planes. And the formation of oxygen vacancies was not only beneficial for the adsorption and activation of biobased carbonyl, but also can improve the migration efficiency of photogenerated carrier, effectively adjusting the semiconductor band structure of the TiO2-based photocatalyst and promoting visible light absorption, and thus improve the overall reduction and transformation process (Fig. 10c).202
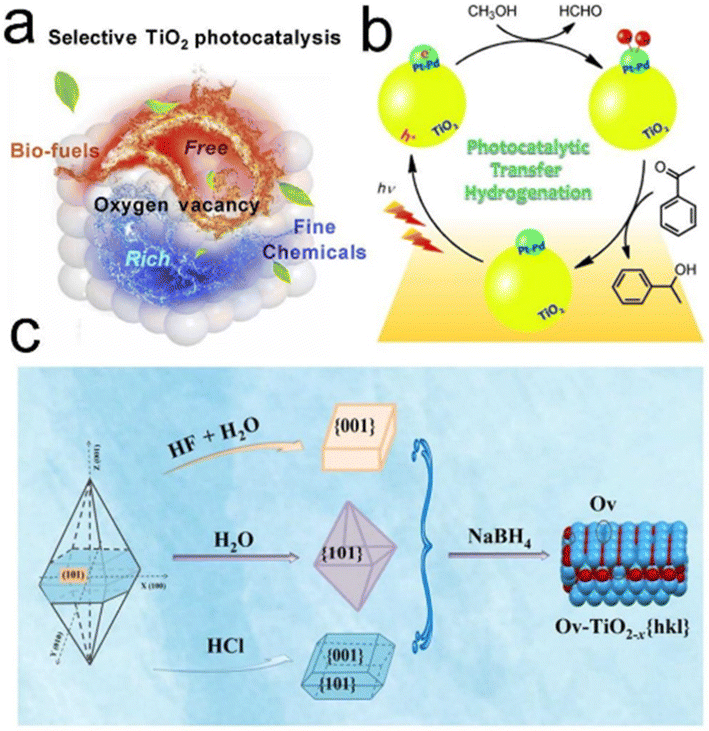 | ||
| Fig. 10 (a) Schematic illustration for the selectivity control by surface engineering of titanium oxide. Reproduced with permission from ref. 200. Copyright 2020 Elsevier. (b) The catalytic reaction mechanism for transfer hydrogenation on the Pd–Pt alloy cocatalyst. Reproduced with permission from ref. 201. Copyright 2014 Wiley-VCH. (c) Preparation schematic of TiO2-based catalysts with different crystal surfaces and specific oxygen vacancy (Ov). Reproduced with permission from ref. 202. Copyright 2022 Elsevier. | ||
4. Conclusion
In this review, we have presented the recent progress in the development of heterogeneous catalysts with excellent catalytic performance for transfer hydrogenation reactions using formic acid and methanol as the hydrogen sources by thermocatalytic and photocatalytic strategies. Many efforts have been made to realize the rational design and synthesis of highly efficient catalysts. However, there are some challenges and opportunities for future work in this area as follows (Scheme 4):For transfer hydrogenation using formic acid as the hydrogen source, the catalyst stability remains the biggest challenge due to the corrosive nature of formic acid solutions, especially for the nonprecious ones. In order to reduce coke deposition and metal leaching, strengthening the interactions between metals and supports or adding some protective layers such as carbon layers will be effective, but it will inevitably affect the catalytic activity. Therefore, it is a key scientific issue to develop new synthetic routes to achieve simultaneous improvement of activity and stability. Given the cost and scarcity, a nonprecious metal catalyst is a better choice, and a single-atom catalyst may be a big breakthrough. Photocatalysis or electrocatalysis technology also needs to be introduced into the system to solve the problem of excessive energy consumption and environmental pollution, but it needs separation and purification technologies.
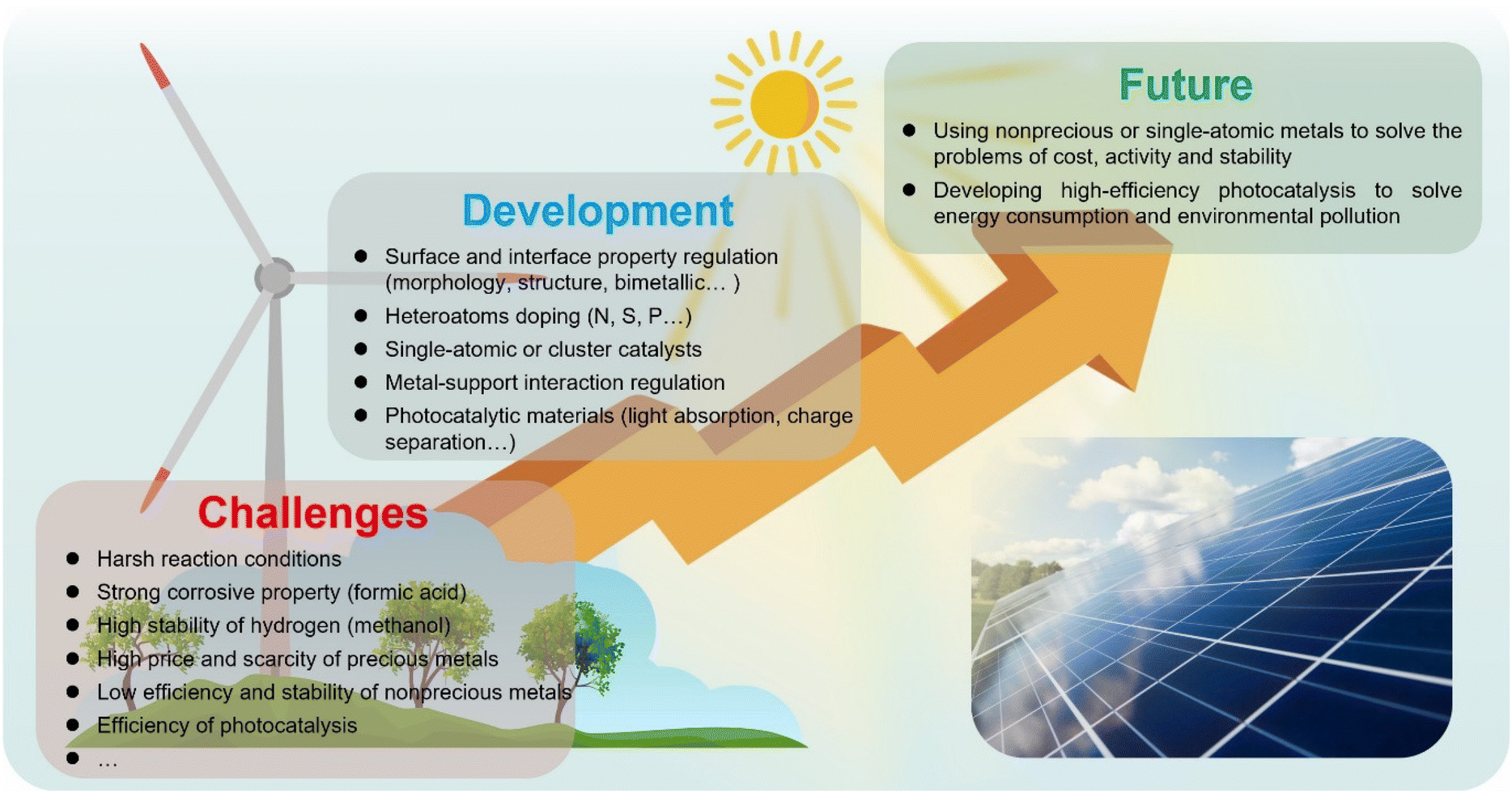 | ||
| Scheme 4 The proposed challenges and opportunities for the development of heterogeneous catalysts in this area. | ||
For transfer hydrogenation using methanol as the hydrogen source, the decomposition of methanol to produce hydrogen is a difficult issue. The methanol systems usually require harsh reaction conditions such as high reaction temperature, which require the design and synthesis of efficient catalysts to make the reaction conditions milder. Recently, photocatalytic hydrogen generation of methanol has been developed rapidly, which means that photocatalytic technology may be a way to achieve this kind of transfer hydrogenation. The catalyst stability is also an important issue for practical applications, and it is a feasible strategy to develop catalysts with a special structure which is conducive to strengthening metal–support interactions.
Finally, we hope that our review will provide a clearer picture for researchers in related fields, as well as serve as a guide for future research direction.
Author contributions
YX, RL and WG conceptualized and designed the review's theme and framework. GC, JM, JL and ZL conducted data collection and literature analysis. GC, JM and WG wrote the original draft. All authors reviewed and edited the draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2020YFA0406103), NSFC (21725102, 51902311, 22122506, 91961106, 22075267, 22232003, and 22109148), the Strategic Priority Research Program of the CAS (XDPB14), the Anhui Provincial Natural Science Foundation (2008085J05), the Youth Innovation Promotion Association of CAS (2019444), the Users with Excellence Program of Hefei Science Center CAS (2020HSC-UE003) and the Fundamental Research Funds for the Central Universities (WK2060000039 and 20720220007).References
- S. Pang, S. Jin, F. Yang, M. Alberts, L. Li, D. Xi, R. G. Gordon, P. Wang, M. J. Aziz and Y. Ji, Nat. Energy, 2023, 8, 1126–1136 CrossRef CAS.
- H. Seo and T. A. Hatton, Nat. Commun., 2023, 14, 313 CrossRef CAS PubMed.
- Y. S. Xia, M. Z. Tang, L. Zhang, J. Liu, C. Jiang, G. K. Gao, L. Z. Dong, L. G. Xie and Y. Q. Lan, Nat. Commun., 2022, 13, 2964 CrossRef CAS PubMed.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- C. J. Hu, Z. W. Jiang, Q. Y. Wu, S. Y. Cao, Q. H. Li, C. Chen, L. Y. Yuan, Y. L. Wang, W. Y. Yang, J. B. Yang, J. Peng, W. Q. Shi, M. L. Zhai, M. Mostafavi and J. Ma, Nat. Commun., 2023, 14, 4767 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- H. Chen, H. Dong, Z. Y. Shi and A. K. SenGupta, Sci. Adv., 2023, 9, eadg1956 CrossRef CAS PubMed.
- J. X. Zhu, J. T. Li, R. H. Lu, R. H. Yu, S. Y. Zhao, C. B. Li, L. Lv, L. X. Xia, X. B. Chen, W. W. Cai, J. S. Meng, W. Zhang, X. L. Pan, X. F. Hong, Y. H. Dai, Y. Mao, J. Li, L. Zhou, G. J. He, Q. Q. Pang, Y. Zhao, C. Xia, Z. Y. Wang, L. M. Dai and L. Q. Mai, Nat. Commun., 2023, 14, 4670 CrossRef CAS PubMed.
- S. Yang, H. Y. An, S. Arnouts, H. Wang, X. Yu, J. de Ruiter, S. Bals, T. Altantzis, B. M. Weckhuysen and W. van der Stam, Nat. Catal., 2023, 6, 796–806 CrossRef CAS.
- T. Sengupta and S. N. Khanna, Commun. Chem., 2023, 6, 53 CrossRef CAS PubMed.
- J. J. Su, C. B. Musgrave, Y. Song, L. B. Huang, Y. Liu, G. Li, Y. E. Xin, P. Xiong, M. M. J. Li, H. R. Wu, M. H. Zhu, H. M. Chen, J. Y. Zhang, H. C. Shen, B. Z. Tang, M. Robert, W. I. I. I. Goddard and R. Q. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- H. Zhou, Z. X. Chen, A. V. López, E. D. López, E. Lam, A. Tsoukalou, E. Willinger, D. A. Kuznetsov, D. Mance, A. Kierzkowska, F. Donat, P. M. Abdala, A. Comas-Vives, C. Copéret, A. Fedorov and C. R. Müller, Nat. Catal., 2021, 4, 860–871 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- J. Lim, A. T. Garcia-Esparza, J. W. Lee, G. Kang, S. Shin, S. S. Jeon and H. Lee, Nanoscale, 2022, 14, 9297–9303 RSC.
- M. Farajzadeh, H. Alamgholiloo, F. Nasibipour, R. Banaei and S. Rostamnia, Sci. Rep., 2020, 10, 18188 CrossRef CAS PubMed.
- C. J. Zhan, X. W. Li, G. X. Lan, E. E. K. Baidoo, Y. K. Yang, Y. Z. Liu, Y. Sun, S. J. Wang, Y. Y. Wang, G. K. Wang, J. Nielsen, J. D. Keasling, Y. Chen and Z. H. Bai, Nat. Catal., 2023, 6, 435–450 CrossRef CAS.
- M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2022, 61, e202112835 CrossRef CAS PubMed.
- P. Hauke, T. Merzdorf, M. Klingenhof and P. Strasser, Nat. Commun., 2023, 14, 4708 CrossRef CAS PubMed.
- A. Parastaev, V. Muravev, E. H. Osta, T. F. Kimpel, J. F. M. Simons, A. J. F. van Hoof, E. Uslamin, L. Zhang, J. J. C. Struijs, D. B. Burueva, E. V. Pokochueva, K. V. Kovtunov, I. V. Koptyug, I. J. Villar-Garcia, C. Escudero, T. Altantzis, P. Liu, A. Béché, S. Bals, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2022, 5, 1051–1060 CrossRef CAS.
- G. Han, G. Li and Y. Sun, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- D. D. Mu, Z. W. Li, S. Y. Yu and S. T. Liu, Catal. Today, 2022, 402, 67–78 CrossRef CAS.
- M. Guo, M. Zhang, R. Liu, X. Zhang and G. Li, Adv. Sci., 2021, 9, 2103361 CrossRef PubMed.
- J. J. Song, Z. F. Huang, L. Pan, K. Li, X. W. Zhang, L. Wang and J. J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- J. J. Wu, Z. Y. Chen, J. H. Barnard, R. Gunasekar, C. Y. Pu, X. F. Wu, S. Y. Zhang, J. W. Ruan and J. L. Xiao, Nat. Catal., 2022, 5, 982–992 CrossRef CAS.
- H. Y. Xu, P. Yang, P. Chuanprasit, H. Hirao and J. R. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5112–5116 CrossRef CAS PubMed.
- Z. H. Zhang, M. Z. Jing, H. Chen, F. Okejiri, J. X. Liu, Y. Leng, H. L. Liu, W. Y. Song, Z. Y. Hou, X. Y. Lu, J. Fu and J. Liu, ACS Catal., 2020, 10, 9098–9108 CrossRef CAS.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- F. J. Zhao, T. X. Xu, G. M. Zhu, K. Wang, X. Xu and L. Liu, Sustainable Energy Fuels, 2022, 6, 1866–1890 RSC.
- Z. D. An and J. Li, Green Chem., 2022, 24, 1780–1808 RSC.
- T. K. Das, A. M. R. Treviño, S. Pandiri, S. Irvankoski, J. H. Siitonen, S. M. Rodriguez, M. Yousufuddin and L. Kürti, Green Chem., 2023, 25, 746–754 RSC.
- K. Azouzi, D. A. Valyaev, S. Bastin and J. B. Sortais, Curr. Opin. Green Sustainable Chem., 2021, 31, 100511 CrossRef CAS.
- S. Mudhulu, Z. J. Gong, H. C. Ku, Y. H. Lu and W. Y. Yu, Mater. Today Sustain., 2022, 19, 100199 CrossRef.
- A. Modak, D. Gill, A. R. Mankar, K. K. Pant, V. Bhasin, C. Nayak and S. Bhattacharya, Nanoscale, 2022, 14, 15875–15888 RSC.
- M. J. Gilkey and B. J. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- R. van Putten, T. Wissink, T. Swinkels and E. A. Pidko, Int. J. Hydrogen Energy, 2019, 44, 28533–28541 CrossRef CAS.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS PubMed.
- D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438–447 CrossRef CAS.
- A. Mahmood, W. H. Guo, H. Tabassum and R. Q. Zou, Adv. Energy Mater., 2016, 6, 1600423 CrossRef.
- S. Zhai, S. Jiang, C. Liu, Z. Li, T. Yu, L. Sun, G. Ren and W. Deng, J. Phys. Chem. Lett., 2022, 13, 8586–8600 CrossRef CAS PubMed.
- S. Keshipour and S. Mohammad-Alizadeh, Sci. Rep., 2021, 11, 16148 CrossRef CAS PubMed.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed.
- J. J. A. Celaje, Z. Y. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- Z. H. Yu, X. B. Lu, L. H. Sun, J. Xiong, L. Ye, X. Y. Li, R. Zhang and N. Ji, ACS Sustainable Chem. Eng., 2021, 9, 2990–3010 CrossRef CAS.
- X. Li, A. E. Surkus, J. Rabeah, M. Anwar, S. Dastigir, H. Junge, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 15849–15854 CrossRef CAS PubMed.
- R. Xu, W. Q. Lu, S. Toan, Z. R. Zhou, C. K. Russell, Z. Sun and Z. Q. Sun, J. Mater. Chem. A, 2021, 9, 24241–24260 RSC.
- S. J. Li, Y. T. Zhou, X. Kang, D. X. Liu, L. Gu, Q. H. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2019, 31, 1806781 CrossRef PubMed.
- S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CrossRef CAS PubMed.
- J. M. Zimbron, M. Dauphinais and A. B. Charette, Green Chem., 2015, 17, 3255–3259 RSC.
- L. Liu, H. Gao, S. Q. Yang, X. C. Chen, Y. Lu, Y. Liu and F. Xia, J. Catal., 2020, 385, 183–193 CrossRef CAS.
- K. Lu, X. Kong, J. M. Cai, S. R. Yu and X. G. Zhang, Nanoscale, 2023, 15, 8084–8109 RSC.
- C. Yu, X. F. Guo, B. Shen, Z. Xi, Q. Li, Z. Y. Yin, H. Liu, M. Muzzio, M. Q. Shen, J. R. Li, C. T. Seto and S. H. Sun, J. Mater. Chem. A, 2018, 6, 23766–23772 RSC.
- X. L. Cui, Y. Long, X. Zhou, G. Q. Yu, J. Yang, M. Yuan, J. T. Ma and Z. P. Dong, Green Chem., 2018, 20, 1121–1130 RSC.
- M. López Granados, J. Moreno, A. C. Alba-Rubio, J. Iglesias, D. Martín Alonso and R. Mariscal, Green Chem., 2020, 22, 1859–1872 RSC.
- R. F. Nie, Y. W. Tao, Y. Q. Nie, T. L. Lu, J. S. Wang, Y. S. Zhang, X. Y. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS.
- R. V. Jagadeesh, D. Banerjee, P. B. Arockiam, H. Junge, K. Junge, M. M. Pohl, J. Radnik, A. Brückner and M. Beller, Green Chem., 2015, 17, 898–902 RSC.
- Y. N. Duan, T. Song, X. S. Dong and Y. Yang, Green Chem., 2018, 20, 2821–2828 RSC.
- Y. H. Han, Z. Y. Wang, R. R. Xu, W. Zhang, W. X. Chen, L. R. Zheng, J. Zhang, J. Luo, K. L. Wu, Y. Q. Zhu, C. Chen, Q. Peng, Q. Liu, P. Hu, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 11262–11266 CrossRef CAS PubMed.
- F. Chen, B. Sahoo, C. Kreyenschulte, H. Lund, M. Zeng, L. He, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6239–6246 RSC.
- M. Yuan, Y. Long, J. Yang, X. W. Hu, D. Xu, Y. Y. Zhu and Z. P. Dong, ChemSusChem, 2018, 11, 4156–4165 CrossRef CAS PubMed.
- A. K. Kar and R. Srivastava, ACS Sustainable Chem. Eng., 2019, 7, 13136–13147 CrossRef CAS.
- I. Sorribes, K. Junge and M. Beller, Chem. – Eur. J., 2014, 20, 7878–7883 CrossRef CAS PubMed.
- Y. T. Chen, Chem. – Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed.
- S. Savourey, G. Lefèvre, J. C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC.
- X. Jiang, C. Wang, Y. W. Wei, D. Xue, Z. T. Liu and J. L. Xiao, Chem. – Eur. J., 2014, 20, 58–63 CrossRef CAS PubMed.
- V. Yadav, G. Sivakumar, V. Gupta and E. Balaraman, ACS Catal., 2021, 11, 14712–14726 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- L. L. Lin, W. Zhou, R. Gao, S. Y. Yao, X. Zhang, W. Q. Xu, S. J. Zheng, Z. Jiang, Q. L. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- J. Zheng, H. Zhou, C. G. Wang, E. Y. Ye, J. W. Xu, X. J. Loh and Z. B. Li, Energy Storage Mater., 2021, 35, 695–722 CrossRef.
- F. Valentini, A. Marrocchi and L. Vaccaro, Adv. Energy Mater., 2022, 12, 2103362 CrossRef CAS.
- Y. Wang, J. F. Han, N. Wang, B. Li, M. Yang, Y. M. Wu, Z. X. Jiang, Y. X. Wei, P. Tian and Z. M. Liu, Chin. J. Catal., 2022, 43, 2259–2269 CrossRef CAS.
- J. B. Zhou, M. B. Gao, J. L. Zhang, W. J. Liu, T. Zhang, H. Li, Z. C. Xu, M. Ye and Z. M. Liu, Nat. Commun., 2021, 12, 17 CrossRef CAS PubMed.
- M. Yang, D. Fan, Y. X. Wei, P. Tian and Z. M. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- E. Ebadzadeh, M. H. Khademi and M. Beheshti, Chem. Eng. J., 2021, 405, 126605 CrossRef CAS.
- A. Hwang and A. Bhan, Acc. Chem. Res., 2019, 52, 2647–2656 CrossRef CAS PubMed.
- I. Lezcano-Gonzalez, E. Campbell, A. E. J. Hoffman, M. Bocus, I. V. Sazanovich, M. Towrie, M. Agote-Aran, E. K. Gibson, A. Greenaway, K. De Wispelaere, V. Van Speybroeck and A. M. Beale, Nat. Mater., 2020, 19, 1081–1087 CrossRef CAS PubMed.
- X. J. Cui, R. Huang and D. H. Deng, EnergyChem, 2021, 3, 100050 CrossRef CAS.
- Y. F. Ma, G. Q. Guan, C. Shi, A. M. Zhu, X. G. Hao, Z. D. Wang, K. Kusakabe and A. Abudula, Int. J. Hydrogen Energy, 2014, 39, 258–266 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- L. López-Rodríguez, D. G. Araiza, D. G. Arcos, A. Gómez-Cortés and G. Díaz, Catal. Today, 2022, 394–396, 486–498 CrossRef.
- T. Tsoncheva, I. Genova, M. Stoyanova, M. M. Pohl, R. Nickolov, M. Dimitrov, E. Sarcadi-Priboczki, M. Mihaylov, D. Kovacheva and K. Hadjiivanov, Appl. Catal., B, 2014, 147, 684–697 CrossRef CAS.
- N. Koivikko, T. Laitinen, S. Ojala, S. Pitkäaho, A. Kucherov and R. L. Keiski, Appl. Catal., B, 2011, 103, 72–78 CrossRef CAS.
- G. C. Behera and K. Parida, Chem. Eng. J., 2012, 180, 270–276 CrossRef CAS.
- J. H. Kim, Y. S. Jang and D. H. Kim, Chem. Eng. J., 2018, 338, 752–763 CrossRef CAS.
- Q. Wang, J. L. Lan, R. Liang, Y. H. Xia, L. Qin, L. W. Chung and Z. P. Zheng, ACS Catal., 2022, 12, 2212–2222 CrossRef CAS.
- G. K. Hu, J. Y. Wang, X. Zhang, D. Liu, B. Yu, T. Huang, M. F. Zhu and H. Yu, Carbon, 2023, 214, 118311 CrossRef CAS.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed.
- L. Li, L. Zhang, L. Gou, S. Wei, X. Hou and L. Wu, Chem. Eng. J., 2023, 454, 140292 CrossRef CAS.
- Y. Tong, X. Yan, J. Liang and S. X. Dou, Small, 2019, 17, 1904126 CrossRef PubMed.
- N. Z. Liu, R. X. Wang, S. J. Gao, R. F. Zhang, F. R. Fan, Y. H. Ma, X. L. Luo, D. Ding and W. Z. Wu, Nano Energy, 2023, 109, 108311 CrossRef CAS.
- J. X. Liu, J. J. Hinsch, H. J. Yin, P. R. Liu, H. J. Zhao and Y. Wang, J. Electroanal. Chem., 2022, 907, 116071 CrossRef CAS.
- J. Chen, M. Ahmad, Y. Zhang, H. Ye, L. Wang, J. Zhang, X.-Z. Fu and J.-L. Luo, Chem. Eng. J., 2023, 454, 140056 CrossRef CAS.
- H. J. Yin, Y. H. Dou, S. Chen, Z. J. Zhu, P. R. Liu and H. J. Zhao, Adv. Mater., 2020, 32, 1904870 CrossRef CAS PubMed.
- Y. Guo, X. Yang, X. Liu, X. Tong and N. Yang, Adv. Funct. Mater., 2022, 33, 2209134 CrossRef.
- Z. H. Li, X. Zhang, J. J. Liu, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. R. Zhang, Adv. Mater., 2021, 33, 2103248 CrossRef CAS PubMed.
- Z. H. Li, J. J. Liu, Y. F. Zhao, R. Shi, G. I. N. Waterhouse, Y. S. Wang, L. Z. Wu, C. H. Tung and T. R. Zhang, Nano Energy, 2019, 60, 467–475 CrossRef CAS.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- X. L. Zhao, S. Chen, H. J. Yin, S. Y. Jiang, K. Zhao, J. Kang, P. F. Liu, L. X. Jiang, Z. J. Zhu, D. D. Cui, P. R. Liu, X. J. Han, H. G. Yang and H. J. Zhao, Matter, 2020, 3, 935–949 CrossRef.
- Z. Li, J. Liu, J. Zhao, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. Zhang, Adv. Funct. Mater., 2022, 33, 2213672 CrossRef.
- Z. C. Zhang, X. C. Tian, B. W. Zhang, L. Huang, F. C. Zhu, X. M. Qu, L. Liu, S. Liu, Y. X. Jiang and S. G. Sun, Nano Energy, 2017, 34, 224–232 CrossRef CAS.
- X. Xu, T. Lan, G. Zhao, Q. Nie, F. Jiang and Y. Lu, Appl. Catal., B, 2023, 334, 122839 CrossRef CAS.
- H. Wang, H. F. Qi, X. Sun, S. Y. Jia, X. Y. Li, T. J. Miao, L. Q. Xiong, S. H. Wang, X. L. Zhang, X. Y. Liu, A. Q. Wang, T. Zhang, W. X. Huang and J. W. Tang, Nat. Mater., 2023, 22, 619–626 CrossRef CAS PubMed.
- L. Wang, Y. Sun, F. Zhang, J. Hu, W. Hu, S. Xie, Y. Wang, J. Feng, Y. Li, G. Wang, B. Zhang, H. Wang, Q. Zhang and Y. Wang, Adv. Mater., 2022, 35, 2205782 CrossRef PubMed.
- X. X. Qin, H. Li, S. H. Xie, K. Li, T. W. Jiang, X. Y. Ma, K. Jiang, Q. Zhang, O. Terasaki, Z. J. Wu and W. B. Cai, ACS Catal., 2020, 10, 3921–3932 CrossRef CAS.
- O. Altan, E. Altintas, S. Alemdar and Ö. Metin, Chem. Eng. J., 2022, 441, 136047 CrossRef CAS.
- J. X. Fan, H. X. Du, Y. Zhao, Q. Wang, Y. N. Liu, D. Q. Li and J. T. Feng, ACS Catal., 2020, 10, 13560–13583 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- M. Bonarowska, Z. Kaszkur, D. Lomot, M. Rawski and Z. Karpinski, Appl. Catal., B, 2015, 162, 45–56 CrossRef CAS.
- L. Zhang, Z. X. Xie and J. L. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed.
- S. Mohanapriya and D. Gopi, Renewable Sustainable Energy Rev., 2021, 148, 111211 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- H. B. Zhang, G. G. Liu, L. Shi and J. H. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- X. B. Fan, G. L. Zhang and F. B. Zhang, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC.
- X. N. Li, X. F. Yang, Y. Q. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- C. Y. Dong, Y. L. Li, D. Y. Cheng, M. T. Zhang, J. J. Liu, Y. G. Wang, D. Q. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- R. Gusain, N. Kumar and S. S. Ray, Coord. Chem. Rev., 2020, 405, 213111 CrossRef CAS.
- Y. Wang, J. Chen, H. Ihara, M. Guan and H. D. Qiu, TrAC, Trends Anal. Chem., 2021, 143, 116421 CrossRef CAS.
- F. Marpaung, M. Kim, J. H. Khan, K. Konstantinov, Y. Yamauchi, M. S. A. Hossain, J. Na and J. Kim, Chem. – Asian J., 2019, 14, 1331–1343 CrossRef CAS PubMed.
- J. C. Ren, Y. L. Huang, H. Zhu, B. H. Zhang, H. K. Zhu, S. H. Shen, G. Q. Tan, F. Wu, H. He, S. Lan, X. H. Xia and Q. Liu, Carbon Energy, 2020, 2, 176–202 CrossRef CAS.
- Y. P. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- Q. Q. Yan, D. X. Wu, S. Q. Chu, Z. Q. Chen, Y. Lin, M. X. Chen, J. Zhang, X. J. Wu and H. W. Liang, Nat. Commun., 2019, 10, 4977 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- W. Y. Ni, J. L. Meibom, N. Ul Hassan, M. Y. Chang, Y. C. Chu, A. Krammer, S. L. Sun, Y. W. Zheng, L. C. Bai, W. C. Ma, S. Lee, S. Jin, J. S. Luterbacher, A. Schüler, H. M. Chen, W. E. Mustain and X. L. Hu, Nat. Catal., 2023, 6, 773–783 CrossRef CAS.
- H. T. Guo, B. W. Wang, P. Z. Qiu, R. X. Gao, M. M. Sun and L. G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 8876–8884 CrossRef CAS.
- J. L. Liu, D. D. Zhu, C. X. Guo, A. Vasileff and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- D. M. Zhang, F. Y. Ye, T. Xue, Y. J. Guan and Y. M. Wang, Catal. Today, 2014, 234, 133–138 CrossRef CAS.
- B. Hu, X. Y. Li, W. Busser, S. Schmidt, W. Xia, G. C. Li, X. B. Li and B. X. Peng, Chem. – Eur. J., 2021, 27, 10948–10956 CrossRef CAS PubMed.
- L. K. Zhang, X. T. Liu, X. Zhou, S. T. Gao, N. Z. Shang, C. Feng and C. Wang, ACS Omega, 2018, 3, 10843–10850 CrossRef CAS PubMed.
- X. L. Xu, J. J. Luo, L. P. Li, D. Zhang, Y. Wang and G. S. Li, Green Chem., 2018, 20, 2038–2046 RSC.
- S. S. Cheng, X. F. Meng, N. Z. Shang, S. T. Gao, C. Feng, C. Wang and Z. Wang, New J. Chem., 2018, 42, 1771–1778 RSC.
- J. Li, S. S. Cheng, T. X. Du, N. Z. Shang, S. T. Gao, C. Feng, C. Wang and Z. Wang, New J. Chem., 2018, 42, 9324–9331 RSC.
- J. R. Yang, X. H. Qi, F. Shen, M. Qiu and R. L. Smith, Sci. Total Environ., 2020, 719, 137534 CrossRef CAS PubMed.
- Y. J. Chou, H. C. Ku, C. C. Chien, C. C. Hu and W. Y. Yu, Catal. Sci. Technol., 2020, 10, 7883–7893 RSC.
- Y. Zhang, L. Chen, Y. Gui and L. Liu, Appl. Surf. Sci., 2022, 592, 153334 CrossRef CAS.
- C. K. P. Neeli, P. Puthiaraj, Y. R. Lee, Y. M. Chung, S. H. Baeck and W. S. Ahn, Catal. Today, 2018, 303, 227–234 CrossRef CAS.
- Z. Liu, W. H. Dong, S. S. Cheng, S. Guo, N. Z. Shang, S. T. Gao, C. Feng, C. Wang and Z. Wang, Catal. Commun., 2017, 95, 50–53 CrossRef CAS.
- Y. You, H. Huang, K. K. Mao, S. Xia, D. Wu, C. Y. Hu, C. Gao, P. Y. M. Liu, R. Long, X. J. Wu and Y. J. Xiong, Sci. China Mater., 2019, 62, 1297–1305 CrossRef CAS.
- S. S. Li, L. Tao, F. Z. R. Wang, Y. M. Liu and Y. Cao, Adv. Synth. Catal., 2016, 358, 1410–1416 CrossRef CAS.
- J. Zhang, Z. J. Wang, M. G. Chen, Y. F. Zhu, Y. M. Liu, H. Y. He, Y. Cao and X. H. Bao, Chin. J. Catal., 2022, 43, 2212–2222 CrossRef CAS.
- C. K. P. Neeli, Y. M. Chung and W. S. Ahn, ChemCatChem, 2017, 9, 4570–4579 CrossRef CAS.
- C. Q. Zhou, Y. Xiao, S. G. Xu, J. M. Li and C. W. Hu, Ind. Eng. Chem. Res., 2020, 59, 17228–17238 CrossRef CAS.
- J. Deng, D. H. Deng and X. H. Bao, Adv. Mater., 2017, 29, 1606967 CrossRef PubMed.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- Y. Wang, J. Hao, Y. Liu, M. Liu, K. Sheng, Y. Wang, J. Yang, J. Li and W. Li, J. Energy Chem., 2023, 76, 601–616 CrossRef CAS.
- D. T. Tran, D. C. Nguyen, H. T. Le, T. Kshetri, V. H. Hoa, T. L. L. Doan, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2021, 115, 100711 CrossRef CAS.
- S. H. Zhou, F. L. Dai, C. Dang, M. Wang, D. T. Liu, F. C. Lu and H. S. Qi, Green Chem., 2019, 21, 4732–4747 RSC.
- L. Jiang, P. Zhou, Z. H. Zhang, S. W. Jin and Q. Chi, Ind. Eng. Chem. Res., 2017, 56, 12556–12565 CrossRef CAS.
- D. Xu, H. Zhao, Z. P. Dong and J. T. Ma, ChemCatChem, 2019, 11, 5475–5486 CrossRef CAS.
- Q. Zhu, X. Sun, H. Zhao, D. Xu and Z. P. Dong, Ind. Eng. Chem. Res., 2020, 59, 5615–5623 CrossRef CAS.
- H. T. Guo, R. X. Gao, M. M. Sun, H. Guo, B. W. Wang and L. G. Chen, ChemSusChem, 2019, 12, 487–494 CrossRef CAS PubMed.
- M. X. Duan, Q. Y. Cheng, M. M. Wang and Y. J. Wang, RSC Adv., 2021, 11, 10996–11003 RSC.
- J. W. Zhao, M. R. Liu, G. L. Fan, L. Yang and F. Li, Ind. Eng. Chem. Res., 2021, 60, 5826–5837 CrossRef CAS.
- P. Yang, Q. Xia, X. Liu and Y. Wang, Fuel, 2017, 187, 159–166 CrossRef CAS.
- L. Xu, R. F. Nie, X. J. Chen, Y. C. Li, Y. X. Jiang and X. Y. Lu, Catal. Sci. Technol., 2021, 11, 1451–1457 RSC.
- L. Xu, X. L. Lyu, Y. X. Jiang, X. W. Wei, R. F. Nie and X. Y. Lu, Green Chem., 2022, 24, 4935–4940 RSC.
- L. Y. Huang, H. Zhang, Y. J. Cheng, Q. D. Sun, T. Gan, Q. He, X. H. He and H. B. Ji, Chin. Chem. Lett., 2022, 33, 2569–2572 CrossRef CAS.
- Z. Gong, Y. Lei, P. Zhou and Z. H. Zhang, New J. Chem., 2017, 41, 10613–10618 RSC.
- F. W. Zhang, J. J. Li, P. Z. Liu, H. Li, S. Chen, Z. H. Li, W. Y. Zan, J. J. Guo and X. M. Zhang, J. Catal., 2021, 400, 40–49 CrossRef CAS.
- H. N. Li, C. Y. Cao, J. Liu, Y. Shi, R. Si, L. Gu and W. G. Song, Sci. China Mater., 2019, 62, 1306–1314 CrossRef CAS.
- C. Liu, B. F. Liang, F. L. Jing and S. Z. Luo, Int. J. Energy Res., 2022, 46, 23682–23697 CrossRef CAS.
- T. Wang, W. Suo, M. M. Qu, F. Li, S. J. Sun, W. Xue and Y. J. Wang, J. Chem. Technol. Biotechnol., 2022, 97, 3172–3182 CrossRef CAS.
- S. Lomate, A. Sultana and T. Fujitani, Catal. Sci. Technol., 2017, 7, 3073–3083 RSC.
- Y. Sun, C. X. Xiong, Q. C. Liu, J. R. Zhang, X. Tang, X. H. Zeng, S. J. Liu and L. Lin, Ind. Eng. Chem. Res., 2019, 58, 5414–5422 CrossRef CAS.
- T. Wang, J. Du, Y. Sun, X. Tang, Z. J. Wei, X. H. Zeng, S. J. Liu and L. Lin, Chin. Chem. Lett., 2021, 32, 1186–1190 CrossRef CAS.
- T. Su and C. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 55568–55576 CrossRef CAS PubMed.
- J. Li, C. Li, S. Feng, Z. Zhao, H. Zhu and Y. Ding, ChemCatChem, 2020, 12, 1546–1550 CrossRef CAS.
- S. Karmakar, S. Barman, F. A. Rahimi, D. Rambabu, S. Nath and T. K. Maji, Nat. Commun., 2023, 14, 4508 CrossRef CAS PubMed.
- X. D. Li, L. Li, G. B. Chen, X. Y. Chu, X. H. Liu, C. Naisa, D. Pohl, M. Löffler and X. L. Feng, Nat. Commun., 2023, 14, 4034 CrossRef CAS PubMed.
- M. Zhou, Z. Q. Wang, A. H. Mei, Z. F. Yang, W. Chen, S. Y. Ou, S. Y. Wang, K. Q. Chen, P. Reiss, K. Qi, J. Y. Ma and Y. L. Liu, Nat. Commun., 2023, 14, 2473 CrossRef CAS PubMed.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. F. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- H. Li, C. K. Cheng, Z. J. Yang and J. J. Wei, Nat. Commun., 2022, 13, 6466 CrossRef CAS PubMed.
- Y. N. Zhang, C. S. Pan, G. M. Bian, J. Xu, Y. M. Dong, Y. Zhang, Y. Lou, W. X. Liu and Y. F. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- X. Q. Yan, M. Y. Xia, H. X. Liu, B. Zhang, C. R. Chang, L. Z. Wang and G. D. Yang, Nat. Commun., 2023, 14, 1741 CrossRef CAS PubMed.
- T. J. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. B. Huang, A. A. Jelle, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521 CrossRef PubMed.
- S. Singh, R. Verma, N. Kaul, J. Sa, A. Punjal, S. Prabhu and V. Polshettiwar, Nat. Commun., 2023, 14, 2551 CrossRef CAS PubMed.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- W. Qiao, X. Fan, W. F. Liu, F. N. Khan, D. S. Zhang, F. Y. Han, B. H. Yue, Y. J. Li, N. Dimitratos, S. Albonetti, X. D. Wen, Y. Yang, F. Besenbacher, Y. W. Li, H. Niemantsverdriet, H. P. Lin and R. Su, J. Am. Chem. Soc., 2023, 145, 5353–5362 CrossRef CAS PubMed.
- X. W. Lu, C. M. Guo, M. Y. Zhang, L. P. Leng, J. H. Horton, W. Wu and Z. J. Li, Nano Res., 2021, 14, 4347–4355 CrossRef CAS.
- K. Tsutsumi, F. Uchikawa, K. Saka and K. Tabata, ACS Catal., 2016, 6, 4394–4398 CrossRef CAS.
- S. H. Dong, Z. Liu, R. H. Liu, L. M. Chen, J. Z. Chen and Y. S. Xu, ACS Appl. Nano Mater., 2018, 1, 4247–4257 CrossRef CAS.
- A. K. Kar, A. Behera and R. Srivastava, ACS Appl. Nano Mater., 2022, 5, 464–475 CrossRef CAS.
- S. Zhang, Y. X. Liu, M. K. Zhang, Y. Y. Ma, J. Hu and Y. Q. Qu, Nat. Commun., 2022, 13, 5527 CrossRef CAS PubMed.
- V. Goyal, J. Gahtori, A. Narani, P. Gupta, A. Bordoloi and K. Natte, J. Org. Chem., 2019, 84, 15389–15398 CrossRef CAS PubMed.
- H.-S. Lee, J. Lee, H. Seo, H. Kang, D. H. Kim and Y.-W. Lee, Fuel Process. Technol., 2021, 218, 106870 CrossRef CAS.
- L. Zhu, S. Ye, J. Wang, J. Zhu, G. He and X. Liu, ChemCatChem, 2022, 14, e202101794 CrossRef CAS.
- V. L. Yfanti and A. A. Lemonidou, Catal. Today, 2020, 355, 727–736 CrossRef CAS.
- X. Tang, Z. Li, X. Zeng, Y. Jiang, S. Liu, T. Lei, Y. Sun and L. Lin, ChemSusChem, 2015, 8, 1601–1607 CrossRef CAS PubMed.
- Z. Zhang, C. Wang, X. Gou, H. Chen, K. Chen, X. Lu, P. Ouyang and J. Fu, Appl. Catal., A, 2019, 570, 245–250 CrossRef CAS.
- I. Z. Awan, G. Beltrami, D. Bonincontro, O. Gimello, T. Cacciaguerra, N. Tanchoux, A. Martucci, S. Albonetti, F. Cavani and F. Di Renzo, Appl. Catal., A, 2021, 609, 117929 CrossRef CAS.
- J. Zhang and J. Chen, ACS Sustainable Chem. Eng., 2017, 5, 5982–5993 CrossRef CAS.
- J. Zhang, C. Li, S. Hu, J. Gu, H. Yuan and Y. Chen, Renewable Energy, 2022, 200, 88–97 CrossRef CAS.
- G. Li, W. Jiao, Z. Sun, Y. Zhao, Z. Shi, Y. Yan, L. Feng, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2018, 6, 4316–4320 CrossRef CAS.
- S. Yao, T. Zhang, X. Tang, D. Li, W. Zhang, D. Lin, R. Li, H. Yan, Y. Liu, X. Feng, X. Chen, X. Zhou and C. Yang, Energy Fuels, 2021, 35, 9970–9982 CrossRef CAS.
- L. Grazia, D. Bonincontro, A. Lolli, T. Tabanelli, C. Lucarelli, S. Albonetti and F. Cavani, Green Chem., 2017, 19, 4412–4422 RSC.
- H. Zhang, X. Tong, Y. Gao, H. Chen, P. Guo and S. Xue, J. Ind. Eng. Chem., 2019, 70, 152–159 CrossRef CAS.
- L. Luo, X. Han and Q. Zeng, Catalysts, 2020, 10, 1096 CrossRef CAS.
- X. Wu, J. Li, S. Xie, P. Duan, H. Zhang, J. Feng, Q. Zhang, J. Cheng and Y. Wang, Chem, 2020, 6, 3038–3053 CAS.
- Y. B. Zhao, F. Pan, H. Li, G. Q. Xu and W. Chen, ChemCatChem, 2014, 6, 454–458 CrossRef CAS.
- Y. M. Jian, Y. Meng, J. Li, H. G. Wu, S. Saravanamurugan and H. Li, J. Environ. Chem. Eng., 2022, 10, 108837 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |