
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevolutionizing cancer therapy: nanoformulation of miRNA-34 – enhancing delivery and efficacy for various cancer immunotherapies: a review
Marola Paula
Fawzy
a,
Hatem A. F. M.
Hassan
 bc,
Nada K.
Sedky
d,
Mohamed S.
Nafie
ef,
Rana A.
Youness
g and
Sherif Ashraf
Fahmy
*ah
bc,
Nada K.
Sedky
d,
Mohamed S.
Nafie
ef,
Rana A.
Youness
g and
Sherif Ashraf
Fahmy
*ah
aDepartment of Chemistry, School of Life and Medical Sciences, University of Hertfordshire Hosted by Global Academic Foundation, R5 New Garden City, New Capital, Cairo 11835, Egypt. E-mail: sheriffahmy@aucegypt.edu
bMedway School of Pharmacy, University of Kent, Central Avenue, Chatham Maritime, Canterbury ME44TB, UK
cDepartment of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, 11562 Cairo, Egypt
dDepartment of Biochemistry, School of Life and Medical Sciences, University of Hertfordshire Hosted by Global Academic Foundation, R5 New Garden City, New Administrative Capital, Cairo 11835, Egypt
eDepartment of Chemistry, College of Sciences, University of Sharjah, (P.O. 27272), Sharjah, United Arab Emirates (UAE)
fChemistry Department, Faculty of Science, Suez Canal University, (P.O. 41522), Ismailia, Egypt
gMolecular Genetics and Biochemistry Department, Molecular Genetics Research Team (MGRT), Faculty of Biotechnology, German International University (GIU), 11835, Cairo, Egypt
hDepartment of Pharmaceutics and Biopharmaceutics, University of Marburg, Robert-Koch-Str. 4, 35037 Marburg, Germany
First published on 20th September 2024
Abstract
Despite recent advancements in cancer therapies, challenges such as severe toxic effects, non-selective targeting, resistance to chemotherapy and radiotherapy, and recurrence of metastatic tumors persist. Consequently, there has been considerable effort to explore innovative anticancer compounds, particularly in immunotherapy, which offer the potential for enhanced biosafety and efficacy in cancer prevention and treatment. One such avenue of exploration involves the miRNA-34 (miR-34) family, known for its ability to inhibit tumorigenesis across various cancers. Dysregulation of miR-34 has been observed in several human cancers, and it is recognized as a tumor suppressor microRNA due to its synergistic interaction with the well-established tumor suppressor p53. However, challenges have arisen with the therapeutic application of miR-34a. These include its susceptibility to degradation by RNase in serum, limiting its ability to penetrate capillary endothelium and reach target cells, as well as reports of immunoreactive adverse reactions. Furthermore, unexpected side effects may occur, such as the accumulation of therapeutic miRNAs in healthy tissues due to interactions with serum proteins on nano-vector surfaces, nanoparticle breakdown in the bloodstream due to shearing stress, and unsuccessful extravasation of nanocarriers to target cells owing to interstitial fluid pressure. Despite these challenges, miR-34a remains a promising candidate for cancer therapy, and other members of the miR-34 family have also shown potential in inhibiting tumor cell proliferation. While the in vivo applications of miR-34b/c are limited, they warrant further exploration for oncotherapy. Recently, procedures utilizing nanoparticles have been developed to address the challenges associated with the clinical use of miR-34, demonstrating efficacy both in vitro and in vivo. This review highlights emerging trends in nanodelivery systems for miR-34 targeting cancer cells, offering insights into novel nanoformulations designed to enhance the anticancer therapeutic activity and targeting precision of miR-34. As far as current knowledge extends, no similar recent review comprehensively addresses the diverse nanoformulations aimed at optimizing the therapeutic potential of miR-34 in anticancer strategies.
1. Introduction
Immunotherapy represents a groundbreaking approach to cancer treatment, diverging from conventional modalities such as radiation and chemotherapy. Its fundamental aim is to bolster the body's immune system, enabling it to combat cancer cells across multiple fronts. This strategy involves dynamically altering the immune response to create an environment conducive to immune cell activation and subsequent recognition and elimination of tumor cells. The primary objective of immunotherapy is to enhance the immune system's capabilities by regulating the immunological microenvironment.1 This entails manipulating factors that influence immune cell function and tumor cell interaction, ultimately leading to tumor eradication. Key to this approach are the genetic abnormalities present in tumor cells and the dynamic nature of the tumor microenvironment (TME), which collectively drive cancer initiation and progression. Genetic alterations in tumor suppressor genes and proto-oncogenes directly contribute to cancer development. Consequently, therapeutic interventions aimed at targeting these aberrations have become pivotal in cancer management. By addressing these genetic abnormalities, immunotherapy aims to exploit the body's natural defenses to combat cancer effectively.21.1. MicroRNAs in cancer therapy
MicroRNAs (miRNAs) have emerged as a promising focal point in immunotherapy, serving as significant predictive and prognostic markers due to their distinctive regulatory functions.3 These molecules, categorized as non-coding RNAs (ncRNAs), consist of short, single-stranded RNA molecules, typically comprising 22 nucleotides, although lengths ranging from 19 to 25 nucleotides have been documented. Their pivotal role lies in modulating gene expression across various cellular processes.4 In the context of cancer, miRNAs exert influence over critical oncogenes and tumor suppressor (TS) genes, thereby orchestrating fundamental processes, including tumor progression, cell proliferation, angiogenesis, invasion, and metastasis.5 Notably, miRNAs exhibit diverse roles in cancer development, functioning as either tumor suppressors or oncogenes depending on their target genes. Predominantly, the dysregulation of miRNAs, leading to their downregulation, instigates various cellular abnormalities such as enhanced cell growth, invasion, metastasis, reduced treatment sensitivity, and increased apoptosis.6In addition, miRNAs have diverse functions in cancer development, acting as TS or oncogenes depending on their targets.5 Luckily, the majority of miRNAs have tumor-suppressive functions in various types of cancer, and their aberrant downregulation causes a variety of cellular abnormalities, including increased cell growth, invasion, metastasis, decreased sensitivity to treatment, and increased apoptosis.6 Some of the reported oncomiRNAs are miR-224 promoting lung cancer,7 miR-21 stimulating the expression of breast cancer, pancreatic adenocarcinoma, and colorectal cancer (CRC);8 and miR-10 was reported to be overexpressed in gastric cancer.9 On the other hand, some of the most well-known TS miRNAs families are miR-34, let-7, miR-29, miR-383, and miR-3174, which are found to be downregulated in various cancer types, such as glioblastoma,10 breast cancer, pancreatic cancer, hepatocellular carcinoma (HCC), lung cancer, head and neck cancer, prostate cancer, colorectal cancer, and esophageal cancer (EC).11–15
2. MicroRNA-34
The miR-34 family has been grabbing a lot of attention in cancer research recently due to its noteworthy tumor-suppressive functions. The family of miR-34 includes three members miR-34a, miR-34b, and miR-34c. While miR-34b and miR-34c are primarily expressed in lung tissue, miR-34a is expressed ubiquitously throughout different tissues, hence, miR-34a is the most recognized in the family.16,17 An increase in miR-34s inhibits apoptosis and senescence, leading to carcinogenesis and cancer progression, henceforth they are essential for controlling the immune microenvironment of several cancers, such as those of the lung, breast, liver, prostate, colorectal, ovarian, and head and neck.182.1. Biosynthesis of miR-34s
The cytoplasm and nucleus are the sites of a multistep process called miR-34 biosynthesis. In the nucleus, miR-34 genes are transcribed by RNA polymerase II or III, producing pri-miRNA, a lengthy molecule structured like a hairpin. Within the nucleus, the pri-miRNA is cleaved by the DROSHA endonuclease, producing pre-miRNAs that are 80–100 nucleotides long. Pre-miR-34s are transported to the cytoplasm by exportin-5, which are further processed by DICER endonuclease to become double-stranded mature miR-34s, around 20–23 nucleotides long.9 The mature miRNAs in question form the RNA-induced silencing complex (RISC) through their association with Argonaute proteins. Whereas the other strand is broken down, one becomes the mature miRNA. The degree of complementarity between the target mRNA binding sites and the miR-34 seed sequence determines whether or not miRNA-mediated gene silencing occurs. Partial complementarity prevents mRNA translation, but full complementarity may cause mRNA destruction. It is important to note that their varying expression levels indicate the diverse and particular roles that miR-34s play in controlling gene expression. We are constantly learning more about the specific processes by which miRNAs influence gene expression, and current studies are helping us understand the complex and dynamic functions that miRNAs play in cellular physiology and the development of illnesses.192.2. miR-34s cancer targets
MiR-34s have an influential role in preventing the progression of several types of cancers, such as breast, prostate, HCC, pancreatic, esophageal and NSCLC, on the other hand, miR-34s might promote the development of other types of cancer, such as bladder cancer. Thus, in this review, we aim to shed more light on the therapeutic role of miR-34 in treating cancer. MiR-34 was reported to target more than 700 oncogenic genes, including the PI3K/AKT, Ras, MAPK, Wnt, Notch, and p53 pathways, which are responsible for the advancement of cancer. Furthermore, miR-34s possess suppressive mechanisms essential to cancer progression, including invasion, migration, and endothelial mesenchymal transition (EMT).20 Research has indicated that the administration of miR-34a systemically, in conjunction with chemotherapy or radiation, might effectively impede the development of tumors.21,22 Thus, in order to create successful cancer treatments, it is crucial to comprehend the regulatory mechanisms of miR-34s and their targets.20 Further investigation into the precise roles and regulatory networks of miR-34s in various cancer types will yield important information for the development of targeted therapies and personalized medicine strategies23 and enhance patient outcomes by using the potential of miR-34s.19 In the following paragraphs, we will highlight the impact of miR-34 downregulation in cancer progression.Moreover, it has been found that non-small-cell lung cancer (NSCLC) expresses miR-34b-3p at a downregulated level. It has been reported that,26 miR-34b targets CDK4 to inhibit NSCLC cell growth and cell cycle progression as well as to cause apoptosis. Likewise, it has been shown that patients with NSCLC who have lower levels of circulating miR-34 family members have worse prognoses, indicating that the miR-34s family may represent a new class of prognostic biomarkers. Metastatic lung cancer samples have been shown to express miR-34s at much lower levels than nonmetastatic samples. The majority of lung adenocarcinomas have been shown to have hypermethylation of the miR-34 promoters, indicating that miR-34 methylation may be used as a prognostic indicator for patients with NSCLC.19,26,27
Furthermore,34 tumor tissues had lower levels of miR-34 than corresponding noncancerous tissues. In HCC, miR-34 downregulation is linked to a poor prognosis. This implies that the decreased expression of miR-34s might play a role in the aggressiveness of HCC and function as an important prognostic indicator for the illness.19 Additionally, research has examined the relationship between polymorphisms in the pri-miR-34b/c promoter area and the risk of HCC. The findings demonstrated the possible involvement of miR-34s in the genetic predisposition to HCC by showing that specific genetic variants in this area are linked to higher susceptibility to HCC.35
Proteolytic enzymes belonging to the MMP family play a crucial role in the remodeling of the extracellular matrix, as its breakdown is necessary for cancer invasion and metastasis, and miRNAs that target MMPs can alter the invasiveness of ESCC cells. Hence, treatment with miR-34a resulted in lower MMP2 and MMP9 protein expression levels. MiR-34a, a p53-downstream miRNA, specifically targeted and inhibited MMP2 and MMP9, which in turn inhibited ESCC cell migration and invasion. It's interesting to note that miR-34a was also discovered to suppress the expression of Yin Yang 1 (YY1), an upstream transcription factor of MMP2 and MMP9, in ESCC cells, resulting in a downregulation of MMP2 and MMP9 levels in ESCC.36
Additionally, one transcription factor belonging to the forkhead box family, FOXM1, is involved in controlling cell division. Angiogenesis, cell cycle acceleration, and metastasis are all significantly impacted by irregular FOXM1. By increasing the expression of MMP-2 and MMP-9, FOXM1 aided in the advancement of cancer. Thus, reduced expression of miR-34a in ESCC tumor tissues as a result of strict regulation of FOXM1 and the expression of its target gene suggest that miR-34a prevents the advancement of ESCC by reducing cancer cells' ability to proliferate and migrate.37
TGFβ1 is a secreted protein that controls a variety of biological processes, including the production of miRNAs, development of EMT, metastases, and chemoresistance. TGFβ1 is responsible for mediating the overexpression of SOX4, a transcription factor that belongs to the SOX (SRY-related HMG-box) family and is known to be implicated in cancer and developmental disorders. Numerous biological processes, including metastasis formation, EMT, apoptosis, and reaction to chemotherapy and radiation, are impacted by SOX4 dysregulation.39
Recent data demonstrated miR-34c's function in NPC chemoresistance and EMT, concluding the downregulation of miR-34c was largely due to overexpression of miR-449b and the ensuing TGFβ1 activity, which led to overexpression of SOX4 and SOX2, which in turn induced EMT and cisplatin resistance. Like other cancer types, miR-34c overexpression made NPC cells more susceptible to cisplatin 40.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 associated deaths.41 Numerous investigations have revealed that the expression level of the miR-34 family was lower in the tissues of CRC patients than in the nearby non-tumor tissues. The downregulation of miR-34a and miR-34c in human colon cancer tissue was related to promoter hypermethylation. Despite the fact that hypermethylation is the reason for the decreased expression of miR-34, it has also been established that SUMOylation controls the amount of miR-34b/c in colon cancer. MiR-34's dysregulation points to a possible biomarker function for it.11 In addition, mir-34a has been identified as a prognostic factor for the recurrence of CRC in stages I–III, as well as for overall survival rates. Moreover, its regulatory actions involve upregulating early growth response protein 1 (EGR1) and inhibiting vimentin, resulting in decreased invasion and migration capabilities. Additionally, modulation of the E2F pathway leads to growth arrest akin to senescence. Recent research findings have elucidated that mir-34a augments EGR1 expression while concurrently impeding vimentin function, thus mitigating invasion and migration tendencies in SW620 cells.42
000 associated deaths.41 Numerous investigations have revealed that the expression level of the miR-34 family was lower in the tissues of CRC patients than in the nearby non-tumor tissues. The downregulation of miR-34a and miR-34c in human colon cancer tissue was related to promoter hypermethylation. Despite the fact that hypermethylation is the reason for the decreased expression of miR-34, it has also been established that SUMOylation controls the amount of miR-34b/c in colon cancer. MiR-34's dysregulation points to a possible biomarker function for it.11 In addition, mir-34a has been identified as a prognostic factor for the recurrence of CRC in stages I–III, as well as for overall survival rates. Moreover, its regulatory actions involve upregulating early growth response protein 1 (EGR1) and inhibiting vimentin, resulting in decreased invasion and migration capabilities. Additionally, modulation of the E2F pathway leads to growth arrest akin to senescence. Recent research findings have elucidated that mir-34a augments EGR1 expression while concurrently impeding vimentin function, thus mitigating invasion and migration tendencies in SW620 cells.42
One of the main inducers of EMT is the transcription factor SNAIL, which is controlled by the miR-34 family. E2F3a is another essential transcription factor that induces cell proliferation. It is a critical activator of the cell cycle, promoting and expediting the G1/S transition. It has been demonstrated that miR-34a also has a negative influence over E2F3a. This was shown in ovarian cancer cells, which resulted in a significant increase in E2F3a expression. Furthermore, it was discovered that miR-34a adversely regulates L1CAM which is a functional membrane glycoprotein that gives tumor cells the ability to migrate and invade and is also essential for EMT. L1CAM overexpression has been linked to a much worse prognosis and worse tumor resectability during initial surgery has been associated with L1CAM expression in ovarian cancer. Moreover, it was shown that the expression of BRCA1/2 mRNA and miR-34b/c, correlated negatively in BRCA wild-type tumors. Given the significant role that the proteins encoded by the tumor suppressor genes BRCA1/2 play in homologous recombination DNA repair, the inverse associations that have been found may be indicative of the more malignant phenotype that is associated with cancers that express fewer members of the miR-34 family and have higher rates of proliferation and DNA replication.44
Primary PCa samples have higher levels of mature miR-34a-5p expression. Several oncogenic signaling pathways, including c-Myc and AR, are activated during PCa development. Upregulated miR-34a, together with increased LRIG1 expression, serve as a feedback inhibitory mechanism to counteract MYC and AR-driven oncogenic signals. Thus, the increased levels of miR-34a found in initial prostate tumors would continue to function as a tumor suppressor. Supporting this finding is the finding that, in PCa samples, miR-34a expression negatively correlates with the tumor (T) stage. Accordingly, the goal of miR-34a replacement treatment is to reintroduce the tumor suppressor miR-34a into tumor cells, restoring its lost function and preventing cancer progression. A therapeutic miR-34a is a fully developed double-stranded duplex that may directly induce gene silencing by penetrating the RNA-induced silencing complex (RISC).46
3. Drawbacks of miRNAs in cancer therapy
Although TS miRNAs have proven to be a beneficial, promising treatment in different cancer types since they promote cell apoptosis, inhibit the endothelial to mesenchymal transition, inhibit cell proliferation, and inhibit oncogene expression, yet they have several drawbacks when it comes to their delivery.47 MiRNA delivery mechanisms are limited due to their poor binding affinity for complementary sequences, their quick clearance from blood,48 their off-target toxicity, the unwanted immune responses,49 inadequate delivery to intended target tissues, insufficient entrapment in the endosome, and lack of penetration of the cell membrane, and their susceptibility to nucleases upon addition into biological systems.50 However, the primary obstacles to the clinical development of packaged miR-34a therapies include vehicle-associated toxicity, decreased stability, and lack of selectivity.46 Thus, in order to overcome these drawbacks, nanoformulation of microRNAs has been arising.4. Nanotherapies of miRNAs
In the last several decades, there has been an inclination toward using nanotechnology in medicine, including safer and more efficient methods of tumor targeting, diagnostics, and therapy. Drug delivery systems based on nanoparticles (NPs) have demonstrated several benefits in cancer treatment, such as improved pharmacokinetics, accurate targeting of tumor cells, decreased side effects, and decreased susceptibility to drug resistance Fig. 1. The size and features of NPs utilized in medication delivery systems are often selected or created in accordance with the tumor's pathophysiology. Nano-carriers in cancer therapy mechanically target tumor cells via the NPs' carrier effect and the target's location upon absorption. The medications are then released onto the tumor cells to initiate apoptosis.51–54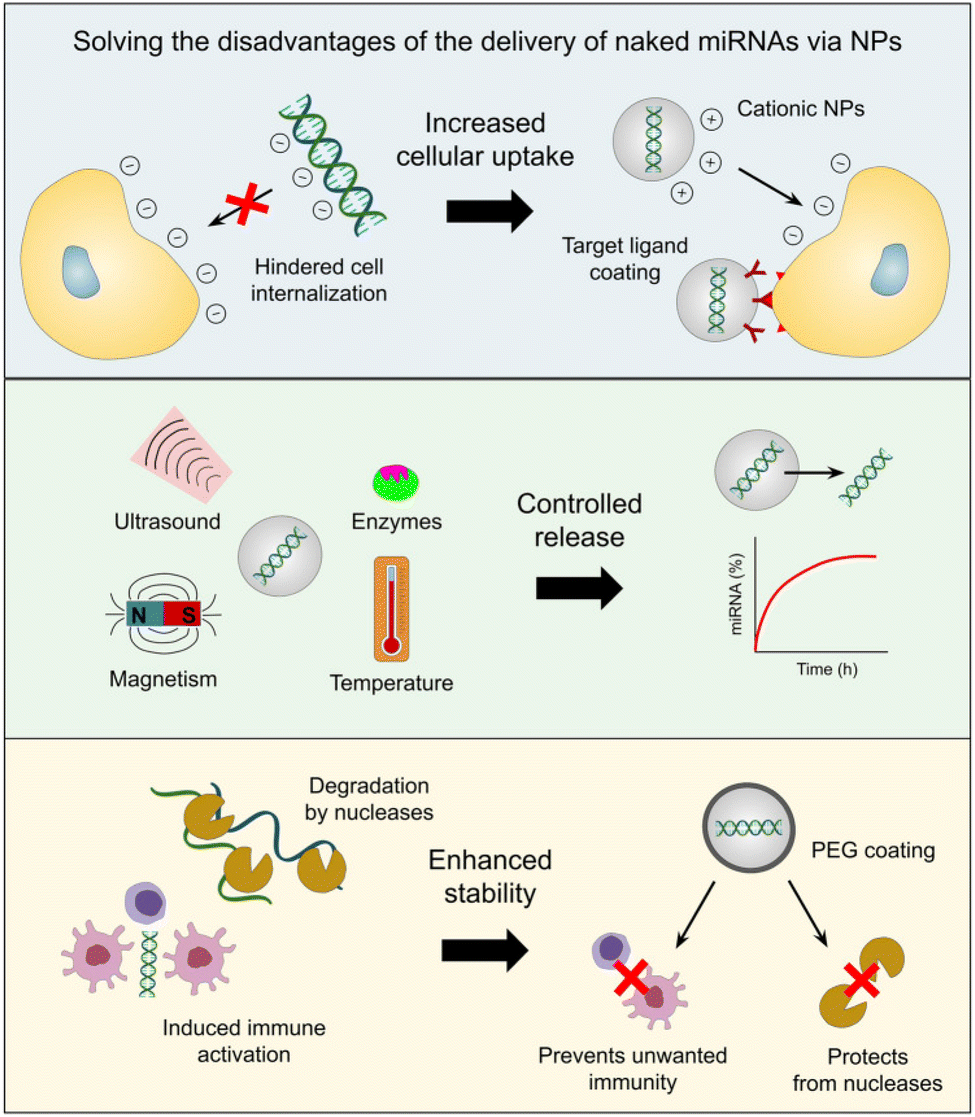 | ||
| Fig. 1 Benefits of miRNA-based medication delivery via NP-mediated means. This figure has been reproduced from ref. 62 with permission from Front. Bioeng. Biotechnol, copyright (2023). | ||
Traditional chemotherapeutic drugs and nucleic acids are among the drugs found inside the nano-carriers, suggesting they may be used in gene therapy and cytotoxic treatments. Moreover, NPs provide a platform that can assist in encapsulating and distributing some poorly soluble medications throughout the body. Nano-carriers can lengthen the half-life of medications and cause their accumulation inside tumor tissues because of the size, surface properties, permeability, and retention-enhancing properties of NPs. Meanwhile, the targeting system lessens the harmful effects of cancer therapy by shielding healthy cells from the cytotoxicity of medications. In addition, the new generation of intelligent NPs may also carry various sorts of medications, such as small molecules, peptides and proteins, nucleic acids, and living cells, in contrast to conventional NPs that are employed to deliver chemotherapeutic agents.51–56
Throughout the past decade, significant progress has been achieved in the creation of nanotechnology-based approaches for the administration of cancer drugs, namely in the cases of glioblastoma,57 lung cancer,58 and breast cancer.59 The delivery of ncRNA, such as miRNAs and small interfering RNAs (siRNAs), into cancer stem cells and metastatic tumors through NP-mediated delivery has produced many positive experimental results that have improved and optimized cancer treatment options.60 Furthermore, the approval of the distribution of nanotechnology platforms for the delivery of anti-cancer drugs, including Mepact (mifamurtide), NanoTherm (Fe2O3), DaunoXome (daunorubicin), and Caelyx and Myocet (doxorubicin),61 suggests a promising future for the NP-driven delivery of miRNA-based medications.62
Liposomes,63 exosomes, dendrimers, mesoporous silica NPs (MSN), quantum dots, gold NPs (AuNPs), iron oxide NPs (IONPs), and core–shell nanomaterials are a few of the main nanocarriers used in miRNA-based cancer treatments.64 When used as delivery methods for miRNA mimics and antagomirs, these nanotechnological tools provide a variety of benefits. Because cationic NPs, in particular, can easily interact with the negatively charged surface of the cell membrane, ncRNAs encapsulated in them have been shown to exhibit increased uptake from target cells.65,66 In contrast, the negative charge and molecular weight of miRNAs would impede their ability to move across the cell membrane.50 Tumor-specific targeting ligands can also be coated on the nanocarrier to promote cell uptake. This can lessen the unintended off-target effects caused by miRNA mimics or antagomirs on normal cells.67,68
The ability to release drugs under controlled conditions under various stimuli, including pH, redox potential, temperature, reactive oxygen species (ROS), hypoxia, ultrasound, magnetic fields, electrical fields, light at various wavelengths (UV, visible, and near-infrared), or the presence of particular enzymes, is another benefit of cancer nanomedicine.69,70 Additionally, artificial miRNAs are protected by nanocarriers from breakdown by nucleases and are more stable, the right concentrations of these molecules can be delivered to cancer cells to have the desired therapeutic impact.71,72
Conversely, miRNA mimics and inhibitors administered through nanotechnological platforms are less likely to cause the unfavorable immunogenic activity of tumor-associated immune cells (e.g. macrophages and monocytes) because artificial miRNAs can induce immune responses characterized by the secretion of inflammatory cytokines and type I interferons (IFNs).73,74 In fact, it has been demonstrated that immune cell activation is inhibited when NPs' surfaces are coated with a stealth coating, such as polyethylene glycol [PEG].75 Consequently, these benefits (shown in Fig. 2) raise the prospect of a notable interest in developing, enhancing, pre-clinical, and clinical testing protocols for administering miRNA drugs via nanotechnology.62 In the following paragraphs, we will discuss the nanoformulation of miR-34 in an attempt to upregulate its levels intercellularly to achieve the goal of cancer treatment.
 | ||
| Fig. 2 Different types of polymeric nanocarriers are available for drug delivery. This figure is reproduced from ref. 78 with permission from MDPI, copyright (2020). | ||
5. miR-34 nano-delivery using nanoparticles
Using nanocarrier systems has the advantage of extending the drug's half-life in blood and increasing its stability. Additionally, nanodelivery devices can be customized with particular tumor-targeted ligands for cell surface receptors to enable active targeting. Particle sizes of 1 to 1000 nm are frequently observed in nanocarriers, which are composed of a variety of materials, including polymers, metals, hybrids, etc. Certain characteristics, including enhanced cellular uptake, biodegradability, biocompatibility, affordability, low immunogenicity, endosomal escape, selective accumulation at the tumor site, and simplicity of manufacture, are characteristics of seamless NPs.76,77 In this review, there is a special focus on the latest advancements and uses nanocarriers in order to deliver miR-34-based cancer treatments.5.1. Polymeric nanoparticles
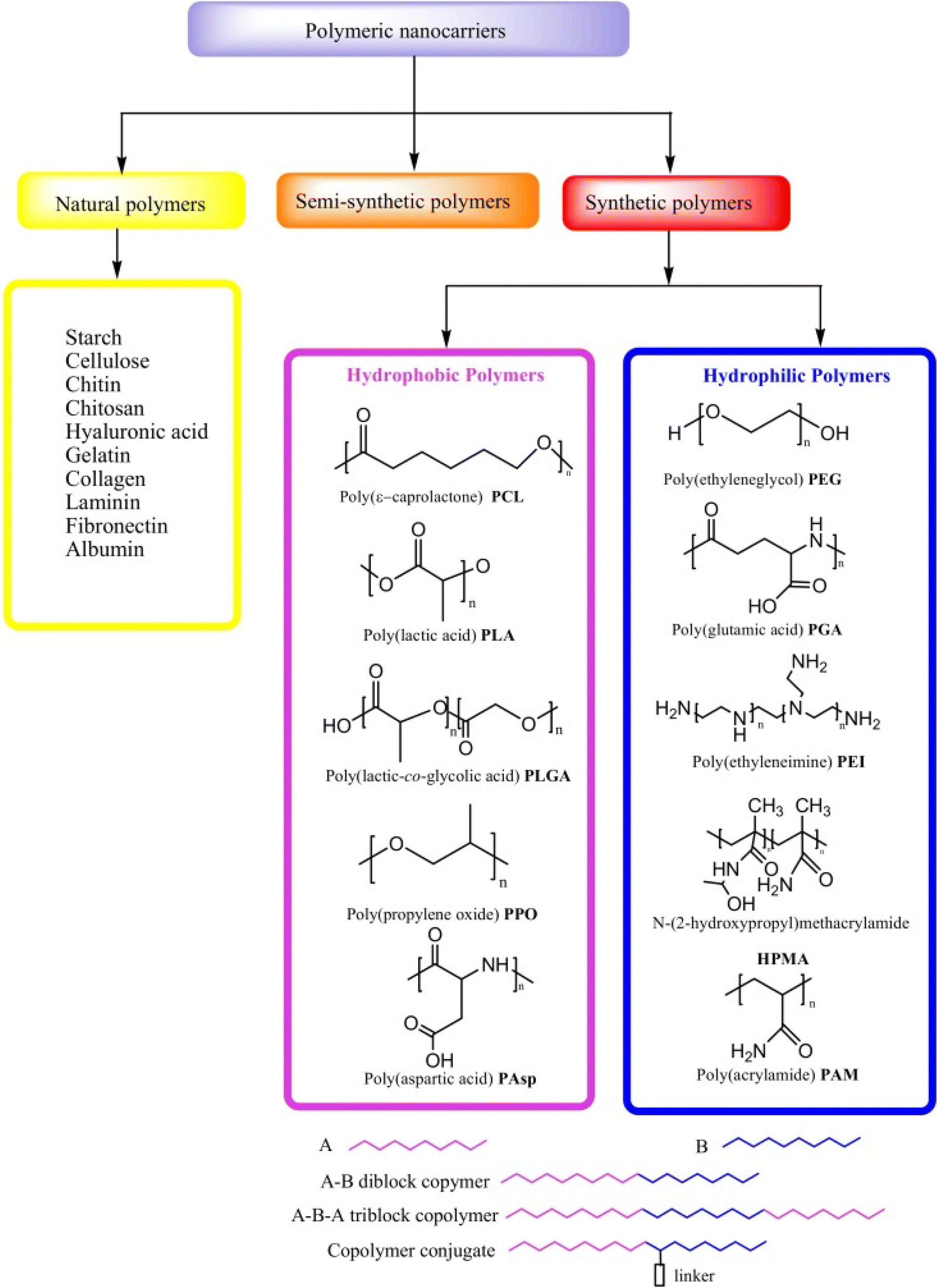
Biopolymers, also known as natural polymers, encompass various polysaccharides and proteins. Their inherent biocompatibility and biodegradability render them particularly suitable for diverse medical applications, including gene therapy, tissue engineering, and cell-based transplantation. Natural polymers can be combined with synthetic molecules through chemical modifications of their functional groups, giving rise to semi-synthetic polymers that mimic human tissue components Fig. 2. However, synthetic polymers hold greater prominence in the development of controlled drug delivery systems (DDSs) due to their versatility in structural design and physicochemical property manipulation. Synthetic polymeric micelles, formed by tailoring the core-forming segments of block copolymers, offer a platform for incorporating a wide array of bioactive compounds such as messenger RNAs (mRNAs), plasmid DNA, proteins, antisense oligonucleotides, small interfering ribonucleic acids (siRNAs), and photosensitizers.78The significant potential of synthetic polymers as carriers for pharmaceutical agents has garnered considerable attention recently, particularly due to their capacity to facilitate the development of DDSs capable of targeted and sustained/controlled release of medications. Enhanced drug delivery efficacy is achieved by encapsulating anticancer drugs within polymeric micelles tailored for cancer targeting and triggered release mechanisms. Synthetic polymers employed in DDSs must exhibit not only biocompatibility and biodegradability but also site-specific activity, stability in systemic circulation, low toxicity and immunogenicity, and the ability to protect drugs from degradation prior to reaching the target tissue. Additionally, it is imperative that polymer nanocarriers for DDSs are facile to assemble and devoid of contaminants.8 Some of the most often used synthetic polymers are poly(ethylene glycol) (PEG), polyglutamic acid (PGA), poly(lactic-co-glycolic acid), (PLGA), polyethyleneimine (PEI) and polylactic acid (PLA), however, natural polymers such as collagen, gelatin, dextran, and chitosan are also used.78–81
Polymers are macromolecules that are produced when one or more monomers combine covalently to form a linear or branching chain. As long as these monomers have two functional groups or more, they may react with another monomer and form a variety of structures. Ideally, a polymer may be produced to achieve specific characteristics by choosing the appropriate type of monomers. In addition, polymers show a high degree of synthetic adaptability that enables their tailoring. Polymeric design might be applied directly to biopolymers through chemical derivatization to achieve specific features. Making synthetic polymers from their corresponding monomers is an additional choice that can result in various forms and uses.80,82
PEG emerges as a synthetic polymer highly esteemed for its remarkable tolerance, biocompatibility, and pronounced solubility in aqueous environments, rendering it an attractive candidate for biomedical endeavors. The endorsement of PEG-conjugated medications by the FDA for human use underscores its safety and utility in therapeutic applications. Consequently, PEG has garnered widespread adoption in various biological realms, encompassing tissue engineering, drug delivery, bioconjugation, biosensing, and imaging modalities. In the domain of bioconjugation and drug administration, PEG plays a pivotal role in enhancing the in vivo stability and solubility of pharmaceutical agents while mitigating their rapid clearance from circulation, thereby optimizing therapeutic efficacy. PEGylation, the process of conjugating PEG directly with pharmaceuticals or attaching it to the surface of drug-encapsulating nanomaterials, has emerged as a transformative strategy. Notably, PEGylation confers augmented in vivo stability upon various nanocarrier systems, including micelles, liposomes, dendrimers, gold nanoshells, quantum dots, and polymeric nanoparticles, consequently enhancing their therapeutic potency.83,84
PEGylated NPs acquire hydrophilicity and near-zero zeta potential, attributes that impede the attachment of opsonins, thereby evading recognition by the mononuclear phagocyte system and subsequent phagocytosis. Furthermore, the substantial hydration levels of PEG chains contribute to the increased hydrodynamic size of PEGylated NPs, affording protection against renal clearance and hindering access to proteolytic enzymes and antibodies. Consequently, PEGylation confers significantly prolonged circulation lifetimes upon NPs, thereby extending these benefits to encapsulated drugs within PEG-based delivery systems. The flexible hydrophilic nature of PEG chains facilitates the rapid diffusion of PEG-modified NPs through mucin fibers, facilitating effective local drug release. The impact of PEG content, molecular weight, NP core characteristics, and timing of administration on achieving optimal therapeutic concentrations while evading immune surveillance and prolonging circulation times has been previously reported. Furthermore, various methods for quantifying PEG density, encompassing both direct and indirect techniques, were delineated. Considering NP PEGylation within the contemporary biomedical landscape underscores its potential to enhance systemic NP delivery, aligning with existing research findings.83
PGA has garnered scientific interest owing to its environmentally friendly characteristics and biodegradability. Illustrated in Fig. 1, PGA represents the fundamental linear aliphatic polyester distinguished by a predominantly crystalline structure with approximately 55% amorphous composition. Notably, PGA exhibits a melting temperature (Tm) within the range of 224 to 227 °C, coupled with a glass transition temperature (Tg) typically ranging from 35 to 40 °C. These thermal properties underscore its suitability for various applications. Given its degradable nature, PGA has captured the attention of clinicians for potential utilization in medical contexts. As elucidated earlier, the primary focus of biodegradable materials lies in their viability as implant materials for surgical interventions.85
PLGA is a copolymer that has garnered significant attention due to its approval by regulatory bodies such as the FDA (U.S. Food and Drug Administration) and EMA (European Medicines Agency), making it suitable for biomedical applications. PLGA possesses desirable characteristics, including biodegradability, biocompatibility,86 and extensive research exploration. Its versatile nature allows for customization through adjustments in copolymer ratios and molecular weights, and it is commercially available for various applications. Also, PLGA exhibits exceptional water solubility, facilitating its utilization in pharmaceutical formulations, in addition to its controlled release capabilities, which can be tailored to suit specific medication requirements and offer flexibility in therapeutic delivery. Additionally, PLGA can be fabricated into diverse shapes and sizes, enhancing its adaptability for different biomedical purposes. Understanding its pharmacokinetic and biodistribution profiles is crucial, as they exhibit dose-dependent and nonlinear behavior, influencing the efficacy and safety of PLGA-based formulations.87,88
PEI stands as a cationic polymer characterized by ethylene (CH2CH2) segments interspersed with recurring primary amino groups. Widely employed in drug delivery systems, PEI serves as both a nanocarrier and transfection reagent, augmenting the efficacy of targeted medications and gene therapy interventions. Notably, a Phase I clinical investigation has explored the use of PEI in delivering a DNA vaccine, highlighting its potential in clinical settings. However, despite its promising applications, research into PEI's clinical utility in cancer management remains scarce, necessitating meticulous safety evaluations prior to its clinical deployment. PEI emerges as a pertinent polymer for encapsulation in the realm of combined gene therapy and anticancer medication delivery. Variations in PEI's properties can significantly influence drug transport capabilities, thus underscoring the importance of understanding its characteristics for optimized therapeutic outcomes. While studies have reported on the use of polymer-based nanocarriers for co-delivering anticancer drugs and gene-targeted therapy, there exists a paucity of research specifically investigating PEI's potential for co-delivery in cancer treatment, particularly in addressing specific subtypes such as breast cancer. This gap underscores the need for further exploration to elucidate PEI's role in advancing cancer therapeutics.89
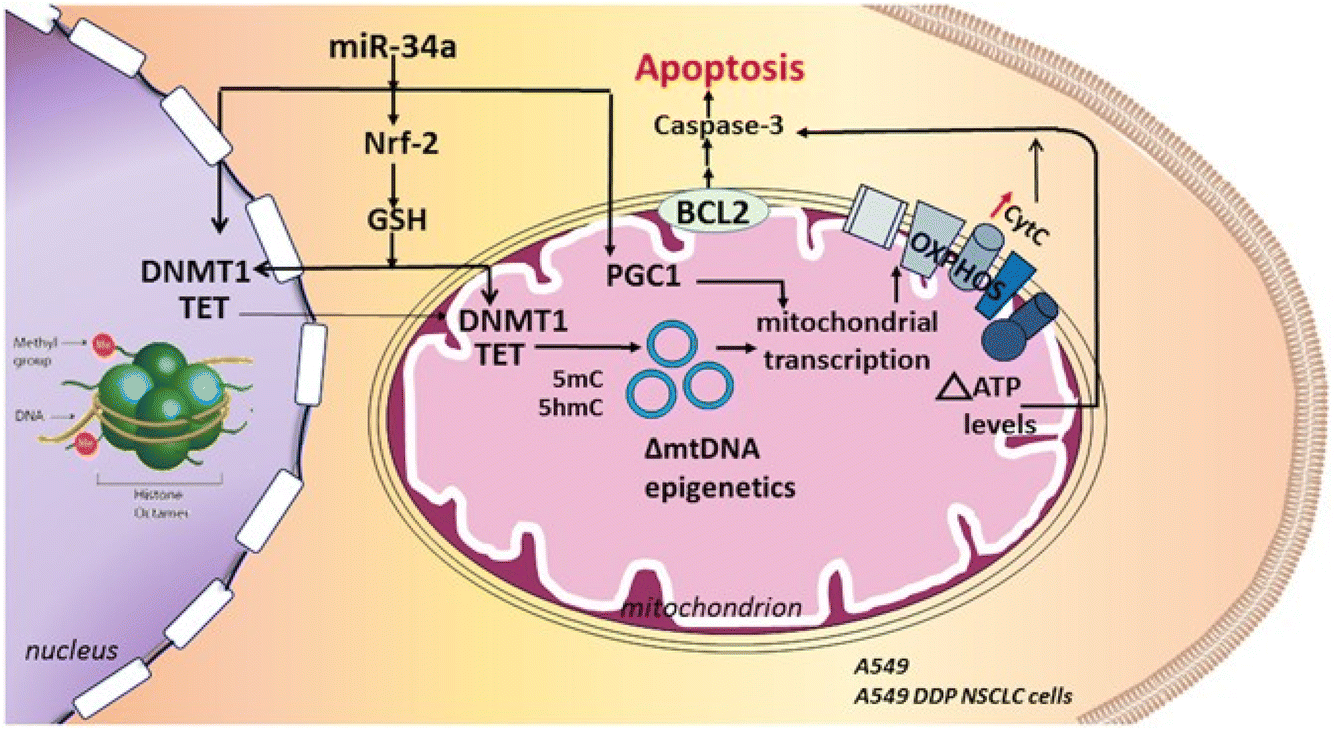 | ||
| Fig. 3 By blocking the Nrf-2 pathway and lowering glutathione (GSH) levels, the miR34a delivery utilizing hyaluronic acid-NPs can cause apoptosis in non-small cell lung cancer cell lines that are susceptible and resistant to cisplatin. This figure is reproduced from ref. 90 with permission from Nature, copyright 2017. | ||
Another study prepared transferrin-decorated thymoquinone-loaded PEG-PLGA nanoparticles (TF-PEG-PLGA-TQ-NPs) for improved TQ administration to NSCLC cells (A549). TF-PEG-PLGA-TQ-NPs results concluded drug loading (% DL) and EE% of 3.5% and 93%, respectively. Dynamic light scattering (DLS) analysis of PEG-PLGA-TQ-NPs indicated a hydrodynamic diameter of 77.50 nm ± 6.35, with a polydispersity index (PDI) of 0.327 ± 0.02. Additionally, Tf-TQ-Np exhibited a time-dependent cytotoxic effect, leading to approximately 55% cell death after 24 hours at a concentration of 5 μg mL−1. This apoptotic mechanism was done via the triggering of p53/ROS feedback loop, activating miR-34a and miR-16 in turn, lowering of Bcl2 and significantly inducing apoptosis in the NSCLC cells. Concurrently, TF-TQ-Np's stimulation of p53-mediated miR-34a inhibited the migration of NSCLC cells.91
A more recent study aimed to develop a nanodelivery system with the intention of attenuating tumor growth subsequent to Microwave Ablation (MWA). This was to be achieved by co-delivering Arsenic Trioxide (As2O3) and miR-34a, thereby eliciting synergistic effects. In this regard, the double emulsion technique was used to blend amphiphilic triblock copolymer (mPEG-PLA-PAE), miR-34a-DSPE, mPEG-DSPE, and arsenic trioxide was utilized to fabricate the nanodelivery system. The sustained release of the drugs and prolonged circulation time within the organism was facilitated by creating protective shells formed by the hydrophilic mPEG fragments encompassing the nanoparticles. The nanoparticles exhibited notable dispersion and assumed a diminutive, nearly spherical morphology. Under physiological conditions (pH 7.4), their size measured approximately 150 nm, which decreased to an average of 110 nm at pH 6.5. Notably, nanoparticles manifested a negative charge of ∼−5 under normal physiological circumstances, transitioning to a positive charge ∼+15 at pH 6.5. These observations suggest that the drug delivery system could augment drug absorption within mildly acidic environments through charge reversal, consequently bolstering drug accumulation at cancerous sites due to extended circulation durations. Moreover, the resultant nanocarrier possessed enhanced cell internalization and absorption in acidic TME, and improved stability, in addition to better biocompatibility down-expressing c-met and up-expressing cyt-c with subsequent prevention of HCC progression.92
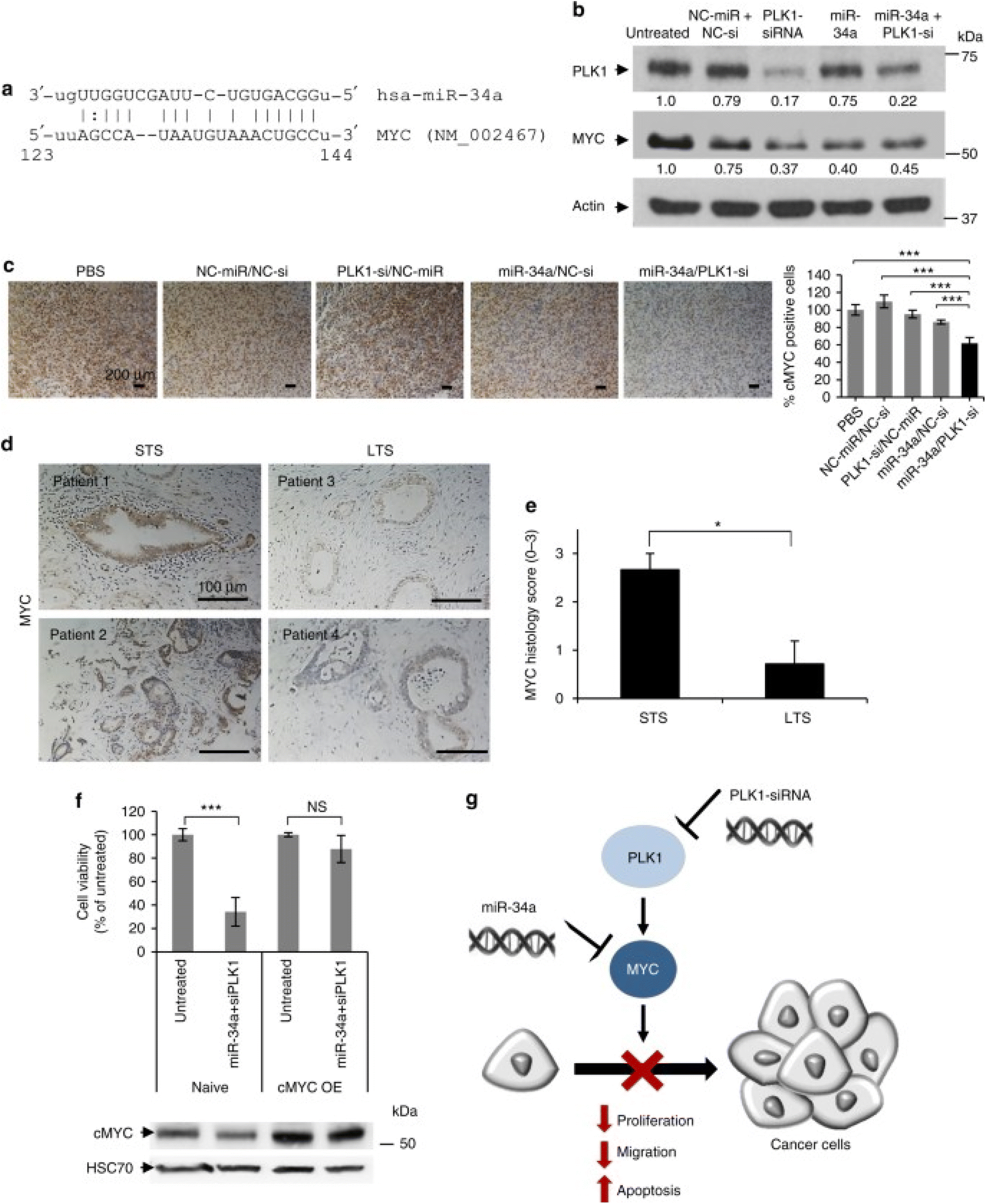 | ||
| Fig. 4 Schematic representation of the mechanism of action of PGA polyplexes loaded miR34a mimics and PLK1-siRNA. (a) A miR-34a binding site located in the MYC 3′-UTR. (b) Analysis of PLK1 and MYC protein levels in MiaPaCa2 cells treated with miRNA and siRNA monotherapies, as well as their combination. (c) Immunostaining of MYC in tumors from various in vivo treatment conditions. (d) MYC immunostaining of short-term and long-term formalin-fixed, paraffin-embedded (FFPE) pancreatic ductal adenocarcinoma (PDAC) specimens. (e) Quantification of MYC immunostaining using histology scores, with a scale of 0 to 3 (0 – none, 1 – weak, 2 – moderate, 3 – high). (f) Cell viability assessment of cMYC-overexpressing MiaPaCa2 cells versus naive cells after 48 hours of combined treatment, with corresponding MYC immunoblotting shown below the graph. (g) Proposed model of synergistic interaction through MYC as a shared target for miR-34a and PLK1. STS indicates short-term survivors, LTS indicates long-term survivors, and cMYC OE denotes cMYC overexpression. Data are expressed as mean ± SD. This figure is reproduced from ref. 93 with permission from Nature, copyright (2018). | ||
A more recent study reported the development of PLGA-poly-L-histidine (PLGA-poly-L-His) NPs containing a moderate cationic charge nanocarrier to deliver miR-34a-5p productively mimics both in vitro and in vivo to NSCLC cancer cell lines (A549). They were formulated as uniform spherical particles with approximately 200 nm in size, a PDI of 0.10–0.20, a surface negative zeta charge of −21 ± 5.60 mV, and EE of 80–100%. The study concluded that PLGA-poly-L-His as a nanocarrier system exhibited better cellular distribution, good stability, and encapsulation of miR-34. The cellular delivery of miR-34 induced p53 upregulation whilst downregulating SIRT1 protein, which plays roles in tumor destruction and regulation of apoptosis, respectively, consequently, inhibiting cancer progression as presented in Fig. 5.95
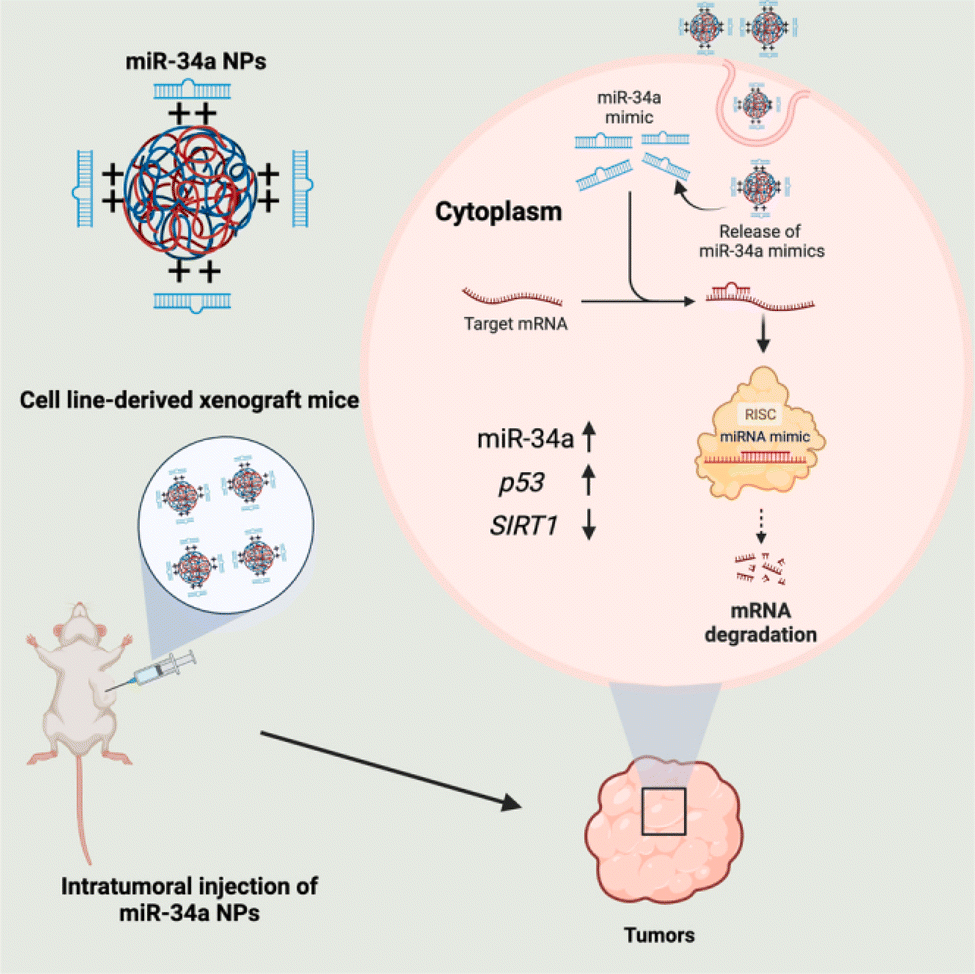 | ||
| Fig. 5 Internalization of miR-34 via PLGA-poly-L-His NPs. This figure was reproduced from ref. 95 with permission from Molecular Therapy, copyright (2023). | ||
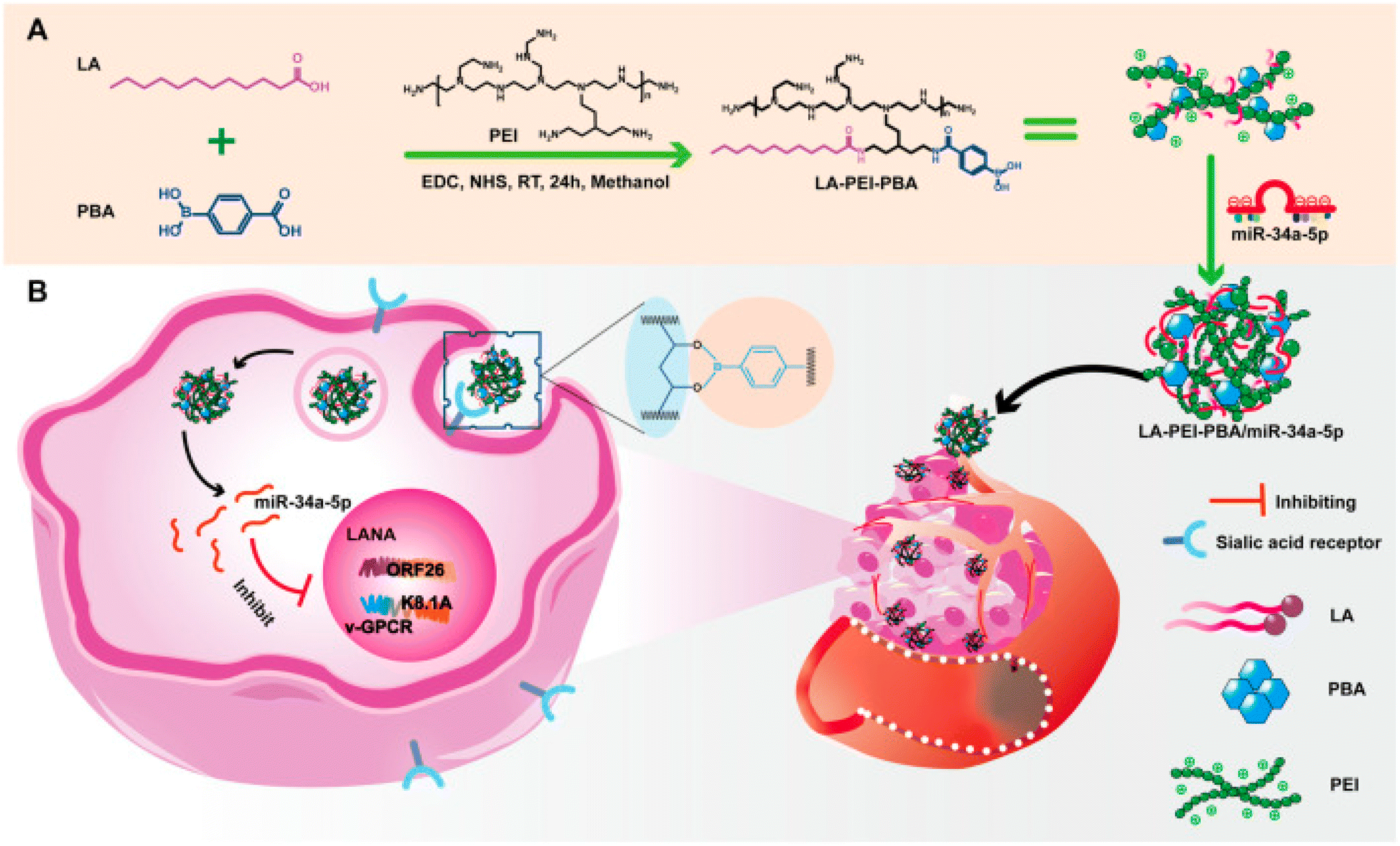 | ||
| Fig. 6 An outline of the synthesis procedure of LA-PEI-PBA and a schematic diagram illustrating the anti-KSHV treatment involving LA-PEI-PBA/miR-34a-5p. (A) Diagram illustrating the synthesis process of LA-PEI-PBA. (B) Schematic representation of the LA-PEI-PBA/miR-34a-5p anti-KSHV treatment protocol. This figure is reproduced from ref. 96 with permission from Front. Bioeng. Biotechnol, copyright (2024). | ||
A very recent study introduced a novel nonviral vector termed PEI-SPDP-Man constructed from polymeric micelles (PSM), engineered to target both intracellular responsive release of miR-34a and cellular uptake pathways simultaneously in TNBC cell lines (MDA-MB-231). PSM is synthesized by linking mannitol (Man) and branching PEI using a glutathione (GSH)-sensitive disulfide bond (succinimidyl 3-(2-pyridyldithio)propionate) (SPDP) after which miR-34a was loaded in. The characterization of PSM/miR-34a exhibited a spherical morphology with a particle size of 148 nm, PDI of 0.096 and a positive zeta potential of 41.2 mV. The resultant PSM/miR-34a gene delivery system employs caveolae-mediated endocytosis to penetrate tumor cells, thereby diminishing miR-34a degradation within lysosomes. To this end, miR-34a is liberated upon detecting the disulfide bond under high GSH concentrations within tumor cells, leading to the downregulation of Bcl-2 and CD44 expression, thus impeding tumor cell growth and invasion, as presented in Fig. 7.97
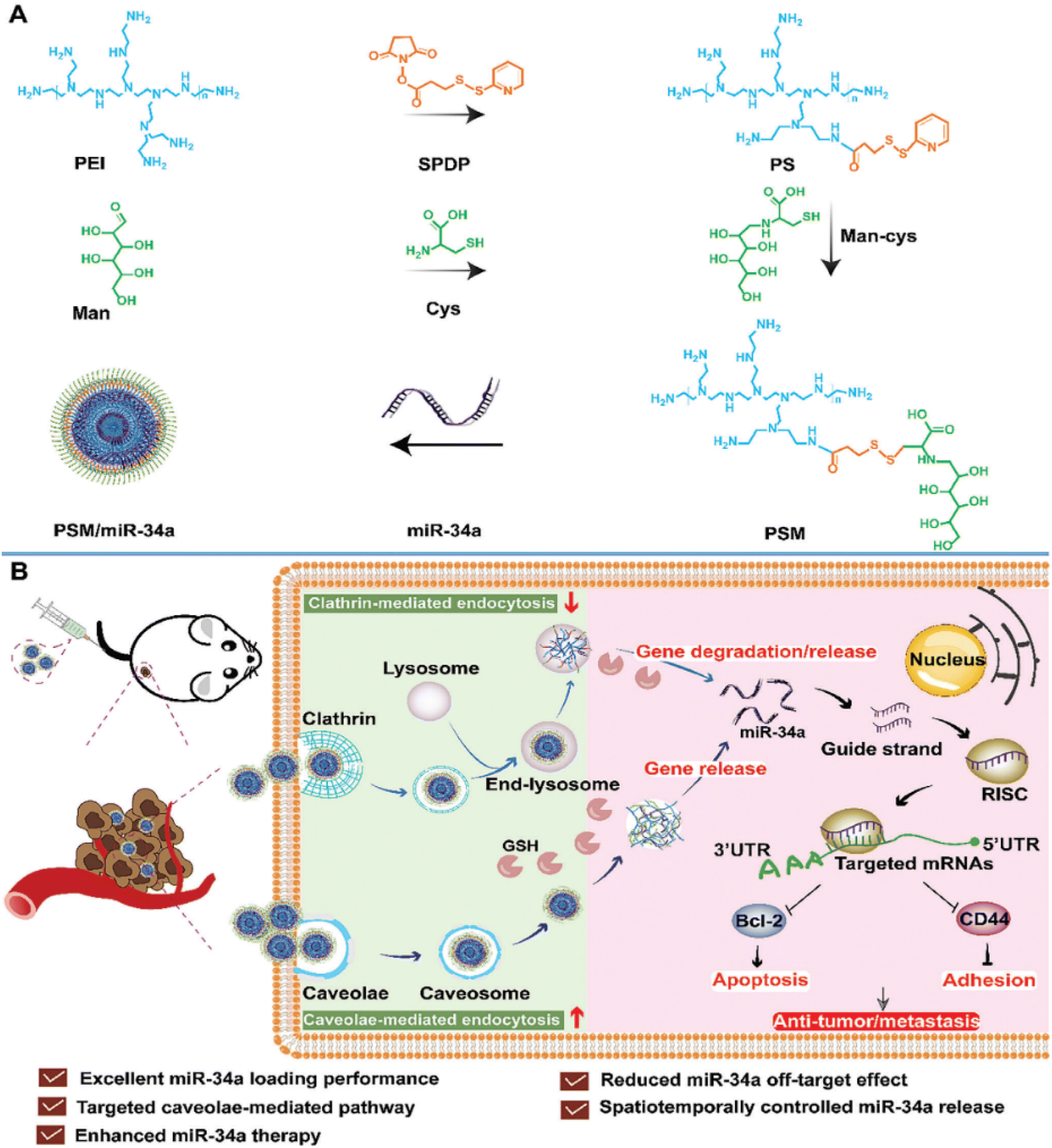 | ||
| Fig. 7 The development of a GSH-responsive-miR-34a delivery platform utilizing PSM termed PSM/miR-34a, coupled with an illustration into its endocytic uptake and intracellular uptake mechanisms. (A) Preparation of the glutathione (GSH)-responsive miR-34a delivery system, PSM/miR-34a. (B) Mechanism of endocytosis and transport designed to enhance the antitumor and anti-metastasis effects in the treatment of orthotopic triple-negative breast cancer. This figure was reproduced from ref. 97 with permission from Wiley Online Library, copyright (2023). | ||
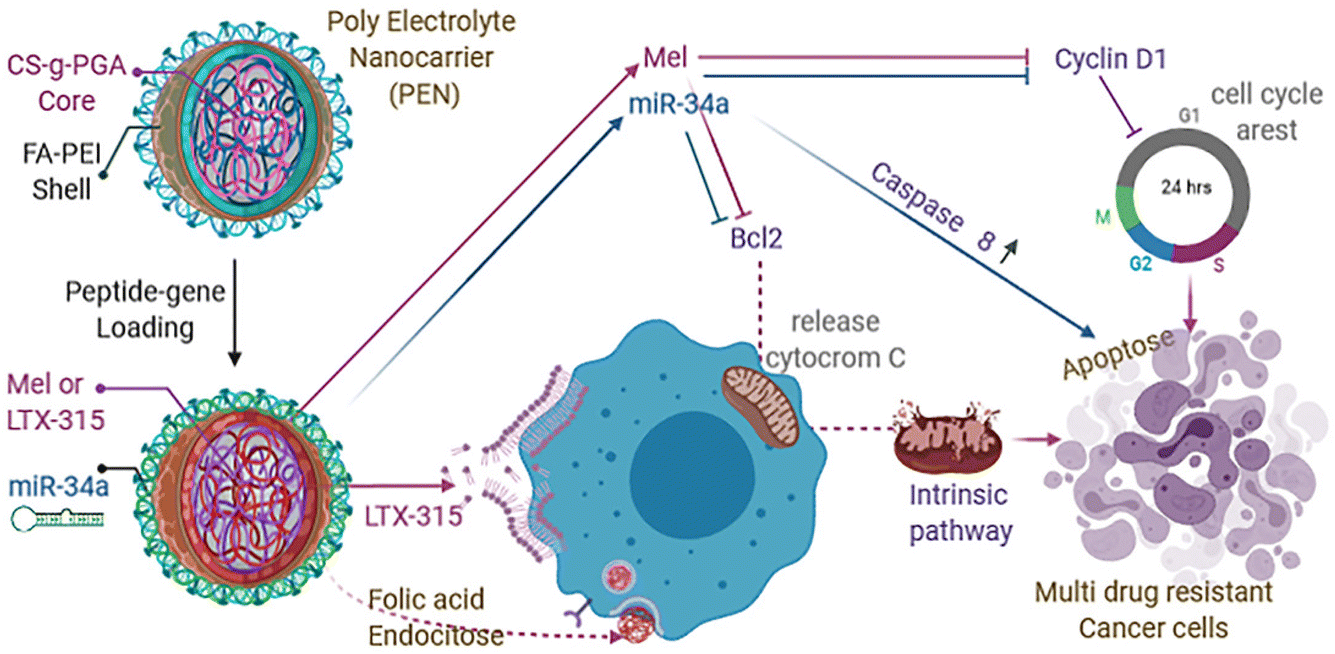 | ||
| Fig. 8 Schematic representation of apoptosis mechanism of action of miR34a, Mel, LTX-315 on TNBC cell line. This figure is reproduced from ref. 98 with permission from ELSEVIER, copyright (2021). | ||
In a more recent study, Chattopadhyay et al., reported the creation of a chitosan-PLGA NPs, which is a chitosan-PLGA-based gene delivery system to deliver capase8 (CASP8), tumor suppressors miR29A-B1 and miR34a for the of treatment NSCLC cell lines (A549). This was done by employing a stabilizer blend comprising chitosan and polyvinyl alcohol (PVA), via emulsion diffusion and evaporation techniques. The inclusion of this stabilizer effectively prevented self-aggregation of the PLGA nanoparticles by serving as a barrier between the PLGA moieties, thereby facilitating the formation of an oil-in-water emulsion. The characterization of these NPs revealed a hydrodynamic diameter of 139 nm and PDI of 0.2, indicating a uniform size distribution. In addition, the presence of amine groups in the chitosan molecule imparts a net positive surface charge to the PVA-chitosan mixture. At acidic pH values, these amine groups undergo protonation, further enhancing the positive charge density of the blend. Additionally, the negatively charged carboxylic groups of PLGA can electrostatically interact with the negatively charged head groups on the cell membrane under acidic pH conditions. This interaction facilitates the binding of the nanoparticles to the membrane and their subsequent internalization within the cells. This nanocarrier resulted in noteworthy biocompatibility, low toxicity, and induced apoptosis, in addition to the suppression of BCL2 and SIRT1 target genes by miRs.99
5.2. Exosomes
Extracellular vesicles (EVs) are a novel structure form that has recently gained attention as a potential delivery system.100 EVs, often referred to as micro- or nanovesicles, are derived from the cell membrane. All prokaryotic and eukaryotic cells can produce these structures in an evolutionarily conserved way.101 They were initially believed to be waste products of cells or entities created when cells were damaged. Nevertheless, additional research on EVs has demonstrated that they are significant cellular constituents and have essential biological roles. Based on their size, origin, and location, EVs may be divided into a number of categories.102 Some of the most well-known EVs are Exosomes, microparticles, shedding vesicles, apoptotic bodies, tolerosomes, proteasomes, and prominosomes. The synthesis of EVs occurs through two distinct processes whether originating directly from cell membrane budding in the first process or as a byproduct of the endocytosis system, namely during the exocytosis of multivesicular bodies. EVs have a significant impact in pathological circumstances and are involved in biological processes in cells. They are a form of communication and have the ability to transport different substances between cells.103Exosomes are double-membrane vesicles released by various cell types, typically exhibiting a size ranging from 30 to 150 nm, with an average diameter of approximately 100 nm. The functional attributes of exosomes are intricately linked to their cellular origin. Specifically, exosomes derived from tumor cells play crucial roles in mediating intercellular communication, particularly in processes such as invasion and migration.104
The phospholipid membrane of exosomes, reflective of their parent cell's composition, harbors a diverse array of proteins and lipids. Notable lipid constituents include sphingomyelin, phosphatidylcholine, phosphatidylethanolamines, phosphatidylinositol, and phosphatidylserine. The composition and abundance of these lipid molecules largely dictate the distinctive characteristics of exosomes. Notably, the high levels of phosphatidylinositol and sphingomyelin contribute to the remarkable stability of exosomes in bodily fluids and across various pH environments. As a result, exosomes benefit from protection against degradation by lipolytic or proteolytic enzymes, courtesy of these lipid molecules. Within the phospholipid membrane of exosomes, lipid rafts harbor proteins such as tyrosine kinase Src and glycosylphosphatidylinositol-containing proteins.103
The protein composition within exosomes is notably intricate. It is understood that exosomes encompass both generic and specialized proteins. Among the generic or nonspecific proteins present in all cell types are CD63, tetraspanins, CD81, and CD9. Conversely, specific proteins are discernible in exosomes derived from distinct cell types, such as HER2 in breast cancer-derived exosomes and EGFR in glioma-derived exosomes. Notably, nonspecific proteins play indispensable roles in exosome functionality. Tetraspanins, as an example of nonspecific proteins, have the capability to form complexes with MHC or integrin molecules through interactions. Moreover, exosomes have the potential to contain ncRNAs, circRNAs, lncRNAs, and miRNAs.103,105
Exosomes hold promise as potential biomarkers for detecting and diagnosing various disorders due to their presence in diverse bodily fluids. For instance, exosomes generated from breast cancer inhibit the growth of T cells and reduce the cytotoxicity of natural killer cells to facilitate immune evasion. On the other hand, exosomes produced from lung cancer cells could activate EMT by upregulating vimentin. HCC cell exosomes can trigger ERK signaling, which in turn causes ZEB1/2 overexpression to facilitate EMT and ultimately cause cancer spread. Additionally, exosomes produced from pancreatic cancer direct macrophage M2 polarization to inhibit immunological response against cancer cells.103 Exosomes generated from ovarian cancer cells may contribute to the development of malignant TME by encouraging fibroblast migration106 and stimulating migration and angiogenesis by upregulating VEGF Fig. 9.107
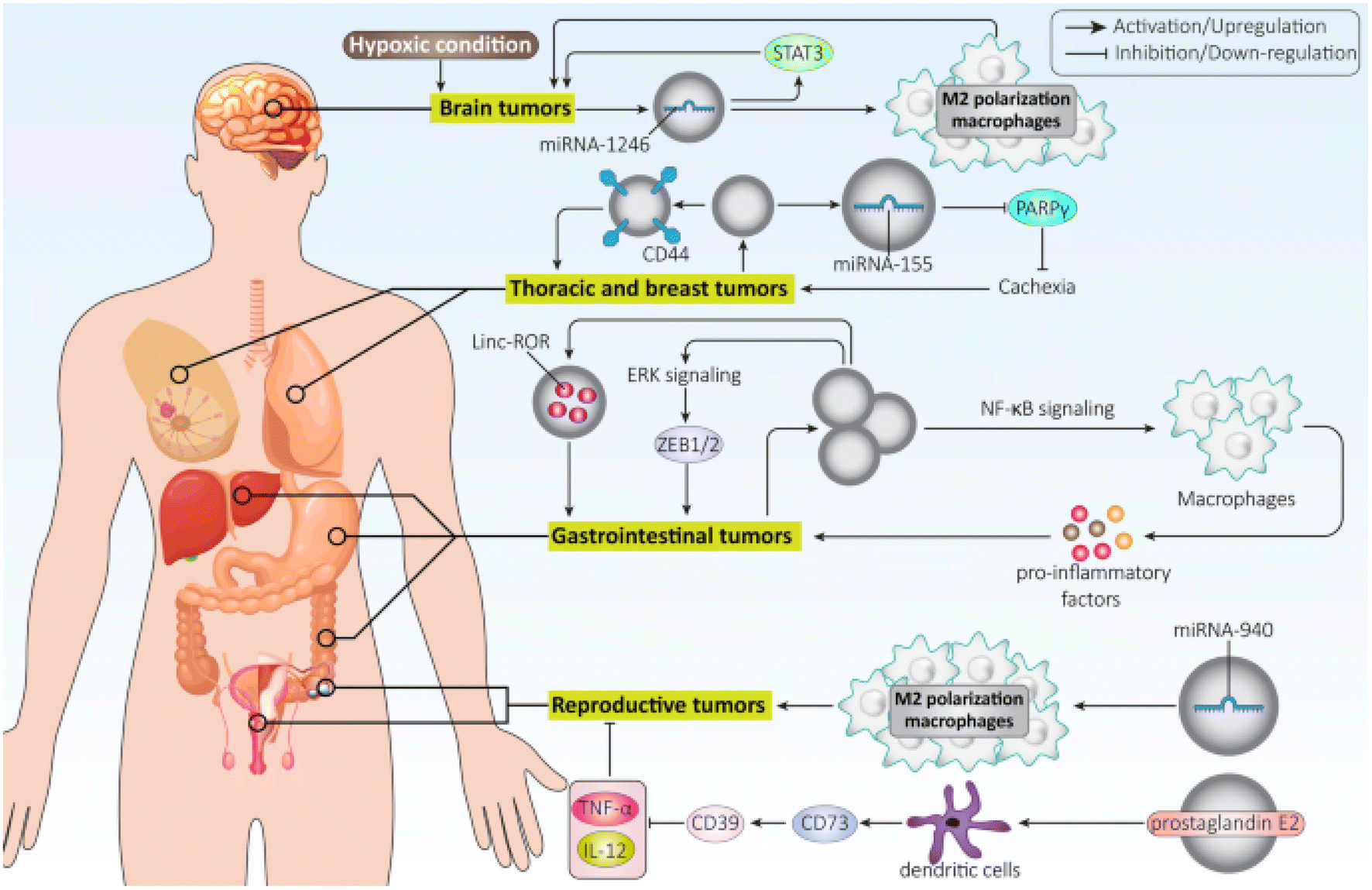 | ||
| Fig. 9 Role of tumor-derived exosomes in cancer progression. This figure is reproduced from ref. 103 with permission from BMC, copyright (2022). | ||
Treatment of pancreatic cancer with tumor suppressor gene miR-34a is restricted due to the absence of a reliable delivery mechanism. Hence, a paper published by Zuo et al., synthesized exosomes-coated miR-34a (exomiR-34a) isolated from the conditioned medium of HEK293 cells via an ultrasound approach. The cell lines utilized in the study include human embryonic kidney (HEK293) cells, normal human pancreatic epithelial ductal cells (HPDE6-C7), as well as human pancreatic cancer cell lines Miapaca-2 and Panc28. The characterization results concluded that exomiR-34a had a diameter of approximately 85.42 nm. According to agarose gel electrophoresis analysis, the most significant value of 64.8 ± 3.5% was attained by the RNA loading rate in exosomes when the ratio of exosomes to miR-34a was 5:1. The results of this study concluded that exomiR-34a effectively crossed the cell membrane and suppressed the expression of Bcl-2. In addition, exomiR-34a treatment dramatically slowed the advancement of pancreatic cancer cells, and the nanoparticles also caused cancer cells to undergo apoptosis by altering the expression of pro-apoptotic proteins such as Bax and p53.108
Another study conducted by Huang et al., demonstrated the role of exosomes in the transmission of low levels of tumor suppressor microRNA-34c-5p (miR-34c) in the advancement of NSCLC by upregulating integrin α2β1. The study characterized exosomes isolated from A549 cells and examined their effects on tumor invasion and metastasis. Exosomes, ranging from 30 to 120 nm, and NSCLC-derived exosomes were shown to promote invasion and migration of NSCLC cells in a dose- and time-dependent manner, while exosomes from normal bronchial epithelial cells inhibited these processes. Further experiments demonstrated direct targeting of integrin α2β1 by miR-34c-3p, inhibition of miR-34c-3p promoting cell migration, and miR-34c-3p-depleted exosomes enhancing cell migration. In vivo experiments in mice confirmed that A549-derived exosomes promoted lung metastases of NSCLC cells in a dose-dependent manner, correlating with increased expression of integrin α2β1. These findings suggest that miR-34c-3p depletion in NSCLC exosomes contributes to tumor progression by upregulating integrin α2β1, thereby promoting invasion and metastasis.109
In nasopharyngeal carcinoma (NPC), malignant behavior and radioresistance pose significant challenges to radiation therapy (RT) efficacy and patient prognosis. Thus, a study published by Wan et al., investigates the tumor suppressor miR-34c and its impact on NPC cell lines (CME-2, 5–8F, and 6–10B) development and radioresistance. Screening methods identified miR-34c as associated with NPC occurrence and radiation resistance. In vitro and in vivo experiments revealed that miR-34c overexpression inhibited NPC malignant behavior by targeting β-Catenin, reducing invasion, migration, proliferation, and epithelial–mesenchymal transition (EMT). Moreover, miR-34c overexpression or β-catenin knockdown alleviated radioresistance in NPCs. Exosomes derived from miR-34c-transfected mesenchymal stem cells (MSCs) attenuated NPC progression and enhanced radiation-induced apoptosis. The transfected MSCs transfer miR-34c via exosomes showed a particle size of approximately 100 nm. Clinical data confirm that overexpressing miR-34c inhibits NPC cell proliferation, migration, and invasion by targeting β-catenin and enhances radiosensitivity by inducing apoptosis and reducing resistance to radiotherapy. MSC-derived exosomes carrying miR-34c inhibit NPC development and radioresistance in vitro and in vivo.110
Another study conducted by Vakhshiteh et al., looked into the possibility of inhibiting the growth of BC cells (MDA-MB-231) using genetically modified dental pulp MSCs (DPSCs) to produce modified exosomes that might be used as a vehicle for miR-34a, whilst comparing exosomes to liposomes. The findings indicated that exosomes had a cup-shaped morphology with a slightly negative zeta potential and mean particle sizes of 77 ± 5 and 65 ± 9 in naïve exosomes and 34a-Exosomes, respectively. On the other hand, the liposomes exhibited a consistent particle size of 100 nm and a spherical shape. When miRNA was added to the liposomes, the particle sizes did not significantly increase (111.4 ± 2.4 nm and 112.2 ± 6.7 nm) but achieved effective miRNA encapsulation and an ideal size for the transportation of genes. The introduction of miR-34a exosomes led to the downregulation of c-MET and Bcl2. Exosomes containing miR-34a exhibited a dose-dependent cytotoxic effect on BC cells. Moreover, compared to untreated exosomes, those carrying miR-34a have significantly attenuated the migratory capacity of MDA-MB-231 cells by 15%. Similarly, therapy involving miR-34a-loaded exosomes markedly suppressed cancer cell invasiveness by 8.5%.111
A recent study conducted by Hosseini et al., reported the nanodelivery of miR-34a delivery to CRC cell line (CT-26) by encapsulation within tumor-derived exosomes (TEXs). The characterization outcomes revealed a predominantly spherical morphology of the particles, characterized by an average TEX size of less than 100 nm and a zeta potential of −19.7 ± 12.12, both falling within the established range for exosome dimensions. Furthermore, the study unveiled that TEX-miR-34a administration exhibited a dose-dependent induction of apoptosis in CRC cell lines, leading to a notable reduction in tumor cell viability. Concurrently, downregulation was observed in PD-L1 expression alongside an enhancement in immunogenicity. Additionally, the investigation highlighted the responsiveness of CRC cells to interleukin-6 (IL-6), which prompted increased expression of IL-6 receptor (IL-6R). This upregulation, mediated through activating the oncogenic transcription factor STAT3, facilitated epithelial–mesenchymal transition (EMT) and subsequent invasion.112
5.3. Metallic nanoparticles
Metal nanoparticles (MNPs) have garnered considerable attention for their multifunctional properties within the realm of nanomedicine. Various types of MNPs, including gold, silver, iron, iron oxide, zinc, titanium, cerium oxide, nickel, copper, magnesium, barium, calcium, and bismuth, have been explored for their potential in cancer therapy. Among these, gold nanoparticles (AuNPs) have exhibited auspicious characteristics, followed by silver and magnetic nanoparticles (MNPs).113 Noble metals-based nanoparticles, such as gold and silver, have been extensively investigated for their applications in cancer treatment Fig. 10. For instance, surface functionalization or coating of AuNPs can enhance their anti-tumor efficacy, rendering them suitable for diagnostic, therapeutic, bioimaging, and prognostic purposes. On the other hand, silver nanoparticles (AgNPs) exert their effects primarily through oxidative stress, reactive oxygen species (ROS) generation, and induction of DNA damage. While ROS plays crucial roles in cellular signaling and homeostasis, an overabundance induced by AgNPs can lead to toxicity by destroying DNA, lipids, and proteins.114 In contrast, zinc oxide nanoparticles (ZnO NPs) belong to the category of non-noble MNPs. Various chemical modifications, including metal doping, polymer coating, and organic photosensitizer utilization, can enhance their photocatalytic activity and ROS production. This heightened ROS generation contributes to their augmented antibacterial and anticancer efficacy.113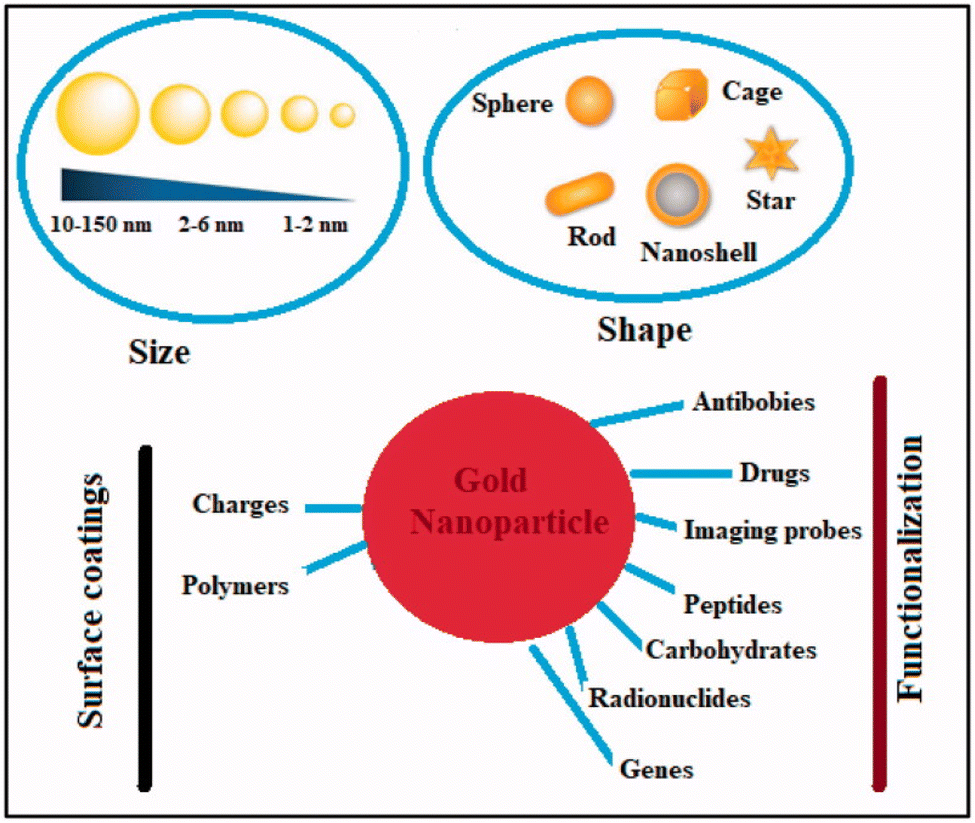 | ||
| Fig. 10 Physical–chemical characteristics of gold nanoparticles and their ability to combat cancer. This figure is reproduced from ref. 113 with permission from Taylor & Francis, copyright (2022). | ||
In another research published by Alden et al., a novel delivery method was introduced utilizing functionalized core-shell-shell (CSS) nanoparticle systems comprising gold–silver–gold layers, modified with FDA Diels–Alder molecular linkers, as a spatiotemporally controlled delivery strategy for synthetic miR-34a mimics (CSS-FDA-miR34a) to be tested in vivo using a xenograft mouse model of human esophageal squamous carcinoma cells (TE10). This approach showed promise for tumor-specific selectivity and effective delivery of miRNA mimics by precisely controlling the spatiotemporal administration of tiny nucleic acid therapies. Through regulated chemical breakage and release of the miRNA mimic payload, the light-inducible particles take advantage of the photothermal heating of metal nanoparticles caused by local surface plasmonic resonance. The characterization concluded that the CSS particles had a spherical morphology with an average diameter size of ∼91.3 ± 5.4 nm, a PDI of 0.187 and a zeta potential of approximately −36 mV. The biology testing concluded that TE10 cells subjected to CSS-FDA-miR34a mimic showed a significant reduction in the expression of ROCK1, STAT3, MYC, and TGIF2 as they are direct targets of miR-34a-5p and a notable increase in the levels of active CASP-3, a key apoptotic signaling protein.116
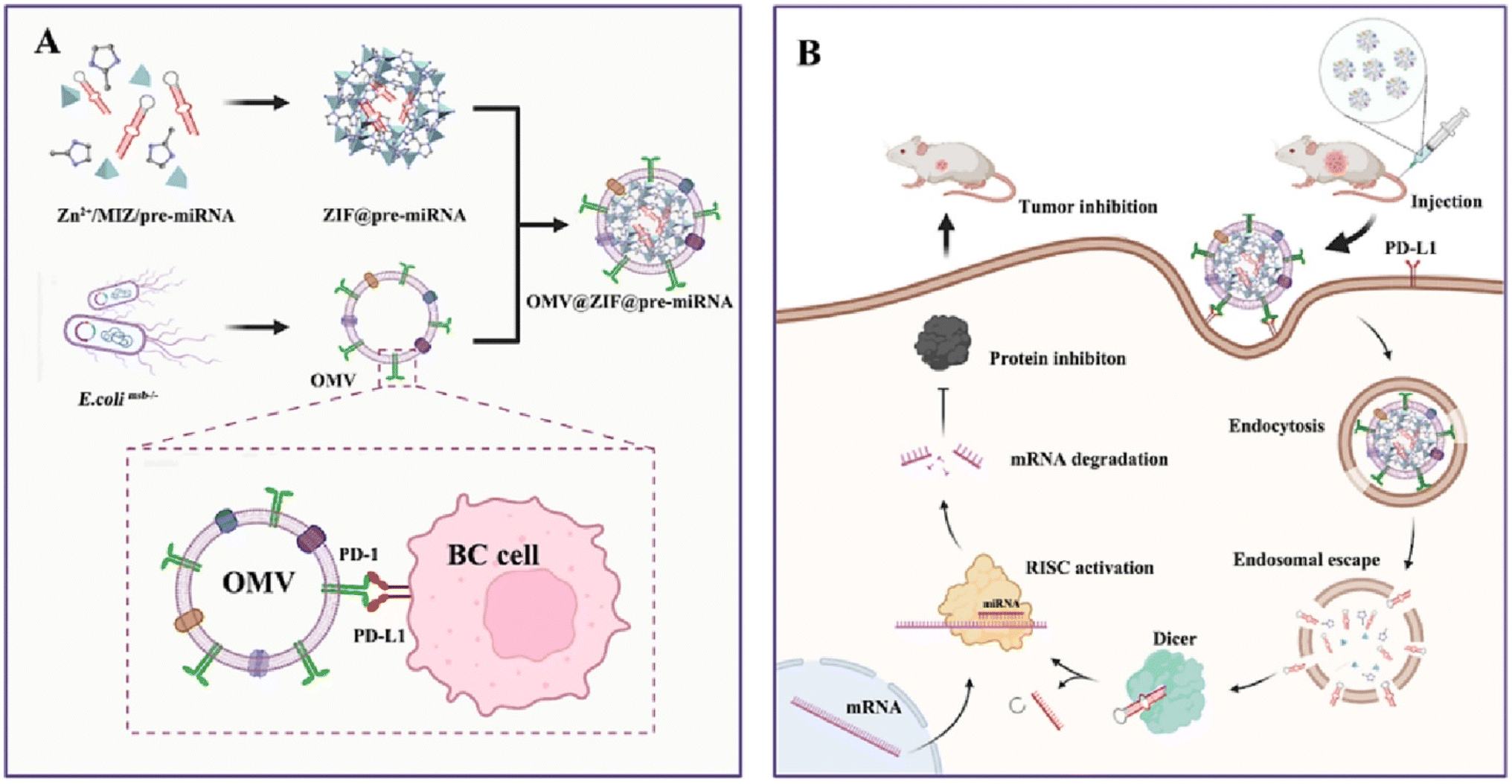 | ||
| Fig. 11 The preparation and mechanism of action of OMV@ZIF-8@miR-34. (A) Preparation process of OMV-PD1@ZIF-8@miRNA. (B) Schematic representation illustrating the targeted delivery and therapeutic efficacy of OMV-PD1@ZIF-8@miRNA. This figure is reproduced from ref. 117 with permission from ELSEVIER, copyright (2023). | ||
5.4. Miscellaneous nanoparticles
However, to achieve optimal outcomes for cancer patients, it may be imperative to utilize MSNs for purposes beyond imaging or treatment alone. Therapeutic benefits cannot be solely attained through imaging, and monitoring of drug distribution, release, and efficacy cannot be adequately accomplished solely through treatment interventions. Additionally, using separate nanoparticles for therapeutic and imaging applications may introduce complexity and heighten the potential for unfavorable outcomes. Therefore, there is a growing emphasis on integrating the dual functionalities of therapy and imaging within a single nanoparticle system. This integration is encapsulated by the term “theranostic,” which merges “therapeutics” and “diagnostics”.121
Panecbianco et al., explore the capability of silicon dioxide nanoparticles, denoted as SiO2NPs, to serve as carriers for biologically active miRNAs. Through experimentation involving both in vitro and in vivo model systems while investigating the efficacy of SiO2NPs in transporting miR-34a into a Claudine-low triple-negative breast cancer (SUM159 pt, human). SiO2NPs were synthesized via a ternary method without microemulsion, yielding uniformly spherical nanoparticles with a negative zeta potential of −10.7 ± 4.8 mV. These SiO2NPs demonstrated effective intra-tumoral delivery and biocompatibility for biologically active miR-34, along with enhanced organoid internalization, without eliciting significant adverse effects. The molecular impact of miR-34a delivered by SiO2NPs revealed a notable downregulation of the Notch1 signaling pathway and upregulation of p53, consequently inducing apoptosis.122
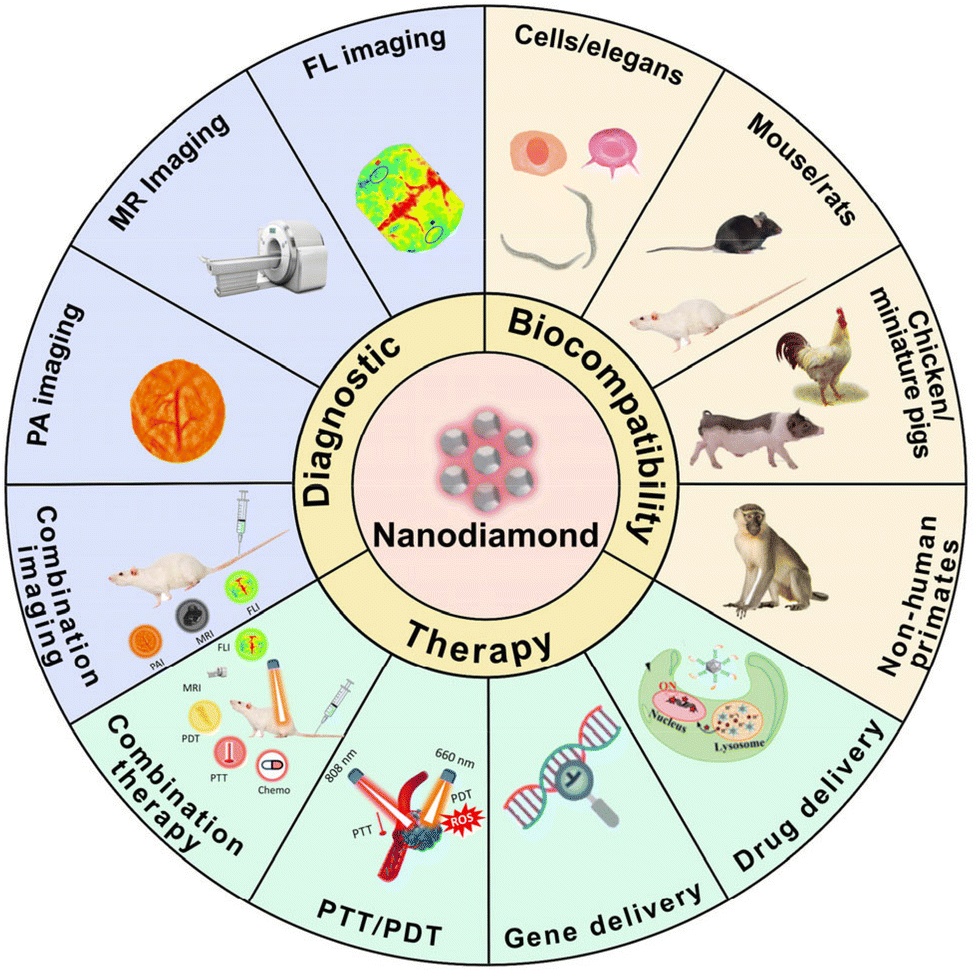 | ||
| Fig. 12 A schematic overview highlighting diverse applications of nanodiamonds (NDs). This figure is reproduced from ref. 123 with permission from ScienceDirect, copyright (2023). | ||
In a recent investigation, Abate et al. evaluated and optimized nanodiamonds modified with PEI to deliver miR-34a, designated as ND-PEI-miR-34, as presented in Fig. 13. Analysis of the physicochemical properties of the nanodiamonds revealed consistent size (304.5 ± 31.33 nm), a sub-globular structure, a polydispersity index (PDI) of 0.43 ± 0.014, and a negative zeta potential of −26.4 ± 2.50 mV. Moreover, the cytotoxicity of the nanodiamonds was assessed on MCF7 and MDA-MB-231 breast cancer cell lines, demonstrating exceptional biocompatibility, significant internalization, and markedly reduced cell migration and proliferation. This effect was attributed to ND-PEI-miR-34 inducing the accumulation of acetylated p53 and downregulation of sirtuin 1 (SIRT1) in MCF7 and MDA-MB-231 cells. Subsequently, ND-PEI-miR-34 impeded tumor progression and mitigated the effects of caspase 3 and beclin 1 deficiency, leading to the induction of apoptosis and autophagy.124
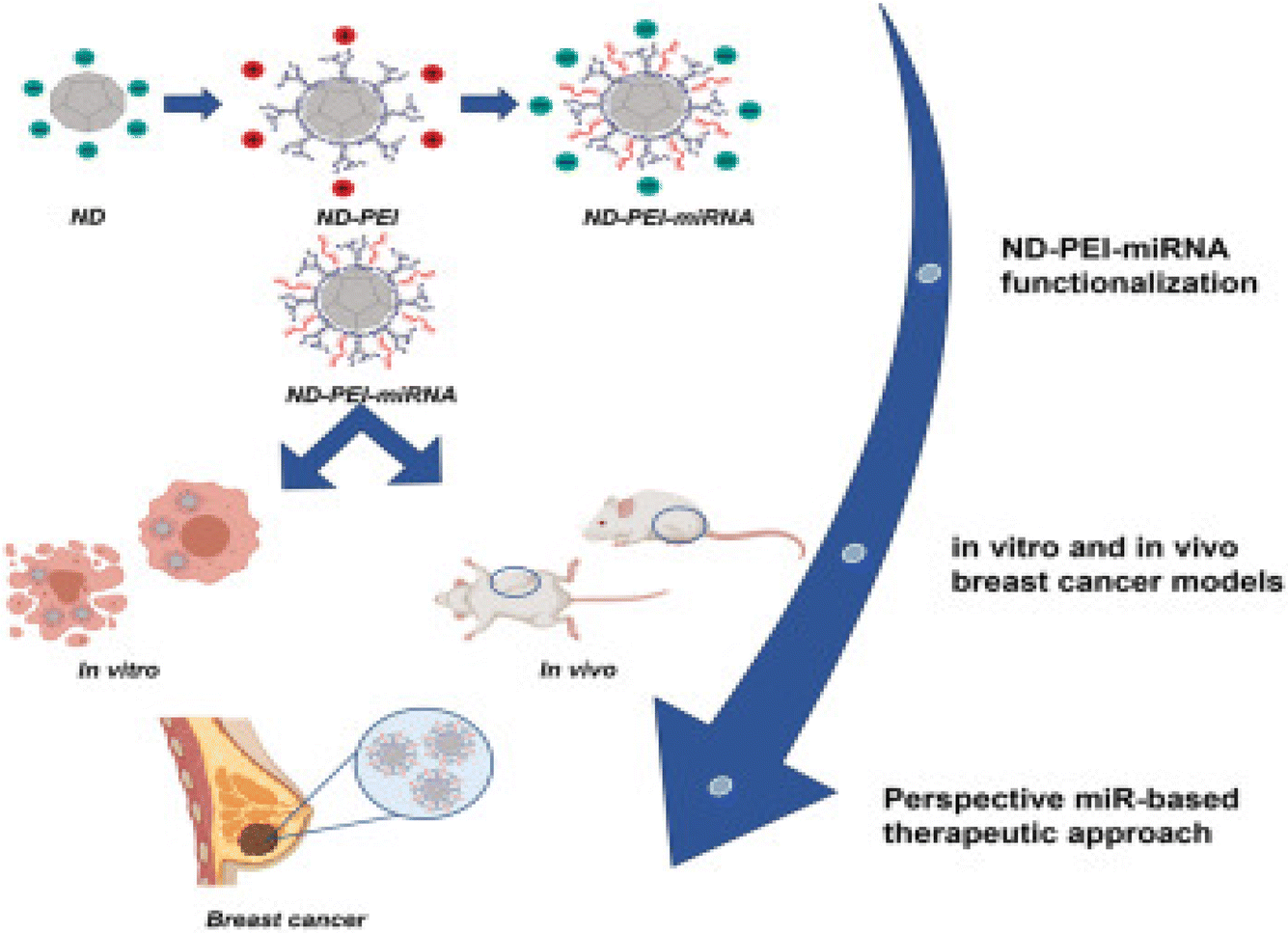 | ||
| Fig. 13 Presenting the steps of formulating modified nanodiamonds. This figure is reproduced from ref. 124 with permission from Molecular Therapy, copyright (2023). | ||
Hybrid systems offer several advantages over non-hybrid counterparts. They have demonstrated superior loading efficiency, release kinetics, cellular uptake, and cytotoxicity both in vitro and in vivo. The morphology of core–shell HNPs provides an optimal delivery platform for co-delivery of multiple drug substances, drugs with biologics, and drugs alongside imaging agents. In gene delivery to cancer cells, lipid-polymer hybrid nanoparticles (LPHNPs) exhibit enhanced cellular delivery efficacy compared to lipoplexes. Hybridizing metal nanocarriers with endogenous substances or lipids reduces toxicity by limiting metal-cell interactions, thus enhancing nanocarrier stability. Additionally, combining inorganic nanocarriers with polymers possessing antioxidant properties decreases reactive oxygen species (ROS) generation by the inorganic nanocarriers. However, hybrid nanocarriers face challenges in predicting and precisely controlling their physical and biological characteristics, as well as reproducibility issues. In addition, it was observed that coating a shell layer over a core increases particle size, affecting the biological fate of the hybrid nanocarrier.125–127
A published paper by Xia et al., was targeting the utilization of ND-based layer-by-layer nanohybrids using a facile self-assembly technique with protamine (PS), and folic acid (FA), resulting in FA/PS/miR-34a/PS@NDs with the goal of delivering miR-34a specifically to TNBC cell lines (MDA-MB-231). The characterization of the FA/PS/miR-34a/PS@NDs nanohybrids presented with a shape of well-defined atomic lattice fringes, which are covered with amorphous materials (PS, miRNA, and FA). Moreover, the size of these nanohybrids was measured to be 210 nm and a negative zeta potential of −25 mV. These results suggest that miR-34a was effectively delivered into MDA-MB-231 cells via FA/PS/miR-34a/PS@NDs nanohybrids, facilitated by a pathway associated with FA, and exhibited anti-tumor effects by inducing apoptosis, inhibiting proliferation, and impeding migration of MDA-MB-231 cells. The internalization and effectiveness of the nanohybrids are due to the inhibition of one of the Activator protein 1 (AP-1) transcription factor and Fra-1 (Fos-related antigen1), a member of transcription factor activator protein (AP-1), consequential in regression of cell motility, growth, migration, and proliferation.128
Goyal et al., reported in a study the employment of layer-by-layer assembled nanoshells (LbL-NS) as a delivery system for miR-34a targeting MDA-MB-231 TNBC cells. The rationale behind this approach was the anticipation that the structured layers would facilitate efficient miR-34a loading, and the positively charged topmost layer composed of poly-L-lysine (PLL) would facilitate cellular uptake through endocytosis. The construction of LbL-NS involved alternating positive PLL and negative miRNA deposition onto negatively charged nanoshells. Particularly, PLL was strategically placed as the outer layer to both shield the miRNA cargo and enhance cellular internalization. LbL-NS was synthesized using nanoshells comprised of 120 nm diameter silica cores and 15 nm thick gold shells. The created nanoshells were sequentially coated with (i) mercaptoundecanoic acid (MUA), (ii) poly-L-lysine (PLL), (iii) miR-34a or control miRNA (miR-co), and (iv) PLL as presented in Fig. 14. The characterization of the formulated NPs showed a spherical morphology with a size of 208 ± 4 nm and a positive zeta potential of + 53 ± 5 mV, promoting their effective penetration through the negatively charged lipid bilayer into cells. This study demonstrated that these NPs presented a distinctive approach for fabricating uniformly dispersed and precisely defined miRNA nanocarriers. In addition, these NPs showed internalization via endocytosis and exhibited intracellular distribution similar to polyplexes. It was shown that these particles may effectively deliver miRNA into MDA-MB-231 cells, as evidenced by the downregulation of SIRT1 and Bcl-2. Furthermore, the observation that miR-34a-LbL-NS-treated cells demonstrate reduced metabolic activity and proliferation compared to control-exposed cells provides additional support for this assertion.129
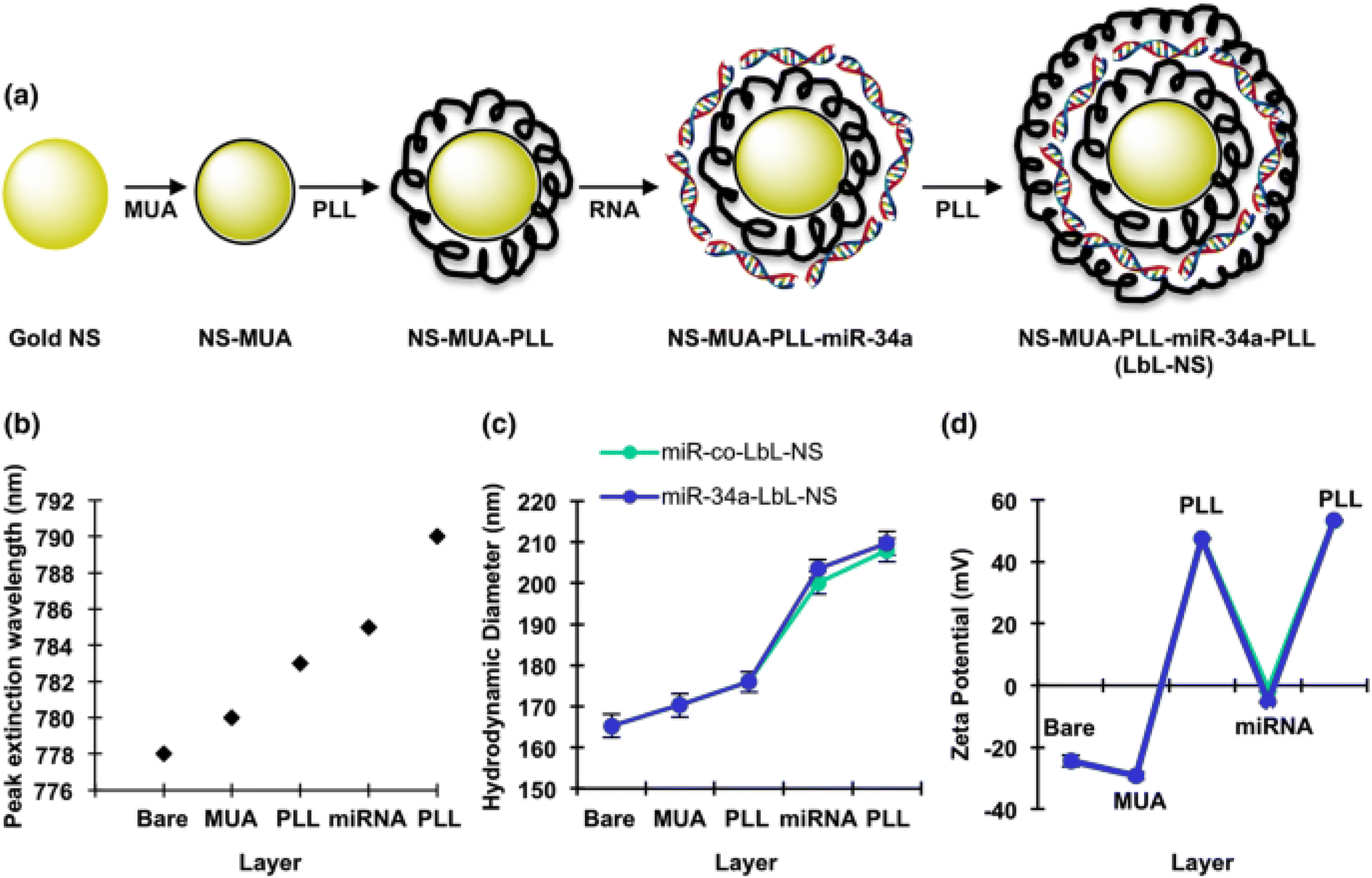 | ||
| Fig. 14 Schematic representation of the production of layer-by-layer gold nanoshells. (a) Schematic diagram of the synthesis process for layer-by-layer assembled nanoshells (LbL-NS). (b) Peak extinction wavelength of the nanoparticles following each coating step. (c) Hydrodynamic diameter and (d) zeta potential of the nanoparticles after the addition of each layer. Data are presented as the averages of three independent experiments, each performed in triplicate. The legend in panel (c) also applies to panel (d). This figure is reproduced from ref. 129 with permission from SPRINGER LINK, copyright (2018). | ||
Thus, researchers have incorporated miRNA-34a into PEGylated niosomal formulation to enhance efficacy against MCF-7 and T47D human breast adenocarcinoma cells. These NPs' formulation resulted in a spherical shape with a diameter of ∼100 nm, outstanding positive zeta potential charge of approximately + 24 mV, and a high EE of almost 100%. The in vitro antitumor effects revealed no observable toxicity. This suggests that any observed anti-proliferative effects are attributed solely to the encapsulated miRNA-34a. Notably, the miRNA-34a/niosome formulation exhibited a concentration-dependent inhibition of cancer cell growth. In addition to showing improved cellular cytotoxicity, enhanced stability, and increased cellular uptake, hence safer delivery of miR-34.131
CDs are naturally occurring cyclic oligosaccharides composed of D-glucopyranose units linked by α1–4 glycosidic bonds. They possess a characteristic truncated cone shape. The three main types of CDs, namely α-, β-, and γ-CDs, each consist of 6, 7, and 8 glucose units, respectively, and exhibit differences in diameter, melting temperatures, and solubility. Structurally, the hydroxyl groups are oriented towards the exterior of the cone, while the hydrogen and glycosidic oxygen linkages are positioned inward, rendering the exterior hydrophilic and the interior hydrophobic. CDs serve as hosts for molecules with appropriate hydrophobic regions and dimensions, allowing them to form inclusion complexes while remaining soluble in aqueous solutions. This process is known as inclusion complex formation.133
CDs play a crucial role in enhancing the perceived aqueous solubility and dissolution rate of medications with low water solubility, potentially reducing side effects and adverse reactions such as gastrointestinal or ocular discomfort. They contribute to improved palatability, handling, and chemical stability in formulations while also facilitating permeability across biological membranes and reducing evaporation while stabilizing flavors. CDs have been extensively utilized to enhance the solubility of class II and IV drugs, which exhibit limited aqueous solubility, for treating various conditions, including cancer and parasitic diseases. However, it's worth noting that while CDs have been employed to enhance stability and mask flavors and odors, there have been reports of complex formation with class I and III medications as well.133,134 However, the utilization of CDs as drug carriers has been impeded by several factors. These include the limited solubility of native CDs, constraints on the range of molecules that can be encapsulated, the relatively short half-life in the bloodstream following in vivo administration, and the challenge of controlling drug release during transport. The release kinetics are dependent on various factors such as host–guest interactions, pH, and the specific biological species present in the surrounding environment.133,135
A recent study introduced folic acid (FA)-modified β-cyclodextrin (CD)-grafted PEI (β-CD-PEI-FA) copolymer nanocarriers as a strategy to combat BCBL-1 cells (KSHV-infected lymphoma cells), and SK-RG cells (KSHV-infected neuroblastoma cells). The β-CD-PEI-FA copolymer was engineered to enhance the stability of nucleic acid medication, specifically miR-34a-5p, for targeted delivery to KSHV-infected cells while diminishing the cytotoxicity associated with PEI. Gel electrophoresis retardation experiments revealed that β-CD-PEI-FA could form an electrostatic adsorption complex with miR-34a-5p at a mass ratio below 1.5, resulting in the formation of a drug-loaded nanocomplex termed β-CD-PEI-FA/miR-34a-5p as presented in Fig. 15. This complex effectively shielded miR-34a-5p from degradation by serum and nucleases. Additionally, as the mass ratio increased progressively from 1:1 to 5:1, the particle size of the drug-loaded nanocomplex decreased to 203.13 ± 0.41 nm, while its surface potential decreased to 27.02 ± 0.72 mV. These changes in particle size and surface potential, particularly the size reduction, enhanced the suitability of the nanocomplex for cellular uptake via endocytosis. In vitro biosafety assessments revealed excellent biocompatibility of the vector and subsequent cell studies demonstrated that β-CD-PEI-FA/miR-34a-5p not only efficiently transported miR-34a-5p but also accelerated the G2-phase process of BCBL-1 and SK-RG cells following KSHV infection, thereby suppressing cell growth.136
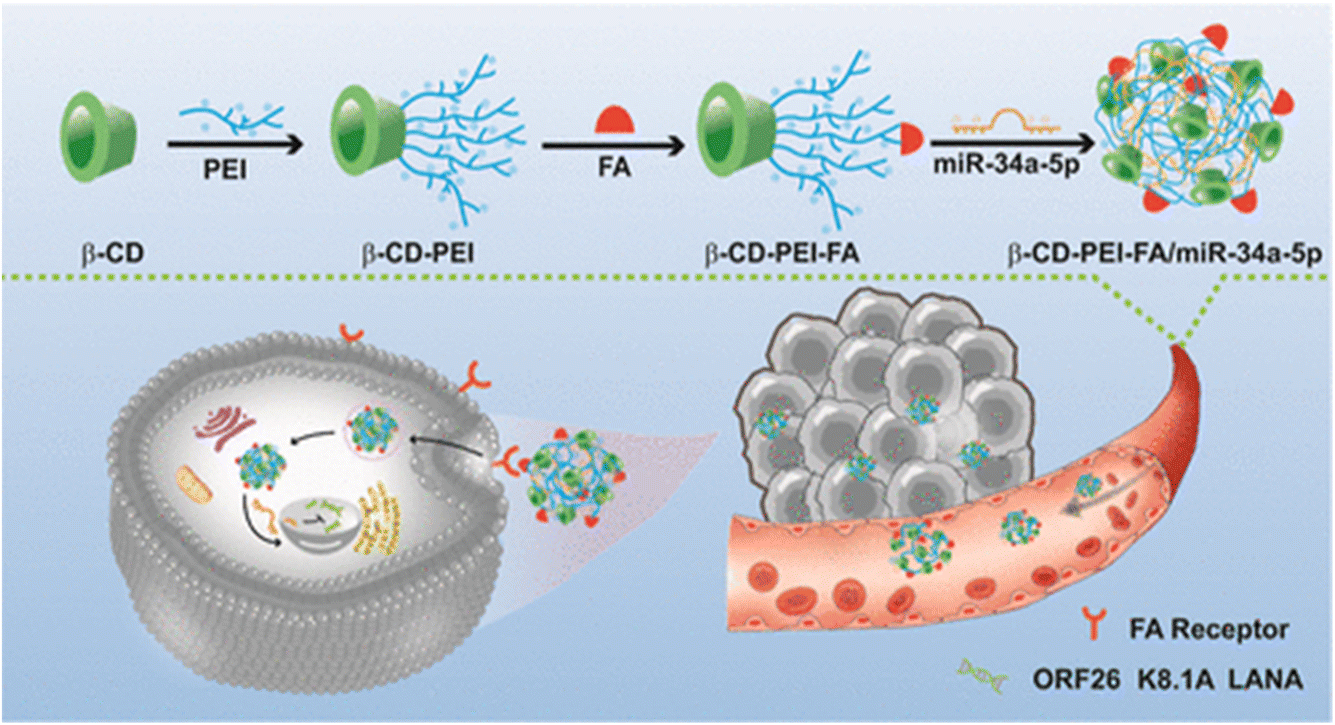 | ||
| Fig. 15 Synthesis of β-CD-PEI-FA. This figure is reproduced from ref. 136 with permission from ACS Publications, copyright (2023). | ||
5.5. Adjuvant nano-chemotherapy
Nanomedicine has revolutionized chemotherapy, offering potent anti-tumor efficacy. However, conventional chemotherapy often falls short of completely eradicating cancer cells, leaving patients vulnerable to recurrence.137 Consequently, the research focus has shifted towards multimodal synergistic cancer treatments, leveraging advancements in nanomedicine and an enhanced understanding of tumor biology and the TME.138 Intelligent nano-platforms not only serve as efficient drug carriers but also enable the integration of additional treatments, such as photothermal therapy (PTT), chemodynamic therapy (CDT), and photodynamic therapy (PDT) alongside chemotherapy (Fig. 16). These platforms can precisely adapt to physiological factors within the tumor and respond to external stimuli such as light, magnetism, ultrasound, and X-rays to achieve enhanced anti-cancer effects.139,140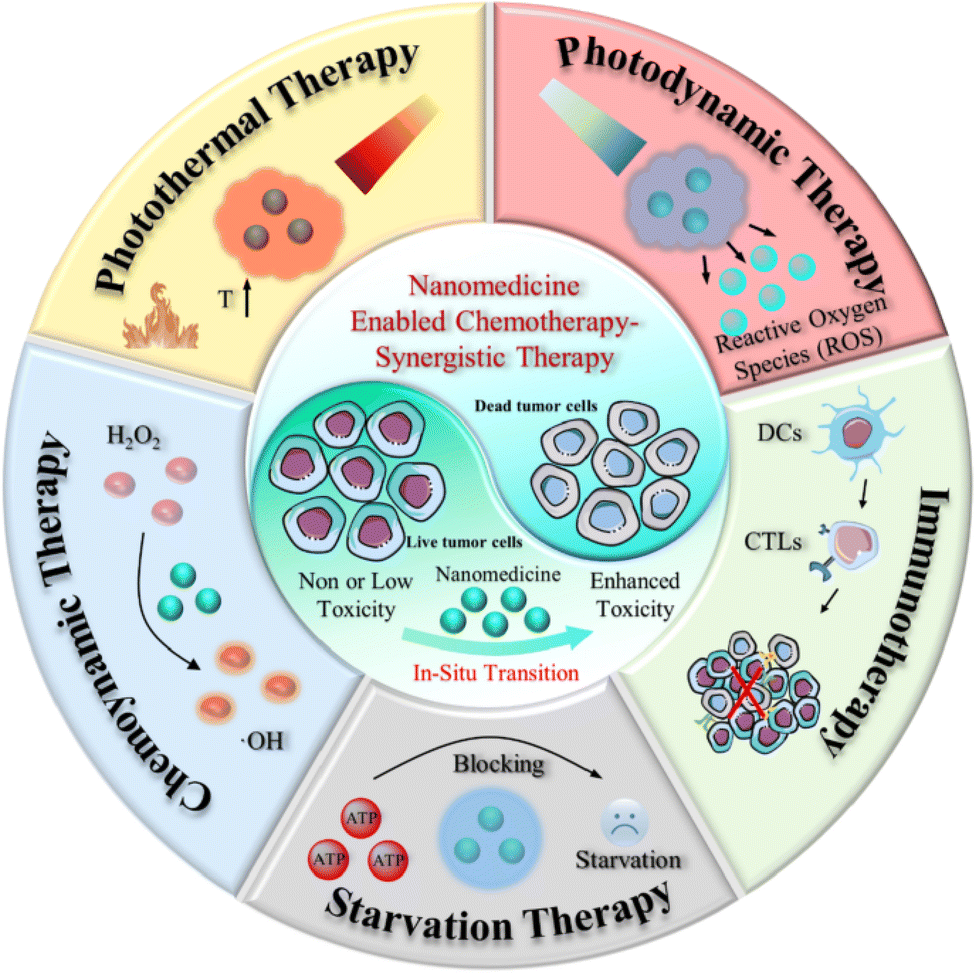 | ||
| Fig. 16 Schematic summary of synergistic cancer treatment generated from nanomedicine and its application in chemotherapy. This figure is reproduced from ref. 139 with permission from BMC, copyright (2022). | ||
Therefore, combinatorial therapies emerged as a promising strategy in cancer research due to their numerous advantages, including heightened efficacy, reduced cytotoxicity, and the ability to utilize NPs and chemotherapeutic drugs at lower concentrations. Combined chemotherapy often yields superior treatment responses and enhanced survival rates compared to single-agent therapy. Nanoformulations (NFs) play a pivotal role in combination therapy by encapsulating multiple therapeutic agents with distinct physicochemical properties into a single carrier. This enables the circumvention of challenges associated with different drug pharmacokinetics and biodistribution within the body, ensuring sustained delivery of an optimal synergistic drug ratio to target cancer cells. Consequently, combination therapy holds great promise as a novel and effective approach to cancer treatment.141
One derivative of paclitaxel is docetaxel (DXT), which is one member of the taxane family's second generation of chemotherapy agents. DXT is indicated for breast cancer, NSCLC, prostatic cancer, advanced gastric cancer, and locally advanced squamous cell head/neck cancer. Also, it works by binding to beta-tubulin, promoting its growth and maintaining its structure; by doing this, cell cycling during G2/M is stopped as it prevents microtubules from correctly assembling into the mitotic spindle as well as lowering the expression of the anti-apoptotic Bcl-2 gene.142 However, DTX's weak water solubility, quick clearance rate, unfavorable side effects, poor tumor penetration, and likelihood of drug resistance severely restrict its clinical use.
Co-delivery of microRNAs and chemotherapeutic agents presents an attractive strategy for synergistic breast cancer treatment, exploiting their complementary mechanisms of action. Thus, Zhang et al., designed a core–shell nanocarrier coated with cationic albumin to concurrently deliver docetaxel (DTX) and miRNA-34a to metastatic BC cells (4T1), aiming for improved treatment outcomes. The co-delivery nanocarriers exhibited EE and DL to be 83.46 ± 2.36% and 13.91 ± 0.39%, respectively. The average particle size of DTX and miRNA-34a co-loaded nanocarriers (CNCs) was approximately 183.9 ± 2.8 nm, with a PDI value of less than 0.2 and a spherical morphology. Moreover, the surface positive charge decreased upon loading miRNA-34a, reaching approximately 23 mV for neutralization. These findings indicate successful loading of DTX and miRNA-34a into the nanocarriers, which exhibited stability against aggregation. Importantly, these nanocarriers facilitated enhanced intracellular delivery by traversing the cytosol through a caveolae-mediated pathway, thereby evading entrapment within endosomes or lysosomes. In vitro studies demonstrated that the co-delivery nanocarriers induced significant cell apoptosis and cytotoxicity, attenuated tumor cell migration, and downregulated the transcriptional and protein expression of Bcl-2, CD44, SIRT1, CDK6, and Notch1 (Fig. 17). In 4T1-tumor-bearing mouse models, the co-delivery nanocarriers exhibited significant suppression of tumor growth and metastasis while also prolonging the blood circulation of DTX in vivo. Additionally, they resulted in increased accumulation of the cargo within tumors.143
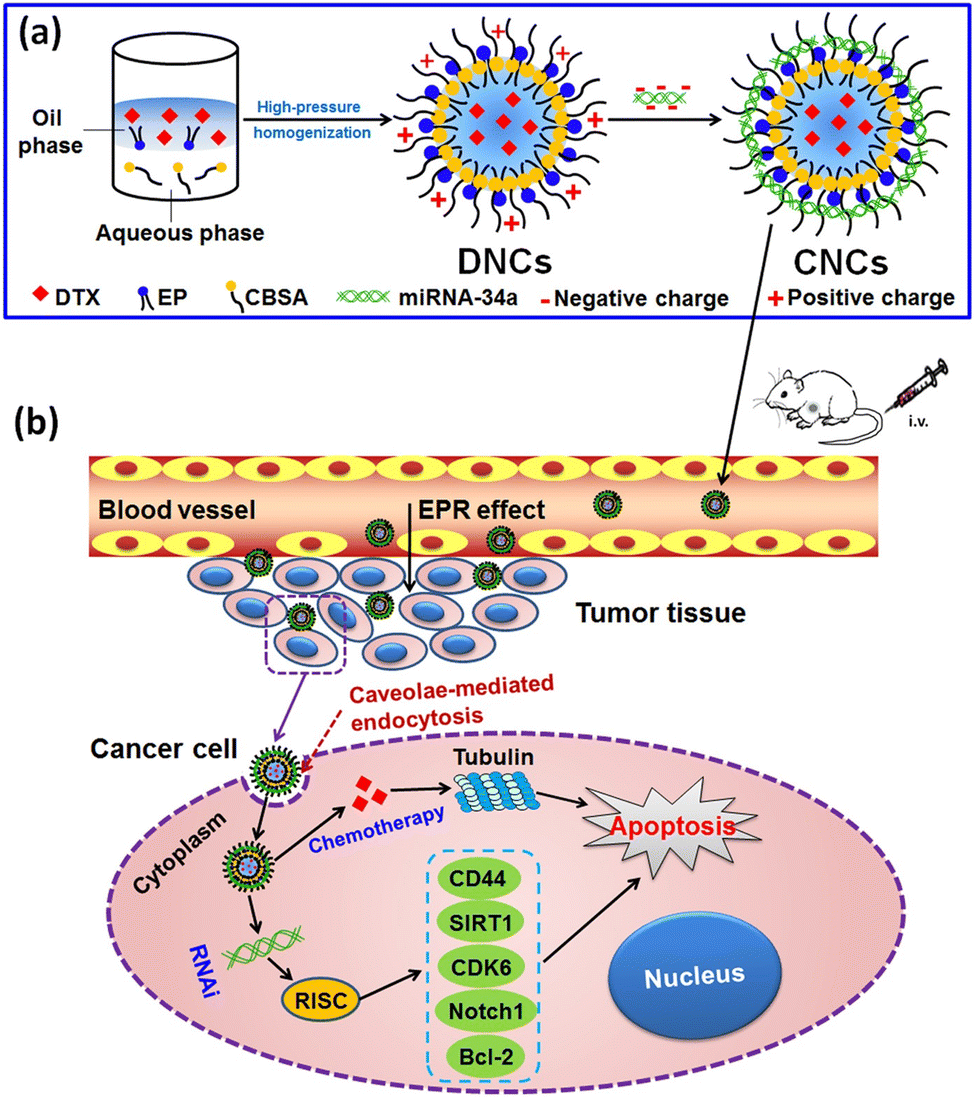 | ||
| Fig. 17 Graphical representation of core–shell nanocarriers for DTX and miRNA-34a co-delivery through a caveolae-mediated pathway. (a) Preparation of the CNCs. (b) Proposed mechanism of cancer cell killing: CNCs are administered systemically and enter the bloodstream. They accumulate in tumor tissue via the enhanced permeability and retention (EPR) effect, are internalized by cancer cells, achieve cytosolic delivery, and subsequently induce apoptosis through the combined anticancer effects of DTX and miRNA-34a. This figure is reproduced from ref. 143 with permission from Nature, copyright (2017). | ||
Irinotecan (IRI), derived from the Chinese tree Camptotheca acuminate, served as a chemotherapeutic drug targeting DNA topoisomerase I, primarily impacting the S and G2 phases of the cell cycle. This medication finds application in treating various solid tumors, including lung, pancreatic, ovarian, and colorectal malignancies, often used as a first- or second-line therapy for colorectal cancer in combination with other agents. DNA topoisomerase I plays a crucial role in DNA replication and transcription by relaxing the DNA double helix by creating single-strand breaks, thereby relieving supercoiling. IRI acts as a prodrug inhibiting DNA topoisomerase I, primarily during the S and G2 phases of the cell cycle. Upon activation, typically by hydrolysis mediated by a carboxylesterase, it forms its biologically active metabolite, SN-38. However, the activity of UDP-glucuronyltransferases subsequently inactivates SN-38 into its SN-38G form. The adverse effects of irinotecan primarily stem from its active metabolite, SN-38. These commonly include neutropenia, diarrhea, nausea, vomiting, alopecia, and fatigue. Neutropenia associated with irinotecan is typically transient but can become severe, especially when diarrhea occurs concurrently. Patients carrying the UGT1A1*28 allele of the UDP-glucuronosyltransferase enzyme, responsible for converting SN-38 to its inactive form SN-38G, may experience reduced glucuronidation, leading to increased incidence of diarrhea and neutropenia. This allele's presence compromises the effective inactivation of the active SN-38 form.144
Hence, a study by Li et al., synthesized amphiphilic copolymers, polyethyleneimine-poly (D, L, lactide) (PEI-PLA) and 1,2-distearoyl-sn-glycero-3 phosphoethanolamine-N-[methoxy (polyethyleneglycol) (DSPE-PEG)] and investigated the potential of utilizing PEI-PLA/DSPE-PEG hybrid micelles as a co-delivery system. Specifically, aimed to co-load IRI and miR-34a into these micelles (MINPs) to achieve a chemo-miRNA combination therapy against CRC cells (CT-26). MINPs were meticulously constructed using two-step film dispersion and electronic interaction methods. The generated NPs exhibited a spherical morphology and uniform dispersity, with a negative zeta potential of −11.65 mV. Additionally, the NPs displayed a relatively high EE ranging from 81% to 91%, with mean diameters measured between 170 and 200 nm. In vitro experiments demonstrated successful encapsulation of IRI and miR-34a into MINPs, which were efficiently delivered to CT-26. The intact encapsulation of MINPs resulted in the upregulation of miR-34a expression and modulation of upstream and downstream genes associated with miR-34a, including Bcl-2 and m-TOR (Fig. 18). Consequently, migration and proliferation of CT-26 cells were inhibited, while apoptosis of cancer cells was intensified. Furthermore, in vivo experiments confirmed the preferential accumulation of MINPs at the tumor site following intravenous administration and demonstrated remarkable antitumor efficacy in a xenograft tumor model, attributed to the synergistic effect of IRI and miR-34a combined therapy, along with their good biocompatibility.145
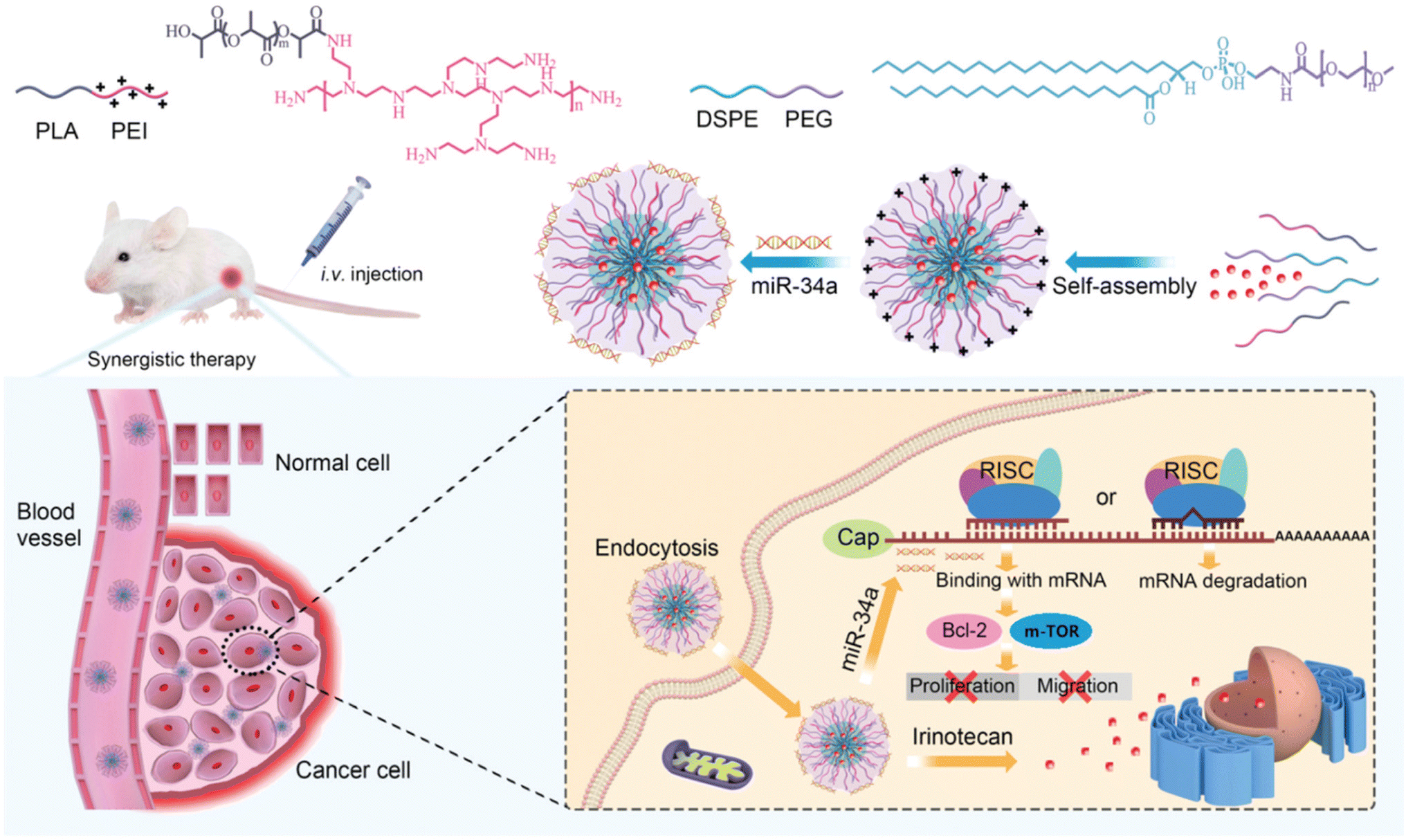 | ||
| Fig. 18 Diagrammatic representation of the self-assembled polymeric hybrid micelles based on PEI-PLA and DSPE-PEG systems for co-delivery of IRI and replenishment of miR-34a that produced combination therapeutic effects by controlling many downstream genes connected to miR-34. This figure is reproduced from ref. 145 with permission from RSC, copyright (2020). | ||
Doxorubicin (DOX), an antibiotic derived from Streptomyces peucetius bacteria, has been a cornerstone of chemotherapy since the 1960s. Belonging to the anthracycline class alongside idarubicin and epirubicin, DOX is extensively helpful in treating various solid tumors across both adult and pediatric populations. Its approved indications span a wide range of cancers, including non-small cell lung cancer (NSCLC), breast cancer, acute myeloblastic leukemia, ovarian cancer, Hodgkin lymphoma, bladder cancer, acute lymphoblastic leukemia, thyroid cancer, and soft tissue and bone sarcomas. The liposomal formulation of doxorubicin, sanctioned by the FDA, is specifically approved for the treatment of multiple myeloma, AIDS-related Kaposi sarcoma, and ovarian cancer in patients unresponsive to platinum-based chemotherapy. DOX exerts its primary mechanism of action by intercalating between DNA base pairs, causing DNA strand breaks and impeding both DNA and RNA synthesis. Furthermore, DOX inhibits topoisomerase II, further damaging DNA and prompting apoptosis, thereby contributing to its potent anti-tumor effects.146–148
Cisplatin (CDDP) stands as one of the most potent chemotherapy drugs employed in treating esophageal cancer (EC). Its mechanism of action involves binding to DNA and forming CDDP-DNA adducts, thereby inhibiting DNA transcription and replication and leading to tumor cell death. However, the efficacy of CDDP is often hindered by inherent or acquired resistance in EC cells, which poses a significant challenge in its clinical use. Both internal and external factors contribute to resistance and variable response rates observed during EC treatment. Internally, mutations in the p53 gene have attracted considerable attention as a major contributor to drug resistance. Externally, the uneven distribution of drugs within the tumor and its subcellular fractions poses a significant concern. This distribution irregularity is primarily attributed to tumor artery malformation and the low rate of transfer of anti-tumor therapies to affected areas. These factors collectively contribute to the complexity of managing EC and underscore the need for novel therapeutic strategies to overcome resistance and improve treatment outcomes.149,150
Therefore, in a study, Fang et al. developed a novel drug formulation comprising miR-34a mimics and DOX-loaded nanomicelles composed of PEG, intending to evaluate its efficacy against CDDP-resistant strains, specifically KYSE-410-CisR (an esophageal squamous cell carcinoma cell line). The nanomicelles exhibited a spherical morphology ranging from 100 to 200 nm, with minimal variation. Introducing miR-34a into the nanomicelles led to a significant enhancement in the inhibition of cell proliferation and apoptosis percentage in KYSE-410-CisR cells. Moreover, results revealed that the anti-tumor effect of miR-34a was primarily attributed to the suppression of SIRT1, rather than the modulation of the p53/p21 anti-tumor pathway, resulting in notable upregulation of the apoptosis-related protein caspase3. Furthermore, the nanomicelles exhibited prolonged retention intervals and facilitated DOX transfer in vivo, potentially due to the inclusion of the TAT-PEG1k-PE complex, consisting of PEG1k, PE, and a TAT protein modified with cysteine, which may enhance cellular uptake via endocytosis. Overall, the nanomicelle formulation significantly increased the apoptosis rate and inhibition of cell growth in KYSE-410-CisR cells, possibly by augmenting DOX concentration within tumor cells and modulating the SIRT1 signaling pathway.149
Fluorouracil (5-FU) is a widely used chemotherapeutic agent effective against various cancers, including gastric, pancreatic, breast, and colorectal adenocarcinomas (CRC), as approved by the FDA. After systemic administration, 5-FU undergoes enhanced transport into cells, which metabolizes into fluorodeoxyuridine monophosphate (FdUMP). FdUMP then forms a complex with thymidylate synthase, inhibiting the synthesis of deoxythymidine monophosphate (dTMP). This disruption in dTMP production leads to an imbalance in intracellular nucleotides, resulting in double-strand breaks in DNA mediated by endonuclease enzymes. In addition to blocking thymidylate synthase, 5-FU acts as a pyrimidine analog, incorporating itself into RNA and DNA instead of uracil or thymine. This interference with nucleic acid synthesis contributes to the extensive damage inflicted on DNA repair mechanisms, ultimately leading to the demise of rapidly proliferating cells. Furthermore, 5-FU demonstrates other mechanisms of action, including upregulation of p53 expression and interference with RNA processing. These multifaceted actions collectively enhance the efficacy of 5-FU in combating cancer growth and progression.151
A recent study introduced a novel approach involving the development of a host–guest-self-assembling nanocarrier comprising adamantane (ADA)-modified TCP1 peptide-targeting ligand (TCP1) as the guest molecule and β-cyclodextrin (CD) attached to quantum dot (QD) as the host molecule (TCP1-CD-QD) (Fig. 19). This resulted in the formation of TCP1-CD-QD, which served as the platform for loading both 5-FU and miR-34a to create TCP1-CD-QDs/5-FU-miR-34a for targeting CRC cell lines, particularly DLD1. This combination treatment strategy aimed to leverage the synergistic effects of nucleotide and chemotherapeutic drugs. The resultant NPs exhibited a compact size range of 7–9 nm and a zeta potential of 10.5 ± 0.9 mV. Moreover, TCP1-CD-QD demonstrated notable loading capacity and entrapment efficiency, measured at 6.4% and 25.8%, respectively, enhancing CRC cell internalization. Consequently, TCP1-CD-QDs/5-FU-miR-34a exhibited a significant increase in miR-34a expression, apoptosis activation, metastasis suppression, and reduced drug resistance and tumor cell migration. These effects were attributed to the silencing of SIRT1, downregulation of CD44, and upregulation of p53. Notably, this study observed a substantial decrease in 5-FU-resistant cells following SIRT1 silencing, suggesting that TCP1-CD-QDs/5-FU-miR-34a may partially overcome CRC cells' resistance to 5-FU, thus presenting a promising therapeutic strategy for combating CRC.152
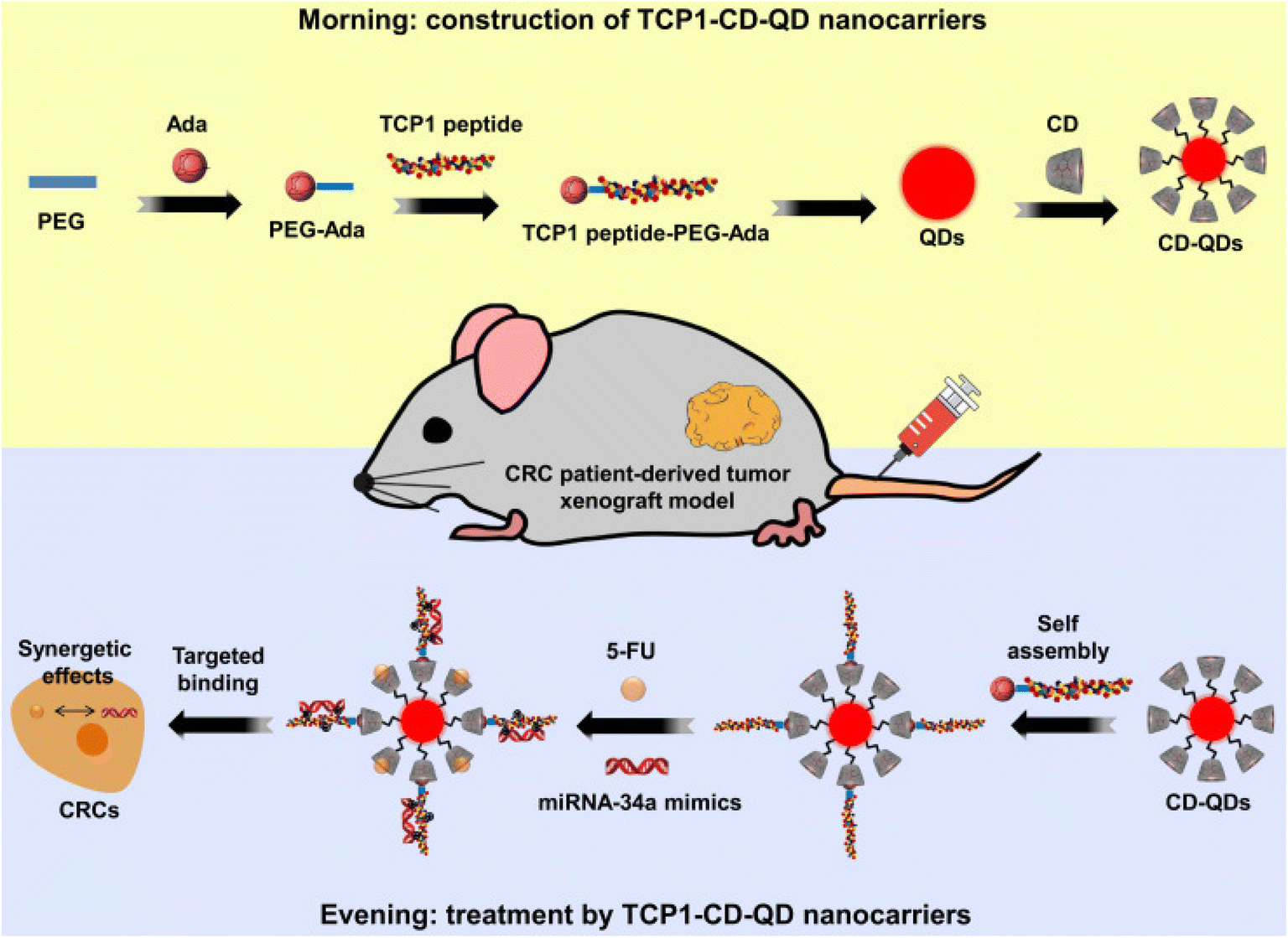 | ||
| Fig. 19 Diagram showing how to create host–guest self-assembled nanocarriers for synergistic treatment of colorectal cancer (CRC) using CD-functionalized QDs (CD-QDs) and the ADA-functionalized targeting peptide (TCP1 peptide-PEG-ADA) loaded with nucleotide drugs (miRNA-34a mimics) and chemotherapeutic drugs (5-FU). This figure is reproduced from ref. 152 with permission from THERANOSTICS, copyright (2021). | ||
The latest research findings regarding the nanodelivery of miRNA-34 have been compiled and presented in Table 1.
| miR34 type (a/b/c) | Nanocarrier | Physicochemical properties | Target cancer cell | Advantages | Safety and efficacy | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Size (nm) | Zeta potential (mV) | EE% | ||||||
| (I) POLYMERS | ||||||||
| miR34a | Self-assembling HA-PEI/HA-PEG | 260–360 | −35 | 94–96% | NSCLC (A549 and A549 DDP) | Increased oxidative stress, decreased GSH, production of apoptotic signals | HA-PEI/HA-PEG were tested on A549 and A549 DDP cell lines, showing consistent uptake and increased miR34a-specific localization | 90 |
| miR34a | Transferrin-decorated thymoquinone-loaded PEG-PLGA nanoparticles (TF-TQ-Np) | 77.50 nm ± 6.35 | — | 93% | NSCLC (A549) | Triggering of p53/ROS feedback loop, lowering of Bcl2 and inhibited the migration of NSCLC cells | TF-TQ-Np were tested on A549 cell line exhibiting synergized stability and targeting parameters. Also, TF-TQ-Np did not generate toxicity on W138 cells whilst displaying notable cellular uptake | 91 |
| miR34a | A blend of an amphiphilic triblock copolymer (mPEG-PLA-PAE), miR-34a-DSPE, mPEG-DSPE, and arsenic trioxide (As/miR34a@NPs) | 110–150 | −5 | — | HCC (H-Huh7) | Enhanced drug accumulation at cancerous sites, extended circulation durations, enhanced cell internalization and absorption in acidic TME, improved stability, better biocompatibility | Testing of As/miR34a@NPs on the H-Huh7 cell line indicates that the drug delivery system may enhance drug absorption in weakly acidic environments via charge reversal and boost drug accumulation at cancerous sites by prolonging circulation times | 92 |
| miR34a | Polyplexes rooted in a PGA encapsulating miR-34a mimics and PLK1-siRNA (APA-miR-34a polyplexes) | 189.79 ± 11 | 4.68 ± 3 | — | Orthotopic pancreatic ductal adenocarcinoma (PDAC) mouse model (MiaPaCa2, Panc1, BxPC3) and the murine pancreatic cancer cell line (Panc02) | No systemic side effects or immunotoxicity, accumulation at the tumor site and internalization | APA-miR-34a polyplexes were tested on MiaPaCa2, showing a significant increase in the expression of miR34a hence, active and potent delivery of miR34a | 93 |
| miR34a | PLGA cores encircled by alternating layers of poly-L-lysine (PLL), miR-34a and PLL (PLGA NPs, PLL, miRNA, and PLL) | 112 ± 8 | +32 | — | TNBC (MDA-MB-231) | Significant cellular binding/uptake, higher Cy5-miR-co signal, inhibition of cell cycle progression, evasion, cell proliferation, apoptosis, and drug sensitivity | PLGA NPs, PLL, miRNA, and PLL were tested on MDA-MB-231 cell lines exhibiting slow release at pH 7.4, high cellular binding and silencing of CCND-1, Notch-1, survivin, and MDR-1 | 94 |
| miR34a | PLGA-poly-L-His NP | 200 | −21 ± 5.60 | 80–100% | NSCLC (A549) | Induced apoptosis, better cellular distribution, good stability and inhibition of cell proliferation | PLGA-poly-L-His NP was tested on A549 cell lines, demonstrating robust cellular absorption with dispersion throughout the cytoplasm and proximity to the nucleus. Also, there has been a noticeable activation of p53, and downregulation of SIRT1, MYC, and BCL2 | 95 |
| miR34a | Lauric acid (LA) and small molecule targeting 4-carboxyphenylboric acid (PBA) grafted onto PEI (LA-PEI-PBA) | 207.3 | 26.74 ± 2.36 | — | KSHV (KMM cells) | Promoting cellular uptake and raised expression of miR-34a-5p, suppresses the expression of KSHV lytic and latent genes and prevents the proliferation and migration of KSHV-infected cells | LA-PEI-PBA presented good biosafety, exceptional targeting capability and the ability to protect miR-34a-5p from degradation, thereby enhancing its stability | 96 |
| miR34a | Polymeric micelles with PEI as the backbone (PEI-SPDP-Man/miR-34a) | 148 | 41.2 | — | TNBC (MDA-MB-231) | Avoiding lysosomal degradation, inducing apoptosis, and enhancing miR34a adhesion | PEI-SPDP-Man/miR-34a presented noted biosafety, stability, biocompatibility, release at the intended location, mitigated gene digestion and degradation of miR-34a by lysosomes | 97 |
| miR34a | A core with PGA grafted chitosan enveloped by a PEI shell | 123 ± 5 | 36 ± 1 | — | TNBC (MDA-MB-231) | Commendable stability, exhibiting high encapsulation efficiency and targeted delivery to specific cells | These NPs presented efficient condensation of miRNA, enhanced permeability and internalization, and inhibition of cell proliferation | 98 |
| miR34a | Chitosan-PLGA | 139 | — | — | NSCLC (A549) | Low toxicity, enhanced cell internalization, biocompatibility, and induced apoptosis | Chitosan-PLGA NPs caused preferable genetic alterations which resulted in the induction of apoptosis and inhibition of cell proliferation. Also, these NPs possessed biocompatibility, significant cell internalization and releasing properties | 99 |
|
||||||||
| (II) Exosomes | ||||||||
| miR34a | Exosomes coated miR34a | 85.42 | — | — | Human embryonic kidney (HEK293) cells, normal human pancreatic epithelial ductal cells (HPDE6-C7), and human pancreatic cancer cell lines Miapaca-2 and Panc28 | Effectively crossed the cell membrane, suppressed the expression of Bcl-2, and altered the expression of pro-apoptotic proteins such as Bax and p53 | Exosomes coated miR34a inhibited the growth of tumor cells substantially by inducing apoptosis | 108 |
| miR34c | Exosomes coated miR34c | 30–120 | — | — | NSCLC (A549) | Downregulation of invasion and metastases | Exosomes coated miR34c showed downregulation of α2β1 protein, relative restoration of luc/R-luc signal, and inhibition of cell migration and invasion | 109 |
| miR34c | Exosomes coated miR34c | ∼100 | — | — | NPC (CME-2, 5–8F and 6–10B) | Inhibition of NPC cell proliferation, migration, invasion and enhanced radiosensitivity by inducing apoptosis and reducing resistance to radiotherapy | Exosomes coated miR34c lower radioresistance, inhibition of EMT and cell proliferation, apoptosis induction, and targeting β-catenin | 110 |
| miR34a | Genetically modified dental pulp MSCs (DPSCs) produced modified exosomes that was used as a vehicle for miR-34a | 65 ± 9 | — | — | TNBC (MDA-MB-231) | Downregulation of c-MET and Bcl2, suppressed migration and invasiveness | Exosomes carrying miR34a showed desirable gene alterations resulting in impaired tumor growth, migration, and invasion | 111 |
| miR34a | Tumor-derived exosomes (TEXs) encapsulating miR34a | 87.13 ± 10.89 | −19.7 ± 12.12 | — | CRC (CT-26) | Downregulation was observed in PD-L1 expression, and immunogenicity was enhanced | TEXs encapsulating miR34a presented decreased tumor cell viability, induced apoptosis, declined invasion and migration, and reduced immune suppression | 112 |
|
||||||||
| Metallic nanoparticles | ||||||||
| miR34a | EGCG-capped AuNPs delivering miR34a | 35 | — | — | HCC cell lines (HepG2) | Enhanced bioavailability and accumulation, inducing cytotoxicity and apoptosis | EGCG-capped AuNPs delivering miR34a presented low antioxidant activity, increased cytotoxicity, increased expression of miR34a, Caspase-3 protein expression, and downregulation of c-Myc protein levels | 115 |
| miR34a | Core–shell–shell (CSS) nanoparticle system, comprising of gold–silver–gold layers delivering miR34a | ∼91.3 ± 5.4 | ∼ −36 | — | Esophageal squamous carcinoma cells (TE10) | Reduction in the expression of ROCK1, STAT3, MYC, and TGIF2 and a notable increase in the levels of active CASP-3, and induced apoptosis | CSS of gold–silver–gold NPs delivering miR34a showed efficiency and biocompatibility, delivering oligonucleotides and siRNA, significant release of miR34a, enhanced intracellular release, high efficiency of tumor cell death, and targeted gene alterations | 116 |
| miR34a | Coating outer membrane vesicles (OMVs) onto zeolitic imidazolate framework-8 (ZIF-8) NPs encapsulating miR-34a | 168 | −20 | ∼75% | BC (4 T1) | Augmented cell targeting and miRNA uptake, obstructing RISC, inhibiting tumor cell proliferation, high safety profile and biocompatibility | OMVs onto ZIF-8 NPs encapsulating miR-34a showed good affinity to tumor cells, enhanced uptake efficiency and intracellular trafficking, successful cytosolic delivery and antitumor effect | 117 |
|
||||||||
| Miscellaneous nanoparticles | ||||||||
| miR34a | Silica | — | −10.7 ± 4.8 | — | Claudine-low TNBC (SUM159 pt, human) | Downregulation of the Notch1 signaling pathway and upregulation of p53, consequently inducing apoptosis | Fluorescent SiO2NPs showed good cell internalization, intra- tumor local delivery miR-34a, which triggered RNA silencing | 122 |
| miR34a | Nanodiamonds with PEI | 304.5 ± 31.33 | −26.4 ± 2.50 | — | TNBC (MCF7 and MDA-MB-231) | Biocompatible, significant internalization, reduced cell migration and proliferation. Inducing p53 and downregulation SIRT1, induction of apoptosis and autophagy | Nanodiamonds with PEI NPs showed induced intracellular miRNA accumulation targeted intracellular localization of NPs, which caused induced cell death and inhibited tumor growth and migration | 124 |
| miR34a | Self-assembly technique with protamine (PS), and folic acid (FA) resulting in FA/PS/miR-34a/PS@NDs | 210 | −25 | — | TNBC (MDA-MB-231) | Induced apoptosis, proliferation, and migration inhibition | FA/PS/miR-34a/PS@NDs presented with efficient significant delivery and cellular uptake. In addition, it suppressed tumor cell migration, proliferation and induction of tumor cell apoptosis | 128 |
| miR34a | Layer-by-layer assembled nanoshells (LbL-NS) of silica core and gold shells resulting in PLL, miR34-co-LbL-NS | 208 ± 4 | + 53 ± 5 | — | TNBC (MDA-MB-231) | Enhanced internalization, intracellular distribution, effective delivery, downregulation of SIRT1 and Bcl-2, reduced metabolic activity and proliferation | PLL, miR34-co-LbL-NS showed safety due to the presence of PLL, good tumor cell internalization, direct regulation of TNBC proliferation, and suppressed expression of SIRT1 and Bcl-2 | 129 |
| miR34a | β-CD-PEI-FA/miR-34a-5p | 203.13 ± 0.41 | 27.02 ± 0.72 | — | KSHV (BCBL-1 cells and SK-RG cells) | Excellent biocompatibility efficiently transportation of miR-34a-5p, accelerated the G2-phase process of BCBL-1 and SK-RG cells thereby suppressing cell growth | β-CD-PEI-FA/miR-34a-5p presented biocompatibility, blood compatibility, relatively stable and unlikely to bind to serum proteins in the blood, thereby avoiding cytotoxic effects. Moreover, they showed efficient delivery of miR-34, inhibition of cell proliferation, G2 phase, and expression of KSHV-related genes | 136 |
|
||||||||
| Adjuvant nano-chemotherapy | ||||||||
| miR34a | DTX encapsulation in a lipid core of nanocarriers, while miRNA-34a was self-assembled in the shell of the nanocarriers via electrostatic interactions with CBSA | 83.46 ± 2.36% | 183.9 ± 2.8 | 23 | BC (4T1) | Evading entrapment within endosomes or lysosomes, induced cell apoptosis and cytotoxicity, attenuated tumor cell migration, and downregulated Bcl-2, CD44, SIRT1, CDK6, and Notch1, suppression of tumor growth and metastasis, prolonged stay in the blood circulation and increased accumulation of the cargo within tumors | These NPs demonstrated protection to mir34a from RNase degradation, hence efficient cellular uptake and internalization to tumor cells, which caused intracellular trafficking and tumor penetration. Additionally, significant reductions in cell viability were noted, indicating that the combined delivery of DTX and miRNA-34a results in enhanced cytotoxicity | 143 |
| miR34a | Self-assembled polymeric hybrid micelles based on PEI-PLA and DSPE-PEG systems encapsulating IRI and miR34a | 170–200 | −11.65 | 81–91% | CRC (CT-26) | Downregulation of Bcl-2 and m-TOR, inhibited migration and proliferation, induced apoptosis, and good biocompatibility | PEI-PLA/DSPE-PEG encapsulating IRI and miR34a demonstrated the capability of efficient delivery of miR-34a to cancer cells and facilitating effective escape from endosomes. Also, there has been a synergistic effect of m-TOR with IRI, which significantly improved the chemotherapeutic efficacy of IRI, impedes the migration of CT-26 cancer cells and minor systemic cytotoxicity | 145 |
| miR34a | DOX-loaded nanomicelles composed of PEG | 100–200 | — | — | Esophageal squamous cancer cell (KYSE-410) | Inhibition of cell proliferation and apoptosis, suppression of SIRT1, upregulation of caspase3, prolonged retention intervals, facilitated DOX transfer, enhance cellular uptake | These nanomicelles induced apoptosis on KYSE-410 cells and elevated the expression of SIRT1 | 149 |
| miR34a | TCP1-CD-QDs/5-FU-miR-34a | 7–9 | 10.5 ± 0.9 | 25.8% | CRC (DLD1) | Enhanced cell internalization, apoptosis activation, and suppression of metastasis, along with a reduction in drug resistance and tumor cell migration | TCP1-CD-QDs/5-FU-miR-34a proved its enhanced internalization, effective simultaneous delivery of 5-FU and miR-34a hence, enhanced apoptotic effects and reduced the metastasis of CRC cells. In addition, these NPs reduced the systemic toxicity of drugs | 152 |
Conclusion
Despite its potential as an immunotherapeutic agent for cancer, miR-34 faces significant obstacles that hinder its translation into clinical practice. These include susceptibility to degradation by RNase and unfavorable pharmacokinetic and biodistribution profiles. This review presents recent advancements in the field, focusing on the formulation of miR-34 into various nano-delivery systems. Several studies have explored the use of nano-delivery systems in cancer-targeted therapies, leveraging their unique properties to improve the efficacy and safety of cancer treatments. These systems can extend the circulation time of therapeutic payloads, enhance their bioavailability and water solubility, and minimize toxic side effects. Additionally, delivering therapeutic agents via nano-particulates can enhance their accumulation in tumor tissues. Despite efforts to develop safe and effective nano-systems for miR-34 delivery, challenges remain in clinical application. These include difficulties in scaling up production methods, complex pharmacokinetics and pharmacodynamics, potential drug leakage leading to low loading and encapsulation efficiencies, and limited understanding of in vivo behavior and immunological responses to different nanoparticles. Future research should prioritize investigating the safety and efficacy of nano-systems in cancer therapy, rather than focusing solely on fabrication techniques and in vitro testing. This holistic approach will provide valuable insights into identifying the safest and most effective nano-carriers, ultimately leading to the development of more advanced cancer treatments.Data availability
This manuscript does not involve any experimental work.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Dr Sherif Ashraf Fahmy acknowledges the financial support and sponsorship received from the Alexander von Humboldt Foundation, Germany.References
- Y. Xing, Z. Wang, Z. Lu, J. Xia, Z. Xie and M. Jiao, et al., MicroRNAs: immune modulators in cancer immunotherapy, Immunother. Adv., 2021, 1(1), 1–13, DOI:10.1093/immadv/ltab006.
- W. W. Tang, K. M. Bauer, C. Barba, H. A. Ekiz and R. M. O’Connell, miR-aculous new avenues for cancer immunotherapy, Front. Immunol., 2022, 13, 1–34, DOI:10.3389/fimmu.2022.929677.
- S. Wang, K. Xie and T. Liu, Cancer Immunotherapies: From Efficacy to Resistance Mechanisms – Not Only Checkpoint Matters, Front. Immunol., 2021, 12 DOI:10.3389/fimmu.2021.690112.
- B. Smolarz, A. Durczyński, H. Romanowicz, K. Szyłło and P. Hogendorf, miRNAs in Cancer (Review of Literature), Int. J. Mol. Sci., 2022, 23(5), 2805, DOI:10.3390/IJMS23052805.
- J. Fu, S. Imani, M. Y. Wu and R. C. Wu, MicroRNA-34 Family in Cancers: Role, Mechanism, and Therapeutic Potential, Cancers, 2023, 15(19, 4723) DOI:10.3390/CANCERS15194723.
- K. Otmani and P. Lewalle, Tumor Suppressor miRNA in Cancer Cells and the Tumor Microenvironment: Mechanism of Deregulation and Clinical Implications, Front. Oncol., 2021, 11 DOI:10.3389/fonc.2021.708765.
- J. Zhou, H. Wang, Q. Sun, X. Liu, Z. Wu and X. Wang, et al., miR-224-5p-enriched exosomes promote tumorigenesis by directly targeting androgen receptor in non-small cell lung cancer, Mol. Ther.–Nucleic Acids, 2021, 23, 1217–1228, DOI:10.1016/J.OMTN.2021.01.028.
- J. Rhim, W. Baek, Y. Seo and J. H. Kim, From Molecular Mechanisms to Therapeutics: Understanding MicroRNA-21 in Cancer, Cells, 2022, 11(18), 2791, DOI:10.3390/cells11182791.
- F. Liu, Y. Shi, Z. Liu, Z. Li and W. Xu, The emerging role of miR-10 family in gastric cancer, Cell Cycle, 2021, 20, 1468, DOI:10.1080/15384101.2021.1949840.
- S. A. Fahmy, A. Dawoud, Y. A. Zeinelabdeen, C. J. Kiriacos, K. A. Daniel and O. Eltahtawy, et al., Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery, Cancers, 2022, 14, 5244, DOI:10.3390/CANCERS14215244.
- T. T. P. Nguyen, K. H. Suman, T. B. Nguyen, H. T. Nguyen and D. N. Do, The Role of miR-29s in Human Cancers—An Update, Biomedicines, 2022, 38(1), 53, DOI:10.1186/s13046-019-1059-5.
- T. T. P. Nguyen, K. H. Suman, T. B. Nguyen, H. T. Nguyen and D. N. Do, The Role of miR-29s in Human Cancers—An Update, Biomedicines, 2022, 10(9, 2121) DOI:10.3390/biomedicines10092121.
- Y. Ma, N. Shen, M. S. Wicha and M. Luo, The Roles of the Let-7 Family of MicroRNAs in the Regulation of Cancer Stemness, Cells, 2021, 10, 2415, DOI:10.3390/CELLS10092415.
- F. Hanif, Y. Zhang, C. Dube, M. K. Gibert, S. Saha and K. Hudson, et al., miR-3174 Is a New Tumor Suppressor MicroRNA That Inhibits Several Tumor-Promoting Genes in Glioblastoma, Int. J. Mol. Sci., 2023, 24, 9326, DOI:10.3390/IJMS24119326/S1.
- A. Jafarzadeh, M. Noori, S. Sarrafzadeh, S. S. Tamehri Zadeh, M. Nemati and N. Chatrabnous, et al., MicroRNA-383: A tumor suppressor miRNA in human cancer, Front. Cell Dev. Biol., 2022, 10, 955486, DOI:10.3389/FCELL.2022.955486/BIBTEX.
- D. Kalfert, M. Ludvikova, M. Pesta, J. Ludvik, L. Dostalova and I. Kholová, Multifunctional Roles of miR-34a in Cancer: A Review with the Emphasis on Head and Neck Squamous Cell Carcinoma and Thyroid Cancer with Clinical Implications, Diagnostics, 2020, 10(8), 563, DOI:10.3390/diagnostics10080563.
- C. Wang, Q. Jia, X. Guo, K. Li, W. Chen and Q. Shen, et al. microRNA-34 family: From mechanism to potential applications., Int. J. Biochem. Cell Biol., 2022, 144, 106168, DOI:10.1016/J.BIOCEL.2022.106168.
- M. Yin, Z. Zhang and Y. Wang, Anti-tumor effects of miR-34a by regulating immune cells in the tumor microenvironment, Cancer Med., 2023, 12, 11602–11610, DOI:10.1002/CAM4.5826.
- J. Fu, S. Imani, M. Y. Wu and R. C. Wu, MicroRNA-34 Family in Cancers: Role, Mechanism, and Therapeutic Potential, Cancers, 2023, 15(19), 4723, DOI:10.3390/cancers15194723.
- S. Li, X. Wei, J. He, Q. Cao, D. Du and X. Zhan, et al., The comprehensive landscape of miR-34a in cancer research, Cancer Metastasis Rev., 2021, 40(3), 925–948, DOI:10.1007/S10555-021-09973-3.
- F. Akad, V. Mocanu, S. N. Peiu, V. Scripcariu, B. Filip and D. Timofte, et al., Mesenchymal Stem Cell-Derived Exosomes Modulate Angiogenesis in Gastric Cancer, Biomedicines, 2023, 11, 1031, DOI:10.3390/BIOMEDICINES11041031.
- Y. Liu, J. Jiang, C. Liu, W. Zhao, Y. Ma and Z. Zheng, et al., Synergistic anti-tumor effect of anti-PD-L1 antibody cationic microbubbles for delivery of the miR-34a gene combined with ultrasound on cervical carcinoma, Am. J. Transl. Res., 2021, 13, 988 CAS.
- Role of miR-34 in gastric cancer: From bench to bedside (Review)n.d., https://www.spandidos-publications.com/10.3892/or.2019.7280?text=fulltext, accessed February 15, 2024.
- K. Dżaman, K. Czerwaty, T. E. Reichert, M. J. Szczepański and N. Ludwig, Expression and Regulatory Mechanisms of MicroRNA in Cholesteatoma: A Systematic Review, Int. J. Mol. Sci., 2023, 24, 12277, DOI:10.3390/ijms241512277.
- W. Lai, Y. Yue and G. Zeng, MicroRNA-34c-5p Reduces Malignant Properties of Lung Cancer Cells through Regulation of TBL1XR1/Wnt/β-catenin Signaling, Curr. Mol. Med., 2023, 24, 114–122, DOI:10.2174/1566524023666230330083819.
- K. Ouyang, D. Xie, H. Liao, Y. He and H. Xiong, Circ_0001786 facilitates gefitinib resistance and malignant progression in non-small cell lung cancer via miR-34b-5p/SRSF1, J. Cardiothorac. Surg., 2024, 19(1), 178 CrossRef PubMed.
- W. Li, Y. Wang, R. Liu, A. L. Kasinski, H. Shen and F. J. Slack, et al., MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic, Front. Cell Dev. Biol., 2021, 9, 640587, DOI:10.3389/FCELL.2021.640587/BIBTEX.
- Y. Tokumaru, E. Katsuta, M. Oshi, J. C. Sporn, L. Yan and L. Le, et al., High expression of miR-34a associated with less aggressive cancer biology but not with survival in breast cancer, Int. J. Mol. Sci., 2020, 2020, 3045, DOI:10.3390/ijms21093045.
- S. M. Yahya, H. K. Nabih, G. H. Elsayed, S. I. A. Mohamed, A. M. Elfiky and S. M. Salem, Restoring microRNA-34a overcomes acquired drug resistance and disease progression in human breast cancer cell lines via suppressing the ABCC1 gene, Breast Cancer Res. Treat., 2024, 204(1), 133–149 CrossRef CAS PubMed.
- N. K. Sedky, N. M. Abdel-Kader, M. Y. Issa, M. M. M. Abdelhady, S. N. Shamma and U. Bakowsky, et al., Co-Delivery of Ylang Ylang Oil of Cananga odorata and Oxaliplatin Using Intelligent pH-Sensitive Lipid-Based Nanovesicles for the Effective Treatment of Triple-Negative Breast Cancer, Int. J. Mol. Sci., 2023, 24, 8392, DOI:10.3390/IJMS24098392/S1.
- N. K. Sedky, M. Braoudaki, N. K. Mahdy, K. Amin, I. M. Fawzy, E. K. Efthimiadou, et al., Box-Behnken design of thermo-responsive nano-liposomes loaded with a platinum(IV) anticancer complex: Evaluation of Cytotoxicity and Apoptotic Pathways in Triple Negative Breast Cancer Cells. 2023. 10.1039/d3na00368j.
- X. P. Xu, X. Q. Peng, X. M. Yin, Y. Liu and Z. Y. Shi, miR-34a-5p suppresses the invasion and metastasis of liver cancer by targeting the transcription factor YY1 to mediate MYCT1 upregulation, Acta Histochem., 2020, 122, 151576, DOI:10.1016/J.ACTHIS.2020.151576.
- X. Zhao, Y. Zhuang, B. Wang, B. Yuan, S. Du and Z. Zeng, The miR-34a-5p-c-MYC-CHK1/CHK2 Axis Counteracts Cancer Stem Cell-Like Properties and Enhances Radiosensitivity in Hepatocellular Cancer Through Repression of the DNA Damage Response, Radiat. Res., 2023, 199, 48–60, DOI:10.1667/RADE-22-00098.1.
- J. Fu, S. Imani, M.-Y. Wu and R.-C. Wu, MicroRNA-34 Family in Cancers: Role, Mechanism, and Therapeutic Potential, Cancers, 2023, 15, 4723, DOI:10.3390/cancers15194723.
- X. Wu, Y. S. L. Cheng, M. Matthen, A. Yoon, G. K. Schwartz and S. Bala, et al., Down-regulation of the tumor suppressor miR-34a contributes to head and neck cancer by up-regulating the MET oncogene and modulating tumor immune evasion, J. Exp. Clin. Cancer Res., 2021, 40(1), 70, DOI:10.1186/s13046-021-01865-2.
- D. Cui and A. L. Cheung, Roles of microRNAs in tumorigenesis and metastasis of esophageal squamous cell carcinoma, World J. Clin. Oncol., 2021, 12, 609, DOI:10.5306/WJCO.V12.I8.609.
- B. Guo, M. He, M. Ma, Z. Tian, J. Jin and G. Tian, Long Non-coding RNA X-Inactive Specific Transcript Promotes Esophageal Squamous Cell Carcinoma Progression via the MicroRNA 34a/Zinc Finger E-box–Binding Homeobox 1 Pathway, Dig. Dis. Sci., 2024, 69(4), 1169–1181 CrossRef CAS PubMed.
- U. Kontny, C. Rodriguez-Galindo, D. Orbach, and M. Casanova, Nasopharyngeal Carcinoma, Pediatric Oncology, 2022, pp. 79–97, DOI:10.1007/978-3-030-92071-5_10.
- D. Grimm, J. Bauer, P. Wise, M. Krüger, U. Simonsen and M. Wehland, et al., The role of SOX family members in solid tumours and metastasis, Semin. Cancer Biol., 2020, 67, 122–153, DOI:10.1016/J.SEMCANCER.2019.03.004.
- P. A. Bissey, M. Teng, J. H. Law, W. Shi, J. P. Bruce and V. Petit, et al., MiR-34c downregulation leads to SOX4 overexpression and cisplatin resistance in nasopharyngeal carcinoma, BMC Cancer, 2020, 20, 597, DOI:10.1186/S12885-020-07081-Z.
- WHO, Colorectal cancer, World Health Organization, 2023, https://www.who.int/news-room/fact-sheets/detail/colorectal-cancer, accessed May 17, 2024.
- A. Mehrgou, S. Ebadollahi, K. Seidi, M. H. Ayoubi-Joshaghani, A. A. Yazdi and P. Zare, et al., Roles of miRNAs in Colorectal Cancer: Therapeutic Implications and Clinical Opportunities, Adv. Pharm. Bull., 2021, 11, 233, DOI:10.34172/APB.2021.029.
- C. Yang, B. R. Xia, Z. C. Zhang, Y. J. Zhang, G. Lou and W. L. Jin, Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant, Front. Immunol., 2020, 11, 577869, DOI:10.3389/FIMMU.2020.577869/BIBTEX.
- H. Welponer, I. Tsibulak, V. Wieser, C. Degasper, G. Shivalingaiah and S. Wenzel, et al., The miR-34 family and its clinical significance in ovarian cancer, J. Cancer, 2020, 11, 1446, DOI:10.7150/JCA.33831.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Cancer Statistics, 2021, Ca-Cancer J. Clin., 2021, 71, 7–33, DOI:10.3322/CAAC.21654.
- W. Li, X. Liu, E. M. Dougherty and D. G. Tang, MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development, Cancers, 2022, 14(18), 4538, DOI:10.3390/cancers14184538.
- K. Otmani and P. Lewalle, Tumor Suppressor miRNA in Cancer Cells and the Tumor Microenvironment: Mechanism of Deregulation and Clinical Implications, Front. Oncol., 2021, 11 DOI:10.3389/fonc.2021.708765.
- J. García-Fernández and M. de la Fuente Freire, Exosome-like systems: nanotechnology to overcome challenges for targeted cancer therapies, Cancer Lett., 2023, 561, 216151 CrossRef PubMed.
- M. Pagoni, C. Cava, D. C. Sideris, M. Avgeris, V. Zoumpourlis and I. Michalopoulos, et al., miRNA-Based Technologies in Cancer Therapy, J. Pers. Med., 2023, 13, 1586, DOI:10.3390/JPM13111586.
- M. Segal and F. J. Slack, Challenges identifying efficacious miRNA therapeutics for cancer, Expert Opin. Drug Discovery, 2020, 15, 987–992, DOI:10.1080/17460441.2020.1765770.
- N. Alrushaid, F. A. Khan, E. A. Al-Suhaimi and A. Elaissari, Nanotechnology in Cancer Diagnosis and Treatment, Pharmaceutics, 2023, 15, 1025, DOI:10.3390/pharmaceutics15031025.
- S. A. Fahmy, E. Preis, A. A. Dayyih, M. Alawak, H. M. El-Said Azzazy, U. Bakowsky and T. Shoeib, Thermosensitive liposomes encapsulating nedaplatin and picoplatin demonstrate enhanced cytotoxicity against breast cancer cells, ACS Omega, 2022, 7(46), 42115–42125 CrossRef CAS PubMed.
- S. Gavas, S. Quazi and T. M. Karpiński, Nanoparticles for Cancer Therapy: Current Progress and Challenges, Nanoscale Res. Lett., 2021, 16, 173, DOI:10.1186/S11671-021-03628-6.
- K. Elumalai, S. Srinivasan and A. Shanmugam, Review of the efficacy of nanoparticle-based drug delivery systems for cancer treatment, Biomed. Technol., 2024, 5, 109–122, DOI:10.1016/J.BMT.2023.09.001.
- Y. Yao, Y. Zhou, L. Liu, Y. Xu, Q. Chen and Y. Wang, et al., Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance, Front. Mol. Biosci., 2020, 7, 558493, DOI:10.3389/FMOLB.2020.00193/BIBTEX.
- L. Sun, H. Liu, Y. Ye, Y. Lei, R. Islam and S. Tan, et al., Smart nanoparticles for cancer therapy, Signal Transduction Targeted Ther., 2023, 8, 1–28, DOI:10.1038/s41392-023-01642-x.
- Y. Liu, M. Zheng, M. Jiao, C. Yan, S. Xu and Q. Du, et al., Polymeric nanoparticle mediated inhibition of miR-21 with enhanced miR-124 expression for combinatorial glioblastoma therapy, Biomaterials, 2021, 276, 121036, DOI:10.1016/J.BIOMATERIALS.2021.121036.
- J. Li, L. Zhu and H. F. Kwok, Nanotechnology-based approaches overcome lung cancer drug resistance through diagnosis and treatment, Drug Resistance Updates, 2023, 66, 100904, DOI:10.1016/J.DRUP.2022.100904.
- S. Raikwar, A. Jain, S. Saraf, P. D. Bidla, P. K. Panda and A. Tiwari, et al., Opportunities in combinational chemo-immunotherapy for breast cancer using nanotechnology: an emerging landscape, Expert Opin. Drug Delivery, 2022, 19, 247–268, DOI:10.1080/17425247.2022.2044785.
- R. Alzhrani, H. O. Alsaab, A. Petrovici, K. Bhise, K. Vanamala and S. Sau, et al., Improving the therapeutic efficiency of noncoding RNAs in cancers using targeted drug delivery systems, Drug Discovery Today, 2020, 25, 718, DOI:10.1016/J.DRUDIS.2019.11.006.
- F. Rodríguez, P. Caruana, N. De la Fuente, P. Español, M. Gámez and J. Balart, et al., Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges, Biomolecules, 2022, 12, 784, DOI:10.3390/BIOM12060784/S1.
- L. A. Bravo-Vázquez, A. Méndez-García, A. L. Rodríguez, P. Sahare, S. Pathak and A. Banerjee, et al., Applications of nanotechnologies for miRNA-based cancer therapeutics: current advances and future perspectives, Front. Bioeng. Biotechnol., 2023, 11, 1208547, DOI:10.3389/FBIOE.2023.1208547/FULL.
- R. A. Youness, A. H. Mohamed, E. K. Efthimiadou, R. Y. Mekky, M. Braoudaki and S. A. Fahmy, A snapshot of photoresponsive liposomes in cancer chemotherapy and immunotherapy: opportunities and challenges, ACS Omega, 2023, 8(47), 44424–44436 CrossRef CAS PubMed.
- S. Boca, D. Gulei, A. A. Zimta, A. Onaciu, L. Magdo and A. B. Tigu, et al., Nanoscale delivery systems for microRNAs in cancer therapy, Cell. Mol. Life Sci., 2020, 77, 1059–1086, DOI:10.1007/S00018-019-03317-9.
- M. B. Silva-Cazares, M. Z. Saavedra-Leos, E. Jordan-Alejandre, S. I. Nunez-Olvera, I. Compean-Martinez and C. Lopez-Camarillo, Lipid-based nanoparticles for the therapeutic delivery of non-coding RNAs in breast cancer (Review), Oncol. Rep., 2020, 44, 2353–2363, DOI:10.3892/OR.2020.7791.
- O. A. Sukocheva, J. Liu, M. E. Neganova, N. M. Beeraka, Y. R. Aleksandrova and P. Manogaran, et al., Perspectives of using microRNA-loaded nanocarriers for epigenetic reprogramming of drug resistant colorectal cancers, Semin. Cancer Biol., 2022, 86, 358–375, DOI:10.1016/J.SEMCANCER.2022.05.012.
- G. Kara, G. A. Calin and B. Ozpolat, RNAi-based therapeutics and tumor targeted delivery in cancer, Adv. Drug Delivery Rev., 2022, 182, 114113, DOI:10.1016/J.ADDR.2022.114113.
- M. J. N. Amaldoss, J. L. Yang, P. Koshy, A. Unnikrishnan and C. C. Sorrell, Inorganic nanoparticle-based advanced cancer therapies: Promising combination strategies, Drug Discovery Today, 2022, 27, 103386, DOI:10.1016/J.DRUDIS.2022.103386.
- V. Jain, H. Kumar, H. V. Anod, P. Chand, N. V. Gupta and S. Dey, et al., A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer, J. Controlled Release, 2020, 326, 628–647, DOI:10.1016/J.JCONREL.2020.07.003.
- F. Moradi Kashkooli, M. Soltani and M. Souri, Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies, J. Controlled Release, 2020, 327, 316–349, DOI:10.1016/J.JCONREL.2020.08.012.
- V. Desantis, I. Saltarella, A. Lamanuzzi, A. Melaccio, A. G. Solimando and M. A. Mariggiò, et al., MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance, Int. J. Mol. Sci., 2020, 21, 3084, DOI:10.3390/IJMS21093084.
- C. Asakiya, L. Zhu, J. Yuhan, L. Zhu, K. Huang and W. Xu, Current progress of miRNA-derivative nucleotide drugs: modifications, delivery systems, applications, Expert Opin. Drug Delivery, 2022, 19, 435–450, DOI:10.1080/17425247.2022.2063835.
- J. Szczepanek, M. Skorupa, J. Jarkiewicz-Tretyn, C. Cybulski and A. Tretyn, Harnessing Epigenetics for Breast Cancer Therapy: The Role of DNA Methylation, Histone Modifications, and MicroRNA, Int. J. Mol. Sci., 2023, 24, 7235, DOI:10.3390/ijms24087235.
- A. Forterre, H. Komuro, S. Aminova and M. Harada, A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies, Cancers, 2020, 12, 1–21, DOI:10.3390/CANCERS12071852.
- B. Roy, S. Ghose and S. Biswas, Therapeutic strategies for miRNA delivery to reduce hepatocellular carcinoma, Semin. Cell Dev. Biol., 2022, 124, 134–144, DOI:10.1016/J.SEMCDB.2021.04.006.
- G. Kara, B. Arun, G. A. Calin and B. Ozpolat, miRacle of microRNA-Driven Cancer Nanotherapeutics, Cancers, 2022, 14, 3818, DOI:10.3390/CANCERS14153818.
- D. Essa, P. P. D. Kondiah, Y. E. Choonara and V. Pillay, The Design of Poly(lactide-co-glycolide) Nanocarriers for Medical Applications, Front. Bioeng. Biotechnol., 2020, 8, 48, DOI:10.3389/FBIOE.2020.00048.
- N. Avramović, B. Mandić, A. Savić-Radojević and T. Simić, Polymeric Nanocarriers of Drug Delivery Systems in Cancer Therapy, Pharmaceutics, 2020, 12, 298, DOI:10.3390/PHARMACEUTICS12040298.
- R. H. A. Yousefi, D. H. Shin and S. Y. Rizi, Polymeric Nanoparticles in Cancer Chemotherapy: A Narrative Review, Iran. J. Public Health, 2022, 51, 226, DOI:10.18502/IJPH.V51I2.8677.
- B. Begines, T. Ortiz, M. Pérez-Aranda, G. Martínez, M. Merinero and F. Argüelles-Arias, et al., Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects, Nanomaterials, 2020, 10, 1–41, DOI:10.3390/NANO10071403.
- A. El-Araby, W. Janati, R. Ullah, S. Ercisli and F. Errachidi, Chitosan, chitosan derivatives, and chitosan-based nanocomposites: eco-friendly materials for advanced applications (a review), Front. Chem., 2024, 11, 1327426 CrossRef PubMed.
- B. Begines, A. Alcudia, R. Aguilera-Velazquez, G. Martinez, Y. He and R. Wildman, et al., Design of highly stabilized nanocomposite inks based on biodegradable polymer-matrix and gold nanoparticles for Inkjet Printing, Sci. Rep., 2019, 9, 16097, DOI:10.1038/S41598-019-52314-2.
- T. T. H. Thi, E. H. Pilkington, D. H. Nguyen, J. S. Lee, K. D. Park and N. P. Truong, The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation, Polymers, 2020, 12, 298, DOI:10.3390/POLYM12020298.
- Q. Xia, Y. Zhang, Z. Li, X. Hou and N. Feng, Red blood cell membrane-camouflaged nanoparticles: a novel drug delivery system for antitumor application, Acta Pharm. Sin. B, 2019, 9, 675–689, DOI:10.1016/J.APSB.2019.01.011.
- S. M. Honmane, P. S. Kumbhar, M. S. Charde, P. B. Chaudhari and A. S. Manjappa, Applications of Biodegradable Polymers in Surgery, in Handbook of Biodegradable Polymers, Jenny Stanford Publishing, 2025, pp. 187–209 Search PubMed.
- D. S. Nady, U. Bakowsky and S. A. Fahmy, Recent advances in brain delivery of synthetic and natural nano therapeutics: Reviving hope for Alzheimer's disease patients, J. Drug Delivery Sci. Technol., 2023, 105047 CrossRef CAS.
- C. V. Rocha, V. Gonçalves, M. C. da Silva, M. Bañobre-López and J. Gallo, PLGA-Based Composites for Various Biomedical Applications, Int. J. Mol. Sci., 2022, 23(4, 2034), 4715–5330, DOI:10.3390/ijms23042034.
- H. M. E. S. Azzazy, A. Abdelnaser, H. Al Mulla, A. M. Sawy, S. N. Shamma and M. Elhusseiny, et al., Essential Oils Extracted from Pistacia sacra Oleo Gum Resin Loaded into PLGA–PCL Nanoparticles: Enhanced Cytotoxic and Apoptotic Effects against Breast Cancer Cells, ACS Omega, 2023, 8, 1017–1025, DOI:10.1021/ACSOMEGA.2C06390/SUPPL_FILE/AO2C06390_SI_001.
- A. I. Fahira, R. Abdulah, M. I. Barliana, V. A. Gatera and R. Amalia, Polyethyleneimine (PEI) as a Polymer-Based Co-Delivery System for Breast Cancer Therapy, Breast Cancer: Targets Ther., 2022, 14, 71, DOI:10.2147/BCTT.S350403.
- M. Trivedi, A. Singh, M. Talekar, G. Pawar, P. Shah and M. Amiji, MicroRNA-34a Encapsulated in Hyaluronic Acid Nanoparticles Induces Epigenetic Changes with Altered Mitochondrial Bioenergetics and Apoptosis in Non-Small-Cell Lung Cancer Cells, Sci. Rep., 2017, 7, 1–17, DOI:10.1038/s41598-017-02816-8.
- P. Upadhyay, S. Sarker, A. Ghosh, P. Gupta, S. Das and M. Ahir, et al., Transferrin-decorated thymoquinone-loaded PEG-PLGA nanoparticles exhibit anticarcinogenic effect in non-small cell lung carcinoma: Via the modulation of miR-34a and miR-16, Biomater. Sci., 2019, 7, 4325–4344, 10.1039/c9bm00912d.
- J. Hu, W. Pei, Z. Jiang and Z. Li, A combined miR-34a and arsenic trioxide nanodrug delivery system for synergistic inhibition of HCC progression after microwave ablation, Cancer Nanotechnol., 2021, 12, 1–14, DOI:10.1186/S12645-021-00105-8/FIGURES/7.
- H. Gibori, S. Eliyahu, A. Krivitsky, D. Ben-Shushan, Y. Epshtein and G. Tiram, et al., Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer, Nat. Commun., 2017, 9, 1–18, DOI:10.1038/s41467-017-02283-9.
- C. H. Kapadia, S. A. Ioele and E. S. Day, Layer-by-layer assembled PLGA nanoparticles carrying miR-34a cargo inhibit the proliferation and cell cycle progression of triple-negative breast cancer cells, J. Biomed. Mater. Res., 2020, 108, 601–613, DOI:10.1002/jbm.a.36840.
- V. Kasina, A. Wahane, C. H. Liu, L. Yang, M. P. Nieh and F. J. Slack, et al., Next-generation poly-L-histidine formulations for miRNA mimic delivery, Mol. Ther.–Methods Clin. Dev., 2023, 29, 271, DOI:10.1016/J.OMTM.2023.03.015.
- F. Li, D. Cao, L. Yao, W. Gu, Z. Liu and D. Li, et al., Targeted delivery of miR-34a-5p by phenylborate-coupled polyethylenimide nanocarriers for anti-KSHV treatment, Front. Bioeng. Biotechnol., 2023, 11, 1343956, DOI:10.3389/fbioe.2023.1343956.
- T. Y. Han, M. L. Huan, Z. Cai, W. He, S. Y. Zhou and B. L. Zhang, Polymer-Initiating Caveolae-Mediated Endocytosis and GSH-Responsive MiR-34a Gene Delivery System for Enhanced Orthotopic Triple Negative Breast Cancer Therapy, Adv. Healthcare Mater., 2023, 12, 2302094, DOI:10.1002/adhm.202302094.
- M. Motiei, F. Aboutalebi, M. Forouzanfar, K. Dormiani, M. H. Nasr-Esfahani and S. Z. Mirahmadi-Zare, Smart co-delivery of miR-34a and cytotoxic peptides (LTX-315 and melittin) by chitosan based polyelectrolyte nanocarriers for specific cancer cell death induction, Mater. Sci. Eng., C, 2021, 128, 112258, DOI:10.1016/J.MSEC.2021.112258.
- S. Chattopadhyay, S. S. Sarkar, S. Saproo, S. Yadav, D. Antil and B. Das, et al., Apoptosis-targeted gene therapy for non-small cell lung cancer using chitosan-poly-lactic-co-glycolic acid-based nano-delivery system and CASP8 and miRs 29A-B1 and 34A, Front. Bioeng. Biotechnol., 2023, 11, 1188652, DOI:10.3389/fbioe.2023.1188652.
- M. K. Jayasinghe, M. Tan, B. Peng, Y. Yang, G. Sethi and M. Pirisinu, et al., New approaches in extracellular vesicle engineering for improving the efficacy of anti-cancer therapies, Semin. Cancer Biol., 2021, 74, 62–78, DOI:10.1016/J.SEMCANCER.2021.02.010.
- J. Weng, X. Xiang, L. Ding, A. L. A. Wong, Q. Zeng and G. Sethi, et al., Extracellular vesicles, the cornerstone of next-generation cancer diagnosis?, Semin. Cancer Biol., 2021, 74, 105–120, DOI:10.1016/J.SEMCANCER.2021.05.011.
- P. Wu, B. Zhang, D. K. W. Ocansey, W. Xu and H. Qian, Extracellular vesicles: A bright star of nanomedicine, Biomaterials, 2021, 269, 120467, DOI:10.1016/J.BIOMATERIALS.2020.120467.
- M. D. A. Paskeh, M. Entezari, S. Mirzaei, A. Zabolian, H. Saleki and M. J. Naghdi, et al., Emerging role of exosomes in cancer progression and tumor microenvironment remodeling, J. Hematol. Oncol., 2022, 15, 1–39, DOI:10.1186/S13045-022-01305-4/TABLES/4.
- A. Deb, S. Gupta and P. B. Mazumder, Exosomes: A new horizon in modern medicine, Life Sci., 2021, 264, 118623, DOI:10.1016/J.LFS.2020.118623.
- J. Jia, S. Guo, D. Zhang, X. Tian and X. Xie, Exosomal-lncRNA DLEU1 Accelerates the Proliferation, Migration, and Invasion of Endometrial Carcinoma Cells by Regulating microRNA-E2F3, OncoTargets Ther., 2020, 13, 8651–8663, DOI:10.2147/OTT.S262661.
- A. H. Lee, D. Ghosh, N. Quach, D. Schroeder and M. R. Dawson, Ovarian Cancer Exosomes Trigger Differential Biophysical Response in Tumor-Derived Fibroblasts, Sci. Rep., 2020, 10, 8686, DOI:10.1038/S41598-020-65628-3.
- M. Ghorbanian, S. Babashah and F. Ataei, The effects of ovarian cancer cell-derived exosomes on vascular endothelial growth factor expression in endothelial cells, EXCLI J., 2019, 18, 899, DOI:10.17179/EXCLI2019-1800.
- L. Zuo, H. Tao, H. Xu, C. Li, G. Qiao and M. Guo, et al., Exosomes-Coated miR-34a Displays Potent Antitumor Activity in Pancreatic Cancer Both in vitro and in vivo, Drug Des., Dev. Ther., 2020, 14, 3495, DOI:10.2147/DDDT.S265423.
- W. Huang, Y. Yan, Y. Liu, M. Lin, J. Ma and W. Zhang, et al., Exosomes with low miR-34c-3p expression promote invasion and migration of non-small cell lung cancer by upregulating integrin α2β1, Signal Transduction Targeted Ther., 2020, 5, 1–13, DOI:10.1038/s41392-020-0133-y.
- F. Z. Wan, K. H. Chen, Y. C. Sun, X. C. Chen, R. B. Liang and L. Chen, et al., Exosomes overexpressing miR-34c inhibit malignant behavior and reverse the radioresistance of nasopharyngeal carcinoma, J. Transl. Med., 2020, 18, 1–19, DOI:10.1186/S12967-019-02203-Z/FIGURES/8.
- F. Vakhshiteh, S. Rahmani, S. N. Ostad, Z. Madjd, R. Dinarvand and F. Atyabi, Exosomes derived from miR-34a-overexpressing mesenchymal stem cells inhibit in vitro tumor growth: A new approach for drug delivery, Life Sci., 2021, 266, 118871, DOI:10.1016/J.LFS.2020.118871.
- M. Hosseini, K. Baghaei, D. Amani and M. Ebtekar, Tumor-derived exosomes encapsulating miR-34a promote apoptosis and inhibit migration and tumor progression of colorectal cancer cells under in vitro condition, Daru, J. Pharm. Sci., 2021, 29, 267–278, DOI:10.1007/S40199-021-00400-0/FIGURES/6.
- J. J. Xu, W. C. Zhang, Y. W. Guo, X. Y. Chen and Y. N. Zhang, Metal nanoparticles as a promising technology in targeted cancer treatment, Drug Delivery, 2022, 29, 664, DOI:10.1080/10717544.2022.2039804.
- N. Jain, P. Jain, D. Rajput and U. K. Patil, Green synthesized plant-based silver nanoparticles: therapeutic prospective for anticancer and antiviral activity, Micro Nano Syst. Lett., 2021, 9, 5, DOI:10.1186/s40486-021-00131-6.
- S. M. Mostafa, A. M. Gamal-Eldeen, N. A. E. Maksoud and A. A. Fahmi, Epigallocatechin gallate-capped gold nanoparticles enhanced the tumor suppressors let-7a and miR-34a in hepatocellular carcinoma cells, An. Acad. Bras. Cienc., 2020, 92, e20200574, DOI:10.1590/0001-3765202020200574.
- N. A. Alden, T. J. Yeingst, H. M. Pfeiffer, N. Celik, J. H. Arrizabalaga and A. M. Helton, et al., Near-Infrared Induced miR-34a Delivery from Nanoparticles in Esophageal Cancer Treatment, Adv. Healthcare Mater., 2024, 2303593, DOI:10.1002/ADHM.202303593.
- C. Cui, H. Qian, J. Wang, J. Kang, W. Ma and Y. Nian, et al., Targeted miR-34a delivery with PD1 displayed bacterial outer membrane vesicles-coated zeolitic imidazolate framework nanoparticles for enhanced tumor therapy, Int. J. Biol. Macromol., 2023, 247, 125692, DOI:10.1016/J.IJBIOMAC.2023.125692.
- M. Gisbert-Garzarán, D. Lozano and M. Vallet-Regí, Mesoporous Silica Nanoparticles for Targeting Subcellular Organelles, Int. J. Mol. Sci., 2020, 21, 9696, DOI:10.3390/IJMS21249696.
- H. Ahmed, S. S. Gomte, E. Prathyusha, P. A, M. Agrawal and A. Alexander, Biomedical applications of mesoporous silica nanoparticles as a drug delivery carrier, J. Drug Delivery Sci. Technol., 2022, 76, 103729, DOI:10.1016/J.JDDST.2022.103729.
- H. Lee, J. Kim, H. H. Kim, C. S. Kim and J. Kim, Review on Optical Imaging Techniques for Multispectral Analysis of Nanomaterials, Nanotheranostics, 2022, 6, 50–61, DOI:10.7150/NTNO.63222.
- Y. Dutta Gupta, Y. Mackeyev, S. Krishnan and S. Bhandary, Mesoporous silica nanotechnology: promising advances in augmenting cancer theranostics, Cancer Nanotechnol., 2024, 15, 1–44, DOI:10.1186/S12645-024-00250-W.
- F. Panebianco, M. Climent, M. A. Malvindi, P. P. Pompa, P. Bonetti and F. Nicassio, Delivery of biologically active miR-34a in normal and cancer mammary epithelial cells by synthetic nanoparticles, Nanomedicine, 2019, 19, 95–105, DOI:10.1016/J.NANO.2019.03.013.
- Y. Li, X. Zhu, H. Zhang, Y. Lu, T. Zeng and H. Liu, et al., Nanodiamond in cancer theranostics, Nano TransMed, 2023, 2, e9130019, DOI:10.26599/NTM.2023.9130019.
- M. Abate, A. Lombardi, A. Luce, M. Porru, C. Leonetti and M. Bocchetti, et al., Fluorescent nanodiamonds as innovative delivery systems for MiR-34a replacement in breast cancer, Mol. Ther.–Nucleic Acids, 2023, 33, 127–141, DOI:10.1016/j.omtn.2023.06.012.
- N. Rajana, A. Mounika, P. S. Chary, V. Bhavana, A. Urati and D. Khatri, et al., Multifunctional hybrid nanoparticles in diagnosis and therapy of breast cancer, J. Controlled Release, 2022, 352, 1024–1047, DOI:10.1016/J.JCONREL.2022.11.009.
- H. Marwah, S. Khare, P. Rawat, S. Singh, P. Kesharwani, M. S. Alam, et al., Strengths, limitations, and regulatory aspects of hybrid drug delivery systems, Hybrid Nanomaterials for Drug Delivery, 2022, pp. 339–355, DOI:10.1016/B978-0-323-85754-3.00011-3.
- J. Ghitman, E. I. Biru, R. Stan and H. Iovu, Review of hybrid PLGA nanoparticles: Future of smart drug delivery and theranostics medicine, Mater. Des., 2020, 193, 108805, DOI:10.1016/J.MATDES.2020.108805.
- Y. Xia, X. Deng, M. Cao, S. Liu, X. Zhang and X. Xiao, et al., Nanodiamond-based layer-by-layer nanohybrids mediate targeted delivery of miR-34a for triple negative breast cancer therapy, RSC Adv., 2018, 8, 13789–13797, 10.1039/c8ra00907d.
- R. Goyal, C. H. Kapadia, J. R. Melamed, R. S. Riley and E. S. Day, Layer-by-Layer Assembled Gold Nanoshells for the Intracellular Delivery of miR-34a, Cell. Mol. Bioeng., 2018, 11, 383–396, DOI:10.1007/S12195-018-0535-X/FIGURES/7.
- S. A. Fahmy, N. K. Sedky, A. Ramzy, M. M. M. Abdelhady, O. A. A. Alabrahim and S. N. Shamma, et al., Green extraction of essential oils from Pistacia lentiscus resins: Encapsulation into Niosomes showed improved preferential cytotoxic and apoptotic effects against breast and ovarian cancer cells, J. Drug Delivery Sci. Technol., 2023, 87, 104820, DOI:10.1016/J.JDDST.2023.104820.
- N. A. Abtahi, J. Z. Reza, T. Mahboob, M. De L. Pereira, R. Hossain Bangabandhu Sheikh Mujibur Rahman, and M. T. Islam Bangabandhu Sheikh Mujibur Rahman, The Novel Method for Delivery miRNA-34a: a New Cationic PEGylated Niosomal Formulation for the Treatment of Breast Cancer, 2021, DOI:10.21203/rs.3.rs-650056/v1.
- V. Aiassa, C. Garnero, M. R. Longhi and A. Zoppi, Cyclodextrin Multicomponent Complexes: Pharmaceutical Applications, Pharmaceutics, 2021, 13, 1099, DOI:10.3390/PHARMACEUTICS13071099.
- D. A. Real, K. Bolaños, J. Priotti, N. Yutronic, M. J. Kogan and R. Sierpe, et al., Cyclodextrin-Modified Nanomaterials for Drug Delivery: Classification and Advances in Controlled Release and Bioavailability, Pharmaceutics, 2021, 13, 2131, DOI:10.3390/PHARMACEUTICS13122131.
- A. García, J. Priotti, A. V. Codina, M. D. Vasconi, A. D. Quiroga and L. I. Hinrichsen, et al., Synthesis and characterization of a new cyclodextrin derivative with improved properties to design oral dosage forms, Drug Delivery Transl. Res., 2019, 9, 273–283, DOI:10.1007/S13346-018-0591-8.
- P. Mura, Advantages of the combined use of cyclodextrins and nanocarriers in drug delivery: A review, Int. J. Pharm., 2020, 579, 119181, DOI:10.1016/J.IJPHARM.2020.119181.
- F. Li, D. Cao, W. Gu, L. Cui, Z. Qiu and Z. Liu, et al., Delivery of miR-34a-5p by Folic Acid-Modified β-Cyclodextrin-Grafted Polyethylenimine Copolymer Nanocarriers to Resist KSHV, ACS Appl. Nano Mater., 2023, 6, 10826–10836, DOI:10.1021/ACSANM.3C02162/ASSET/IMAGES/LARGE/AN3C02162_0011.JPEG.
- N. Yang, H. Guo, C. Cao, X. Wang, X. Song and W. Wang, et al., Infection microenvironment-activated nanoparticles for NIR-II photoacoustic imaging-guided photothermal/chemodynamic synergistic anti-infective therapy, Biomaterials, 2021, 275, 120918, DOI:10.1016/J.BIOMATERIALS.2021.120918.
- L. Sun, F. Shen, L. Tian, H. Tao, Z. Xiong and J. Xu, et al., ATP-Responsive Smart Hydrogel Releasing Immune Adjuvant Synchronized with Repeated Chemotherapy or Radiotherapy to Boost Antitumor Immunity, Adv. Mater., 2021, 33, 2007910, DOI:10.1002/ADMA.202007910.
- W. Wu, Y. Pu and J. Shi, Nanomedicine-enabled chemotherapy-based synergetic cancer treatments, J. Nanobiotechnol., 2022, 20, 1–21, DOI:10.1186/S12951-021-01181-Z/TABLES/1.
- G. Liu, J. F. Lovell, L. Zhang and Y. Zhang, Stimulus-Responsive Nanomedicines for Disease Diagnosis and Treatment, Int. J. Mol. Sci., 2020, 21, 6380, DOI:10.3390/IJMS21176380.
- S. Gurunathan, M. H. Kang, M. Qasim and J. H. Kim, Nanoparticle-Mediated Combination Therapy: Two-in-One Approach for Cancer, Int. J. Mol. Sci., 2018, 19, 3264, DOI:10.3390/IJMS19103264.
- C. K. Phillips, and D. P. Petrylak, Docetaxel, Drug Management of Prostate Cancer, 2022, pp. 133–146, DOI:10.1007/978-1-60327-829-4_12.
- L. Zhang, X. Yang, Y. Lv, X. Xin, C. Qin and X. Han, et al., Cytosolic co-delivery of miRNA-34a and docetaxel with core-shell nanocarriers via caveolae-mediated pathway for the treatment of metastatic breast cancer, Sci. Rep., 2017, 7, 1–16, DOI:10.1038/srep46186.
- G. Reyhanoglu, and T. Smith, Irinotecan. XPharm: The Comprehensive Pharmacology Reference, 2023, pp. 1–5, DOI:10.1016/B978-008055232-3.61952-X.
- Y. Li, F. Jia, X. Deng, X. Wang, J. Lu and L. Shao, et al., Combinatorial miRNA-34a replenishment and irinotecan delivery via auto-fluorescent polymeric hybrid micelles for synchronous colorectal cancer theranostics, Biomater. Sci., 2020, 8, 7132, 10.1039/d0bm01579b.
- K. Johnson-Arbor, and R. Dubey, Doxorubicin. XPharm: The Comprehensive Pharmacology Reference, 2023, pp. 1–5, DOI:10.1016/B978-008055232-3.61650-2.
- R. A. Youness, A. M. Al-Mahallawi, F. H. Mahmoud, H. Atta, M. Braoudaki and S. A. Fahmy, Oral Delivery of Psoralidin by Mucoadhesive Surface-Modified Bilosomes Showed Boosted Apoptotic and Necrotic Effects against Breast and Lung Cancer Cells, Polymers, 2023, 15, 1464, DOI:10.3390/POLYM15061464.
- H. M. E. S. Azzazy, A. M. Sawy, A. Abdelnaser, M. R. Meselhy, T. Shoeib and S. A. Fahmy, Peganum harmala Alkaloids and Tannic Acid Encapsulated in PAMAM Dendrimers: Improved Anticancer Activities as Compared to Doxorubicin, ACS Appl. Polym. Mater., 2022, 4, 7228–7239, DOI:10.1021/ACSAPM.2C01093/ASSET/IMAGES/LARGE/AP2C01093_0008.JPEG.
- J. Fang, Y. Wang, Z. Wang, T. Xie, F. Yan and L. Wang, et al., A nanomicelle with miR-34a and doxorubicin reverses the drug resistance of cisplatin in esophageal carcinoma cells by inhibiting SIRT1 signal pathway, Transl. Cancer Res., 2020, 9, 4131–4140, DOI:10.21037/TCR-19-975.
- I. Ritacco, A. M. Al, M. K. Abd El-Rahman, S. A. Fahmy, N. Russo and T. Shoeib, et al., Hydrolysis in Acidic Environment and Degradation of Satraplatin: A Joint Experimental and Theoretical Investigation, Inorg. Chem., 2017, 56, 6013–6026, DOI:10.1021/ACS.INORGCHEM.7B00945/ASSET/IMAGES/LARGE/IC-2017-00945Z_0013.JPEG.
- B. C. Rudy, and B. Z. Senkowski, Fluorouracil, Analytical Profiles of Drug Substances and Excipients, 2022, vol. 2, pp. 221–44, DOI:10.1016/S0099-5428(08)60041-6.
- J. Xu, G. Zhang, X. Luo, D. Wang, W. Zhou and Y. Zhang, et al., Co-delivery of 5-fluorouracil and miRNA-34a mimics by host–guest self-assembly nanocarriers for efficacious targeted therapy in colorectal cancer patient-derived tumor xenografts, Theranostics, 2021, 11, 2475, DOI:10.7150/THNO.52076.
| This journal is © The Royal Society of Chemistry 2024 |