
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative reagent monitoring in paper-based electrochemical rapid diagnostic tests†
Léonard
Bezinge
 ,
Andrew J.
deMello
,
Chih-Jen
Shih
and
Daniel A.
Richards
*
,
Andrew J.
deMello
,
Chih-Jen
Shih
and
Daniel A.
Richards
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: daniel.richards@chem.ethz.ch
First published on 28th June 2024
Abstract
Paper-based rapid diagnostic tests (RDTs) are an essential component of modern healthcare, particularly for the management of infectious diseases. Despite their utility, these capillary-driven RDTs are compromised by high failure rates, primarily caused by user error. This limits their utility in complex assays that require multiple user operations. Here, we demonstrate how this issue can be directly addressed through continuous electrochemical monitoring of reagent flow inside an RDT using embedded graphenized electrodes. Our method relies on applying short voltage pulses and measuring variations in capacitive discharge currents to precisely determine the flow times of injected samples and reagents. This information is reported to the user, guiding them through the testing process, highlighting failure cases and ultimately decreasing errors. Significantly, the same electrodes can be used to quantify electrochemical signals from immunoassays, providing an integrated solution for both monitoring assays and reporting results. We demonstrate the applicability of this approach in a serology test for the detection of anti-SARS-CoV-2 IgG in clinical serum samples. This method paves the way towards “smart” RDTs able to continuously monitor the testing process and improve the robustness of point-of-care diagnostics.
Introduction
Rapid and reliable diagnostic tools are essential for managing infectious diseases, a reality starkly highlighted by the COVID-19 pandemic.1 Among these tools, rapid diagnostic tests (RDTs) have emerged as a frontrunner, offering quick and affordable testing at the point of need without the necessity for laboratory equipment.2,3 As such, RDTs have become an indispensable component of the track-trace-treat strategy for pandemic management,3,4 as evidenced by their deployment across a wide variety of settings, including clinics, testing centers, pharmacies, and the home.5 The widespread acceptance of RDTs can be credited to their user-friendly nature, in large part made possible by their passive, capillary-driven operation.3 This simplicity ensures accessibility for individuals without specialized training.6,7Recently, the integration of electrodes into RDTs has sought to enhance their functionality by providing quantitative readings through electrochemical signal transduction,8–11 and thus improving on the qualitative nature and error-prone interpretation of colourimetric tests.12,13 Nonetheless, a frequently overlooked limitation of RDTs is their high failure rate,14 which primarily originates from user error or misuse.15,16 This issue is particularly pronounced when performing complex assays, such as serology tests, which require intricate specimen collection and transfer, as well as the use of multiple reagents for dilution, washing, and signal amplification.3,6,17 For instance, a recent study revealed that up to 41% of users struggled to add the correct amount of sample or reagent to a serological HIV rapid test cassette,18 an observation echoed in several other studies.16 To address these challenges, commercial tests commonly provide printed instructions or companion software applications to guide users through the assay procedure.3,19 However, these measures do not guarantee error-free testing, as they cannot detect anomalies or misuse, such as improper sample addition.18 We hypothesized that electrochemical sensors, with their ability to continuously communicate with their reader,20,21 provide an untapped opportunity for real-time monitoring of testing protocols, especially since non-faradic processes are highly sensitive to the local physicochemical environment at the electrode-electrolyte interface, such as the flux of ions.22
Within capillary-driven microfluidic devices, the use of electrodes for flow sensing has been restricted to detecting the flow front through changes in resistance or capacitance.23,24 Although the flow front behaviour may serve as an indicator for RDT failure,25 it cannot provide information on injected volumes or any aspect of multi-injection testing procedures. Several flow sensing solutions have been developed for pressure-driven microfluidic systems,26 involving, for example, deformable membranes,27 microstructures,28,29 or microfabricated thermal sensors;30 however, these microfabrication approaches are incompatible with paper-based systems. Additionally, flow-tracking methods relying on specific redox reporters interfere with electrochemical bioassays.31 As a result, no integrated solution for flow monitoring in paper-based devices has yet been reported.
Herein, we show that the interfacial capacitive properties of an electrode can track fluid movement across its interface without the need for any additives. We demonstrate the applicability of our method in an electrochemical vertical flow immunoassay for anti-SARS-CoV-2 nucleocapsid IgG in serum samples. By actively monitoring flow dynamics, we can identify samples with incorrect volumes or abnormal viscosities. Importantly, for devices already incorporating an electrochemical sensor, the approach requires no additional hardware and can be autonomously performed by the test reader's software. Our method paves the way for the next generation of smart rapid diagnostic tests that monitor assays in real-time to guide the user, ultimately improving testing robustness and confidence.
Results and discussion
Our system comprises an electrochemical vertical flow test connected to a miniaturized USB potentiostat (Fig. 1a and S1†). The vertical flow device has a stacked-layer configuration, comprising a nitrocellulose capture pad, a paper-based electrode layer and an absorbent wicking pad (Fig. 1b and S2†).32 The electrodes are seamlessly patterned in the paper through laser-induced pyrolysis of the cellulose (geometric surface area = 13 mm2 and electrochemical porosity factor η = 6.3 ± 0.3)32 and, owing to their porosity and wettability, support unrestricted capillary-driven flow through the electrochemical sensor.32 The device is designed to capture the target antibodies (anti-SARS-CoV-2 nucleocapsid IgG) from serum on the nitrocellulose pad (functionalized with anti-human IgG secondary antibodies). The captured antibodies are then labelled using a nucleocapsid antigen conjugated to alkaline phosphatase to facilitate selective electrochemical signalling.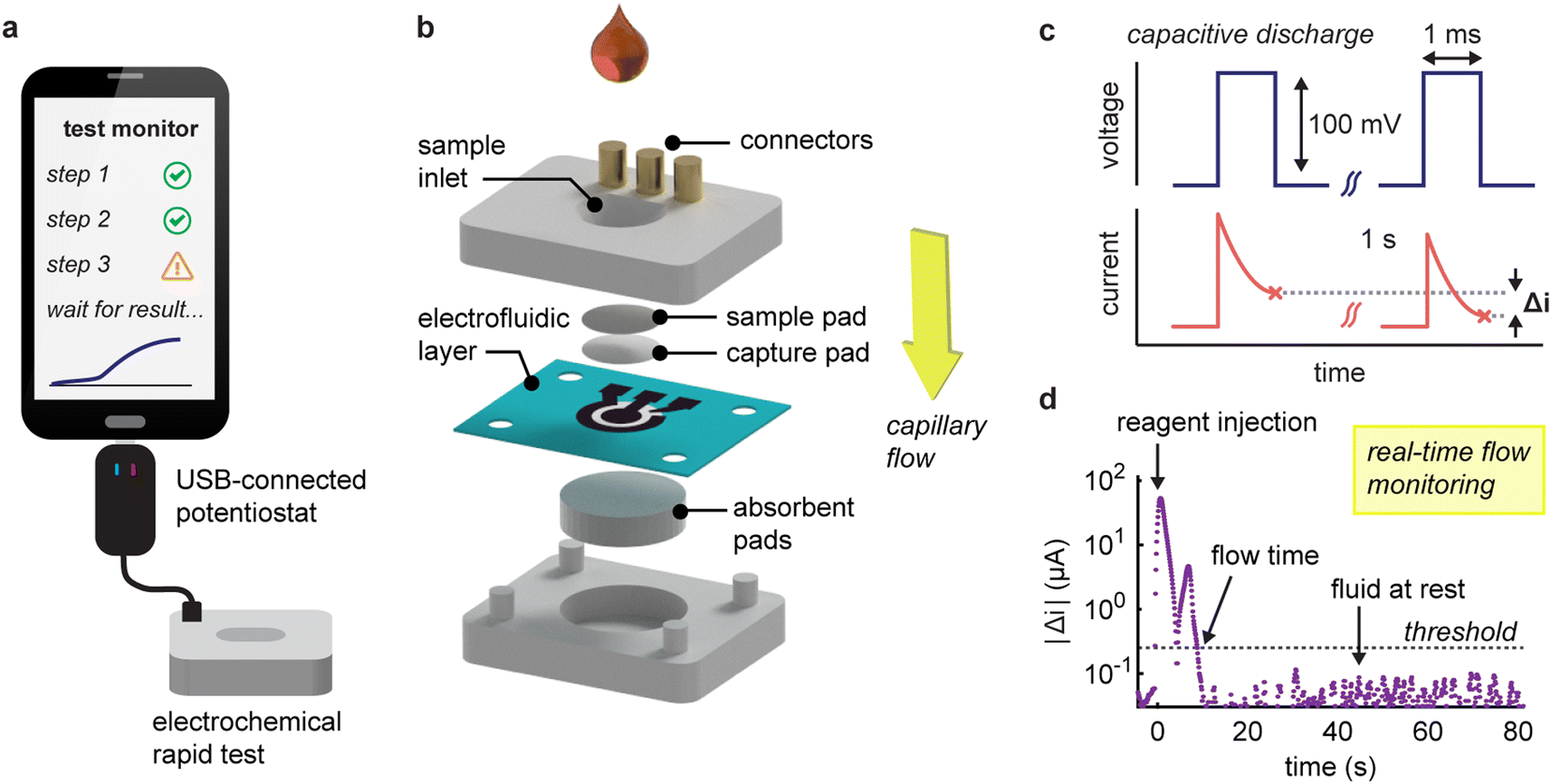 | ||
| Fig. 1 (a) Schematic of the RDT system comprising an electrochemical rapid test connected to a programmable USB-connected potentiostat. (b) Exploded view of the vertical flow device showing the component layers (c) fluid movement through the permeable paper-based electrode is tracked by applying a series of voltage pulses to the working electrode and recording the variations in current response, Δi. (d) Signal variation following the injection of 100 μL of a 20 mM Tris-HCl buffer solution. The absolute magnitude of Δi peaks after the injection, dips during steady flow, and increases again as the flow decelerates and stops. Flow time is determined by comparing these changes to a predetermined threshold. | ||
Our system operates over two distinct phases. During the initial assay phase, the user injects sample and reagents through the sample inlet in a sequential manner, whilst the system tracks fluid movement. Then, following the addition of the last reagent, the device switches its electrode interrogation protocol to quantify captured target antibody by square wave voltammetry.
The fluid tracking method centres on the continuous interrogation of the working electrode using 1-ms pulses to capture current responses (Fig. 1c). This strategy effectively tracks variations in the capacitive discharge characteristics at the electrode-solution interface (Fig. S3†). The magnitude of the non-faradic response scales with the surface area of this interface and its variations with the changes in flow velocity.22 Our method is thus particularly suited to laser-pyrolyzed paper electrodes in a vertical flow format, characterized by a large porous surface that allows rapid flow-through operation, although this concept is extendable to other flow-based electrode systems.32 To enhance reliability, we calculate the differences in current between voltage pulses spaced one second apart and then average these over ten pulses. By plotting the magnitude of these variations over time, distinct flow conditions within the device can be identified. When a solution is injected, we typically observe two peaks in the response signal (Fig. 1d). The first peak arises from a sudden increase in flow rate, and the second peak occurs when the flow decelerates as the liquid is fully drained. It is important to note that absolute current variations are used, as the sign will depend on prior interactions with the electrode. To distinguish between moving and stationary flows, we establish a threshold based on the mean plus ten standard deviations of measurements from a static liquid (equal to 0.25 μA s−1 for solutions in 20 mM Tris·HCl). Furthermore, we incorporate redundancy measures such as a ten-hit minimum for flow detection and a ten second time delay that allows the algorithm to confidently determine when the flow has stopped.
One of the primary appeals of tracking fluidic movement is the ability to verify that appropriate volumes of reagents are being added to the device. To investigate this, we injected increasing volumes of running buffer ranging from 25 to 200 μL and monitored changes in capacitive discharge (Fig. 2a). Prior to each measurement, the electrodes were pre-wetted with water to record flow dynamics in a fully saturated flow regime,33 and avoid the signals originating from the wetting of the electrode itself. At 25 μL, the volume was insufficient to fully reach the electrode (dead volume ≈ 20 μL). This situation resulted in a slow signal drift (reflected by a larger sustained variation) due to ion diffusion in the electrode area. In contrast, volumes between 50 and 200 μL displayed excellent linearity (R2 = 0.90), with a relation between flow time and added volume of 0.19 ± 0.02 s μL−1 (Fig. 2b). These times also correlated linearly with visual absorption times. Interestingly, our method recorded average flow times 1.8 s longer (interquartile range, IQR: −0.1–2.5 s, Fig. S4†). This is perhaps unexpected when we consider that the integrated sensor probes internal dynamics and thus may stabilize slightly later than what is apparent from surface-level inspections. It's noteworthy that, for larger volumes, we occasionally observed intermediate peaks in current variations. These fluctuations are attributed to the non-linear behaviour of capillary flow in vertical flow devices, stemming from less-than-ideal contact between the layers.32
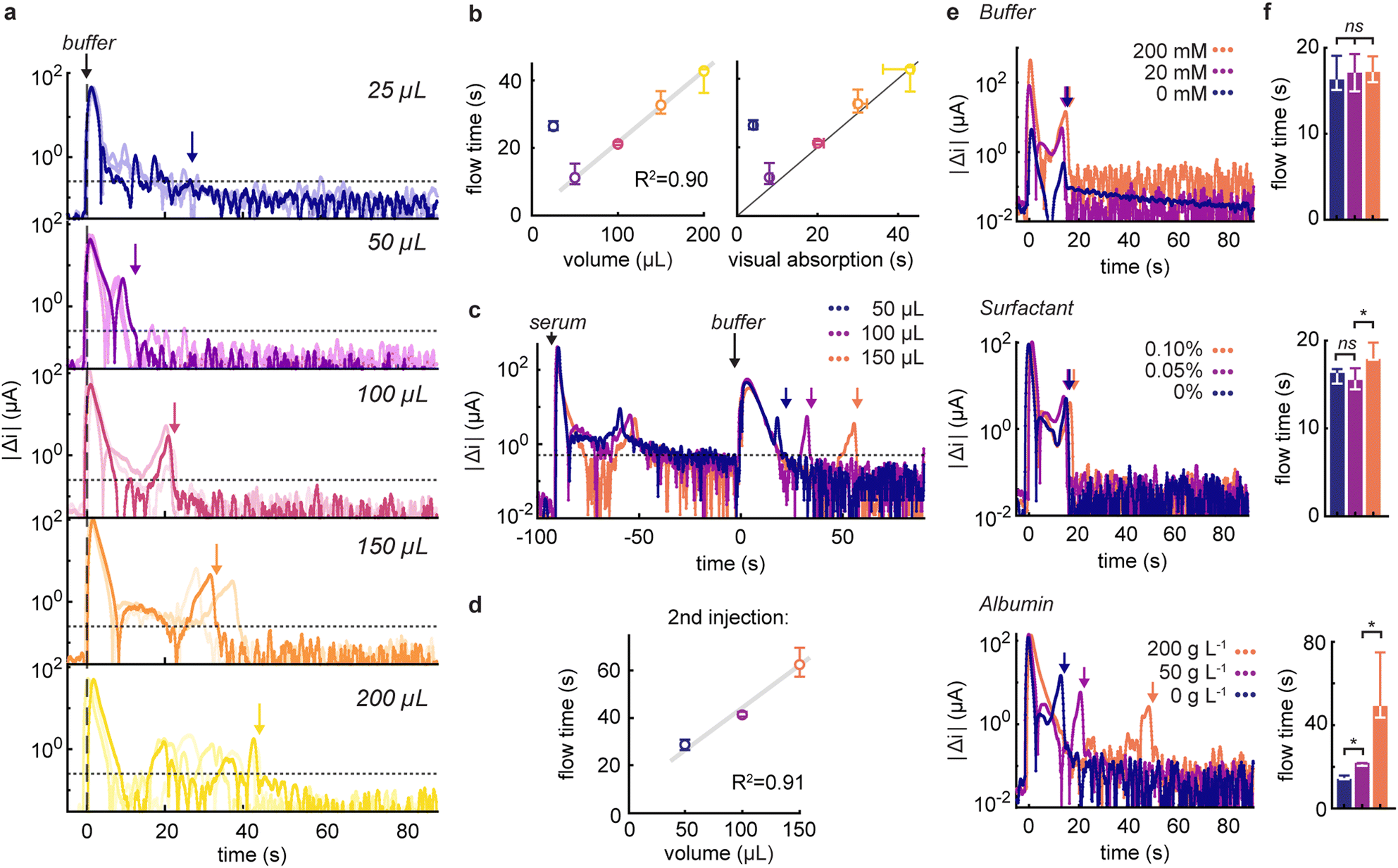 | ||
| Fig. 2 (a) Absolute current variations recorded following the injection of 25 to 200 μL of running buffer (50 g L−1 albumin, 0.05 vol% Tween-20 in 20 mM Tris·HCl pH 7.5), n = 3. The dotted line indicates the threshold at 0.25 μA and the coloured arrows the corresponding flow times. (b) Correlation between the recorded flow times and the injected volume, as well as the visual absorption times (n = 3). (c) Current responses after injection of a 100 μL serum sample followed by 50 to 150 μL of running buffer. (d) Recorded flow times of the running buffer as a function of volume (n = 3). (e) Signals following the injection of 100 μL of buffer containing varying concentrations of Tris·HCl buffer concentrations, Tween-20 surfactant (in vol%), or serum albumin, and (f) the associated flow times (n = 3). ns: non-significant, *: p < 0.05. | ||
Our initial investigations focused on responses from single injections. However, given that rapid tests usually involve the use of multiple reagents, it is critical to confirm that our results are applicable to multi-injection protocols. To this end, we evaluated the ability of our method to accurately measure the volumes of running buffer injected after the addition of 100 μL undiluted serum (Fig. 2c). Notably, we found minimal effect from electrode fouling on signal strength, and linearity between measured flow times and injected volumes was maintained (R2 = 0.91, Fig. 2d). In this scenario, with a scaling factor of 0.36 ± 0.04 s μL−1, we were able to quantify the slower flow in comparison to a pristine device, attributed to the increasingly saturated absorbent pad and in accordance with the principles of capillary flow.34
To better understand the factors that underpin the response of our flow monitor, we measured current responses following the injection of buffer solutions of known composition (Fig. 2e and f). Specifically, we focused on the effect of buffer concentration, surfactant, and serum albumin content at levels that would be expected to be present in reagents from diagnostic test kits.35,36 Upon injecting simple buffer solutions of varying concentration into our system, we observed a strong dependence of signal variation intensities on electrolyte concentration, as expected from the double-layer capacitance dependence on ionic strength.22 For example, current variations at 200 mM buffer were approximately four times higher than those at 20 mM, while the absence of buffer altogether resulted in low and constant signals. Crucially, and despite differences in signal magnitude, the flow times were unaffected by buffer concentrations. Additionally, after the fluid flow tapered off, we observed that background signals and noise levels were also more elevated in high ionic strength conditions, highlighting the need for higher thresholds in salt-rich buffers.
When examining the effect of surfactant, we found consistent responses across different concentrations of non-ionic surfactant. Interestingly, at higher surfactant concentrations, flow dynamics exhibited longer durations, indicative of either a diminished flow rate or slower wetting of the deeper hydrophobic regions of the electrode facilitated by the surfactant.32 Encouragingly, the introduction of protein did not led to electrode fouling or reduction in signals, and allowed us to quantify the increased flow times due to higher viscosity at elevated protein levels, with flow times reduced by factors of 1.5 and 2.5 in the presence of 50 and 200 g L−1 albumin, respectively. Considering the sign of the capactive current variations, we noticed that the variations were positive for increasing flow rates or ionic strength (compared to the previous injection), whereas decelerating flow rates, lower ionic strength or rinsing albumin off the electrodes exhibited distinct negative signals (Fig. S5†).
Building upon the foundation laid above, we integrated the flow tracking method in an electrochemical serological test for the detection of anti-SARS-CoV-2 IgG. This test is contingent on the capture of human IgG in serum on a high-area nitrocellulose pad positioned atop the permeable electrochemical sensor (Fig. 3a). The bound IgG targets are selectively labelled using gold nanoparticle reporters. These nanolabels are co-functionalized with SARS-CoV-2 nucleocapsid antigen and alkaline phosphatase, with their surface composition engineered to balance target recognition and electrochemical signal amplification (Fig. S6 and S7†).37–39
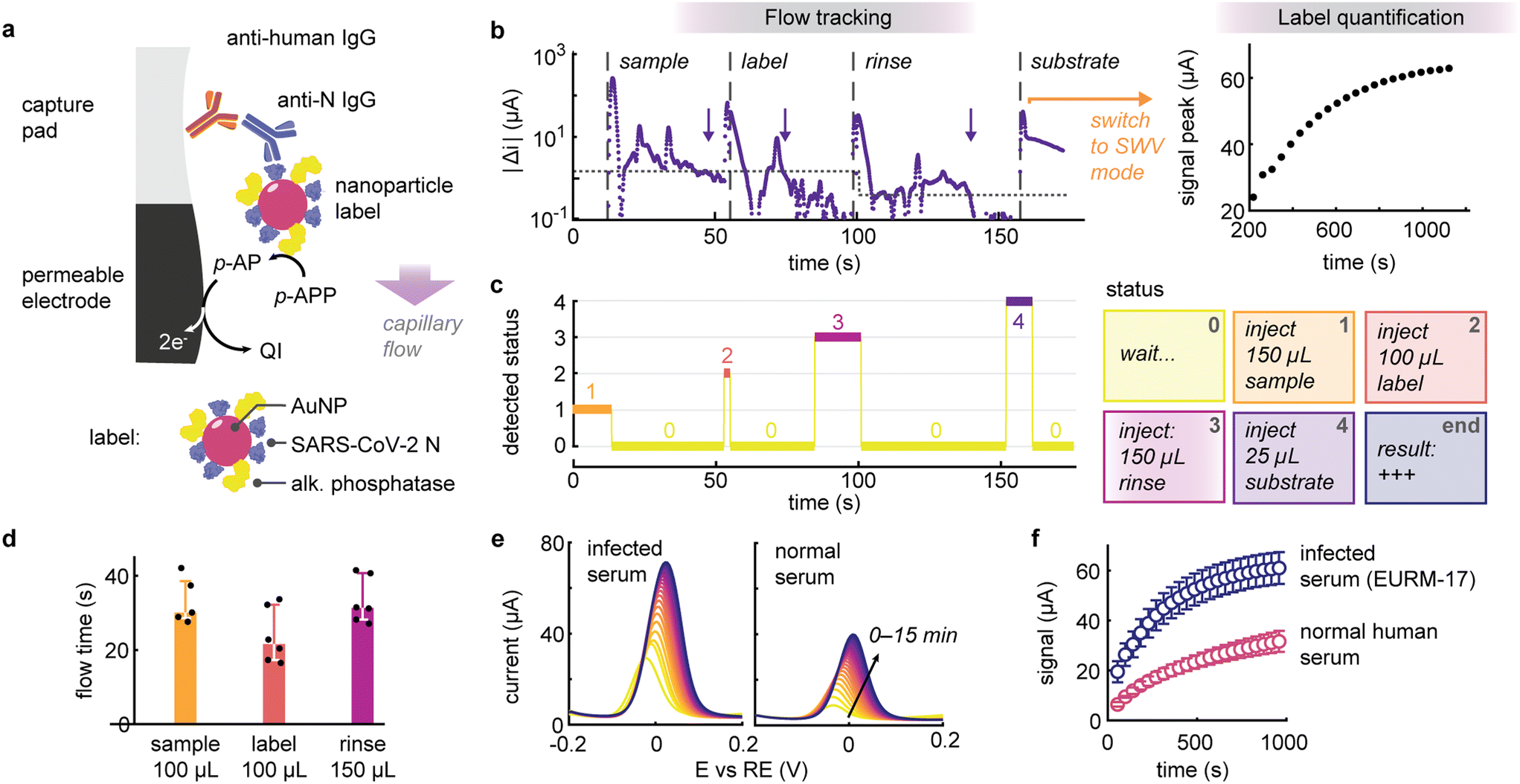 | ||
| Fig. 3 (a) Schematic of the electrochemical serological immunoassay. Anti SARS-Cov-2 nucleocapsid (anti-N) human IgGs are captured on the nitrocellulose pad and labelled with a functional gold nanoparticle. The nanoparticle label is co-functionalized with SARS-CoV-2 nucleocapsid (N) antigen and alkaline phosphatase for signal amplification. p-APP: p-aminophenyl phosphate, p-AP: p-aminophenol, QI: p-quinoneimine. (b) Representative signal acquired during the serology test. First, the flow tracking mode monitors the addition of the reagents, which includes the serum sample, labelling, rinsing and amplification solutions. The dotted line represents the threshold current variation and the arrows detected flow times. When the last reagent is detected, the reader switches to quantification mode to continuously acquire square wave voltammograms to return a result for the presence of target anti-N IgG. (c) In flow tracking mode, the reader classifies the temporal signal into different statuses corresponding to the assay protocol. (d) Detected flow times for the serum sample, labelling and rinsing reagents of various volumes (n = 5 to 6). (e) Square wave voltammogram recorded over time for serum samples with and without prior SARS-CoV-2 infection. (f) Evolution of the peak magnitude for infected or normal serum samples (n = 3). | ||
The reader operates in two distinct stages: initially, it tracks the addition of sample, labelling and rinsing buffers, then automatically switches to result quantification upon addition of the enzymatic substrate for signal amplification (Fig. 3b). Leveraging the connectivity of the compact USB potentiostat, the reader continuously processes and transmits the data (to a laptop or smartphone) to monitor and classify the various injections depending on the capacitive discharge currents (Fig. 3c). For example, upon adding a reagent, the algorithm can prompt the user to wait until the fluid has been completely absorbed and inform them about the next reagent to add. Overall, this process resulted in a hands-on time of less than three minutes. It is worth noting that due to the heterogeneity of the reagents, different thresholds before and after rinsing (1.5 and 0.4 μA s−1, respectively), were implemented to increase the robustness of the flow detection protocol. This enabled us to define expected flow times for this assay (Fig. 3d), i.e., 30 s for the 100 μL serum sample (IQR: 29–39 s, 5 out of 6 sample detected), 22 s for 100 μL labelling solution (IQR 17–32 s, n = 6) and 31 s for 150 μL of rinsing buffer (IQR: 28–41 s, n = 6). Deviation from these normal ranges could be employed to flag erroneous test operations to the user.
Upon addition of the last reagent (the substrate solution), the algorithm switches to quantification mode acquiring square wave voltammograms every 40 seconds up to 15 minutes (Fig. 3e and f). When testing a seropositive serum sample for COVID-19, we observed that the difference in signal was statistically significant on the first measurement, i.e. after approximately 1 minute (p < 0.05, n = 3), suggesting that the entire test workflow could be accomplished in less than 4 minutes. Note that there is still room to further optimize assay performance and reduce nonspecific binding. Although the algorithm currently remains passive, merely processing data and informing the user, this approach lays the groundwork for implementing an automatic stop criterion to deliver results as soon as they become significant, thereby expediting readout, particularly for strongly reactive samples.
Conclusions
In summary, we have demonstrated that capacitive discharge monitoring offers a simple yet powerful method for tracking fluids in electrochemical rapid diagnostic tests, without necessitating the use of additional probes or hardware. By continuously interrogating the electrodes with short voltage pulses, we can determine the flow times of reagents, enabling precise measurement of injected volumes or the assessment of viscosity effects with high accuracy. In this proof-of-concept study, we employed the simplest approach of comparing absolute variations in discharge currents to a predetermined fixed threshold. In its current form, the algorithm is, for example, unable to differentiate between samples with abnormally high viscosity or larger volumes if they exhibit similar flow times. Looking ahead, we believe that, when combined with more sophisticated data-driven techniques such as machine learning, we can better exploit the subtle nuances behind signal variations, leading to a more quantitative approach with direct links to physical flow parameters. This study lays the foundations for a new generation of rapid, connected tests that not only enhance the user experience but also increase user confidence in the results through the integration of real-time digital processing technology.40,41Data and materials availability
All data are available in the main text or the ESI.† The design files for digital fabrication of the devices presented in this study, as well as the code for controlling the potentiostat, are available at DOI: https://doi.org/10.17632/nd9cyw62rk.1.Author contributions
Conceptualization: L. B., D. A. R., A. J. D., C.-J.-S.; investigation: L. B.; formal analysis: L. B.; writing (original draft): L. B., D. A. R., A. J. D., C.-J. S.; writing (review and editing): L. B., D. A. R., A. J. D., C.-J. S.; visualization: L. B.; supervision: L. B., D. A. R., A. J. D., C.-J. S.; resources: D. A. R., A. J. D., C.-J. S.; funding acquisition: D. A. R., A. J. D., C.-J. S.Conflicts of interest
L. B., D. A. R., A. J. D. and C.-J. S. are listed as inventors on a patent application covering part of this work (application no. EP22186139 filed on 21 July 2022 by ETH Transfer).Acknowledgements
D. A. R. acknowledges funding from the ETH Career Seed Grant (SEED-13 21-2) and the ETH4D Research Grant.References
- R. W. Peeling, D. L. Heymann, Y.-Y. Teo and P. J. Garcia, Lancet, 2022, 399, 757–768 CrossRef CAS PubMed.
- B. O'Farrell, in Lateral Flow Immunoassay, ed. R. Wong and H. Tse, Humana Press, Totowa, NJ, 2009, pp. 1–33 Search PubMed.
- J. Budd, B. S. Miller, N. E. Weckman, D. Cherkaoui, D. Huang, A. T. Decruz, N. Fongwen, G.-R. Han, M. Broto, C. S. Estcourt, J. Gibbs, D. Pillay, P. Sonnenberg, R. Meurant, M. R. Thomas, N. Keegan, M. M. Stevens, E. Nastouli, E. J. Topol, A. M. Johnson, M. Shahmanesh, A. Ozcan, J. J. Collins, M. Fernandez Suarez, B. Rodriguez, R. W. Peeling and R. A. McKendry, Nat. Rev. Bioeng., 2023, 1, 13–31 CrossRef.
- N. I. Cheng, P. T.-N. Tabong, P. M. Netongo, B. A. Mensah, C. E. Chu, E.-B. Yaw, J. Y. Enos, K. Malm and C. S. Ahorlu, PLoS One, 2022, 17, e0275976 CrossRef CAS PubMed.
- C. P. Price, BMJ, 2001, 322, 1285–1288 CrossRef CAS PubMed.
- C. J. Atchison, M. Moshe, J. C. Brown, M. Whitaker, N. C. K. Wong, A. A. Bharath, R. A. McKendry, A. Darzi, D. Ashby, C. A. Donnelly, S. Riley, P. Elliott, W. S. Barclay, G. S. Cooke and H. Ward, Clin. Infect. Dis., 2023, 76, 658–666 CrossRef CAS PubMed.
- ECDC, Considerations on the use of self-tests for COVID-19 in the EU/EEA, European Centre for Disease Prevention and Control, 2021 Search PubMed.
- J. Cheng, G. Yang, J. Guo, S. Liu and J. Guo, Analyst, 2022, 147, 554–570 RSC.
- E. J. Maxwell, A. D. Mazzeo and G. M. Whitesides, MRS Bull., 2013, 38, 309–314 CrossRef CAS.
- A. G. Ochoa-Ruiz, G. Parra, D. López-Espinoza, P. Astudillo, D. Galyamin, N. Sabaté, J. P. Esquivel and A. A. Vallejo-Cardona, Electroanalysis, 2023, 35, e202200053 CrossRef CAS.
- P. Nandhakumar, C. Muñoz San Martín, B. Arévalo, S. Ding, M. Lunker, E. Vargas, O. Djassemi, S. Campuzano and J. Wang, ACS Sens., 2023, 8(10), 3892–3901 CrossRef CAS PubMed.
- P. Bwana, L. Ochieng' and M. Mwau, PLoS One, 2018, 13, e0202491 CrossRef PubMed.
- M. M. Serrano, D. N. Rodríguez, N. T. Palop, R. O. Arenas, M. M. Córdoba, M. D. O. Mochón and C. G. Cardona, J. Clin. Virol., 2020, 129, 104529 CrossRef CAS PubMed.
- M. C. Tollånes, A.-M. Bakken Kran, E. Abildsnes, P. A. Jenum, A. C. Breivik and S. Sandberg, Clin. Chem. Lab. Med., 2020, 58, 1595–1600 CrossRef PubMed.
- C. C. Johnson, V. Fonner, A. Sands, N. Ford, C. M. Obermeyer, S. Tsui, V. Wong and R. Baggaley, J. Int. AIDS Soc., 2017, 20, 21755 CrossRef PubMed.
- C. Figueroa, C. Johnson, N. Ford, A. Sands, S. Dalal, R. Meurant, I. Prat, K. Hatzold, W. Urassa and R. Baggaley, Lancet HIV, 2018, 5, e277–e290 CrossRef PubMed.
- J. J. Deeks, J. Dinnes, Y. Takwoingi, C. Davenport, R. Spijker, S. Taylor-Phillips, A. Adriano, S. Beese, J. Dretzke, L. Ferrante Di Ruffano, I. M. Harris, M. J. Price, S. Dittrich, D. Emperador, L. Hooft, M. M. Leeflang, A. Van Den Bruel and Cochrane COVID-19 Diagnostic Test Accuracy Group, Cochrane Database Syst. Rev., 2022,(11), CD013652 Search PubMed.
- D. Y. Lee, J. J. Ong, K. Smith, M. S. Jamil, R. McIver, R. Wigan, K. Maddaford, A. McNulty, J. M. Kaldor, C. K. Fairley, B. Bavinton, M. Chen, E. P. Chow, A. E. Grulich, M. Holt, D. P. Conway, M. Stoove, H. Wand and R. J. Guy, Med. J. Aust., 2022, 217, 149–154 CrossRef PubMed.
- C. Park, H. Ngo, L. R. Lavitt, V. Karuri, S. Bhatt, P. Lubell-Doughtie, A. H. Shankar, L. Ndwiga, V. Osoti, J. K. Wambua, P. Bejon, L. I. Ochola-Oyier, M. Chilver, N. Stocks, V. Lyon, B. R. Lutz, M. Thompson, A. Mariakakis and S. Patel, Proc. ACM Interact. Mob. Wearable Ubiquitous Technol., 2021, 5, 1–26 Search PubMed.
- Z. Nie, F. Deiss, X. Liu, O. Akbulut and G. M. Whitesides, Lab Chip, 2010, 10, 3163 RSC.
- A. Nemiroski, D. C. Christodouleas, J. W. Hennek, A. A. Kumar, E. J. Maxwell, M. T. Fernández-Abedul and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11984–11989 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, Wiley, 2022 Search PubMed.
- Y. Temiz and E. Delamarche, Sci. Rep., 2018, 8, 10603 CrossRef PubMed.
- L. Bezinge, N. Tappauf, D. A. Richards, C.-J. Shih and A. J. deMello, ACS Sens., 2023, 8, 3964–3972 CrossRef CAS PubMed.
- M. Colombo, L. Bezinge, A. R. Tapia, C.-J. Shih, A. J. DeMello and D. A. Richards, Sens. Diagn., 2023, 2, 100–110 RSC.
- C. Cavaniol, W. Cesar, S. Descroix and J.-L. Viovy, Lab Chip, 2022, 22, 3603–3617 RSC.
- M. H. Zarifi, H. Sadabadi, S. H. Hejazi, M. Daneshmand and A. Sanati-Nezhad, Sci. Rep., 2018, 8, 139 CrossRef PubMed.
- A. Alfadhel, B. Li, A. Zaher, O. Yassine and J. Kosel, Lab Chip, 2014, 14, 4362–4369 RSC.
- R. Attia, D. C. Pregibon, P. S. Doyle, J.-L. Viovy and D. Bartolo, Lab Chip, 2009, 9, 1213 RSC.
- H. Berthet, J. Jundt, J. Durivault, B. Mercier and D. Angelescu, Lab Chip, 2011, 11, 215–223 RSC.
- C. Bathany, J.-R. Han, K. Abi-Samra, S. Takayama and Y.-K. Cho, Biosens. Bioelectron., 2015, 70, 115–121 CrossRef CAS PubMed.
- L. Bezinge, J. M. Lesinski, A. Suea-Ngam, D. A. Richards, A. J. deMello and C. Shih, Adv. Mater., 2023, 35, 2302893 CrossRef CAS PubMed.
- S. Whitaker, Transp. Porous Media, 1986, 1, 3–25 CrossRef.
- L. Bezinge, C. Shih, D. A. Richards and A. J. deMello, Small, 2024, 2401148 CrossRef PubMed.
- C. Parolo, A. Sena-Torralba, J. F. Bergua, E. Calucho, C. Fuentes-Chust, L. Hu, L. Rivas, R. Álvarez-Diduk, E. P. Nguyen, S. Cinti, D. Quesada-González and A. Merkoçi, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed.
- EMD Millipore, Rapid Lateral Flow Test Strips, Considerations for Product Development, Merck Millipore, Bedford, MA, 2013, https://www.merckmillipore.com/INTERSHOP/web/WFS/Merck-RU-Site/ru_RU/-/USD/ShowDocument-Pronet?id=2 Search PubMed.
- L. Bezinge, A. Suea-Ngam, A. J. Demello and C. J. Shih, Mol. Syst. Des. Eng., 2020, 5, 49–66 RSC.
- P. Ciaurriz, F. Fernández, E. Tellechea, J. F. Moran and A. C. Asensio, Beilstein J. Nanotechnol., 2017, 8, 244–253 CrossRef CAS PubMed.
- M. S. Tabatabaei, R. Islam and M. Ahmed, Anal. Chim. Acta, 2021, 1143, 250–266 CrossRef CAS PubMed.
- N. Gous, D. I. Boeras, B. Cheng, J. Takle, B. Cunningham and R. W. Peeling, Expert Rev. Mol. Diagn., 2018, 18, 385–397 CrossRef CAS PubMed.
- K. J. Land, D. I. Boeras, X. S. Chen, A. R. Ramsay and R. W. Peeling, Nat. Microbiol., 2019, 4, 46–54 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00390j |
| This journal is © The Royal Society of Chemistry 2024 |