
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental determination of the partitioning of representative organic pollutants to the air–water interface†
Emma M.
McLay
a,
Carole
Abdel Nour
b,
Yao Yan
Huang
a,
Zoë M.
Golay
a,
Pascal
Wong-Wah-Chung
b,
Stéphanie
Rossignol
b and
D. James
Donaldson
 *ac
*ac
aDepartment of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: james.donaldson@utoronto.ca
bAix Marseille Univ, CNRS, LCE, UMR 7376, Marseille, France
cDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, Canada
First published on 8th February 2024
Abstract
Using glancing-angle laser-induced fluorescence (GALIF) spectroscopy as a probe, the partitioning of naphthalene, fluoranthene, pyrene, umbelliferone, phenol red, and bisphenol A from bulk solution to the air–water interface was examined in both pure water and aqueous solutions of 6 mM octanol. Previous studies provided similar Langmuir adsorption isotherms for anthracene and imidazole 2-carboxaldehyde. The surface partitioning behaviour of each compound in both environments was well described using a Langmuir adsorption model; partitioning coefficients were derived from the fits to such isotherms. Only the PAH molecules, naphthalene, fluoranthene and pyrene, saw an enhancement in the surface partitioning in octanol solution compared to pure water. The surface partitioning to pure water surfaces could be fairly well described using a one parameter linear free energy relationship based on either solubility or KOW.
Environmental significanceUnderstanding and predicting the partitioning of organic pollutants to the air–water interface is critical to understanding their chemical reactivity at ocean and freshwater surfaces, their presence in aerosol particles and their global distribution transport and fate. Natural aqueous surfaces are typically coated with an organic microlayer, whose presence may significantly influence this partition. In spite of this importance, few experimental constraints are available to help develop accurate predictive models. In this paper we describe experimental measurements of the partitioning equilibria of several organic species to the air–aqueous interface (both organic coated and “clean”). These measurements allow us to develop a simple linear free energy relationship to predict the partitioning behaviour. |
Introduction
The air–water interface of large bodies of water, including oceans, links the biogeochemical processes of water bodies to chemical processes in the atmosphere.1–3 For example, the composition of the air–water interface of a body of water directly affects the composition of boundary layer aerosols originating from the water, and heterogenous chemistry involving gaseous constituents in the atmosphere and species present in the aqueous phase primarily occurs with compounds present at the interface.1,4 At the ocean surface, this interface is often coined the sea surface microlayer, comprising the top ∼100 micrometers of the water.1 The interfacial zone often differs in solute composition from the bulk water beneath it, often with enhancement of the concentration of amphiphilic (i.e. fatty acids and alcohols) molecules being seen here due to their surface-active properties.1,5–7 This resulting coating on the water can play many roles, including preventing, slowing, or even enhancing transport of species between the atmosphere and the aqueous phase, reacting with gas phase species directly, and acting to enhance the surface concentrations of hydrophobic molecules present in the water.1 This last point is of most interest to this study as it means that compounds that are less soluble in water (i.e. organic hydrophobic molecules) can often more easily solubilize at the interface in a more nonpolar environment.8As mentioned above, the composition of aerosol formed by bodies of water (i.e. sea spray) is primarily influenced by the composition of the water at the interface,1,3,9 so enhancement of organics like polycyclic aromatic hydrocarbons (PAHs) and other potentially toxic chemicals at the interface will result in aerosol containing enhanced concentrations of these molecules, a concern for health.10,11 The presence of such compounds preferentially at the aqueous interface of aerosols or sea surfaces makes them easier to access by gas phase reagents and photons.12 Atmospheric oxidants like ozone, hydroxyl radicals, singlet oxygen, nitrate radicals, and peroxyl radicals are of the most importance in terms of reactivity towards organics.12 Clearly, for a good understanding of the surface reactivity, it is necessary to have a good understanding of the surface-bulk partitioning for organic molecules that may be present in bodies of water, especially if these species are of health or environmental concern and/or have heterogenous reactivity.
There are few direct experimental determinations of partitioning thermodynamics of organic pollutants to the air–water interface, and no reports that we are aware of giving a systematic study of several different compounds. Inverse chromatography (e.g. Raja et al.13) has been used to infer partitioning coefficients, but this assumes that all partitioning is to the air–water interface. A very recent study using molecular dynamics simulations has reported a systematic approach to the air–water interfacial partitioning.14 Nevertheless, it is clear that there remains a clear need for experimental measurements of the water surface partitioning thermodynamics over a range of organic compounds, especially when this surface is modified by organic coating.
Previous work from this group investigating partitioning of organic pollutants to the water surface has used the analytical method of glancing-angle laser-induced fluorescence (GALIF). By using glancing angle impingement of the laser on the sample, probing of the surface region – generally the upper 10's of nanometers – may be achieved.15 The GALIF technique used in these experiments was developed and has been used over the past two decades to investigate surface partitioning, pH and reaction kinetics at a variety of environmentally relevant surfaces.15–20 For example, Mmereki et al.16 measured the surface uptake coefficients of anthracene and pyrene from the gas phase to freshwater surfaces and to octanol- and hexanoic acid-coated surfaces. A 2–3 times enhancement in the surface uptake coefficient was seen when octanol was present at the surface, as compared to when the surface was freshwater.16 This same work also studied the partitioning of anthracene from bulk water to the freshwater surface, as is done here with other compounds. Other work looked at the partitioning of imidazole-2-carboxaldehyde to freshwater and octanol-coated surfaces.21 Other work in this area, using other methods, has indicated a significant enrichment of organics, like PAHs and PCBs, in the sea-surface microlayer.22,23
In this study, we report quantitative experimental determinations of the surface-bulk partitioning of several representative organic compounds and draw connections between these and published physio-chemical properties. “Pure” freshwater surfaces as well as aqueous surfaces with an octanol coating, used as a proxy for the naturally occurring surface microlayer1 were examined. An octanol concentration of 6 mM was chosen, as this is about twice that corresponding to an octanol monolayer on the surface.12
Six organic molecules were chosen to be investigated: naphthalene, fluoranthene, pyrene, umbelliferone (7-hydroxycoumarin), phenol red (phenolsulfonphthalein), and bisphenol A (BPA). A seventh compound (1-methylnaphthalene) gave signal levels too low to use here. Included in the analysis are two additional molecules previously investigated in our group: anthracene and imidazole-2-carboxaldehyde (IC).16,21 The structures of all species studied here are shown in Fig. 1. All eight compounds are fluorescent, which was necessary for detection using GALIF. Fluoranthene, pyrene, naphthalene and anthracene are polycyclic aromatic hydrocarbons (PAHs), which are carcinogenic and mutagenic compounds often emitted from burning fossil fuels like coal.10 Umbelliferone is a pharmacological agent, widely used in sunscreens.24 IC, an N-derivatized imidazole, can photosensitize reactions responsible for secondary organic aerosol (SOA) formation.21 Phenol red and bisphenol A both contain aromatic groups, but are not as highly conjugated as the first four listed. Phenol red is a pH indicator, used extensively to test cell and tissue media,25 that exists in neutral or zwitterionic forms. Bisphenol A is an endocrine disruptor often used in polycarbonate and epoxy plastic manufacturing that can be harmful to human health and aquatic environments especially.26 It is reasonable to assume that these and structurally similar compounds can end up in large bodies of water, like oceans, and thus be present at the air–water interface.
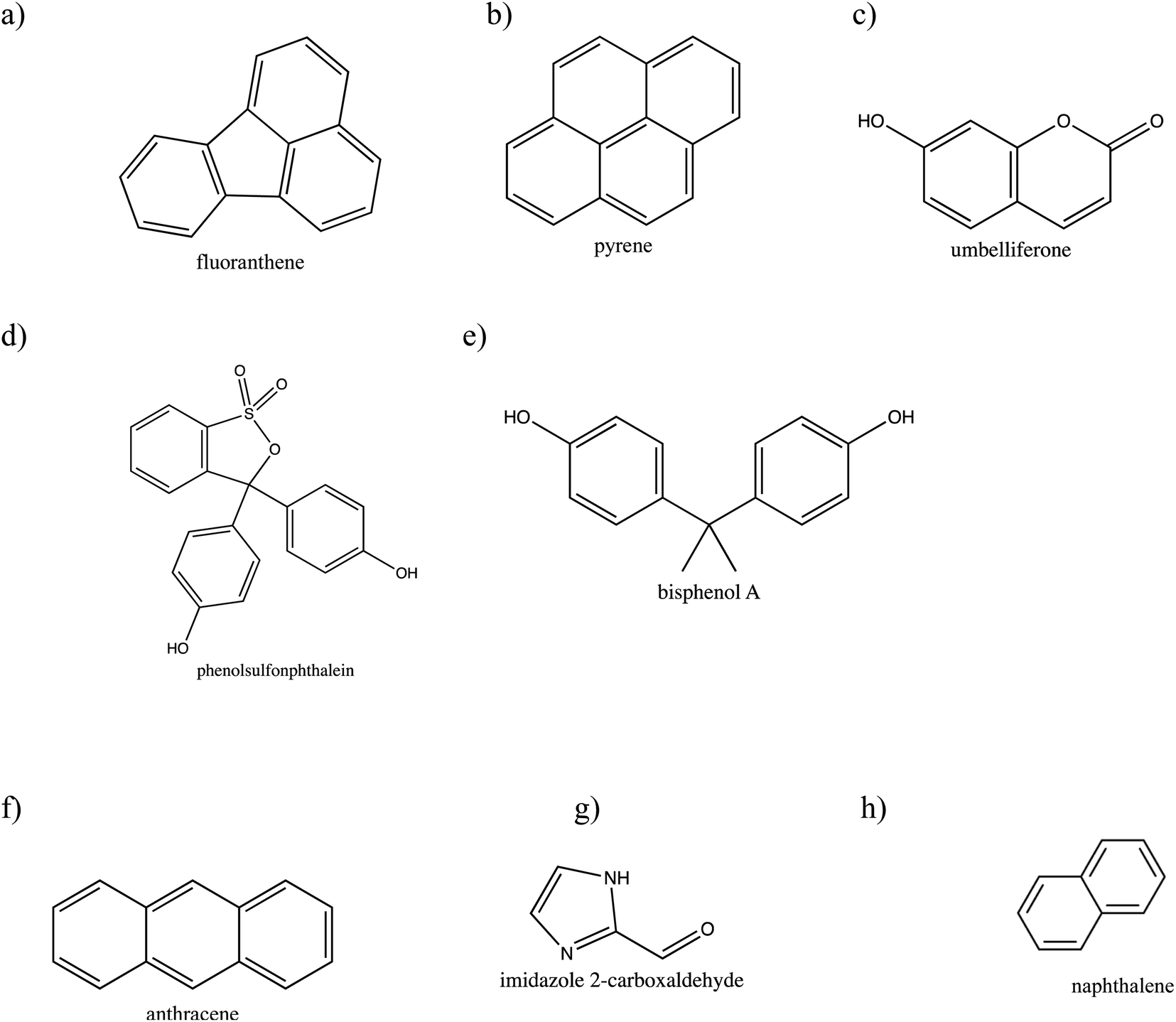 | ||
| Fig. 1 Chemical structures of (a) fluoranthene, (b) pyrene, (c) umbelliferone, (d) phenol red (phenolsulfonphthalein), (e) bisphenol A, (f) anthracene, (g) imidazole 2-carboxaldehyde (IC) (h) naphthalene. | ||
These compounds span a large range of solubilities and octanol–water partitioning coefficients, which will allow us to test how these properties may affect or reflect the partitioning out of solution to the air–water interface. Aqueous solubility describes a compound's propensity to be solvated in water, rather than remain as a single-phase solid – it thus reflects the free energy benefit to remaining in solution. The octanol–water partitioning coefficient is related to the difference in solvation free energy between the polar, high dielectric solvent water, and the much less polar, lower dielectric solvent octanol. Since the air–water interface is a less polar environment than bulk water,7 the KOW may describe partitioning to the air–water interface. In this sense, we will attempt to find single parameter predictors of surface-bulk partitioning coefficients, similar to the use of Linear Free Energy Relationships (LFER),27 and to what was done in Staikova et al. using a somewhat different approach.28 The values for these physico-chemical parameters can be found in Table S1 of the ESI.†
Methods
Instrument setup
The surface partitioning experiments were carried out using glancing-angle laser-induced fluorescence (GALIF) using the setup shown in Fig. 2. It consists of an unfocussed tunable Nd:YAG laser-pumped optical parametric oscillator (OPO) system with a 3 ns pulse operating at a pulse frequency of 20 Hz. Typical pulse energies were ∼0.5 mJ per pulse at 228 nm and ∼1.2 mJ per pulse at 337 nm. The laser output was directed towards a circular sample dish, 6 cm in diameter and about 1 cm deep, using several mirrors. The central part of the beam was selected using irises and directed to the sample dish using an adjustable mirror, so as to impinge the surface of the liquid sample at an angle of around 87° from the surface normal. An optical light guide was positioned perpendicular to the sample surface, exactly at the point where the laser beam hits the surface. It was placed around 5 mm above the surface; fluorescence collected by the guide was sent to a ¼ metre focal length monochromator. The signal passed by the monochromator at a selected wavelength was detected by a photomultiplier tube and visualized with a digital oscilloscope that averaged over 16 laser pulses. The oscilloscope output was sampled and collected by a computer for subsequent analysis. Following a set number of readings, the monochromator wavelength could be changed, and the procedure repeated (for measuring a fluorescence spectrum), or the sample could be changed to measure an adsorption isotherm. A few experiments were carried out in the bulk. In these, the sample dish was replaced with a quartz cuvette, and the light guide was positioned at the side of the cuvette, perpendicular to the laser beam passing through the cuvette. Signal collection and analysis was the same as for the glancing angle experiments.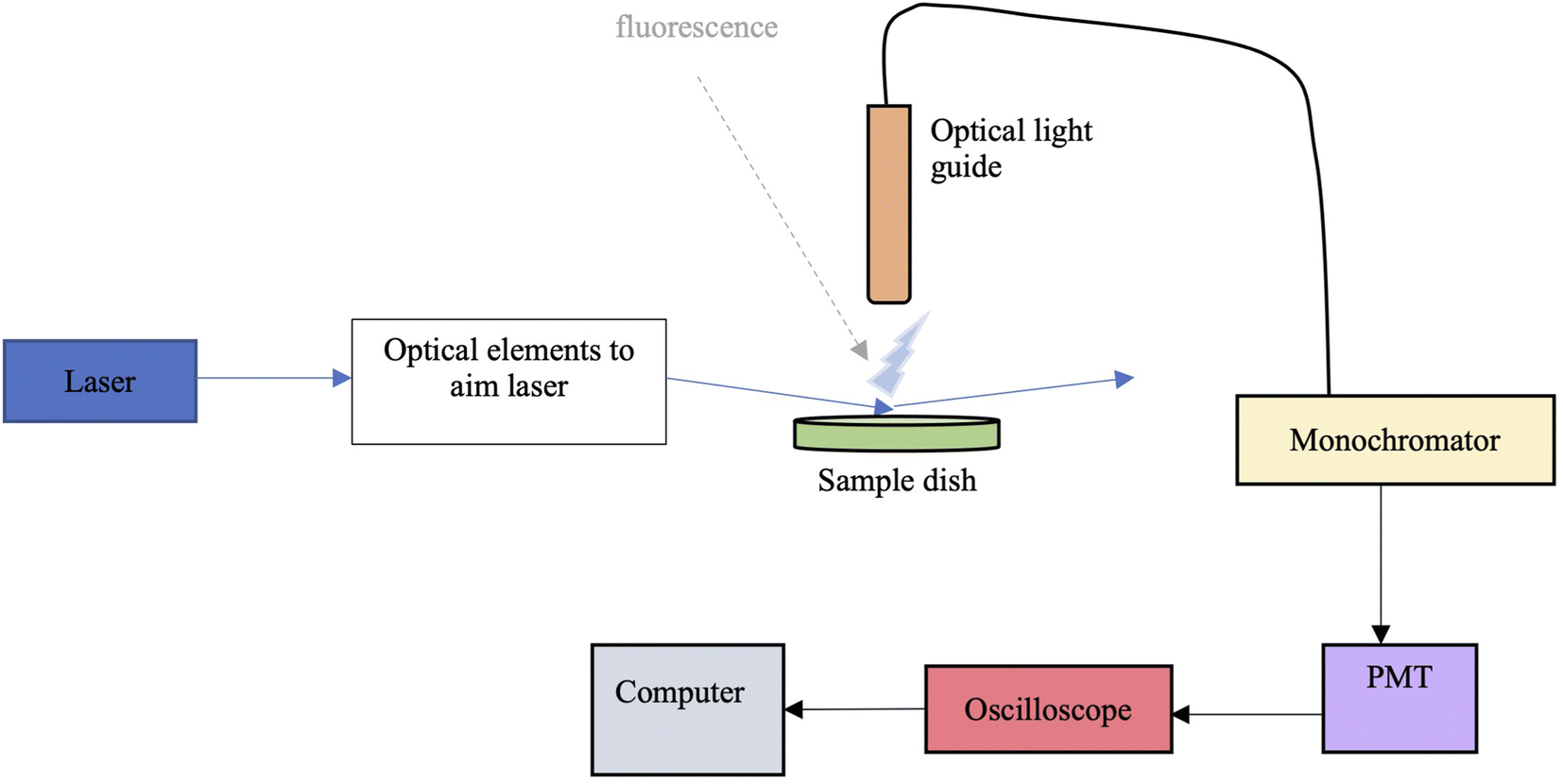 | ||
| Fig. 2 Box diagram of the glancing-angle laser-induced fluorescence instrument setup. | ||
Fluorescence experiment procedures
Fluorescence experiments using samples in pure water were carried out in the bulk and using GALIF. Experiments using samples in octanol solution were only carried out using GALIF. All experiments, except for those with BPA and naphthalene, were carried out using an excitation wavelength of 337 nm, since this was a reasonable excitation wavelength for all these compounds. For BPA and naphthalene experiments, the laser was tuned to 228 nm and 313 nm respectively, near the absorption maximum for these molecules. The monochromator was set at the maximum emission wavelength of whichever compound was being measured, as determined by scanning the fluorescence spectrum of each. The emission wavelengths used here of the compounds at their respective excitation wavelengths can be found in Table S2 of the ESI.† Bulk fluorescence experiments were carried out first to determine the linear range of the bulk fluorescence, where self-absorption and other non-linearities did not affect the observed fluorescence response. For some of the phenol red experiments, 3 maximum emission wavelengths were used, 1 nm apart, to average over the peak maximum. No difference in the results was seen using this procedure vs. taking a single point. There was never any evidence (i.e. loss of signal with time or change in the spectrum) of photodegradation of the probe molecules by the laser in these experiments, either on the surface or in the bulk.For the bulk experiments, a 1 cm path length quartz cuvette was filled with 3 mL of solution at the highest desired concentration for the experiment. Five measurements of the fluorescence intensity were taken, for averaging, before the concentration was changed via dilution with 18 MΩ water. For some of the aqueous phenol red experiments, 2 measurements at each of the 3 emission wavelengths around the emission maximum were taken instead for a total of 6 measurements to be averaged. Dilutions were done by carefully removing the desired amount of solution with an autopipette and replacing it with 18 MΩ water of exactly the same volume. Concentrations of each solution were back calculated using the initial concentration of the first solution and the amount of solution removed each time. Different pipette tips were used for removing solution and adding water to prevent any carry over of solution. In total, 5–15 dilutions were carried out with measurements being taken for each concentration. A fluorescence measurement of pure 18 MΩ water was obtained at the end after washing the cuvette. The determined linear concentration ranges from the bulk experiments can be found in Table S3 of the ESI,† and the plots for each can be seen in Fig. S1 of the ESI.† These concentration ranges were then the starting point concentration ranges used for the GALIF experiments, which were carried out afterwards.
For the glancing angle experiments, the sample dish was filled with 32.0 mL of solution, to fill the dish flush with its top. The same successive dilution process was used for these experiments, with 5–15 dilutions done for each experiment run. For the purely freshwater experiments, 18 MΩ water was used to dilute, and for the octanol experiments, a 6 mM octanol solution was used to ensure a constant concentration of octanol throughout the experiment. Fluorescence measurements for pure 18 MΩ water or 6 mM octanol solution were obtained at the end after washing the dish.
Except for BPA, one freshwater bulk experiment, at least three freshwater GALIF experiments, and at least three GALIF experiments with 6 mM octanol were successfully completed for each compound. Experiments using BPA were only done in freshwater. In some cases, more than three experiments were necessary to obtain a reasonable set of results.
Solution preparation
For fluoranthene (Sigma Aldrich, 99%), pyrene (Alfa Aesar, 98%), naphthalene (Fisher Scientific 99%) and BPA (TCI, 99.0%), saturated aqueous solutions were made by adding at least twice the required amount of solid to achieve saturation to 125 mL or 250 mL of 18 MΩ water. Fluoranthene, pyrene, and naphthalene solutions were stirred for at least 12 hours with at least 2 hours of gentle heating before being fully cooled and filtered using 0.22 μm PTFE filters. BPA solutions were stirred and gently heated for 1.5 hours before being cooled and immediately filtered using 0.22 μm PTFE filters. Only the required amount of BPA solution was filtered each time, allowing the rest to remain heating and stirring until more was needed. All solutions were covered in aluminum foil while heating and stirring to minimize contact with ambient light. Fluoranthene, pyrene, and naphthalene stock solutions were assumed to be 100% saturated (1.29 μM for fluoranthene, 0.67 μM for pyrene, and 249 μM for naphthalene based on experimental solubilities of the compounds as in Table S1 of the ESI†). BPA stock solution was assumed to be 2.95 mM as determined by UV-Vis absorbance measurements (shown in Fig. S2 of the ESI†) taken immediately after cooling and filtering of the solution. This value exceeds somewhat the solubility of BPA listed in Table S1,† likely due to the timing of the heating and filtering process immediately prior to making the measurement. The concentration was maximized this way to make up for the low quantum yield of BPA and obtain a sizeable signal.29 These stock solutions were used to make up solutions of various concentrations depending on what was required for each experiment.For umbelliferone (Alfa Aesar, 98%) and phenol red (VWR Life Science, purity unknown), 10.0 μM and 50.0 μM stock solutions, respectively were made using 18 MΩ water. These stock solutions were then used to make solutions of various concentrations for the experiments. It is important to note that the phenol red was in its protonated form for these experiments as indicated by its yellow colour.
When making solutions containing octanol, the stock solutions were used along with pure octanol (Sigma-Aldrich, ≥99%) to make up new solutions with the desired organic compound concentration and 6 mM of octanol. All solutions were stored for up to one week in glass containers wrapped in foil, except for BPA solutions which were only used the day of their making for reasons elaborated on above.
Data analysis
Average fluorescence intensity values measured using the GALIF setup were plotted against bulk concentration for each experiment. In some cases, the measurement for pure water or pure 6 mM octanol was not close to zero; this baseline value was subtracted from all of the measurements for that particular experiment. For each experiment, these plots were fit using a hyperbolic equation of the form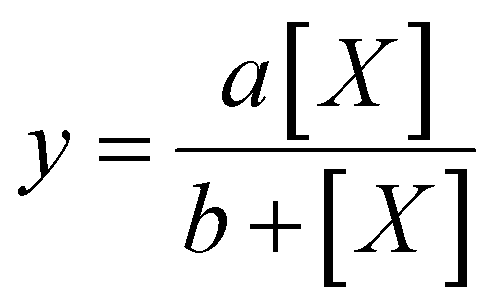 | (1) |
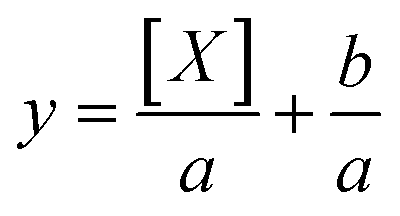 | (2) |
Hyperbolic fits were also performed on datasets constructed by normalizing each isotherm such that all isotherms for the three (or more) experiments done using each compound in each type of solution spanned the same intensity range, then combining these into a single set. This process gave rise to a common (and arbitrary, as it depended on the normalization choice) a value for the combined set; the resulting b values, along with their errors, and the r2 value for the fit were recorded as seen in Tables S4 and S5 of the ESI.†
Results
For each of fluoranthene, pyrene, umbelliferone, BPA, phenol red, and naphthalene Langmuir adsorption isotherms were obtained according to the procedures outlined above. As well, data from our previous reports16,21 of anthracene and IC partitioning were fit as described above to obtain their respective a and b values. Fitting the combined data produced results with generally better correlation coefficients and similar fit parameters to fitting isotherms individually. The correlation coefficients and fit parameters can be seen in Tables S4 and S5 of the ESI.† The b values obtained from this method of fitting were those used to calculate KLang as the inverse of b. Table 1 displays the values of log(KLang), for aqueous and octanol-coated surfaces. A common value of the uncertainty on the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KLang values was taken that spanned the maximum of those generated by the fit to the combined data. Fig. 3 depicts the fluoranthene isotherms derived from combined data for the aqueous and 6 mM-octanol coatings. Similar plots for the other compounds are provided in Fig. S3 of the ESI.†Table 1 also includes values for anthracene as determined from results from Mmereki et al.16 and imidazole-2-carboxaldehyde as determined from results from Fu et al.21 No anthracene or bisphenol A data from experiments using octanol-coated surfaces was available and no data using 6 mM octanol-coated surfaces was available for imidazole 2-carboxaldehyde. Fig. S4 in the ESI† displays the fitted data from these earlier reports.
KLang values was taken that spanned the maximum of those generated by the fit to the combined data. Fig. 3 depicts the fluoranthene isotherms derived from combined data for the aqueous and 6 mM-octanol coatings. Similar plots for the other compounds are provided in Fig. S3 of the ESI.†Table 1 also includes values for anthracene as determined from results from Mmereki et al.16 and imidazole-2-carboxaldehyde as determined from results from Fu et al.21 No anthracene or bisphenol A data from experiments using octanol-coated surfaces was available and no data using 6 mM octanol-coated surfaces was available for imidazole 2-carboxaldehyde. Fig. S4 in the ESI† displays the fitted data from these earlier reports.
| A | B | C | D | E | F | G | H | |
|---|---|---|---|---|---|---|---|---|
| Pure water log(KLang) | 4.28 ± 0.05 | 7.71 ± 0.20 | 7.59 ± 0.10 | 8.54 ± 0.15 | 5.77 ± 0.05 | 4.31 ± 0.20 | 5.14 ± 0.15 | 4.52 ± 0.15 |
| Octanol-coated log(KLang) | 5.55 ± 0.15 | 8.95 ± 0.15 | N/A | 10.85 ± 0.20 | 5.47 ± 0.15 | 4.16 ± 0.05 | N/A | 4.51 ± 0.15 |
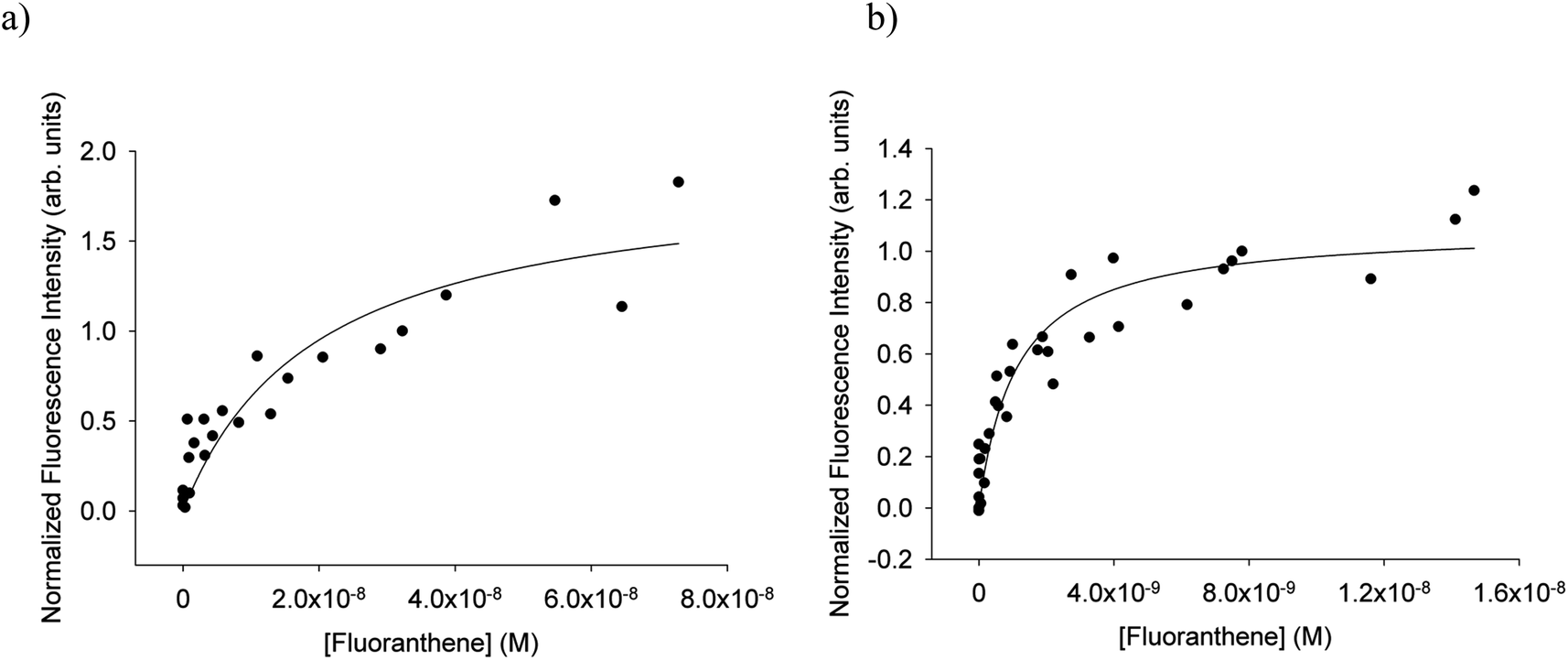 | ||
| Fig. 3 Normalized isotherms for (a) fluoranthene in freshwater and (b) fluoranthene in 6 mM octanol from all successful experiments. The fits shown are those for all of the normalized isotherms combined. The correlation coefficients and b values are: (a) b = (1.97 ± 0.70) × 10−8, r2 = 0.83; (b) b = (1.12 ± 0.23) × 10−9, r2 = 0.90. | ||
Discussion
Two features of the results presented in Table 1 are immediately apparent: first, all of the compounds studied here show significant partitioning to the air–aqueous interface spanning 4–5 orders of magnitude. The PAH compounds generally show the strongest partitioning behaviour, with the others displaying significantly less surface activity at both the coated and uncoated surface. Comparing the octanol-coated and pure water result for each compound reveals the second interesting feature: the PAH compounds show markedly enhanced partitioning to an octanol coated interface, while the others show little or no difference between the coated and uncoated cases. These two features will be discussed separately below.Partitioning to the water–air interface
Chemical intuition based on the structures illustrated in Fig. 1, suggests a relationship may exist between the observed partitioning strength and the compounds' solubilities, and/or octanol–water partitioning coefficients (KOWs). These physico-chemical parameters relate to the propensity for a compound to be solvated in aqueous solution compared to remaining in a condensed state (solubility) or partitioning to the much less polar organic phase (KOW). We test this idea by constructing LFERs using solubility and KOW values given in Table S1 of the ESI.†In order to make the LFERs meaningful outside our specific surface fluorescence measurements, we must first transform the KLang values presented in Table 1 into “true” thermodynamic equilibrium constants. As mentioned above, the b constant in eqn (1) is given by the ratio of rate constants for surface desorption to adsorption, b = kdes/kads.30 This represents the inverse of a phenomenological “kinetic equilibrium constant” (referred to here as KLang), allowing comparison with other, similarly-determined values, as we have done above. However, it does not directly provide a true thermodynamic equilibrium constant, Kads, from which thermochemical quantities such as 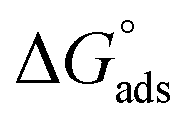 may be derived.
may be derived.
In several earlier papers we have discussed this issue and demonstrated ways in which this connection can be made.31–33 To do this, we start with the thermodynamic definition of the equilibrium constant:
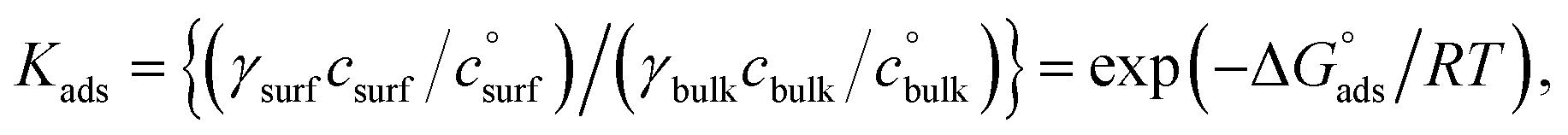 | (3) |
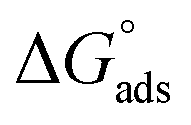 relates the free energy change between the two standard states represented by
relates the free energy change between the two standard states represented by 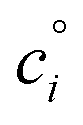 . In order to use the Langmuir fit parameters to extract thermodynamic parameters, these standard states (often implicit!) must be well defined.31–33 In the present case, we choose the bulk and surface standard states to be an ideal solution of 1 mol L−1 for the bulk phase and an ideal 1 mol L−1 solution in a 5 Å thick surface layer, corresponding to a surface density of 3.01 × 1013 molecules cm−2 for the surface.31 The relationship between the Langmuir-derived and true thermodynamic equilibrium constants is then:21
. In order to use the Langmuir fit parameters to extract thermodynamic parameters, these standard states (often implicit!) must be well defined.31–33 In the present case, we choose the bulk and surface standard states to be an ideal solution of 1 mol L−1 for the bulk phase and an ideal 1 mol L−1 solution in a 5 Å thick surface layer, corresponding to a surface density of 3.01 × 1013 molecules cm−2 for the surface.31 The relationship between the Langmuir-derived and true thermodynamic equilibrium constants is then:21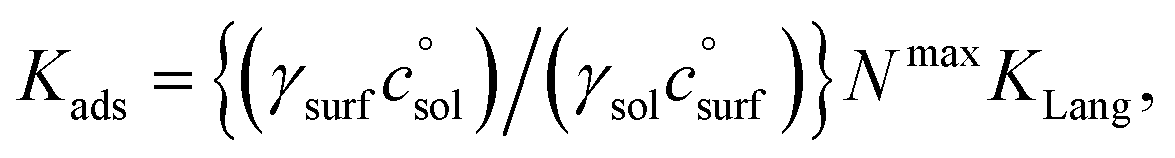 | (4) |
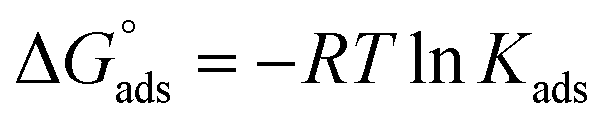 may be obtained, if desired for use in thermochemical cycles. We will assume that the concentration dependence of the activity coefficients is the same at the interface and in the bulk, meaning the ratio of activity coefficients is unity. The values of Nmax for the compounds of interest here are not experimentally available. In ref. 5 we estimated it to be 1 × 1013 molecules cm−2 for anthracene; as an estimate here, we take a range between 1 × 1013 and 1 × 1014 molecules cm−2 to be appropriate for all 7 species studied here.
may be obtained, if desired for use in thermochemical cycles. We will assume that the concentration dependence of the activity coefficients is the same at the interface and in the bulk, meaning the ratio of activity coefficients is unity. The values of Nmax for the compounds of interest here are not experimentally available. In ref. 5 we estimated it to be 1 × 1013 molecules cm−2 for anthracene; as an estimate here, we take a range between 1 × 1013 and 1 × 1014 molecules cm−2 to be appropriate for all 7 species studied here.
Fig. 4a shows a plot of log(Kads) for the freshwater surface versus the logarithm of solubility for all of the compounds. For each compound, we plot the range of Kads values (with uncertainties) corresponding to the range of Nmax values chosen. With the solubility (S) expressed in μmole L−1, a negative relationship is seen with a correlation coefficient of 0.72:
| logKads = (7.34 ± 0.47) − (0.60 ± 0.15)logS | (5) |
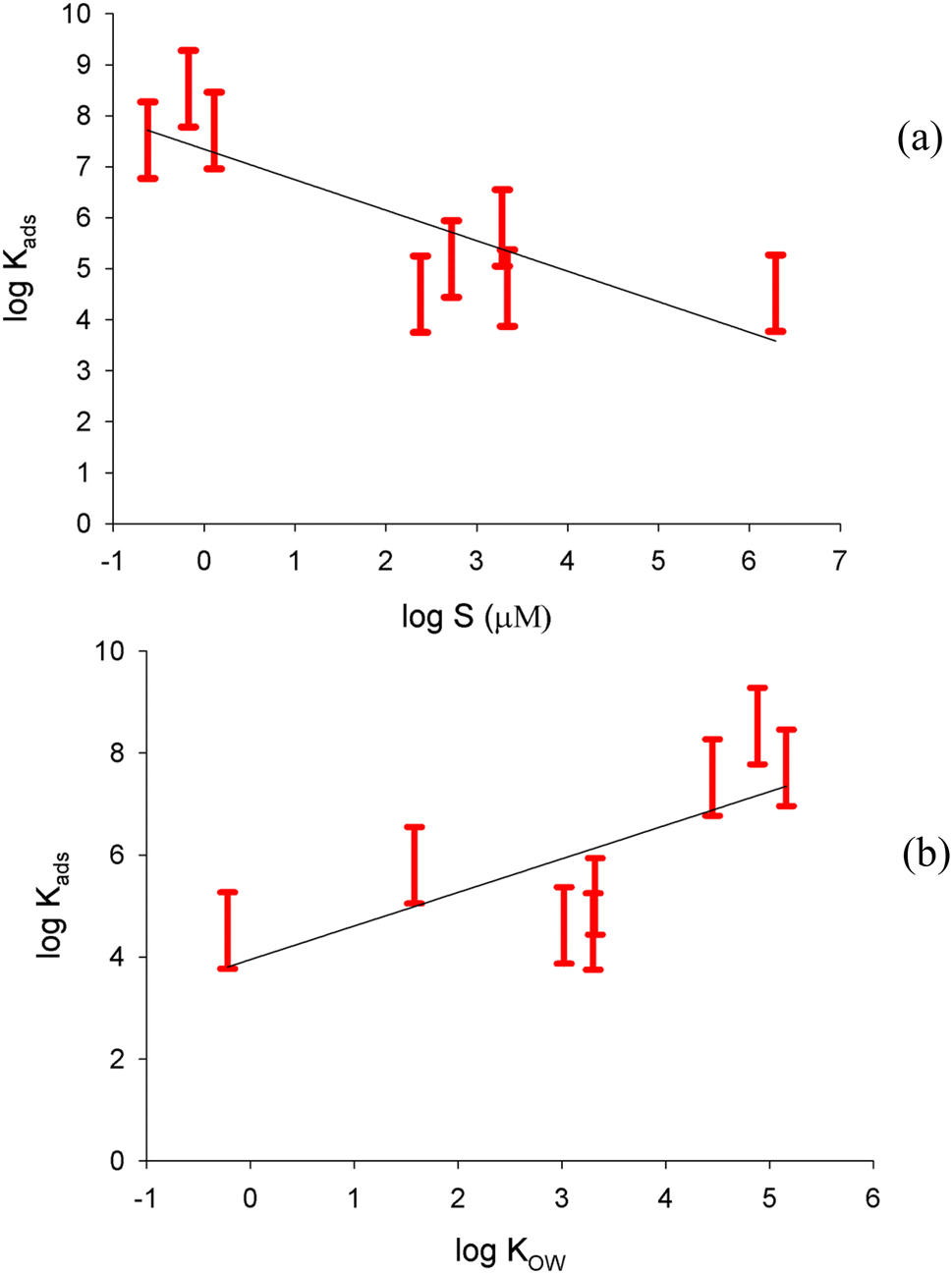 | ||
| Fig. 4 Plots of logKads as determined here against (a) logS, with the solubility expressed in μmol L−1; (b) logKOW. The red bars indicate the range of logKads values calculated with different assumptions of the maximum surface coverage, including experimental uncertainties. The black lines show a linear regression, representing an LFER. | ||
This confirms the hypothesis that the less soluble compounds have more preference for the air–water interface than the more soluble compounds, which show more preference to remain dissolved in the bulk solution. Plotting logKads against logKOW for the freshwater surface, as displayed in Fig. 4b, shows a positive relationship with correlation coefficient of 0.53:
| logKads = (3.9 ± 0.9) + (0.66 ± 0.25)logKOW | (6) |
This somewhat less robust LFER suggests that compounds that are more soluble in the less polar octanol environment are also more likely to partition to the air–water interface, which is less polar than the bulk.7
Being able to predict the partitioning of a compound to the surface is important for estimating surface concentrations of organics in bodies of water or aerosol droplets; these are key to understanding processes involving the air–water interface.1 The LFERs presented in eqn (3) and (4) above provide confirmation that a simple, general expression can reasonably describe partitioning from solution to the freshwater surface for a variety of organic compounds. Some seven orders of magnitude in solubility and five orders of magnitude in KOW are captured here, with a somewhat better fit using solubility as the descriptor. The compounds represented in this study show a wide range of chemical structures as well, from aromatic hydrocarbons to N-containing aldehydes and multiply hydroxylated species. The good predictive ability over this wide range in chemical structures and physico-chemical properties lends some confidence to the LFERs derived here.
Other approaches to predicting the partitioning of organics to the air–water interface have used polyparameter free energy relationships,18 the use of commercial software packages,25 and molecular dynamics simulations.14 The report by Lemay et al.14 gives three compounds which are also determined here (pyrene, fluoranthene and bisphenol A); that of Goss27 has only one (naphthalene). The different choice of standard states in these earlier works makes a direct comparison with our predictions and measurements not straightforward. However, the simulations of Lemay et al.14 predict that pyrene and bisphenol A should have the same air–water partitioning constants, both being about three times smaller than that of fluoranthene. This feature is not what is observed experimentally in the present work.
Although single-parameter relationships such as eqn (3) and (4) are often quite crude predictors of properties, in some cases they perform well, without the introduction of multiple fitting parameters. For example, Staikova et al.28 derived a very successful quantitative single-parameter relationship relating the vapour pressure and KOA of 159 halogenated organics to Density Functional Theory-calculated molecular polarizabilities. Of course, such single parameter relationships may not account for all potential interactions of a chemical that affect the property of interest.27 However, in the absence of a comprehensive experimental database, they can act as a starting point to estimate the surface partitioning behaviour of organic compounds in aqueous solution.
Partitioning to the octanol-coated surface
The smaller dataset available for the octanol-coated surface makes a detailed analysis challenging. From Table 1, it is certainly evident that there are two distinct classes of compound represented: fluoranthene, naphthalene, and pyrene see increased partitioning to the surface when octanol is present there, and the other compounds show little or no such increase. Dividing the KLang for the octanol-coated surface by the KLang for the freshwater surface, shows an enhancement in surface partitioning of around 20 times for fluoranthene and naphthalene, and 200 times for pyrene. This enhancement indicates that the PAH compounds were more easily solubilized in octanol, hence their preference for partitioning there. Given the nonpolar nature of these PAHs, and the higher KOW values associated with them, this makes sense, and it is highly likely that anthracene, another very nonpolar PAH would behave similarly in this regard. Umbelliferone, phenol red, BPA, and IC did not display this enhancement, in spite of their KOW values being above unity (see Table S1†). As shown in Fig. 1, each of these molecules contain additional functional groups (i.e. ketones, alcohols, amines) not present in the PAHs, increasing their solubility in water relative to octanol.Other than the present report, there are few previous measurements of this type in the literature. Vaida and co-workers34 studied partitioning of a small series of aromatic acids to the air–water and the air–phospholipid–water interfaces using a combination of surface tension measurements and molecular dynamics simulations. Counterintuitively, the hydrophobic behavior seemed to play no role in the observed results, with all partitioning apparently governed by interactions between the aromatic probe molecules. No large enhancement of partitioning was seen for the phospholipid-coated surface over the bare water.34 Earlier, Mmereki et al.16 also determined surface uptake coefficients of pyrene to the air–water interface from gas phase, using the same GALIF technique used in these experiments. Fig. 5 of that work shows that surface uptake coefficients of anthracene and pyrene were 2–3 times higher for 6 mM octanol-coated surfaces than for freshwater surfaces.16 Although quantitatively quite different from the enhancement factors of 20–200 for pyrene, naphthalene and fluoranthene seen here, partitioning from the gas phase to the water surface may well be weaker than from solution, especially for relatively high vapor pressure PAHs like pyrene and anthracene. It may also be that the thickness and identity of the organic surface coating plays an important role in the surface partitioning enhancement of molecules such as those studied to date. Further research may shine some light on this issue.
Conclusions
In this study, the partitioning of six organic compounds (fluoranthene, naphthalene, pyrene, umbelliferone, phenol red, and bisphenol A) in aqueous and octanol environments, to the air–water interface from bulk solution was investigated using glancing-angle laser-induced fluorescence (GALIF). The partitioning behaviour of all of the compounds in both environments showed Langmuir adsorption isotherms. Only the three PAHs, fluoranthene, naphthalene, and pyrene, saw enhanced partitioning to the surface in octanol solution compared to aqueous solution. Including Langmuir adsorption isotherms for anthracene and imidazole 2-carboxaldehyde determined previously, LFERs were derived relating the surface partition equilibrium constant to solubility and KOW. This work may be helpful to better understand and predict the composition of the air–water interface of aqueous aerosols and bodies of water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERCC, to whom we are grateful. Funding for the laser system was partially supplied by the University of Toronto. Another part of this research was funded by the Agence Nationale de la Recherche ANR through the project PhotoSeaSMiL.References
- D. J. Donaldson and C. George, Sea-Surface Chemistry and Its Impact on the Marine Boundary Layer, Environ. Sci. Technol., 2012, 46(19), 10385–10389, DOI:10.1021/es301651m.
- A. Engel, H. W. Bange, M. Cunliffe, S. M. Burrows, G. Friedrichs, L. Galgani, H. Herrmann, N. Hertkorn, M. Johnson, P. S. Liss, P. K. Quinn, M. Schartau, A. Soloviev, C. Stolle, R. C. Upstill-Goddard, M. van Pinxteren and B. Zäncker, The Ocean’s Vital Skin: Toward an Integrated Understanding of the Sea Surface Microlayer, Frontiers in Marine Science, 2017, 4, 165, DOI:10.3389/fmars.2017.00165.
- M. van Pinxteren, S. Zeppenfeld, K. W. Fomba, N. Triesch, S. Frka and H. Herrmann, Amino Acids, Carbohydrates, and Lipids in the Tropical Oligotrophic Atlantic Ocean: Sea-to-Air Transfer and Atmospheric in Situ Formation, Atmos. Chem. Phys., 2023, 23(11), 6571–6590, DOI:10.5194/acp-23-6571-2023.
- L. Tinel, T. J. Adams, L. D. J. Hollis, A. J. M. Bridger, R. J. Chance, M. W. Ward, S. M. Ball and L. J. Carpenter, Influence of the Sea Surface Microlayer on Oceanic Iodine Emissions, Environ. Sci. Technol., 2020, 54(20), 13228–13237, DOI:10.1021/acs.est.0c02736.
- N. Triesch, M. van Pinxteren, S. Frka, C. Stolle, T. Spranger, E. H. Hoffmann, X. Gong, H. Wex, D. Schulz-Bull, B. Gašparović and H. Herrmann, Concerted Measurements of Lipids in Seawater and on Submicrometer Aerosol Particles at the Cabo Verde Islands: Biogenic Sources, Selective Transfer and High Enrichments, Atmos. Chem. Phys., 2021, 21(6), 4267–4283, DOI:10.5194/acp-21-4267-2021.
- T. Barthelmeß, F. Schütte and A. Engel, Variability of the Sea Surface Microlayer across a Filament’s Edge and Potential Influences on Gas Exchange, Frontiers in Marine Science, 2021, 8, 718384, DOI:10.3389/fmars.2021.718384.
- M. Cunliffe, A. Engel, S. Frka, B. Gašparović, C. Guitart, J. C. Murrell, M. Salter, C. Stolle, R. Upstill-Goddard and O. Wurl, Sea Surface Microlayers: A Unified Physicochemical and Biological Perspective of the Air–Ocean Interface, Prog. Oceanogr., 2013, 109, 104–116, DOI:10.1016/j.pocean.2012.08.004.
- D. J. Donaldson and V. Vaida, The Influence of Organic Films at the Air–Aqueous Boundary on Atmospheric Processes, Chem. Rev., 2006, 106(4), 1445–1461, DOI:10.1021/cr040367c.
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 2015, 115(10), 4383–4399, DOI:10.1021/cr500713g.
- J. Kongpran, W. Kliengchuay, S. Niampradit, N. Sahanavin, W. Siriratruengsuk and K. Tantrakarnapa, The Health Risks of Airborne Polycyclic Aromatic Hydrocarbons (PAHs): Upper North Thailand, Geohealth, 2021, 5(4), e2020GH000352, DOI:10.1029/2020GH000352.
- G. Casas, A. Martínez-Varela, J. L. Roscales, M. Vila-Costa, J. Dachs and B. Jiménez, Enrichment of Perfluoroalkyl Substances in the Sea-Surface Microlayer and Sea-Spray Aerosols in the Southern Ocean, Environ. Pollut., 2020, 267, 115512, DOI:10.1016/j.envpol.2020.115512.
- D. J. Donaldson and K. T. Valsaraj, Adsorption and Reaction of Trace Gas-Phase Organic Compounds on Atmospheric Water Film Surfaces: A Critical Review, Environ. Sci. Technol., 2010, 44(3), 865–873, DOI:10.1021/es902720s.
- S. Raja, F. S. Yaccone, R. Ravikrishna and K. T. Valsaraj, Thermodynamic Parameters for the Adsorption of Aromatic Hydrocarbon Vapors at the Gas–Water Interface, J. Chem. Eng. Data, 2002, 47(5), 1213–1219, DOI:10.1021/je025520j.
- A. C. Lemay, E. J. Sontarp, D. Martinez, P. Maruri, R. Mohammed, R. Neapole, M. Wiese, J. A. R. Willemsen and I. C. Bourg, Molecular Dynamics Simulation Prediction of the Partitioning Constants (KH, Kiw, Kia) of 82 Legacy and Emerging Organic Contaminants at the Water–Air Interface, Environ. Sci. Technol., 2023, 57(15), 6296–6308, DOI:10.1021/acs.est.3c00267.
- B. T. Mmereki and D. J. Donaldson, Laser Induced Fluorescence of Pyrene at an Organic Coated Air–Water Interface, Phys. Chem. Chem. Phys., 2002, 4(17), 4186–4191, 10.1039/B204754C.
- B. T. Mmereki, S. R. Chaudhuri and D. J. Donaldson, Enhanced Uptake of PAHs by Organic-Coated Aqueous Surfaces, J. Phys. Chem. A, 2003, 107(13), 2264–2269, DOI:10.1021/jp027361g.
- T. F. Kahan, N.-O. A. Kwamena and D. J. Donaldson, Heterogeneous Ozonation Kinetics of Polycyclic Aromatic Hydrocarbons on Organic Films, Atmos. Environ., 2006, 40(19), 3448–3459, DOI:10.1016/j.atmosenv.2006.02.004.
- T. F. Kahan and D. J. Donaldson, Photolysis of Polycyclic Aromatic Hydrocarbons on Water and Ice Surfaces, J. Phys. Chem. A, 2007, 111(7), 1277–1285, DOI:10.1021/jp066660t.
- B. T. Mmereki and D. J. Donaldson, Direct Observation of the Kinetics of an Atmospherically Important Reaction at the Air–Aqueous Interface, J. Phys. Chem. A, 2003, 107(50), 11038–11042, DOI:10.1021/jp036119m.
- K. J. Morenz Korol, I. M. Kumayon, T. F. Kahan and D. J. Donaldson, Chemical Morphology Controls Reactivity of OH Radicals at the Air–Ice Interface, J. Phys. Chem. A, 2021, 125(40), 8925–8932, DOI:10.1021/acs.jpca.1c06434.
- H. Fu, R. Ciuraru, Y. Dupart, M. Passananti, L. Tinel, S. Rossignol, S. Perrier, D. J. Donaldson, J. Chen and C. George, Photosensitized Production of Atmospherically Reactive Organic Compounds at the Air/Aqueous Interface, J. Am. Chem. Soc., 2015, 137(26), 8348–8351, DOI:10.1021/jacs.5b04051.
- L. Manodori, A. Gambaro, R. Piazza, S. Ferrari, A. M. Stortini, I. Moret and G. Capodaglio, PCBs and PAHs in Sea-Surface Microlayer and Sub-Surface Water Samples of the Venice Lagoon (Italy), Mar. Pollut. Bull., 2006, 52(2), 184–192, DOI:10.1016/j.marpolbul.2005.08.017.
- Y.-J. Huang, B.-S. Lin, C.-L. Lee and P. Brimblecombe, Enrichment Behavior of Contemporary PAHs and Legacy PCBs at the Sea-Surface Microlayer in Harbor Water, Chemosphere, 2020, 245, 125647, DOI:10.1016/j.chemosphere.2019.125647.
- O. Mazimba, Umbelliferone: Sources, Chemistry and Bioactivities Review, Bull. Fac. Pharm., 2017, 55(2), 223–232, DOI:10.1016/j.bfopcu.2017.05.001.
- A. Morgan, D. Babu, B. Reiz, R. Whittal, L. Y. K. Suh and A. G. Siraki, Caution for the Routine Use of Phenol Red – It Is More than Just a PH Indicator, Chem.-Biol. Interact., 2019, 310, 108739, DOI:10.1016/j.cbi.2019.108739.
- Y. Q. Huang, C. K. C. Wong, J. S. Zheng, H. Bouwman, R. Barra, B. Wahlström, L. Neretin and M. H. Wong, Bisphenol A (BPA) in China: A Review of Sources, Environmental Levels, and Potential Human Health Impacts, Environ. Int., 2012, 42, 91–99, DOI:10.1016/j.envint.2011.04.010.
- K.-U. Goss and R. P. Schwarzenbach, Linear Free Energy Relationships Used to Evaluate Equilibrium Partitioning of Organic Compounds, Environ. Sci. Technol., 2001, 35(1), 1–9, DOI:10.1021/es000996d.
- M. Staikova, F. Wania and D. J. Donaldson, Molecular Polarizability as a Single-Parameter Predictor of Vapour Pressures and Octanol–Air Partitioning Coefficients of Non-Polar Compounds: A Priori Approach and Results, Atmos. Environ., 2004, 38(2), 213–225, DOI:10.1016/j.atmosenv.2003.09.055.
- E. N. Bocharnikova, O. N. Tchaikovskaya, O. K. Bazyl, V. Y. Artyukhov and G. V. Mayer, Theoretical Study of Bisphenol A Photolysis, 2020, pp. 191–217, DOI:10.1016/bs.aiq.2019.12.001.
- A. W. Adamson, Physical Chemistry of Surfaces, John Wiley & Sons, Inc., 5th edn, 1990 Search PubMed.
- D. J. Donaldson, Experimental Confirmation of H2O2 Adsorption at the Water–Air Interface, J. Phys. Chem. A, 2022, 126(33), 5647–5653, DOI:10.1021/acs.jpca.2c04373.
- D. J. Donaldson, M. Ammann, T. Bartels-Rausch and U. Pöschl, Standard States and Thermochemical Kinetics in Heterogeneous Atmospheric Chemistry, J. Phys. Chem. A, 2012, 116(24), 6312–6316, DOI:10.1021/jp212015g.
- D. J. Donaldson, Adsorption of Atmospheric Gases at the Air–Water Interface. I. NH3, J. Phys. Chem. A, 1999, 103(1), 62–70, DOI:10.1021/jp9833247.
- R. J. Perkins, K. Kukharchuk, P. Delcroi, R. K. Shoemaker, M. Roeselová, L. Cwiklik and V. Vaida, The Partitioning of Small Aromatic Molecules to Air–Water and Phospholipid Interfaces Mediated by Non-Hydrophobic Interactions, J. Phys. Chem. B, 2016, 120(30), 7408–7422, DOI:10.1021/acs.jpcb.6b05084.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00394a |
| This journal is © The Royal Society of Chemistry 2024 |