
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring cobalt spinel oxide with site-specific single atom incorporation for high-performance electrocatalysis†
Kangjae
Lee‡
ab,
Jaehyuk
Shim‡
ab,
Hyunsoo
Ji‡
ab,
Jungho
Kim
f,
Hyeon Seok
Lee
ab,
Heejong
Shin
ab,
Megalamane S.
Bootharaju
ab,
Kug-Seung
Lee
 c,
Wonjae
Ko
ab,
Jaewoo
Lee
ab,
Kang
Kim
ab,
Seungwoo
Yoo
ab,
Sungeun
Heo
ab,
Jaeyune
Ryu
ab,
Seoin
Back
*f,
Byoung-Hoon
Lee
*de,
Yung-Eun
Sung
*ab and
Taeghwan
Hyeon
*ab
c,
Wonjae
Ko
ab,
Jaewoo
Lee
ab,
Kang
Kim
ab,
Seungwoo
Yoo
ab,
Sungeun
Heo
ab,
Jaeyune
Ryu
ab,
Seoin
Back
*f,
Byoung-Hoon
Lee
*de,
Yung-Eun
Sung
*ab and
Taeghwan
Hyeon
*ab
aCenter for Nanoparticle Research, Institute for Basic Science (IBS), Seoul 08826, Republic of Korea
bSchool of Chemical and Biological Engineering, and Institute of Chemical Processes, Seoul National University, Seoul 08826, Republic of Korea
cPohang Accelerator Laboratory (PAL), Pohang University of Science and Technology (POSTECH), Pohang, Republic of Korea
dKU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul 02481, Republic of Korea
eDepartment of Integrative Energy Engineering, College of Engineering, Korea University, Seoul 02481, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Institute of Emergent Materials, Sogang University, Seoul 04107, Republic of Korea
First published on 25th April 2024
Abstract
Universal incorporation of metals into cobalt spinel oxide (CSO) has emerged as a versatile and promising strategy to enhance catalytic performance. However, the uncontrolled reactivity of early transition metal and metalloid precursors with water has presented a significant challenge in achieving atomic-scale metal incorporation within CSO. This study presents a groundbreaking approach for the atomic-scale integration of diverse dopants, including Hf, Ta, W, Ti, Pd, Ga, and Ge, while elucidating the atomic stabilization sites for these metal cations within CSO. Notably, certain metals, such as Ta, W, and Ge exhibit greater stability at the surface rather than within the core of CSO, resulting in a Co2+-enriched surface that serves as a both catalytically active and protective shell. Exploiting these remarkable features, Ta-doped Co3O4 demonstrates the lowest overpotential, registering a mere 378 mV at 10 mA cm−2, while maintaining its catalytic activity for over 140 hours in acidic electrolyte.
Broader contextSpinel oxides with AB2O4 crystal structure have attracted significant attention due to their versatility in accommodating various metal compositions and the corresponding range of physicochemical properties. Cobalt spinel oxide, for instance, shows promise as a cost-effective catalyst for oxygen evolution reaction (OER). However, incorporation of metal elements into cobalt spinel oxide has been predominantly limited to first-row transition metal elements. Herein, we incorporate a wide range of elements into cobalt spinel oxide from early-transition metals to metalloids (including Hf, Ta, W, Ti, Ga, Ge, and Pd), which offers new possibilities. In addition, our research pinpoints specific stabilization sites for these metal elements within cobalt spinel oxide, with Ta, W, and Ge predominantly stabilized on the surface, resulting in a significant increase in surface Co2+ species. This dopant-enriched shell serves both as an active layer and a protective layer, enhancing the performance of acidic OER. Leveraging these remarkable features, Ta-doped Co3O4 demonstrates the lowest overpotential, registering a mere 378 mV at 10 mA cm−2, while maintaining its catalytic activity for over 140 hours in acidic electrolytes. |
Introduction
The performance of heterogeneous catalysts critically depends on surface characteristics,1–3 underscoring the imperative need for meticulous control over electronic structure and chemical composition.4–6 In this context, spinel oxides, featuring the AB2O4 crystal structure, have attracted significant attention as catalysts owing to their tunable physicochemical properties enabled by precise control over the composition of cationic A and B sites.7–12 In particular, incorporation of various metal cations into cobalt spinel oxide (CSO, Co3O4) has emerged as a potent approach for precisely tailoring the catalytic properties of CSOs, in the context of a wide range of electrocatalytic reactions.13–16Designing catalytic systems for the oxygen evolution reaction (OER) in acidic conditions is a subject of great interest.17–21 The primary challenge in advancing acidic OER lies in the slow kinetics and the reliance on precious metals like Ru and Ir.22–25 Notably, the CSO system has showcased substantial potential as a non-noble metal catalyst for acidic OER, offering a promising alternative to scarce and expensive noble metals.26–28 A key strategy to address these challenges involves the deliberate incorporation of metal cations into a specific cobalt oxide platform.29 Despite the suggested approach, current research has been predominantly limited to the integration of first-row transition metal ions, such as Fe, Mn, Ni, Cu, and Zn, which limits the range and scope of potential enhancements in the catalytic performance and durability of Co3O4.30–34 Establishing a universal synthetic protocol enabling the incorporation of a diverse range of metal cations into the CSO system would usher in a new realm of possibilities for exploring novel catalytic materials.35,36
However, the process of identifying and effectively incorporating suitable cations for substitution in CSO remains elusive, primarily due to the limited understanding of the physicochemical characteristics governing the integration of a diverse range of metal cations into CSOs. Moreover, the inclusion of third-row early transition metals, such as Hf, Ta, and W, poses an even more intricate challenge, given their uncontrolled reaction with water, often leading to the undesired formation of heterogeneous oxides.37,38 This chemical reaction obstructs the precise atomic-scale incorporation of these metals into Co3O4,39,40 preventing the synthesis and catalytic applications of single atom catalysts based on third-row early transition metal doped in CSO.41
In this work, we provide a comprehensive understanding of the incorporation of metal cations into CSO. First, we have developed a versatile synthetic method that enables synthesizing various metal-doped CSO materials, including metal cations from early transition metals and metalloids. The metal organic framework (MOF) played a pivotal role in the successful synthesis of Hf, Ta, W, Ti, Pd, Ga, and Ge-incorporated CSO without the unwanted formation of hetero-metal-oxide species. Secondly, we have elucidated that each metal species occupies distinct stabilization sites within the cobalt spinel structure. Ta, W, and Ge tend to stabilize at the octahedral sites on the surface of CSO, significantly increasing the surface Co2+ species. This dopant-rich shell and the high density of surface Co2+ provide active and protective layers, resulting in high-performance in acidic OER. For instance, Ta-doped Co3O4 exhibited the lowest overpotential of 378 mV at 10 mA cm−2, and maintained its activity for over 140 hours in acidic electrolytes. Our in situ X-ray absorption spectroscopy (XAS) and inductively coupled plasma (ICP) analysis indicate that this protective shell effectively suppresses over-oxidation and the dissolution of Co species during the reaction.
Results and discussion
Catalyst design and characterization
To synthesize metal-doped CSO, ZIF-67 MOF was selected as the source of cobalt. The dopant precursor is metal ethoxide, which is stabilized in methanol to mitigate the formation of hetero-metal-oxides. The dopant precursor was introduced during the rapid formation of ZIF-67 at room temperature, effectively confining the metal ethoxide within the pores of ZIF-67. Our encapsulation approach proved highly successful in preventing the generation of hetero-metal oxide byproducts, even after drying the as-synthesized material (Fig. S1, ESI†). Furthermore, this encapsulation method preserves the precursor in its single-atom-state, and the subsequent oxidation in a muffle furnace causes the MOF structure to rapidly disintegrate (Fig. S2, ESI†), resulting in the production of single-atom-doped CSO (Fig. 1a).42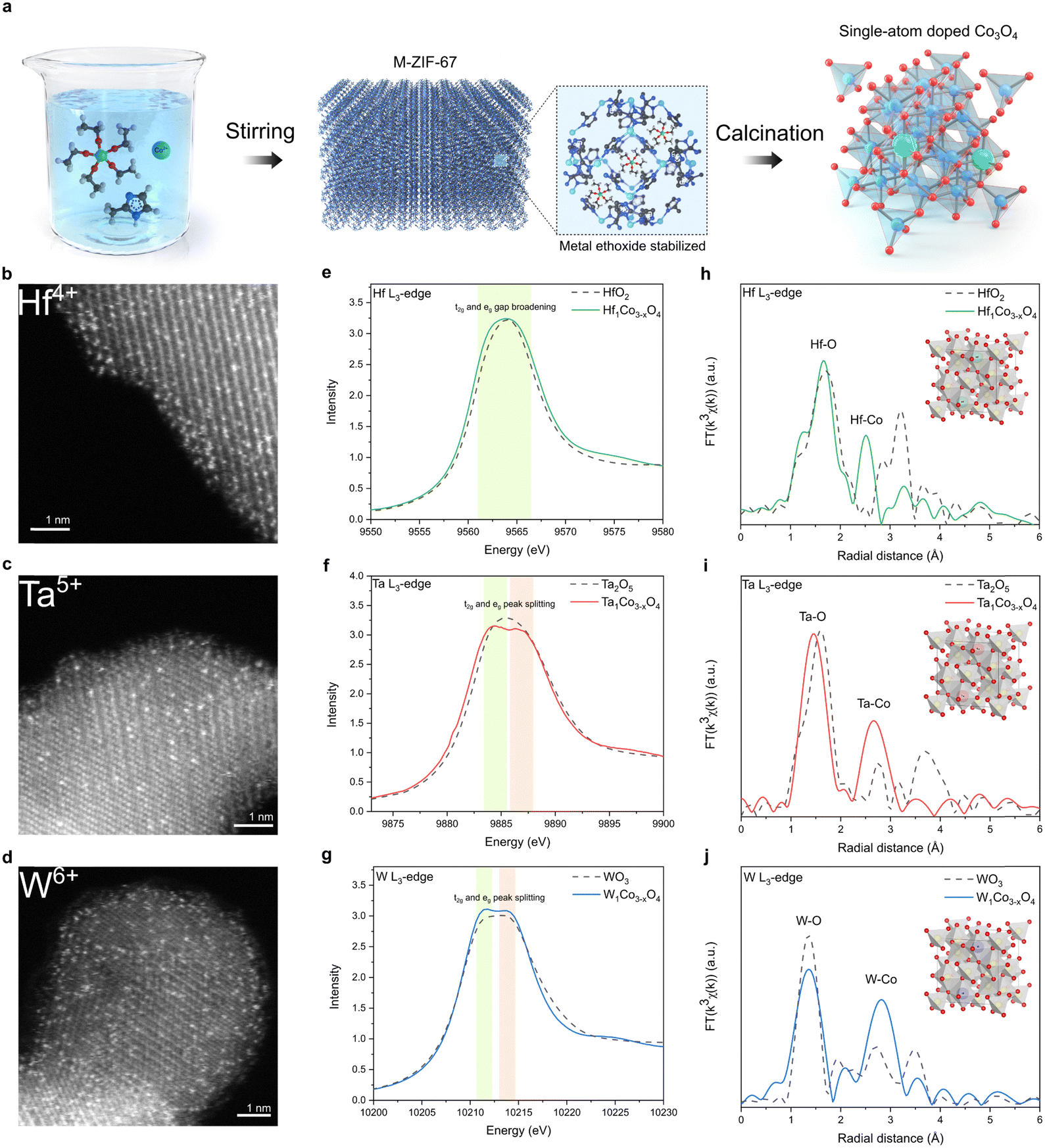 | ||
| Fig. 1 Synthesis scheme and structural characterization. (a) Schematic illustration of synthetic process. HAADF-STEM images of (b) Hf1Co3−xO4, (c) Ta1Co3−xO4, (d) W1Co3−xO4. The metal dopants were atomically incorporated in CSO crystal structure. L3-edge XANES of (e) Hf in Hf1Co3−xO4, (f) Ta in Ta1Co3−xO4, and (g) W in W1Co3−xO4. The d orbital broadening was observed. L3-edge EXAFS of (h) Hf in Hf1Co3−xO4, (i) Ta in Ta1Co3−xO4, and (j) W in W1Co3−xO4. | ||
Powder X-ray diffraction (XRD) analysis revealed the presence of peaks corresponding exclusively to CSO crystal planes, indicating that no secondary heterogeneous metal phases were formed during the oxidation process (Fig. S3, ESI†). Atomic-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) clearly showed the atomic-scale incorporation of metal dopants throughout the CSO (Fig. 1b–d). In conjunction with the STEM images, we conducted XAS to gain deeper insights into the local atomic structure of single-atom dopants. According to the X-ray absorption near edge spectroscopy (XANES) and X-ray photoelectron spectroscopy (XPS) data, the single-atom metal dopants possessed oxidation states similar to their metal oxide counterparts (Fig. 1e–g and Fig. S4, ESI†). However, the peak shape of the white line for the dopants differentiates from that of their respective metal oxide references. Specifically, upon the incorporation of metal dopants into the cobalt spinel structure, only peak broadening was observed for Hf, whereas peak splitting into two different peaks was observed for Ta and W in L3-edge XAS spectra. The Hf, Ta, and W L3-edge XAS spectra illustrate the transition of core electrons from the occupied 2p to the vacant 5d states of the respective metals. The observed peak splitting in the L3-edge spectrum results from the increased energy gap between the t2g and eg states within the 5d orbitals. This phenomenon can be attributed to the robust octahedral hybridization of orbitals due to enhanced metal-oxygen coordination. These results reveal that the Hf, Ta, and W atoms are effectively stabilized in the octahedral sites of CSO.43 This arrangement would foster enhanced orbital hybridization between the metal dopants and CSO, leading to the broadening of the 5d orbital energy gap. Furthermore, the extended X-ray absorption fine structure (EXAFS) analyses indicate the atomic dispersion of metal dopants, as the bond length between metal and oxygen slightly decreases upon the incorporation of dopants into the CSO structure (Fig. 1h–j).
In addition to early transition metals from the sixth row of the periodic table, including Hf, Ta, and W, we successfully incorporated Pd, Ga, Ge, and Ti dopants into the CSO using the same synthetic procedure. The characterization data, including STEM, energy-dispersive X-ray spectroscopy (EDS), and EXAFS, consistently confirm the atomic dispersion of single-atom dopants within the CSO (Fig. S5–S8, ESI†).
Stabilization sites of single-atom dopants
To gain atomic-level fundamental insights into the effect of incorporated dopants on the physicochemical properties of CSO, we conducted Co 2p XPS, Co L-edge XAS and Co K-edge XAS. XPS and Co L-edge XAS, known for their surface sensitivity, offer detailed information about the surface cobalt metal species.44,45 The average oxidation state of Co changes (Fig. 2a) depending on the type of metal dopants, indicating that these dopants uniquely modify the electronic structure of CSO.46,47 For a more comprehensive understanding, Co 2p XPS spectra were deconvoluted, displaying Co3+ 2p3/2 peak at 779.2 eV and Co2+ 2p3/2 peak at 780.6 eV (Table S1, ESI†). We calculated the ratio of Co2+ to Co3+, which is an indicator of the oxidation state of surface cobalt after the incorporation of metal cations. Notably, this ratio exhibited significant variations depending on the type of single-atom dopants, ranging from 0.959 (Pd) to 1.615 (Ge), displaying a maximum peak shift of 0.7 eV from the pristine CSO. To further elucidate the local coordination structure of the surface, we performed Co L-edge XAS. We observed that the spectra of all synthesized metal doped CSO's have the same shape with the pristine CSO, demonstrating the absence of change in the surface structure after the metal incorporation (Fig. S9, ESI†).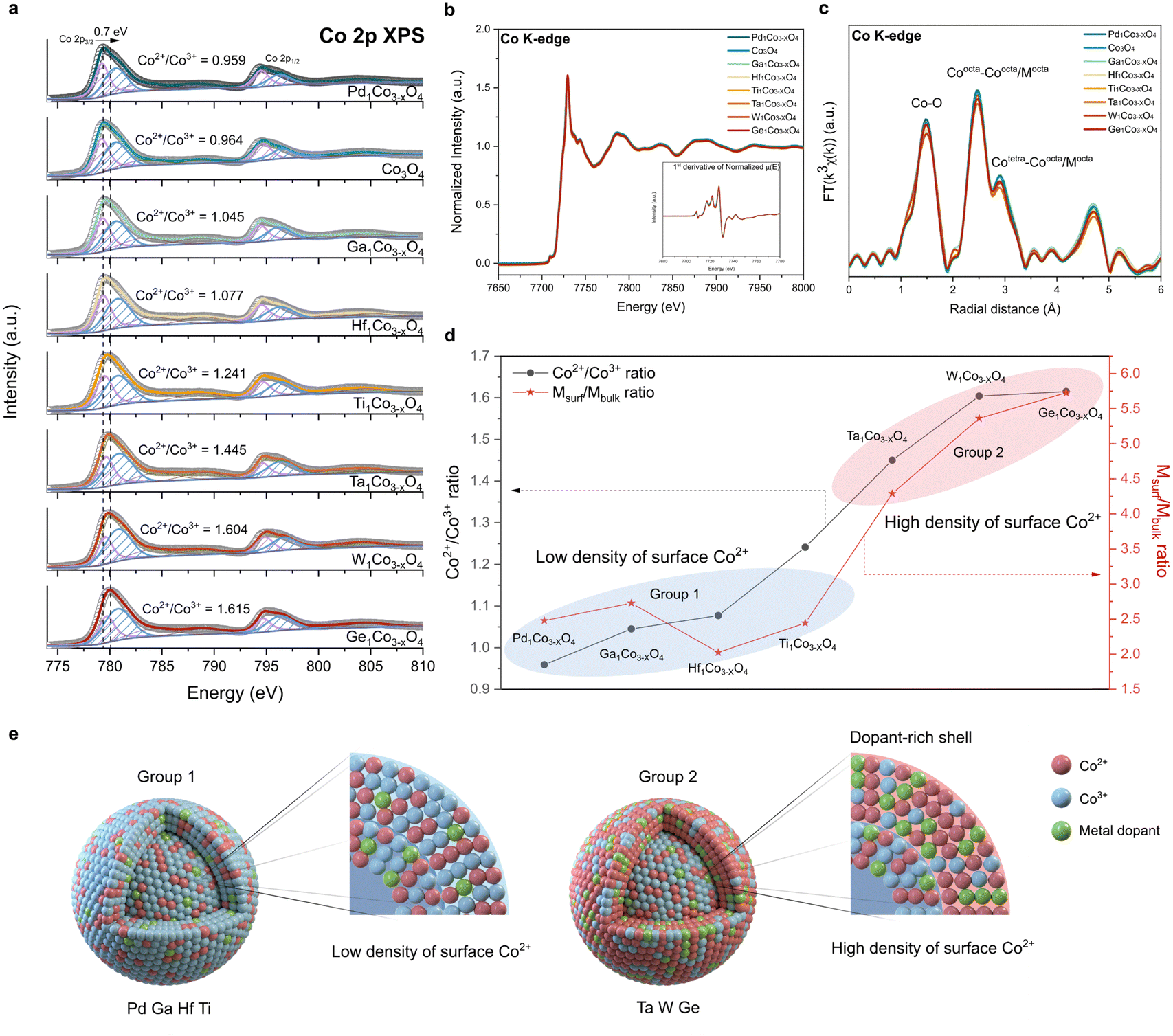 | ||
| Fig. 2 Electronic structure of metal-doped Co3O4. (a) Co 2p XPS, (b) Co K-edge XANES, and (c) Co K-edge EXAFS spectra of metal-doped Co3O4. Inset of (b): 1st derivative of normalized μ(E). (d) Co2+/Co3+ ratio and Msurf/Mbulk ratio of metal-doped Co3O4. The blue and red ellipses indicate group 1 and group 2 metals, respectively. (e) Schematic illustration of group 1 and group 2 metal-doped Co3O4. A dopant-rich shell is shown in the latter. | ||
Unlike XPS, which offers surface-specific information, the Co K-edge XAS data provide insights into the bulk properties. As shown in Fig. 2b, all the metal-doped CSOs exhibit a uniform XANES structure and oxidation state, indicating that the Co oxidation state within the bulk oxide remains nearly unchanged. Moreover, the Co K-edge EXAFS, as shown in Fig. 2c, revealed no discernible differences in the local structure of the bulk CSO. Therefore, in contrast to the cobalt species on the surface, it appears that the bulk cobalt species remain unaffected by the incorporation of various types of metal ions. These combined data from XANES and XPS reveal that the dopants are primarily stabilized on the surface rather than within the bulk of CSO.
To characterize the specific sites of the stabilized metal dopants, we calculated the atomic ratio between metal dopants and cobalt on both the surface and the bulk of CSO. The atomic ratio of the surface (Msurf = (atomic percent of metal dopants by XPS)/(atomic percent of Co by XPS)) was derived from the XPS results (Table S2, ESI†) and the atomic ratio of the bulk (Mbulk = (atomic percent of metal dopants by ICP-AES)/(atomic percent of Co by ICP-AES)) was calculated based on the ICP-AES data (Table S3, ESI†). A high Msurf/Mbulk ratio (group 2) implies that the metal dopants are predominantly stabilized on the surface of CSO, whereas a low Msurf/Mbulk ratio (group 1) suggests that the metal dopants are evenly distributed throughout both the surface and bulk sites. Notably, Ta, W, and Ge exhibit high Msurf/Mbulk ratios, exerting a considerable impact on the surface electronic structure and leading to a high density of surface Co2+ species (Fig. 2d). On the other hand, Pd, Ga, Hf and Ti demonstrate low Msurf/Mbulk ratios, which have minimal influence on the surface properties of CSO. We further calculated the stabilization energies of two slab structures: one with a dopant located in the first layer and the other with the dopant in the third layer. We assume that these positions correspond to the surface and the bulk position, respectively (Fig. S10 and S11, ESI†). The qualitative results are generally in agreement with the experimental observations, where Ta, Ge and W with large energy differences between two layers show a preference for surface positions, whereas Ga, Pd, Ti and Hf tend to prefer bulk positions.
From all these data, we conclude that the choice of metal dopants significantly impacts the surface characteristics of CSO, as illustrated in Fig. 2e. Notably, Ta, W, and Ge, categorized as group 2 metals, result in CSOs with dopant-rich shells that contain a high density of Co2+ species on the surface. This result is corroborated by energy-filtered transmission electron microscopy (EF-TEM) and XPS depth profile. EF-TEM images demonstrate the uniform coverage of the surface with Ta single atoms (Fig. S12, ESI†). XPS depth profile shows that Ta is more stabilized on the near surface rather than in the bulk (Fig. S13, ESI†). Because increased content of Ta species near the surface affects the peripheral Co electronic structure, Co species located closer to the surface tends to be more reduced, which is supported by Co 2p angle resolved-XPS (AR-XPS) spectra (Fig. S14, ESI†). Moreover, we analyzed the Bader charges of Co atoms before and after the Ta doping (Fig. S15, ESI†). The Bader charge values of Co atoms near the Ta dopant decrease by 0.15, which is consistent with Co 2p XPS. Additionally, scanning electron microscopy (SEM) analysis reveals that the dopant-rich shell serves as a protective layer,45 preserving the dodecahedron morphology of ZIF-67 during calcination in the air (Fig. S16, ESI†).
Catalytic performance
We next examined how the foregoing modifications of surface electronic structure and composition affect both catalytic activity and durability. To establish the correlation between these surface properties and catalytic performance, we evaluated the OER performance for all the synthesized catalysts using a rotating disk electrode (RDE) in a 0.05 M H2SO4 electrolyte. The recorded linear sweep voltammetry (LSV) curves for all the synthesized catalysts (Fig. 3a and b) reveal a noticeable enhancement in OER performance compared to bare Co3O4. By systematically varying the Ta content, we identified an optimal composition. The samples with varying amounts of Ta were designated as Ta1Co3−xO4 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) and Ta1Co3−xO4 (10:1), representing higher and lower Ta content compared to Ta1Co3−xO4, respectively (as detailed in Table S4, ESI†). In both cases, while the activity exhibited a decrease compared to Ta1Co3−xO4, it remained superior to that of the bare Co3O4. We then investigated the OER catalytic activity of Ta1Co3−xO4, where Co1 is Co atom located right next to the Ta dopant, while Co2 is the one far from the Ta single atom (Fig. S17, ESI†). The pristine Co3O4 is known to bind OER adsorbates weakly, making the *OH adsorption a rate-determining step (RDS) with the ηOER of 0.554 VRHE.10 Once the Ta is doped, the binding affinity of OH* increased, changing the RDS to *O and *OOH formation for Co1 and Co2, respectively. ηOER decreased to 0.334 VRHE for Co1 and 0.489 VRHE for Co2. In contrast, the Ta single atom exhibited a high ηOER of 0.933 VRHE with the *OOH formation step being the RDS. This indicates that incorporation of Ta within Co3O4 could enhance the OER activity, primarily by activating surface Co species. In Fig. 3c, we provide a summary of the overpotentials required to achieve a current density of 10 mA cm−2 for all the measured samples. Notably, the most active Ta1Co3−xO4 catalyst demonstrated an overpotential of 378 mV, 88 mV lower than that of Co3O4. Taken together, these studies demonstrate the significance of precise and controlled metal incorporation as a powerful tool for optimizing the OER performance of CSO systems (Table S5, ESI†).
:5) and Ta1Co3−xO4 (10:1), representing higher and lower Ta content compared to Ta1Co3−xO4, respectively (as detailed in Table S4, ESI†). In both cases, while the activity exhibited a decrease compared to Ta1Co3−xO4, it remained superior to that of the bare Co3O4. We then investigated the OER catalytic activity of Ta1Co3−xO4, where Co1 is Co atom located right next to the Ta dopant, while Co2 is the one far from the Ta single atom (Fig. S17, ESI†). The pristine Co3O4 is known to bind OER adsorbates weakly, making the *OH adsorption a rate-determining step (RDS) with the ηOER of 0.554 VRHE.10 Once the Ta is doped, the binding affinity of OH* increased, changing the RDS to *O and *OOH formation for Co1 and Co2, respectively. ηOER decreased to 0.334 VRHE for Co1 and 0.489 VRHE for Co2. In contrast, the Ta single atom exhibited a high ηOER of 0.933 VRHE with the *OOH formation step being the RDS. This indicates that incorporation of Ta within Co3O4 could enhance the OER activity, primarily by activating surface Co species. In Fig. 3c, we provide a summary of the overpotentials required to achieve a current density of 10 mA cm−2 for all the measured samples. Notably, the most active Ta1Co3−xO4 catalyst demonstrated an overpotential of 378 mV, 88 mV lower than that of Co3O4. Taken together, these studies demonstrate the significance of precise and controlled metal incorporation as a powerful tool for optimizing the OER performance of CSO systems (Table S5, ESI†).
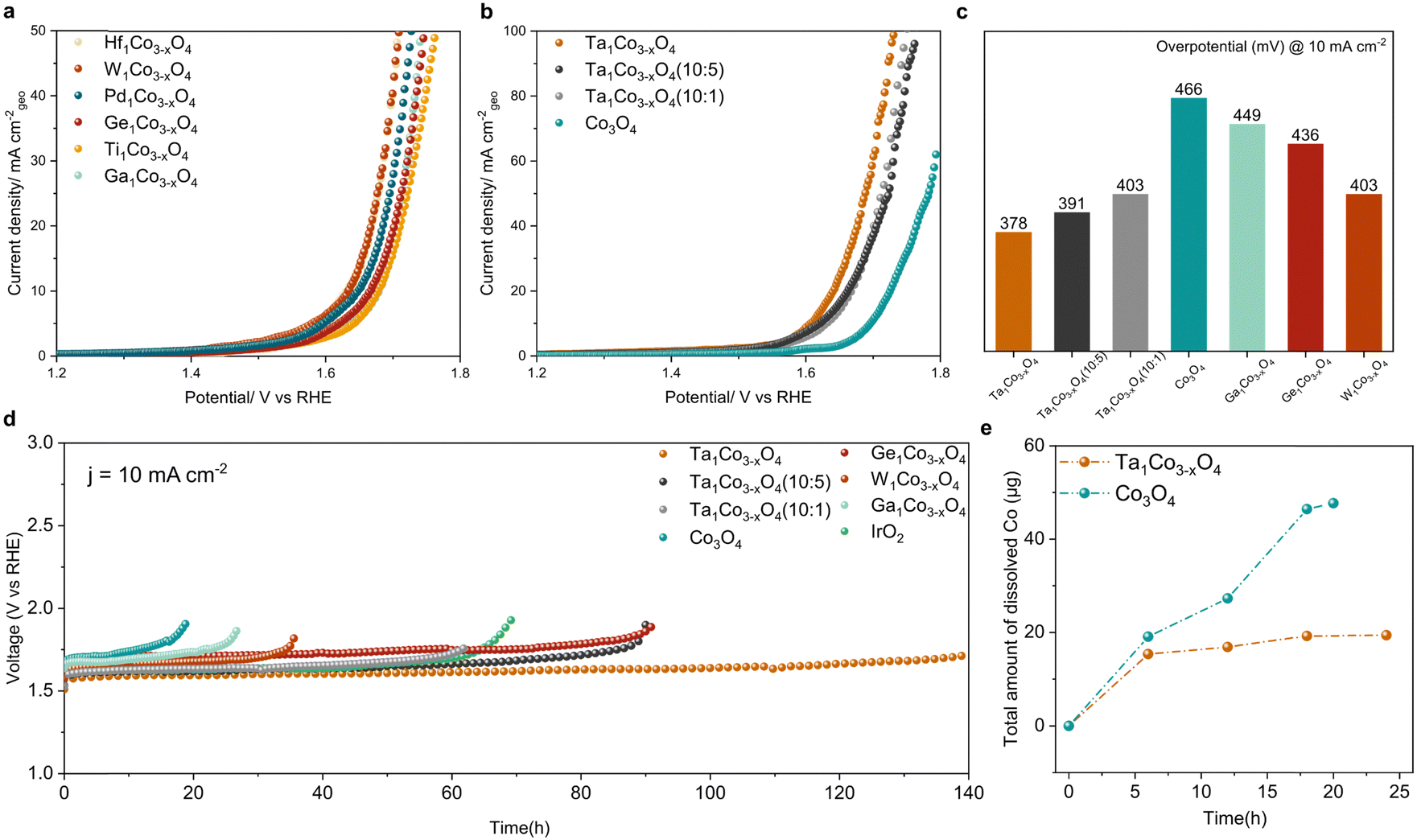 | ||
| Fig. 3 Electrocatalytic acidic OER performance. (a) Linear sweep voltammetry (LSV) curves of Co3O4 doped with Hf, W, Pd, Ge, Ti, and Ga in 0.05 M H2SO4 electrolyte. (b) LSV curves of Co3O4 with varying Ta amounts. (c) Overpotential bar graph of catalysts in panels (a) and (b) at 10 mA cm−2. (d) Chronopotentiometry curves of the synthesized catalysts and IrO2 at 10 mA cm−2. (e) Amount of dissolved cobalt ion measured by ICP-MS analysis during the stability test shown in panel (d). | ||
We further investigated the durability trends across all the samples by comparing the increases in their overpotentials at a current density of 10 mA cm−2 (Fig. 3d). The metals with the high Msurf/Mbulk ratios (Ta, W, and Ge) showed a striking enhancement in acidic OER stability, suggesting that the dopant-rich shell could effectively function as a protective barrier in the face of harsh oxidative conditions. Among these, Ta1Co3−xO4 exhibited the most remarkable catalytic stability with no discernible decay in its activity observed over a 140-hour period, recording a degradation rate of 1.04 mV h−1. In contrast, Co3O4 exhibited a rapid deterioration in activity after 19 hours, with a degradation rate of 11.67 mV h−1, 11 times higher than that of Ta1Co3−xO4. Additionally, we assessed the stability of Ga1Co3−xO4 as a representative of metals belonging to the low Msurf/Mbulk group (encompassing Ga, Hf, Ti, and Pd), which displayed minimal progress in terms of oxidative stability.
To further analyze the catalytic stability of Ta1Co3−xO4 and Co3O4, we quantified the degree of cobalt ion dissolution following 24 hours of operation in an acidic OER environment (Fig. 3e).48 The ICP results reveal that the introduction of Ta to the Co3O4 surface effectively mitigated the dissolution of cobalt species. Once the surface was stabilized during the reaction, Ta1Co3−xO4 displayed a notably stabilized trend in dissolution, in stark contrast to the continuous dissolution observed in Co3O4. We calculated S-number based on the ICP results at 6-hour and 18-hour intervals (Fig. S18, ESI†). For Ta1Co3−xO4, the S-number values were approximately 2.148 × 103 at the 6-hour mark and 5.138 × 103 at the 18-hour mark. In contrast, the S-number values for Co3O4 were approximately 1.730 × 103 at 6 hours and 2.130 × 103 at 18 hours. These values differed significantly, with Ta1Co3−xO4 exhibiting a 1.24-fold at 6 hours and a 2.41-fold at 18 hours compared to Co3O4. This discrepancy shows the enhanced stability achieved through the incorporation of Ta on the surface, with the difference in stability becoming more pronounced as time progresses.
Durability of catalyst
To demonstrate durability of the catalysts, we performed the post-OER characterization of Ta1Co3−xO4. HAADF-STEM image and STEM-EDS mapping images of Ta1Co3−xO4 reveal that atomically dispersed Ta metals maintained after OER. This result indicates that the Ta single atoms incorporated in CSO crystal structure remain stable during acidic OER (Fig. 4a and b). XRD patterns of Ta1Co3−xO4 on carbon paper after OER showed the presence of peaks corresponding exclusively to CSO crystal planes, indicating no hetero-metal phase formed during the reaction (Fig. 4c).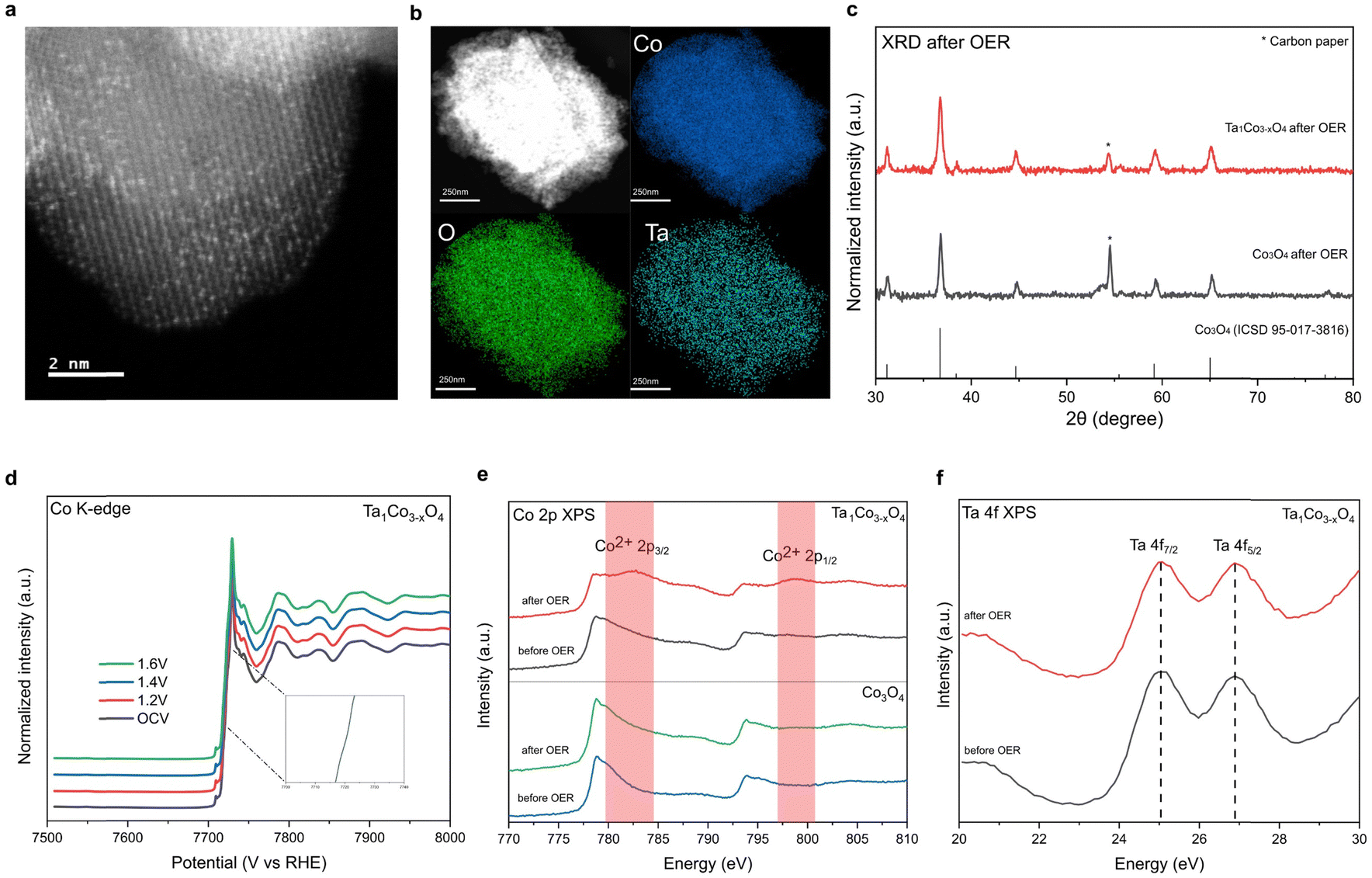 | ||
| Fig. 4 Post-OER characterization of Ta1Co3−xO4. (a) HAADF-STEM image of Ta1Co3−xO4 after OER. (b) STEM-EDS mapping images of Ta1Co3−xO4 after OER. The atomically dispersed Ta atoms maintained without aggregation after OER. (c) XRD patterns of Ta1Co3−xO4 on carbon paper after OER. The patterns indicated that Co3O4 structure maintained after OER. (d) In situ XANES spectra of Ta1Co3−xO4 at different applied potentials in 0.05 M H2SO4. (e) Co 2p XPS spectra of Ta1Co3−xO4 and Co3O4 before and after the OER stability test. The surface Co2+ remained even after long-term acidic OER. (f) Ta 4f XPS of Ta1Co3−xO4 after OER showed that the oxidation state of surface Ta maintained after OER. | ||
Furthermore, an in situ XAS study was conducted on Ta1Co3−xO4 to investigate changes in the electronic structure during electrochemical reaction (Fig. 4d). Remarkably, in situ XANES spectra of Ta1Co3−xO4 showed negligible alteration in oxidation state as the applied voltage increased in acidic electrolytes, demonstrating the dopant-rich shell and high density of surface Co2+ suppresses the overoxidation of Co species during OER.
Additionally, ex situ investigations of XPS were carried out on post-OER samples (Fig. 4e).49 The distinct Co2+ XPS peaks of Ta1Co3−xO4 were observed after OER, indicating that the Co2+ species in the form of Ta–O–Co are significantly more stable compared to Co3+ species in oxidative condition. The stability of the Ta single atom, as evidenced by the HAADF-STEM image, along with the absence of hetero-phase formation, supports the conclusion that the Co metal species in close proximity to the Ta single atom remain stable during OER. During the course of the reaction, the less stable surface Co3+ species dissolve, leaving behind Co2+ species alongside Ta dopants, which play a pivotal role in enhancing the durability of Ta1Co3−xO4 in acidic electrolytes. Conversely, the Co 2p XPS spectra of Co3O4 control exhibit minimal changes after OER, which can be attributed to the ongoing dissolution of surface Co species during the OER process. This observation is consistent with the findings from the aforementioned 24-hour ex situ ICP analysis, which revealed a persistent dissolution of Co ions in the Co3O4 control, while no further dissolution of Co was observed in Ta1Co3−xO4 after the initial 5 hours (Fig. 3e). Ta 4f XPS of Ta1Co3−xO4 showed that the oxidation state of Ta maintained after OER (Fig. 4f). We also calculated dissolution potentials (Udiss) of tetrahedral Co2+ atoms in both pure Co3O4 and Ta1Co3−xO4 to evaluate the stability of Co2+ species after the incorporation of Ta (Fig. S19, ESI†). A higher Udiss indicates higher stability against the dissolution during acidic OER. We found that the Udiss of tetrahedral Co2+ increased from 1.25 VSHE to 1.58 VSHE upon doping with Ta, signifying that Ta dopants effectively prevent the dissolution of tetrahedral Co2+. Collectively, the dopant-rich shell serves to preserve the presence of surface Co2+ species throughout the reaction, effectively inhibiting the dissolution of surface Co species.
To investigate the possibility of a newly constructed surface structure contributing to enhanced OER performance as Co3+ species dissolve, we conducted CS-TEM analysis on the catalyst surface after OER. It turned out that lattice near the surface maintained the initial CSO structure without forming new amorphous or crystalline surface structures (Fig. S20, ESI†). Moreover, the Co L-edge XAS spectra of post-OER catalysts consistently show an absence of alteration in the surface structure after acidic OER (Fig. S21, ESI†).
Conclusion
In summary, we present a comprehensive understanding of the crystal structure of single-atom-doped CSO and introduce a general synthetic method applicable to a wide spectrum of metal elements, extending beyond the first-row transition metals. The encapsulation method effectively stabilizes water-sensitive metal precursors, including early-transition metal and metalloid, thus enabling the precise incorporation of diverse metal elements at the atomic scale within CSO. Our experimental results demonstrate the successful atomic distribution of metal dopants in CSO, with no discernible formation of hetero-metal-oxide species. Furthermore, we unveiled the metal-specific stabilization sites in CSO, revealing distinctive surface physicochemical properties for each metal. These finely controlled surface properties have the potential to translate into improved catalytic performance and enduring stability during acidic OER. Our discovery in the general synthesis of incorporating multiple metals into CSO and controlling resulting surface properties has promising applications across a broad spectrum of catalytic processes in sustainable energy storage and conversion systems.Experimental methods
Materials and methods
Synthesis of ZIF-67. Synthetic method was slightly modified to improve its extensibility of various elements incorporation.27 To synthesize ZIF-67, 40 mL of MeOH (solution A) solution which fully dissolved 1313.6 mg of 2-methylimidazole was prepared. Another 40 mL of MeOH which dissolved 580.1 mg of Co(NO3)2·6H2O was prepared and directly added to solution A with vigorous stirring at room temperature. After 18 hours, ZIF-67 precipitate was obtained by centrifugation, which was followed by washing with MeOH. The precipitate was dried for several hours under vacuum (60 °C).
Synthesis of Co3O4. As-synthesized violet powders were heated at 400 °C for 4 h under air in a muffle furnace with a ramping rate of 5 °C min−1. After cooling, black powder was acquired.
Synthesis of Ta1Co3−xO4. Ta1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ta(CH3CH2O)5 was used during the synthesis of ZIF-67. 0.6 mmol of Ta(CH3CH2O)5 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Ta1Co3−xO4 (10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :1).
Ta1Co3−xO4 (10:1) was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ta(CH3CH2O)5 was used during the synthesis of ZIF-67. 0.2 mmol of Ta(CH3CH2O)5 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
:1).
Ta1Co3−xO4 (10:1) was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ta(CH3CH2O)5 was used during the synthesis of ZIF-67. 0.2 mmol of Ta(CH3CH2O)5 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Ta1Co3−xO4 (10
:5).
Ta1Co3−xO4 (10:5) was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ta(CH3CH2O)5 was used during the synthesis of ZIF-67. 1 mmol of Ta(CH3CH2O)5 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Hf1Co3−xO4. Hf1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Hf(CH3CH2O)4 was used during the synthesis of ZIF-67. 0.2 mmol of Hf(CH3CH2O)4 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of W1Co3−xO4. W1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that W(CH3CH2O)6 was used during the synthesis of ZIF-67. 0.2 mmol of W(CH3CH2O)6 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Ti1Co3−xO4. Ti1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ti(CH3CH2O)4 was used during the synthesis of ZIF-67. 0.2 mmol of Ti(CH3CH2O)4 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Ga1Co3−xO4. Ga1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ga(CH3CH2O)3 was used during the synthesis of ZIF-67. 0.6 mmol of Ga(CH3CH2O)3 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Ge1Co3−xO4. Ge1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Ge(CH3CH2O)4 was used during the synthesis of ZIF-67. 0.6 mmol of Ge(CH3CH2O)4 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Synthesis of Pd1Co3−xO4. Pd1Co3−xO4 was synthesized using the same procedure employed for the synthesis of Co3O4, except that Pd(acac)2 was used during the synthesis of ZIF-67. 0.2 mmol of Pd(acac)2 was well dispersed in MeOH solution. Maintaining the total volume of the solution at 80 mL, all solutions were mixed together with vigorous stirring to obtain violet solution.
Characterization
Atomic-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was conducted at 200 kV with a spherical aberration-corrected JEM ARM-200F (Cold FEG Type, JEOL) installed at the National Center for Inter-University Research Facilities at Seoul National University. CS-TEM was conducted at 200 kV with a spherical aberration-corrected JEM ARM200F (Cold FEG Type, JEOL) equipped with OneView camera (25 fps at full 4k × 4k resolution) and Orius SC200D camera (2k × 2k resolution) at the National Center for Inter-University Research Facilities at Seoul National University. XANES and EXAFS spectra were acquired using beamline 8C nano XAFS and 10C wide XAFS installed at Pohang light source II in Pohang accelerator laboratory. All XAS measurements were carried out with the transmission mode. Calibration of E0 values of each sample was performed based on the simultaneously measured E0 value of each metal reference foils. In situ XAFS data at the Co K-edge were acquired at the 8c beamline of the Pohang Accelerator Laboratory in Korea, employing the fluorescence mode, while the measurements were conducted within an electrolysis cell. Software ATHENA was used to reduce data to get XANES and EXAFS spectra. XPS measurements were performed by K-Alpha+ XPS system (Thermo Fisher Scientific) using X-ray source as microfocused monochromated Al-Kα (1486.6 eV) at the Korea Basic Science Institute in Busan. XPS was conducted with high resolution of step size 0.1 eV in the energy range where the peaks were located. All spectra were calibrated by C 1s binding energy of graphite. XPS depth profile and AR-XPS were conducted by K-alpha (Thermo Scientific Inc., U.K.) using X-ray source (12 kV, 3 mA) as monochromated Al-Kα (1486.6 eV) at the Yonsei cooperative center for research facilities. Argon etching (500 eV) was employed to etch the surface of the pellet and XPS was measured every 30 s. AR-XPS was conducted with a high resolution of a step size 0.1 eV. All spectra were calibrated using the C 1s binding energy. Powder XRD patterns were collected using a SmartLab (Rigaku) diffractometer with Cu Kα radiation. EDS mapping and corresponding STEM images were obtained using STEM mode equipped with a single drift detector (X-MAXN, Oxford Instruments). Energy filtered transmission electron microscopy (EF-TEM) images were obtained using GIF mode of TEM (JEM ARM-200F) equipped with 965 GIF Quantum ER. SEM images were collected using SIGMA (Carl Zeiss). To prepare SEM samples, platinum was sputtered on the samples using sputter coater EM ACE200 (Leica). The bulk composition of Co and other metal elements was collected using ICP-AES (OPTIMA 8300) except that Ge and W which are hardly decomposed so that they need to be dissolved in hydrofluoric acid. Their composition was measured using Agilent 5110, treated with hydrofluoric acid. Dissolved cobalt ions during the reaction were subjected to small fractional extraction at specified intervals, followed by subsequent ICP-MS analysis (Varian 820-MS). Thermogravimetric analysis (TGA) was conducted by an SDT-Q600 (TA instruments) ramping to 600 °C with ramping rate 10 °C min−1 and air purging (100 mL min−1). IR spectroscopy was performed using VERTEX 70v (Bruker) at room temperature under vacuum. IR spectra was obtained averaging 300 scans per spectra at a spectral resolution of 0.5 cm−1 after background measurement.Electrochemical analysis
Electrochemical measurements were conducted under controlled conditions in a 0.05 M H2SO4 electrolyte solution (pH = 1.0) at 25 °C. A three-electrode system was employed, utilizing an Autolab potentiostat (PGSTAT302N) for data acquisition. The working electrode consisted of a rotating disk electrode (RDE) paired with a glassy carbon electrode (surface area of 0.1965 cm2). An Ag/AgCl electrode served as the reference electrode, while a platinum sheet functioned as the counter electrode. All potentials were recalibrated using the following formula: ERHE = EAg/AgCl + 0.1976 + 0.0591 × pH = EAg/AgCl + 0.2567. In the preparation of the catalyst ink on the glassy carbon electrode, a mixture of 5 mg of the catalyst and 1.67 mg of Vulcan Carbon was dispersed in a 1 mL solution composed of 970 μl of deionized (DI) water and isopropanol in a volume ratio of 2:8, along with 30 μl of a 5% Nafion solution. Subsequently, the resulting mixtures underwent ultrasonication for approximately 1 hour to achieve uniform ink dispersion. Using a spin-coating technique, 17.64 μl of the dispersed ink was deposited onto a glassy carbon electrode, resulting in a catalyst loading of 0.45 mg cm−2. Subsequently, linear sweep voltammetry (LSV) with an 100% iR correction was conducted at an ambient temperature of 25 °C, employing a scan rate of 0.01 V s−1 and a working electrode rotating at 1600 rpm. For stability assessments on carbon papers, 5 mg of the catalyst was dispersed in a 1 mL solution consisting of 970 μl isopropanol and deionized (DI) water in a volume ratio of 8:2, complemented by the addition of 30 μl of a 5% Nafion solution. Following approximately 1 hour of ultrasonication, the resulting ink was evenly deposited onto a carbon paper substrate via air spraying, achieving a loading of 3 mg cm−2. To ensure accurate mass loading, the carbon paper's weight was measured both before and after the air spraying process. In the case of the commercial IrO2 catalyst, the loading was adjusted to 1 mg cm−2.
The S-number values were calculated from the catalyst on a carbon paper during the stability test. The S-number was calculated according to eqn (1),
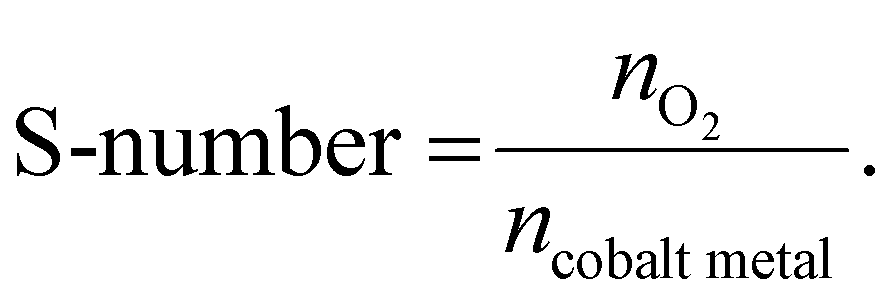
Computational detail
Spin-polarized density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP) version 5.4.4.50,51 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional and the projector augmented wave (PAW) pseudopotential were employed.52–54 A Hubbard U correction method was applied with the effective Ueff (U − J) value of 3.32 eV for Co to take into account strong on-site Coulombic interactions of localized d-orbitals.55 The optimized magnetic moments of the octahedral and tetrahedral Co atoms were 0.00 and 2.75, which correspond to low-spin Co3+ and high-spin Co2+, respectively.9 The metallic bulk Co structure (mp-102) was calculated without this U correction. A kinetic energy cutoff of 500 eV was set for all calculations. Convergence for forces and energies was considered reached when they become smaller than 10−4 eV and 0.05 eV Å−1 for slab structures and 10−5 eV and 0.01 eV Å−1 for bulk structures, respectively. Monkhorst–Pack k-points were set at 3 × 3 × 1 for slab structures and 4 × 4 × 4 for bulk structures.56A four-layered cobalt spinel (001) slab structure was modelled using the corresponding optimized bulk structure (mp-18748). A vacuum space of 15 Å was added in the z-direction and the bottom two layers were fixed to their bulk positions. Starting with this optimized slab structures, transition metal doped structures were created by substituting the octahedral Co in the first and the third layer with dopant atoms. We note that the positions of the dopant in the first and third layers represent the surface and bulk positions, respectively. To calculate the dissolution potentials, defective structures were generated by removing octahedral or tetrahedral Co atoms from the first layer.
The theoretical overpotential for OER was calculated using the computational hydrogen electrode (CHE) method.57 This method equates the chemical potentials of the proton-electron pair (H+ + e−) with half of H2 gas under standard conditions. Following the adsorbate evolution mechanism (AEM),58 which involves four consecutive proton–electron transfers (* + H2O → *OOH → *O + H2O → *OH → * + O2), the Gibbs free energy change for each step was calculated as follows:
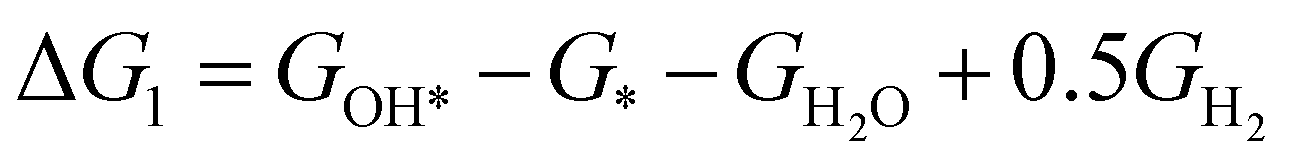 | (1) |
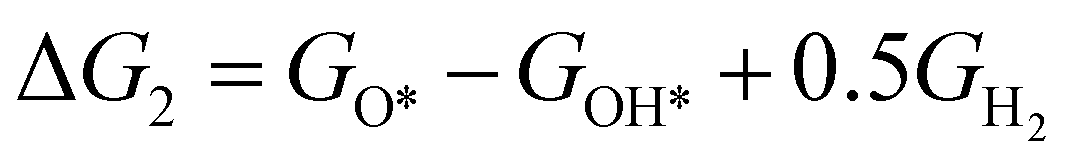 | (2) |
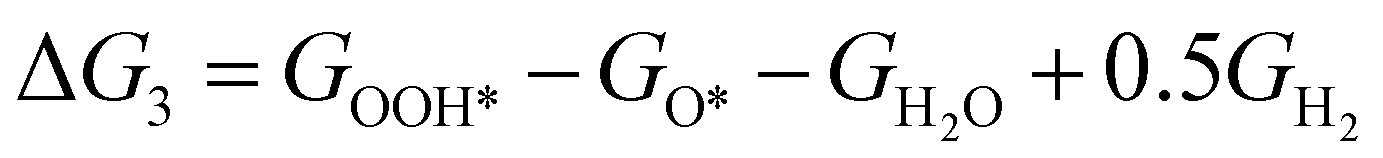 | (3) |
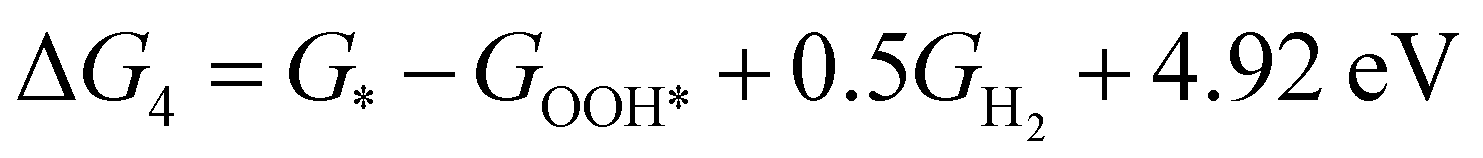 | (4) |
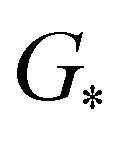 ,
, 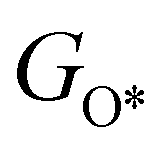 ,
, 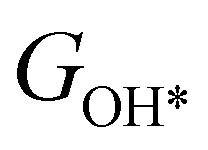 ,
, 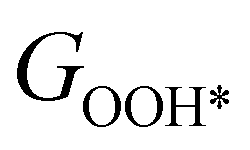 , GH2, GH2O are the Gibbs free energies of bare surface, O, OH, OOH-adsorbed surfaces, H2 and H2O gas, respectively. The Gibbs free energies were calculated by adding free energy correction values to DFT energies, where the correction term consists of zero-point energies, enthalpic and entropic contributions. These correction values were calculated using the Harmonic oscillator and the Ideal gas approximation for adsorbates and gas molecules, respectively, as implemented in atomic simulation environment (ASE).59 The theoretical overpotential was determined as the difference between the equilibrium potential of 1.23 V and the largest Gibbs free energy change, i.e., ηOER = max(ΔG1,ΔG2,ΔG3,ΔG4)/e − 1.23 V.
, GH2, GH2O are the Gibbs free energies of bare surface, O, OH, OOH-adsorbed surfaces, H2 and H2O gas, respectively. The Gibbs free energies were calculated by adding free energy correction values to DFT energies, where the correction term consists of zero-point energies, enthalpic and entropic contributions. These correction values were calculated using the Harmonic oscillator and the Ideal gas approximation for adsorbates and gas molecules, respectively, as implemented in atomic simulation environment (ASE).59 The theoretical overpotential was determined as the difference between the equilibrium potential of 1.23 V and the largest Gibbs free energy change, i.e., ηOER = max(ΔG1,ΔG2,ΔG3,ΔG4)/e − 1.23 V.
The electrochemical stability of Co against the dissolution was investigated considering the following reaction:
| Cox−1Oy + Coz+ + ze− ⇌ CoxOy | (5) |
| ΔGdiss = ECoxOy − ECox−1Oy − ECoz++ze− | (6) |
| = (ECoxOy − ECox−1Oy − ECo) + (ECo − ECoz++ze−) | (7) |
| = ΔGform,Co,Spinel + ΔGCo | (8) |
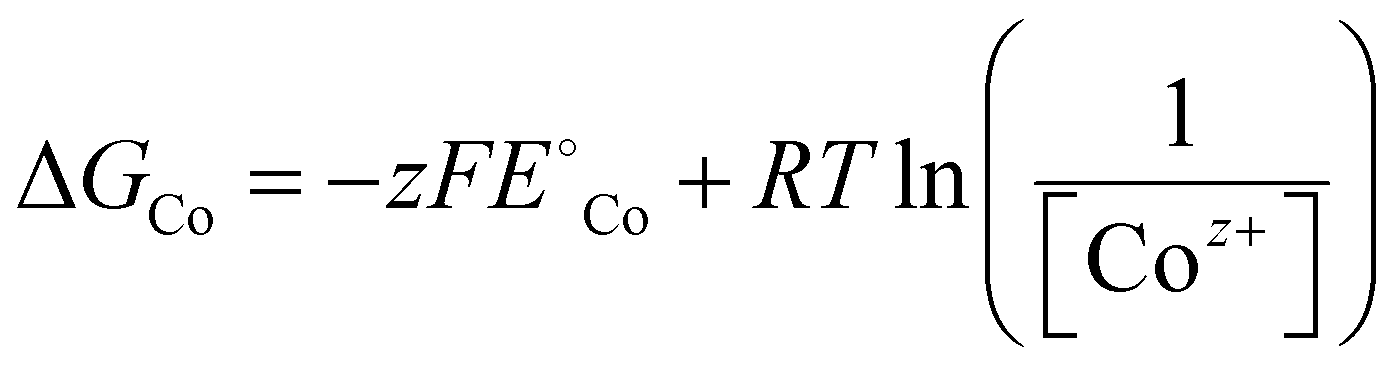 | (9) |
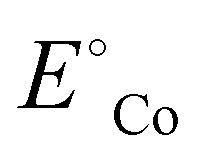 is the standard reduction potential of Co, F is the Faraday constant, R is the gas constant and T is the absolute temperature, which was set to 298.15 K. [Coz+] is the concentration of Coz+, which was assumed to be 10−6 M.60 We note that the dissolution of Co as Co2+ was considered (Co2+ + 2 e− ⇌ Co, E° = −0.28 V), which is known to be more favorable than the formation of Co3+ (Co3+ + 3 e− ⇌ Co, E° = +0.45 V). The dissolution potential of Co (Udiss) is given by
is the standard reduction potential of Co, F is the Faraday constant, R is the gas constant and T is the absolute temperature, which was set to 298.15 K. [Coz+] is the concentration of Coz+, which was assumed to be 10−6 M.60 We note that the dissolution of Co as Co2+ was considered (Co2+ + 2 e− ⇌ Co, E° = −0.28 V), which is known to be more favorable than the formation of Co3+ (Co3+ + 3 e− ⇌ Co, E° = +0.45 V). The dissolution potential of Co (Udiss) is given by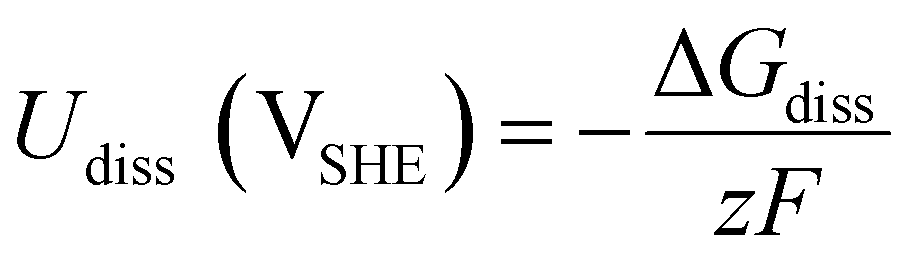 | (10) |
Data availability
All relevant data are included in the main text and its ESI.†Author contributions
K. L., J. S., B.-H. L., Y.-E. S., and T. H. conceived and designed the experiments. K. L. and H. J. synthesized the catalysts. K. L., J. S., H. S. L., H. J. and H. S. performed XAS characterization. J. L., W. K., M. S. B., K. K., and K.-S. L. helped with XAS and XPS data analysis. J. S., S. H., and S. Y. performed electrochemical characterizations. J. R., H. S. and H. S. L. helped with the data processing. J. K. and S. B. performed DFT calculations. K. L., J. S., H. J., B.-H. L., Y.-E. S., and T. H. co-wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
T. H. acknowledges the financial support by the Research Center Program of the IBS (IBS-R006-D1) in Korea. Y.-E. S. acknowledges the financial support by the IBS (IBS-R006-A2). M. S. B. acknowledges the IBS for the Young Scientist Fellowship (IBS-R006-Y2). K.-S. L. acknowledges the financial support by the National Research Foundation of Korea (2019M3D1A1079309). S. B. acknowledges the support from the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1A6A1A03012845) and generous supercomputing time from KISTI.References
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2019, 120, 1184–1249 CrossRef PubMed.
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- J. Cao, B. Wu, R. Chen, Y. Wu, Y. Hui, B. W. Mao and N. Zheng, Adv. Mater., 2018, 30, 1705596 CrossRef PubMed.
- N. Tahsini, A.-C. Yang, V. Streibel, B. Werghi, E. D. Goodman, A. Aitbekova, S. R. Bare, Y. Li, F. Abild-Pedersen and M. Cargnello, J. Am. Chem. Soc., 2022, 144, 1612–1621 CrossRef CAS PubMed.
- M. H. Oh, M. G. Cho, D. Y. Chung, I. Park, Y. P. Kwon, C. Ophus, D. Kim, M. G. Kim, B. Jeong and X. W. Gu, Nature, 2020, 577, 359–363 CrossRef CAS PubMed.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- J. Kim, W. Ko, J. M. Yoo, V. K. Paidi, H. Y. Jang, M. Shepit, J. Lee, H. Chang, H. S. Lee and J. Jo, Adv. Mater., 2022, 34, 2107868 CrossRef CAS.
- J. Jo, J. M. Yoo, D. H. Mok, H. Y. Jang, J. Kim, W. Ko, K. Yeom, M. S. Bootharaju, S. Back and Y.-E. Sung, Nano Lett., 2022, 22, 3636–3644 CrossRef CAS PubMed.
- M. H. Oh, T. Yu, S.-H. Yu, B. Lim, K.-T. Ko, M.-G. Willinger, D.-H. Seo, B. H. Kim, M. G. Cho and J.-H. Park, Science, 2013, 340, 964–968 CrossRef CAS PubMed.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef CAS.
- Y. Sun, X. Ren, S. Sun, Z. Liu, S. Xi and Z. J. Xu, Angew. Chem., Int. Ed., 2021, 133, 14657–14665 CrossRef.
- C. Mu, J. Mao, J. Guo, Q. Guo, Z. Li, W. Qin, Z. Hu, K. Davey, T. Ling and S. Z. Qiao, Adv. Mater., 2020, 32, 1907168 CrossRef CAS PubMed.
- Y. Jiang, Y. Zheng and S.-Z. Qiao, Chem, 2023, 9, 1628–1630 CAS.
- K.-R. Yeo, K.-S. Lee, H. Kim, J. Lee and S.-K. Kim, Energy Environ. Sci., 2022, 15, 3449–3461 RSC.
- L. A. King, M. A. Hubert, C. Capuano, J. Manco, N. Danilovic, E. Valle, T. R. Hellstern, K. Ayers and T. F. Jaramillo, Nat. Nanotechnol., 2019, 14, 1071–1074 CrossRef CAS PubMed.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu and Y. Qian, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- S. Ge, R. Xie, B. Huang, Z. Zhang, H. Liu, X. Kang, S. Hu, S. Li, Y. Luo, Q. Yu, J. Wang, G. Chai, L. Guan, H.-M. Cheng and B. Liu, Energy Environ. Sci., 2023, 16, 3734–3742 RSC.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H.-J. Peng, L. Gu, Y. Zheng and T. Song, Nat. Commun., 2023, 14, 354 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim and D. Myers, Angew. Chem., Int. Ed., 2014, 53, 14016–14021 CrossRef CAS PubMed.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen and T.-W. Song, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- T. Zhu, S. Liu, B. Huang, Q. Shao, M. Wang, F. Li, X. Tan, Y. Pi, S.-C. Weng, B. Huang, Z. Hu, J. Wu, Y. Qian and X. Huang, Energy Environ. Sci., 2021, 14, 3194–3202 RSC.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han and M. Jaroniec, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu and X.-M. Lin, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- J. Baek, M. D. Hossain, P. Mukherjee, J. Lee, K. T. Winther, J. Leem, Y. Jiang, W. C. Chueh, M. Bajdich and X. Zheng, Nat. Commun., 2023, 14, 5936 CrossRef CAS PubMed.
- C. Li, X. Han, F. Cheng, Y. Hu, C. Chen and J. Chen, Nat. Commun., 2015, 6, 7345 CrossRef CAS PubMed.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- K. Chakrapani, G. Bendt, H. Hajiyani, T. Lunkenbein, M. T. Greiner, L. Masliuk, S. Salamon, J. Landers, R. Schlögl and H. Wende, ACS Catal., 2018, 8, 1259–1267 CrossRef CAS.
- M. Zhao, J. Deng, J. Liu, Y. Li, J. Liu, Z. Duan, J. Xiong, Z. Zhao, Y. Wei and W. Song, ACS Catal., 2019, 9, 7548–7567 CrossRef CAS.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- K.-C. Kao, A.-C. Yang, W. Huang, C. Zhou, E. D. Goodman, A. Holm, C. W. Frank and M. Cargnello, Angew. Chem., Int. Ed., 2021, 60, 7971–7979 CrossRef CAS PubMed.
- A. Holm, E. D. Goodman, J. H. Stenlid, A. Aitbekova, R. Zelaya, B. T. Diroll, A. C. Johnston-Peck, K.-C. Kao, C. W. Frank, L. G. M. Pettersson and M. Cargnello, J. Am. Chem. Soc., 2020, 142, 14481–14494 CrossRef CAS PubMed.
- L. Deng, S. F. Hung, Z. Y. Lin, Y. Zhang, C. Zhang, Y. Hao, S. Liu, C. H. Kuo, H. Y. Chen and J. Peng, Adv. Mater., 2023, 2305939 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, J. Fang, M. Li, M. G. Sendeku, X. Wang, H. Wu, Y. Li, J. Ge and Z. Zhuang, Joule, 2023, 7, 558–573 CrossRef CAS.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2022, 123, 5948–6002 CrossRef PubMed.
- A. Aitbekova, C. Zhou, M. L. Stone, J. S. Lezama-Pacheco, A.-C. Yang, A. S. Hoffman, E. D. Goodman, P. Huber, J. F. Stebbins, K. C. Bustillo, P. Ercius, J. Ciston, S. R. Bare, P. N. Plessow and M. Cargnello, Nat. Mater., 2022, 21, 1290–1297 CrossRef CAS PubMed.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2008, 112, 6869–6879 CrossRef CAS.
- J. Peng, B. Chen, Z. Wang, J. Guo, B. Wu, S. Hao, Q. Zhang, L. Gu, Q. Zhou and Z. Liu, Nature, 2020, 586, 390–394 CrossRef CAS PubMed.
- D. Lu, Y. Zhang, M. Lai, A. Lee, C. Xie, J. Lin, T. Lei, Z. Lin, C. S. Kley and J. Huang, Nano Lett., 2018, 18, 6967–6973 CrossRef CAS PubMed.
- Z. Liu, G. Wang, X. Zhu, Y. Wang, Y. Zou, S. Zang and S. Wang, Angew. Chem., Int. Ed., 2020, 132, 4766–4772 CrossRef.
- W. Qu, Z. Tang, S. Tang, H. Wen, J. Fang, Q. Lian, D. Shu and C. He, Adv. Funct. Mater., 2023, 33, 2301677 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. Zhu, Nat. Energy, 2020, 5, 222–230 CrossRef.
- M. Kim, J. Park, H. Ju, J. Y. Kim, H.-S. Cho, C.-H. Kim, B.-H. Kim and S. W. Lee, Energy Environ. Sci., 2021, 14, 3053–3063 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer and C. Hargus, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- K. A. Persson, B. Waldwick, P. Lazic and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235438 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00058g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |